

Abstract
Tissue homeostasis of skin is sustained by epidermal progenitor cells localized within the basal layer of the skin epithelium. Post‐translational modification of the proteome, such as protein phosphorylation, plays a fundamental role in the regulation of stemness and differentiation of somatic stem cells. However, it remains unclear how phosphoproteomic changes occur and contribute to epidermal differentiation. In this study, we survey the epidermal cell differentiation in a systematic manner by combining quantitative phosphoproteomics with mammalian kinome cDNA library screen. This approach identified a key signaling event, phosphorylation of a desmosome component, PKP1 (plakophilin‐1) by RIPK4 (receptor‐interacting serine–threonine kinase 4) during epidermal differentiation. With genome‐editing and mouse genetics approach, we show that loss of function of either Pkp1 or Ripk4 impairs skin differentiation and enhances epidermal carcinogenesis in vivo. Phosphorylation of PKP1's N‐terminal domain by RIPK4 is essential for their role in epidermal differentiation. Taken together, our study presents a global view of phosphoproteomic changes that occur during epidermal differentiation, and identifies RIPK‐PKP1 signaling as novel axis involved in skin stratification and tumorigenesis.
Keywords: differentiation, epidermal progenitor cell, skin, tumorigenesis
Subject Categories: Cancer, Development & Differentiation, Signal Transduction
Introduction
Mammalian skin provides an essential barrier that protects us from various environmental damages (Fuchs, 2008). Homeostasis of skin is sustained by epidermal progenitor cells that localize at the basal layer of skin epidermis. In adult skin, these cells periodically move upward from their niche at the basement membrane and undergo terminal differentiation to replenish lost skin cells in a process called epidermal stratification (Fuchs, 2008; Lopez‐Pajares et al, 2013; Perdigoto et al, 2014). Aberrant epidermal differentiation contributes to the development of various skin diseases including skin cancers, such as SCC (squamous cell carcinoma).
Despite our knowledge of the morphogenetic and transcriptional changes that occur during epidermal differentiation (Fuchs, 2008; Lopez‐Pajares et al, 2013; Perdigoto et al, 2014), much remains to be learned about the signaling networks underlying this process. Protein phosphorylation and dephosphorylation are important post‐translational modifications of the cellular proteome and are extensively involved in cellular signal transduction (Fischer, 2013). Recent development of SILAC (stable isotope labeling by amino acids in cell culture) technology offers us an effective approach to quantitatively compare the modified proteome in skin stem cells (Ong et al, 2003). Our systematic evaluation of the phosphoproteome in epidermal progenitor cells during differentiation revealed significant changes in the phosphorylation of desmosomal proteins. Desmosomes are intercellular junctions abundantly present in epithelia and cardiac muscle (Getsios et al, 2004; Garrod & Chidgey, 2008; Nekrasova & Green, 2013). In addition to their well‐documented role to provide tissues with the essential mechanical strength, accumulating evidence shows that desmosomes can also act as signaling sensors in response to environmental and cellular cues, and participate in fundamental processes such as cell proliferation, differentiation, and tissue morphogenesis (Green & Gaudry, 2000; Kitajima, 2014; Nitoiu et al, 2014; Broussard et al, 2015).
Our proteomic results identified multiple differentiation‐dependent phosphorylation sites located in the head domain of a desmosome protein, Pkp1 (plakophilin‐1). Pkp1 is a ~80 kDa (kilodalton) protein and a member of the Armadillo protein family (Schmidt & Jager, 2005). Pkp1 mediates the interaction between desmosomal cadherin proteins with desmoplakin and keratin intermediate filaments. Structurally, Pkp1 contains an N‐terminal head domain, nine armadillo repeats, and a short C‐terminal tail domain. Loss‐of‐function mutations of Pkp1 in human lead to ectodermal dysplasia/skin fragility (EDSF) syndrome (McGrath et al, 1997; Sprecher et al, 2004). Patients suffer from hyperkeratosis in the epidermis of soles and palms, reduced hair density or alopecia, and blistering with erosions of their skin upon mechanical stress. Although the role of Pkp1 in skin stratification has not been studied, EDSF patients' skin is significant thicker and Krt‐14 (keratin 14)‐positive basal cells show aberrant localization in suprabasal layers (McGrath et al, 1997). In this report, with CRISPR (clustered regularly interspaced short palindromic repeats)‐mediated deletion of Pkp1, we presented compelling evidence that Pkp1 is critically involved in skin differentiation, and its role in epidermal differentiation requires its phosphorylation at the head domain.
To search for potential kinases that are responsible for Pkp1 phosphorylation, we developed a human kinome cDNA library (Yang et al, 2011) and carried out a kinome‐wide screening. We found that RIPK4 phosphorylates Pkp1 at the head domain. RIPK4 was initially cloned as a PKC‐interacting protein by yeast two‐hybrid screens (Bhr et al, 2000; Chen et al, 2001). In mice, deletion of RIPK4 leads to perinatal lethality (Holland et al, 2002). The mutant animals display striking abnormality in skin differentiation. The outermost cornified layers (terminally differentiated layer) are absent in RIPK4 KO (knockout) animals, and the KO skin becomes thicker with marked hyperplasia of both spinous and granular layers. In humans, two recent studies identified RIPK4 mutations as the cause for autosomal‐recessive form of popliteal pterygium syndrome, which is also known as Bartsocas‐Papas syndrome (BS; Kalay et al, 2012; Mitchell et al, 2012). BS is a rare but frequently lethal congenital disorder, characterized by aberrant skin, craniofacial, and genital development. Aberrant RIPK4 function is also associated with tumorigenesis, as recent systematic exome sequencing revealed multiple mutations of RIPK4 in human head and neck SCC (Stransky et al, 2011). Collectively, all the evidence suggests a critical role of RIPK4 in skin development and SCC oncogenesis.
Despite the potential significance of RIPK4 in skin, little is known about how it functions to regulate epidermal differentiation and tumorigenesis at the molecular level. In this study, we generated a conditional knockout (cKO) model of RIPK4 and demonstrated that RIPK4 is essential for skin development during embryogenesis and epidermal homeostasis in adult animals. Loss of RIPK4 in skin epidermis greatly increases the susceptibility of skin to carcinogenesis. Additionally, deletion of RIPK4 leads to a profound change in epidermal phosphoproteome, and phosphorylation of Pkp1 is essential for skin epidermal differentiation. Taken together, our results revealed global changes in the phosphoproteome upon epidermal differentiation and illuminated an important molecular mechanism whereby differentiation of skin somatic stem cells is regulated by the phosphorylation of desmosomal proteins.
Results
Quantitative phosphoproteomics identify significant changes of desmosome protein phosphorylation during epidermal differentiation
In order to uncover how changes in the phosphoproteome regulate self‐renewal and differentiation of epidermal stem/progenitor cells, we applied SILAC technology (Chahrour et al, 2015) by metabolically labeling two populations of epidermal keratinocytes with either regular amino acids (light) or amino acids labeled with heavy isotopes (heavy) (Fig 1A). Cells were maintained in the labeling medium for more than 10 doublings, leading to a labeling efficiency greater than 98% (Fig EV1A). Heavy isotope labeling of epidermal progenitor cells did not affect cell proliferation or cell differentiation (Fig EV1B). Labeling did not lead to significant changes in cell death, either (data not shown).
Figure 1. Quantitative phosphoproteomics reveals potential role of desmosomal protein phosphorylation in epidermal differentiation.
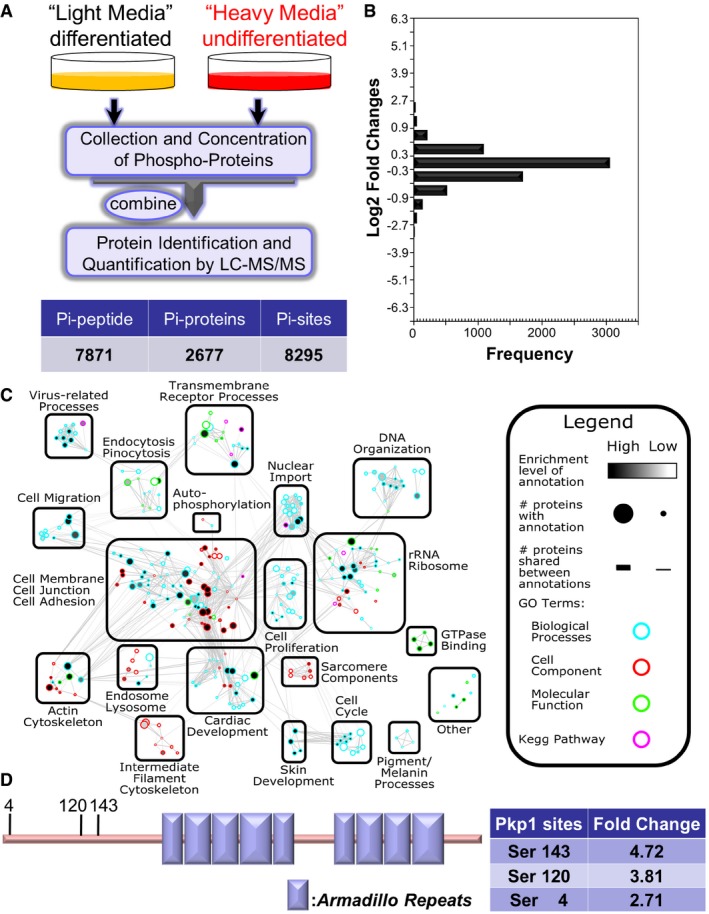
- Diagram of SILAC‐MS workflow. Undifferentiated cells (labeled with regular arginine and lysine) and differentiated cells (labeled with arginine and lysine containing heavy isotope) were mixed together and examined by LC‐MS/MS. Number of identified phosphorylation (Pi) peptides, proteins, and sites are listed in the table below.
- The distribution of normalized log2‐transformed ratios of phosphopeptides enriched in undifferentiated cells compared to differentiated epidermal progenitor cells.
- Mapped enrichment results from proteomics analysis. Proteins of phosphosites with at least twofold increase or decrease in differentiated cell vs. undifferentiated cells were analyzed for GO term and KEGG pathways enrichment using g:Profiler. Visualization of results was performed with Cytoscape and EnrichmentMapApp. Nodes represent a gene set with shared enrichment annotation. Diameter of nodes correspond to the amount of proteins found associated with enrichment term. Outer node color corresponds to GO term or KEGG pathways enrichment terms. Inner node color (gray scale) corresponds to FDR q‐value enrichment significance for given enrichment term. Proteins shared among nodes are denoted as lines between nodes (edges). Edge thickness corresponds to number of proteins shared between two enrichment terms. Terms visualized have a cutoff P‐value and FDR q‐value cutoff of < 0.05.
- Schematic view of Pkp1 (plakophilin) protein structure. Pkp1 contains a large N‐terminal head domain region, nine armadillo domains (boxes), and a small C‐terminal tail region. Three phosphosites enriched in differentiated cells in the head domain region are highlighted with the corresponding fold change listed in the table.
Figure EV1. Quantitative analysis of phosphoproteome upon epidermal differentiation (related to Fig 1).
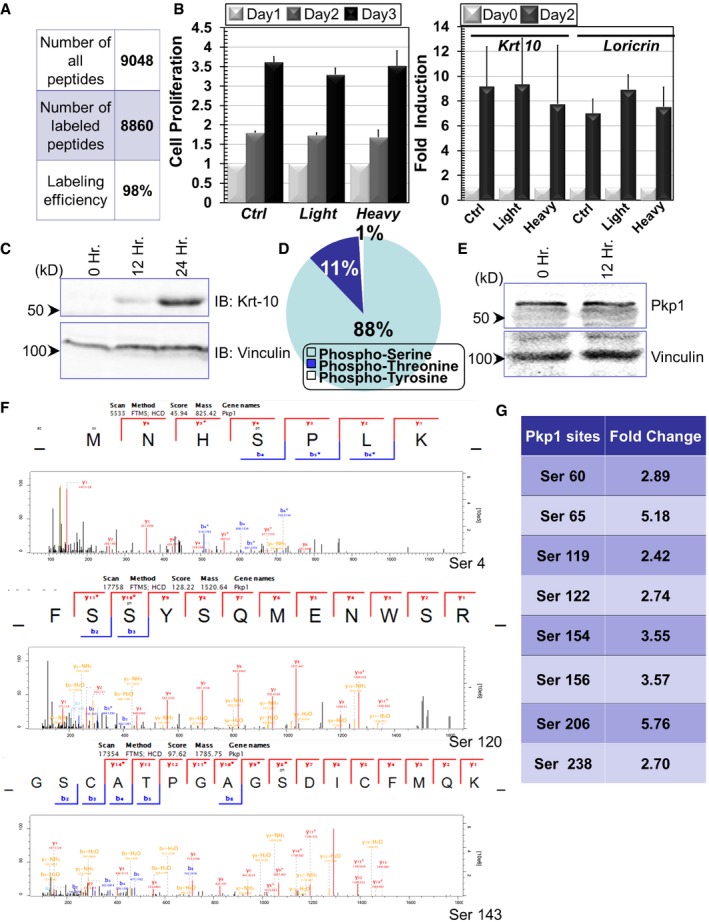
- Mass spectrometry quantification indicates the efficiency of SILAC labeling in cultured epidermal basal cells.
- Cell proliferation of control (Ctrl) and SILAC labeled cells (light or heavy isotope). Fold increase of cell numbers is quantified for all cell types (left panel). In the right panel, induction of cell differentiation after calcium shift is quantified for control and SILAC labeled cells. Error bars represent SD, n = 3.
- Cell lysates were prepared from undifferentiated or differentiated keratinocytes (12 or 24 h post‐calcium shift) and subjected to immunoblots with different antibodies as indicated. Hr: hour. Numbers on left side indicate molecular weight markers. kD: kilodalton.
- Percentage of distribution of different phosphorylated residues in phosphoproteome of epidermal stem cells.
- Cell lysates were prepared from undifferentiated or differentiated keratinocytes (12 h post‐calcium shift) and subjected to immunoblots with different antibodies as indicated.
- Annotated spectra of the three Pkp1 phosphorylation sites at the N‐terminal head domain.
- Other potential phosphosites in Pkp1 head domain that were identified in our proteomics analysis are listed in the table.
With one population maintained as undifferentiated progenitor cells (heavy), we induced differentiation of the other population of epidermal progenitor cells by a well‐established calcium switch protocol. Cells (light) were incubated in high calcium‐containing medium for 12 h, because initiation of differentiation and the expression of early differentiation markers can be readily detected at this time point (Fig EV1C). Proteins were isolated from the two cell populations and combined for analysis by liquid chromatography coupled with tandem mass spectrometry (LC‐MS/MS). Our search identified 8,295 phosphorylation sites across 2,677 phosphoproteins in the light and heavy samples (Figs 1A and EV1D, and Dataset EV1). 6,910 of the 8,295 phosphorylation sites were quantified. Among them, 242 phosphorylation sites were down‐regulated (fold change > 2) in differentiated progenitor cells, whereas 611 phosphorylation sites were up‐regulated. The frequency of phosphorylation site fold change and its cumulative percentage are illustrated in Fig 1B.
GO term and KEGG enrichment analysis of proteins with altered phosphosites revealed a variety of enriched protein clusters following keratinocyte differentiation (Fig 1C), including annotations pertaining to cell membrane/junction/adhesion, epidermis/skin development, cytoskeleton organization, rRNA ribosomes, and transmembrane receptor processes. Interestingly, clusters with the highest enrichment (P‐value < 0.000000429) are cell junction proteins. In particular, desmosomal proteins were identified as a significant part of this enriched group of phosphorylated proteins during differentiation (Fig 1C). Of these desmosomal proteins, Pkp1 seems to be one of the most modified proteins with multiple upregulated phosphorylation sites upon differentiation. The changes were not due to altered expression level of Pkp1, as immunoblot analysis revealed comparable level of Pkp1 in both undifferentiated and differentiated (12 h) keratinocytes (Fig EV1E). The head domain of Pkp1 is functionally critical for maintaining Pkp1 interactions with other desmosomal components (Schmidt & Jager, 2005). Ten potential phosphorylation sites in Pkp1 were identified at the N‐terminal head domain, which were increased upon calcium shift, including serine 4, 120, and 143 (Figs 1D and G, and EV1F). Mutations of Pkp1 lead to EDSF syndrome in human (McGrath et al, 1997; Sprecher et al, 2004). Interestingly, in EDSF patients' skin, Krt‐14‐positive basal cells show aberrant localization in suprabasal layers (McGrath et al, 1997). Consistently, a recent study with complete KO of Pkp1 in mice shows profound cell junctional aberrancy and expanded expression of Krt‐5 and Krt‐14 toward suprabasal layers in the KO skin (Rietscher et al, 2016). Together, it strongly suggests that Pkp1 could play a critical role in epidermal differentiation, potentially through its phosphorylation at the head domain.
Deletion of Pkp1 in skin progenitor cells with CRISPR leads to aberrant epidermal differentiation
To investigate the role of Pkp1 in epidermal differentiation, we first took advantage of CRISPR‐Cas9 (CRISPR associated protein 9) system (Hsu et al, 2014) and deleted Pkp1 in cultured mouse epidermal progenitor cells. Lentivirus encoding both Cas9 and gRNA (guide RNA) that targets exon 1 of Pkp1 was developed. Infection of primary epidermal cells with resultant virus led to efficient deletion of endogenous Pkp1. Loss of Pkp1 protein was verified by Western blot and immunofluorescence staining (Fig 2A and B). Ablation of Pkp1 in skin epidermal cells did not affect cell proliferation in vitro (Fig EV2A). However, when induced to differentiate in vitro, Pkp1‐deficient cells exhibited markedly reduced expression of early (Krt‐10, keratin 10) and late (loricrin) epidermal differentiation marker compared to WT (wild type) cells infected with control lentiviral vector (Figs 2C and EV2B).
Figure 2. Pkp1 regulates skin stratification and tumorigenesis.
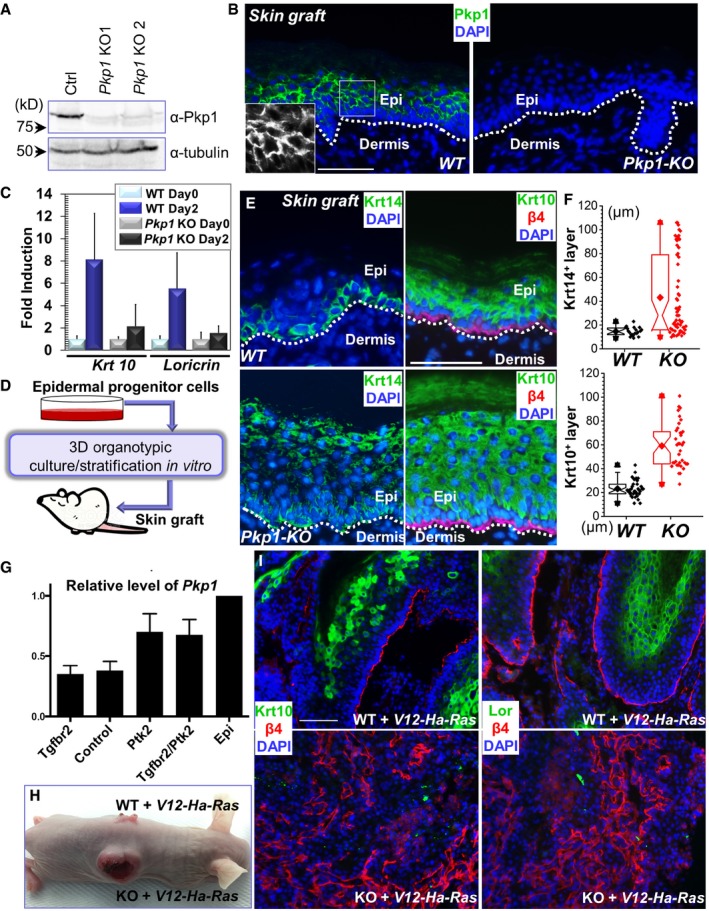
- Western blot analysis reveals loss of Pkp1 expression in the CRISPR KO (knockout) cells. Numbers on left side indicate molecular weight markers. kD: kilodalton.
- WT and Pkp1 KO cells were grafted onto nude mice, and grafted tissue was collected and subjected to immunofluorescence staining with antibody against Pkp1. DAPI: nuclear stain. The dashed line denotes the basement membrane that separates dermis and epidermis (Epi). Scale bar = 50 μm. Boxed areas are magnified as insets that show only Pkp1 staining.
- Expression of early (Krt10) and late (loricrin) differentiation marker in WT and KO keratinocytes upon calcium shift was determined by densitometry and quantified. Error bars represent SD, n = 3.
- Diagram demonstrating the procedure for skin engraftment. Cultured epidermal progenitor cells were plated on top of acellularized dermis, and then exposed to air/liquid interphase to induce stratification in vitro. The resultant skin organoids were transplanted to nude mice and epidermal differentiation was examined in vivo.
- WT and Pkp1 KO cells were grafted onto mice, and grafted tissue was collected and subjected to immunofluorescence staining with different antibodies as indicated. Krt14: keratin 14; β4: CD104, β4‐integrin. Scale bar = 50 μm.
- Deletion of Pkp1 led to thickened epidermis. Thickness of Krt14‐positive layer and Krt10‐positive layer was quantified and showed as box‐and‐whisker plots. The plots indicate the mean (solid diamond within the box), 25th percentile (bottom line of the box), median (middle line of the box), 75th percentile (top line of the box), 5th and 95th percentile (whiskers), 1st and 99th percentile (solid triangles) and minimum and maximum measurements (solid squares). The difference between WT and KO is statistically significant (P < 0.05, Mann–Whitney U‐test) for both Krt14 and Krt10 layers. Each data point in the graft represents average thickness of one field in the skin section, n > 15.
- Bar graphs indicate mean Pkp1 expression levels in a6hib1hi SCC cells (N = 4 per genotype) compared to a6hib1hi epidermal progenitor cells (N = 2, serve as baseline for comparison). Error bars indicate SD. Epi: epidermal progenitor cells.
- WT and Pkp1‐deficient cells were infected with lentivirus encoding Ha‐Ras mutant. Infected cells were transplanted to nude mice for tumorigenesis analysis.
- WT and Pkp1 KO tumors were collected and subjected to immunofluorescence staining with different antibodies as indicated. Lor: loricrin. Loss of Pkp1 leads to enhanced carcinogenesis in skin. The KO tumors display reduced level of epidermal differentiation markers, and a disorganized basal cell marker (β4‐integrin). Scale bar = 50 μm.
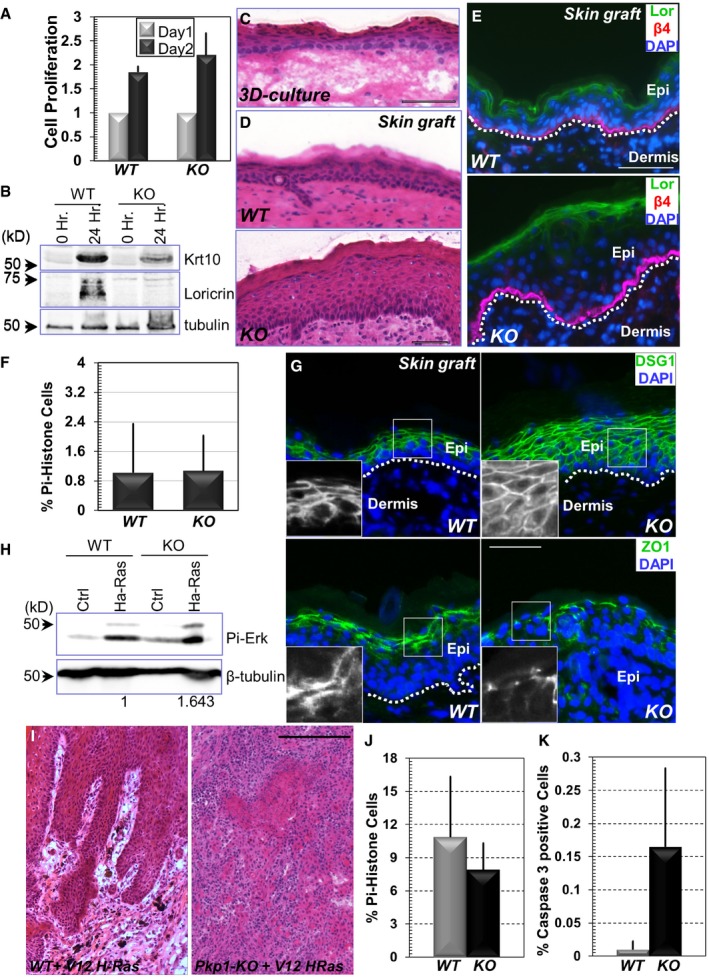
Figure EV2. Loss of Pkp1 inhibits epidermal differentiation and promotes skin tumorigenesis (related to Figure 2).
-
ACell proliferation of WT and Pkp1 KO cells. Fold increase of cell numbers is quantified for both cell types. Error bars represent SD, n = 3.
-
BWestern blot analysis of early (Krt10) and late (loricrin) differentiation marker expression in WT and KO keratinocytes upon calcium shift. Band intensity was determined by densitometry and quantified (Fig 2C).
-
CH/E (hematoxylin and eosin) staining of skin organoid culture in vitro (WT cells). Scale bar = 100 μm.
-
DH/E staining of grafted skin (WT or Pkp1 KO) after skin transplantation. Scale bar = 100 μm.
-
EWT and Pkp1 KO cells were grafted to mice, and grafted tissue was collected and subjected to immunofluorescence staining with different antibodies as indicated. Lor: loricrin; β4: CD104, β4‐integrin; DAPI: nuclear stain. The dashed line denotes the basement membrane that separates dermis and epidermis (Epi). Scale bar = 50 μm.
-
FQuantification of cell proliferation in vivo. Percentage of phospho‐histone H3‐positive cells in WT or Pkp1 KO skin grafts was quantified and shown as bar graph. Error bars represent SD, n = 8.
-
GGrafted skins were stained with different antibodies as indicated. Boxed areas are magnified as insets that show only DSG1 or ZO1 staining. DSG1: desmoglein 1. Scale bar = 50 μm.
-
HImmunoblot of WT and Pkp1 KO cells infected with virus encoding Ha‐Ras mutant. Note increased phospho (Pi)‐Erk level upon expression of Ras mutant. Loss of Pkp1 leads to a further increase in the phospho‐Erk level. The numbers below indicate relative Pi‐Erk level in both WT and KO cells upon expression of Ha‐Ras mutant, as quantified by densitometry.
-
IH/E staining of skin tumors generated from WT and Pkp1 KO cells. Scale bar = 500 μm.
-
J, KQuantification of proliferative (phospho‐histone) and apoptotic (active caspase‐3 staining) cells in WT and Pkp1 KO tumors (infected with virus encoding Ha‐Ras mutant). Error bars represent SD. Apoptosis is significantly increased (P < 0.05, Student's t‐test) and proliferation is mildly but significantly reduced (P < 0.05, Student's t‐test) in the KO skin, n > 10.
Although approaches of exploring epidermal differentiation in vitro are readily available, it remains challenging to develop mouse models and examine skin phenotypes in vivo. The established protocol for culturing skin epidermal progenitor cells makes it possible to generate a functional skin tissue from engineered progenitor cells, which can carry defined genetic alterations to investigate the consequence in epidermal differentiation in vivo. However, direct engraftment of passaged epidermal progenitor cells via the traditional skin progenitor cell technique was proved unsuccessful. To resolve this issue, we first employed organotypic culture of epidermal keratinocytes in vitro by culturing the cells on top of an acellularized dermis. After initial submerged culture, exposure to the air/liquid interphase can induce stratification of the cultured cells to generate a skin‐like organoid in vitro (Figs 2D and EV2C; Prunieras et al, 1983). Transplantation of this cultured skin organoid onto nude host led to efficient skin engraftments (Liu et al, 2015b; Yue et al, 2016).
To determine whether Pkp1 is involved in skin stratification and differentiation in vivo, we grafted WT and Pkp1‐deficient cells onto nude mice (Figs 2E and EV2D). Upon engraftment, WT cells gave rise to a skin epidermis with differentiation indistinguishable from the host skin (Figs 2E and EV2D and E). By contrast, Pkp1‐deficient cells generated a significantly thicker epidermis in vivo. Krt‐14, the genuine marker for epidermal basal cells, was present throughout the stratified epidermis in Pkp1 KO skin (Fig 2E and quantification in Fig 2F), resembling aberrant localization of skin basal cells in EDSF patients (McGrath et al, 1997). In addition, we also saw a marked increase of the thickness of the spinous layer, as determined by staining with Krt‐10 (Fig 2E and quantification in Fig 2F). Granular layer was present in the Pkp1 KO skin, as indicated by loricrin staining. The thickness of the granular layer was not significantly altered in the mutant skin (Fig EV2E). Consistent with in vitro analysis, cell proliferation was not significantly altered in grafted Pkp1 KO skin in vivo (Fig EV2F).
Pkp1 is a desmosomal structural molecule. Mutation of Pkp1 in EDSF patients leads to severe cell junctional abnormality (McGrath et al, 1997). With desmoglein 1 (DSG1) staining, both KO and WT skin grafts exhibited intercellular junctional localization of DSG1 (Fig EV2G). However, loss of Pkp1 led to more diffusive DSG1 staining in the regenerated skin, suggesting potential defects in desmosome formation (Fig EV2G). Tight junctions also appeared to be abnormal in the KO skin. ZO‐1 (zonula occudens‐1) staining in Pkp1 KO skin was significantly reduced and discontinuous comparing with the WT counterpart (Fig EV2G). These results are also consistent with the recent report that complete KO of Pkp1 in mice leads to profound cell junctional aberrancy in vivo (Rietscher et al, 2016).
Aberrant epidermal differentiation marker expression is a characteristic of squamous cell carcinomas (SCC). Recent in vivo proteomics survey has demonstrated suppressed expression of desmosomal proteins, such as DSG1 and DSG2 in both mouse and human cutaneous SCCs, suggesting a potential role of the desmosomal proteins in skin carcinogenesis (Zanivan et al, 2013). To test for a role of Pkp1 in skin carcinogenesis, we first explored its relative expression levels on published microarray data sets that compared transcriptomes of tumor propagating cancer cells (TPCs) isolated from moderately to poorly differentiated control and Tgfbr2 KO SCC and well differentiated Ptk2 KO and Tgfbr2 KO /Ptk2 KO SCCs to normal skin epithelial stem and progenitor cells (Schober & Fuchs, 2011). Pkp1 (mRNA) is consistently reduced in TPCs compared to normal skin epithelial stem and progenitor cells with the lowest expression level in highly tumorigenic Tgfbr2 KO TPCs (Fig 2G). These data suggest that reduced Pkp1 expression might accelerate skin carcinogenesis and progression. To test this hypothesis, we infected WT and Pkp1 KO cells with lentivirus encoding a G12V mutant of Ha‐Ras. Exogenous expression of Ha‐Ras mutant led to significant increase of phospho‐Erk level in WT cells. Interestingly, loss of Pkp1 resulted in a further increase in the phospho‐Erk level (Fig EV2H). Upon engraftment to nude mice, control grafts from mutant Ha‐Ras infected WT cells exhibited visible and morphological signs of benign papilloma. In striking contrast, grafts from Pkp1 KO cell displayed overt aberration and developed large, aggressive tumors, which often developed signs of ulceration and necrosis in the center of the tumor, consistent with the features of skin SCCs (Figs 2H and EV2I). When tumor samples from WT or Pkp1 KO cells were subjected to immunostaining, Pkp1 deletion led to a dramatic decrease of expression of differentiation‐associated markers, including Krt10 and loricrin (Fig 2I), whereas apoptosis was increased and cell proliferation was mildly decreased in the KO tumor (Fig EV2J and K). Together, these results strongly suggest that enhanced tumorigenesis and progression in Pkp1 KO skin is due to aberrant epidermal differentiation.
Taken together, our data presents compelling evidence that Pkp1 is essentially involved in epidermal differentiation and loss of Pkp1 promotes the development of SCC in vivo.
RIPK4 phosphorylates head domain of Pkp1
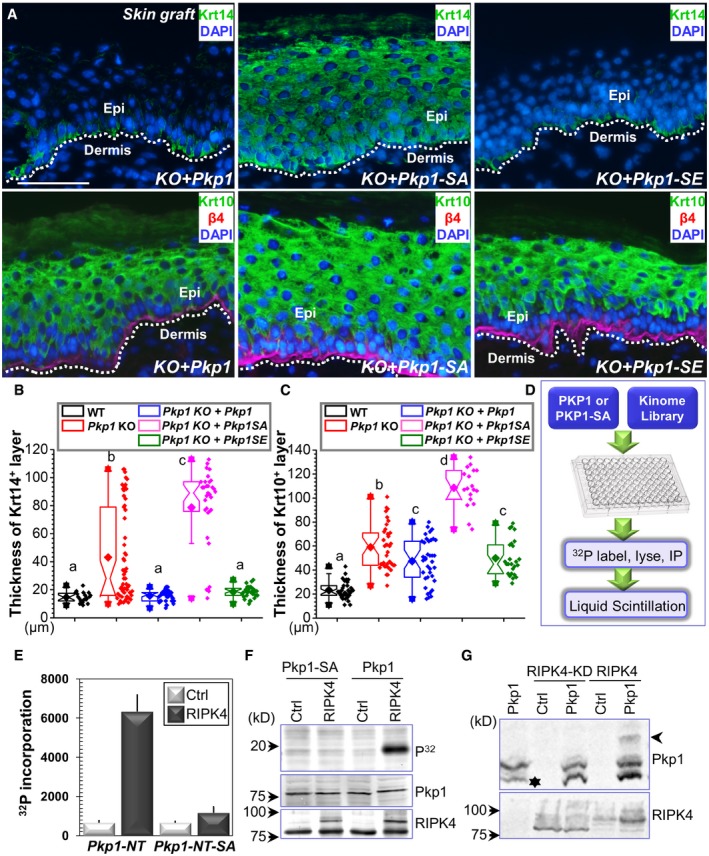
Three phosphorylation sites (serine 4, serine 120, and serine 143) at the head domain of Pkp1 show significant changes over epidermal differentiation (Fig 1D). To determine the relevance of Pkp1 phosphorylation on these sites, we generated Pkp1 mutants that convert the three phosphorylation sites to either a kinase‐refractory version harboring serine to alanine mutations (SA) or a phosphomimetic version, containing serine to glutamate mutations (SE). Pkp1 KO cells were infected with lentivirus encoding either WT Pkp1 or Pkp1 SA or SE mutants. Silent mutations were introduced to exogenously expressed Pkp1 to ensure no interference from CRISPR gRNA. Stable expression clones were isolated, and exogenous Pkp1 expression was verified by Western blot (Fig EV3A). Cells were then engrafted to nude mice to test skin differentiation in vivo. Expression of WT or SE mutant of Pkp1 in the KO cells restored normal epidermal stratification, as indicated by a significantly thinner spinous layer (Krt‐10 staining) and restricted localization of Krt‐14‐positive cells at the basal layer (Fig 3A and quantification in Fig 3B). By contrast, expression of SA mutant of Pkp1 failed to rescue the differentiation defects (Fig 3A and quantification in Fig 3B), strongly suggesting that phosphorylation of Pkp1 head domain is essential for skin epidermal differentiation. Loss of Pkp1 leads to diffusive staining of DSG1 in skin epidermal cells (Fig EV2G). Expression of WT and SE mutant, but not SA mutant of Pkp1 can restore cell junctional localization of DSG1 in the KO skin (Fig EV3B). Our MS analysis revealed multiple phosphorylation sites in the head domain of Pkp1 (Fig EV1G). However, unlike these three sites, mutations on other potential sites did not lead to significant changes in epidermal differentiation.
Figure EV3. Exogenous expression of Pkp1 or its mutants in Pkp1 KO cells (related to Fig 3).
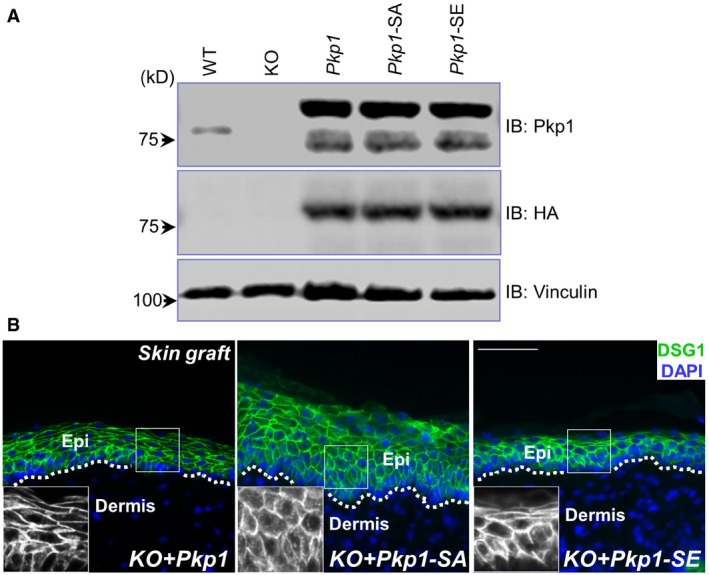
- Cell lysates were collected from KO and rescued cells, and examined by immunoblots with different antibodies as indicated.
- Skin sections from different grafted skin were immunostained with antibodies as indicated. Boxed areas are magnified as insets that show only DSG1 staining. Scale bar = 50 μm.
Figure 3. Phosphorylation of Pkp1 is required for skin differentiation.
-
APkp1 KO cells were rescued with different constructs expressing WT Pkp1 or Pkp1 SA or SE mutants. Rescued cells were grafted to nude mice, and skin stratification was determined by immunofluorescence staining with different antibodies as indicated. Scale bar = 50 μm.
-
B, CThickness of Krt14 (B) and Krt10 (C) layers in different skin grafts were quantified and shown as box‐and‐whisker plots. The plots indicate the mean (solid diamond within the box), 25th percentile (bottom line of the box), median (middle line of the box), 75th percentile (top line of the box), 5th and 95th percentile (whiskers), 1st and 99th percentile (solid triangles) and minimum and maximum measurements (solid squares). One‐way ANOVA test was used for statistical analysis (data with different superscript letters are significantly different, P < 0.05), n > 15.
-
DDiagram of kinome library screening to identify potential kinase responsible for Pkp1 phosphorylation.
-
ELiquid scintillation analysis shows that WT Pkp1 but not Pkp1 SA mutant can be efficiently phosphorylated by RIPK4. Error bars represent SD, n = 3.
-
FIn vitro kinase assay demonstrates direct phosphorylation of Pkp1 NT but not its SA mutant by RIPK4. Phospho‐protein was determined by phosphoradiography. Protein samples were also examined by immunoblots with different antibodies as indicated.
-
GPkp1 was co‐expressed with WT RIPK4 or RIPK4 kinase dead (KD) mutant. Phosphorylation of Pkp1 was determined by Phos‐tag gel analysis. The arrowhead denotes phosphorylated Pkp1 band which exhibits reduced mobility in Phos‐tag gel. Exogenously expressed Pkp1 exhibits two bands in immunoblots. The lower band (star) likely represents degradation product of the over‐expressed protein.
To search for the potential kinases responsible for Pkp1 phosphorylation in an unbiased manner, we constructed a kinome library (560 kinases) that can be efficiently expressed in vitro with gateway cloning. Plasmid encoding WT Pkp1 or Pkp1 SA mutant was co‐transfected with each individual kinase to HEK293T cells, and the cells were then labeled with [32P]‐orthophosphate. Pkp1 protein was isolated from cell lysate with magnetic beads, and potential phosphorylation was determined by the quantification of 32P incorporation with liquid scintillation (Fig 3D). With a near kinome‐wide screening, we identified RIPK4 as a novel kinase that phosphorylates Pkp1 at its head domain (Fig 3E). To confirm Pkp1 phosphorylation by RIPK4, we carried out in vitro kinase assay. When purified WT Pkp1 head domain was incubated with RIPK4 kinase, it led to significant incorporation of radioactively labeled ATP in vitro (Fig 3F). As expected, phosphorylation was abolished when SA mutant of Pkp1 is used (Fig 3F). In addition, when co‐expressed in cells, WT RIPK4 but not RIPK4 kinase dead (KD) mutant resulted in significant phosphorylation of Pkp1, as determined by Phos‐tag gel analysis (Fig 3G).
The head domain of Pkp1 contains many serine and threonine residues, and some of them also display differentiation‐dependent change in phosphorylation level. Consistently, our kinome analysis identified several other potential kinases that can phosphorylate Pkp1, including casein kinase 2, and GSK3A. Their potential role in regulation of Pkp1 function and epidermal differentiation will be investigated in the future.
Loss of RIPK4 leads to aberrant epidermal differentiation
RIPK4 has been implicated in skin development during embryogenesis (Holland et al, 2002). To further investigate the role of RIPK4 in epidermal differentiation and potentially skin carcinogenesis, we generated a skin cKO mouse model of RIPK4. By homologous recombination, a targeting cassette containing loxP sites were inserted into the RIPK4 locus in mouse chromosome 16. To conditionally target RIPK4 in skin, we bred RIPK4 fl/fl mice with K14‐Cre recombinase transgenic mice, which efficiently excised floxed exons by embryonic day E15.5 (Wu et al, 2008; Fig EV4A and B). Western blot analysis confirms the specific loss of RIPK4 protein in cKO skin epidermis (Figs 4A and EV4C). Neonatal mice genotypic for K14‐Cre and RIPK4 fl/fl alleles (cKO) were born in the expected Mendelian numbers and grew to adulthood. However, the cKO animals are smaller in size and usually exhibit patchy hair loss (Fig 4B).
Figure EV4. Generation of RIPK4 skin cKO strain (related to Fig 4).
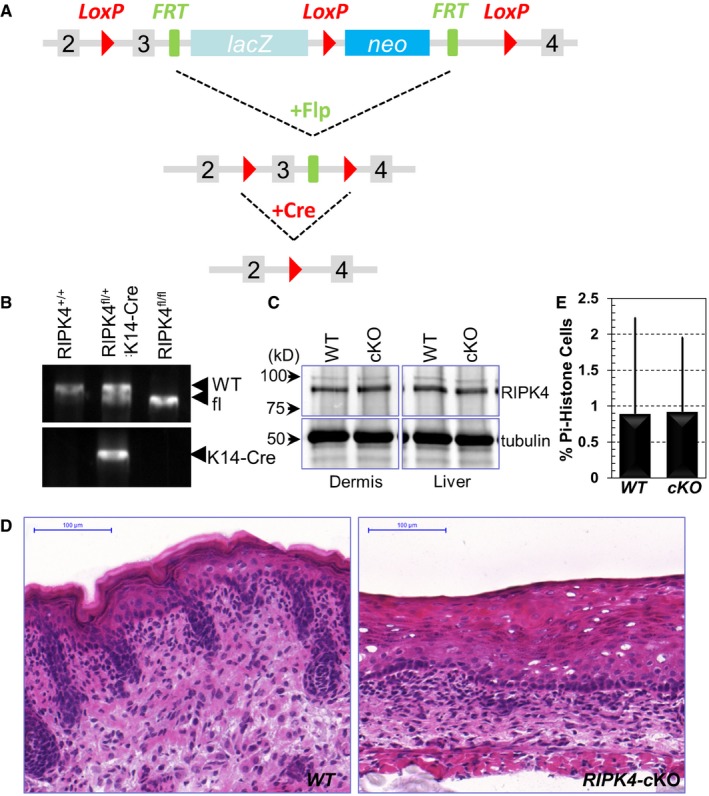
- Diagram of the strategy for generation of RIPK4 skin cKO strain.
- Typical genotyping results for RIPK4 cKO.
- Lysates were prepared from skin dermis and liver from WT and RIPK4 cKO animals. Lysates were immunoblotted with different antibodies as indicated.
- H/E staining of WT and RIPK cKO skin (E18.5). Scale bar = 100 μm.
- Quantification of proliferative (phospho‐histone) cells in WT and RIPK4 cKO skin. Error bars represent SD, n > 10.
Figure 4. RIPK4 regulates skin development and tissue homeostasis.
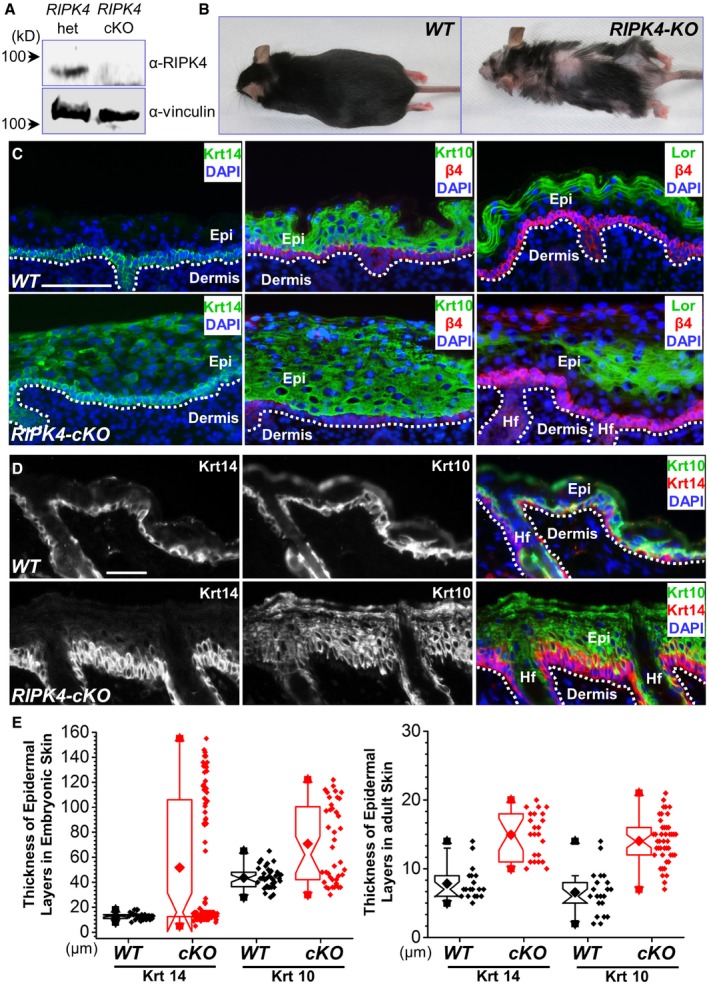
- Western blot analysis reveals loss of RIPK4 expression in the cKO (conditional knockout) skin epidermis.
- RIPK4 skin cKO mice can survive to adulthood, but display patchy hair loss.
- WT and RIPK4 cKO skins were collected from E18.5 embryos. Skin stratification was examined by immunofluorescence staining with different antibodies as indicated. Dashed lines denote basement membrane. HF: hair follicles. Scale bar = 50 μm.
- WT and RIPK4 cKO skins were collected from adult mice. Skin stratification was examined by immunofluorescence staining with different antibodies as indicated. Dashed lines denote basement membrane. Scale bar = 50 μm.
- Thickness of Krt14 and Krt10 layers in different skins were quantified and shown as box‐and‐whisker plots. The plots indicate the mean (solid diamond within the box), 25th percentile (bottom line of the box), median (middle line of the box), 75th percentile (top line of the box), 5th and 95th percentile (whiskers), 1st and 99th percentile (solid triangles) and minimum and maximum measurements (solid squares). The difference between WT and KO is statistically significant (P < 0.05, Mann–Whitney U‐test) for both Krt14 and Krt10 layers, n > 15.
To first investigate the role of RIPK4 in skin development, we analyzed WT and RIPK4 cKO skin at embryonic day E18.5. Consistent with the previous report (Holland et al, 2002), RIPK4 cKO skin is significantly thicker than WT skin (Figs 4C and EV4D). Unlike WT skin, where Krt‐14‐positive cells localize only in the basal layer, loss of RIPK4 leads to the presence of Krt‐14 cells in the suprabasal layers throughout the epidermis (Fig 4C and quantification in Fig 4E). In addition, RIPK4 cKO skin exhibits significantly thickened spinous layer as indicated by Krt‐10 staining (Fig 4C and quantification in Fig 4E). Loricrin staining reveals granular layer in the cKO skin with aberrant morphology and varied thickness (Fig 4C). Overall, the aberrant skin stratification phenotype in RIPK4 cKO animals resembles what we have seen in Pkp1 KO skin (Fig 2E).
Adult skin of RIPK4 cKO mice exhibits similarly thickened epidermis compared with the WT counterpart. Loss of RIPK4 in the adult skin also leads to expansion of spinous layer and basal cells (Krt‐14‐positive) (Fig 4D and quantification in Fig 4E). Cell proliferation is not significantly affected by deletion of RIPK4 (Fig EV4E). Together, our results strongly suggest that RIPK4 regulates embryonic skin development and maintains normal skin tissue homeostasis in adult animals through the regulation of epidermal differentiation.
RIPK4 deficiency enhances skin carcinogenesis
Loss‐of‐function mutations of RIPK4 have been identified through exome sequencing in human head and neck SCC (Stransky et al, 2011). However, the role of RIPK4 in tumorigenesis remains elusive, as studies also suggest that RIPK4 can potentially enhance tumor development by promoting Wnt signaling (Huang et al, 2013; Liu et al, 2015a). While maintaining the RIPK4 cKO mouse, we detected spontaneous tumorigenesis in the facial region of cKO animals (Fig 5A). Quantification shows that ~60% of RIPK4 cKO mice developed spontaneous tumor when aged, whereas none of the WT littermates did (Fig 5B).
Figure 5. RIPK4 suppresses skin carcinogenesis.
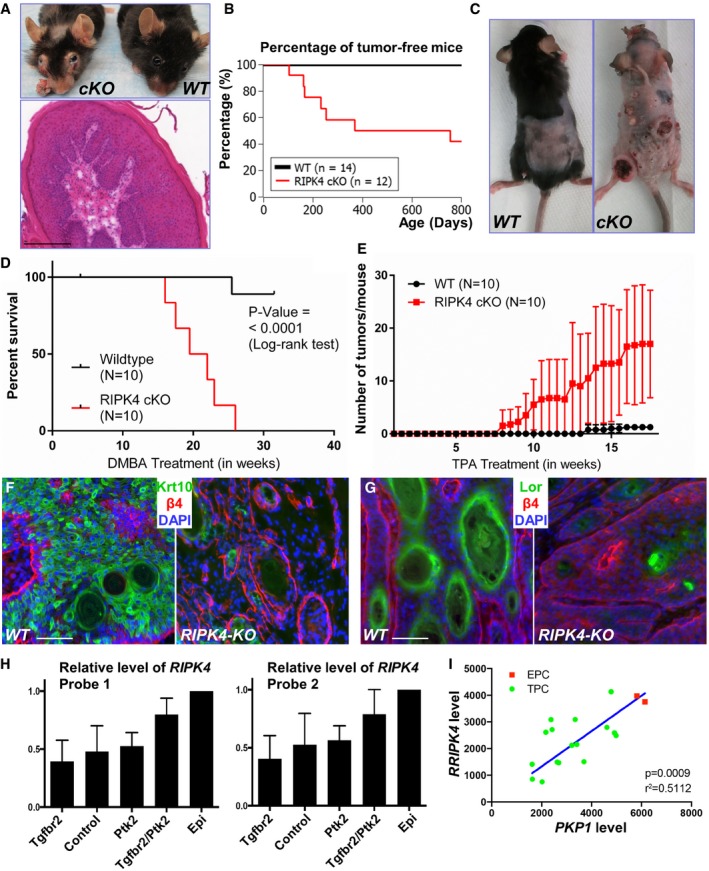
-
ARIPK4 cKO mice but not WT littermates develop spontaneous tumors in the facial region. An H/E staining of the tumor from cKO mice is displayed below. Scale bar = 200 μm.
-
BKaplan–Meier survival plot for skin tumorigenesis in WT and RIPK4 cKO mice.
-
CWhen subjected to two‐step skin carcinogenesis model, RIPK4 cKO mice develop significantly more and worse skin lesions compared with WT littermates.
-
DKaplan–Meier survival curve for WT and RIPK4 cKO mice upon application of two‐step skin carcinogenesis model. The difference between WT and cKO is statistically significant (P < 0.0001, log‐rank test).
-
EQuantification of tumor number per mouse upon DMBA/TPA treatment. Error bars represent SD.
-
F, GWT and RIPK4 cKO tumors were collected and subjected to immunofluorescence staining with different antibodies as indicated. Lor: loricrin. Scale bar = 50 μm.
-
HBar graphs indicate mean RIPK4 expression levels for two independent probe sets in a6hib1hi SCC cells (N = 4 per genotype) compared to a6hib1hi epidermal progenitor cells (N = 2). Error bars indicate SD.
-
IScatter plot indicating the correlation between RIPK4 and Pkp1 expression levels in a6hib1hi SCC and epidermal progenitor cells. Pearson correlation coefficient indicates significant positive correlation between RIPK4 and Pkp1 expression levels. EPC: epidermal progenitor cells; TPC: tumor propagating cancer cells.
To verify the potential role of RIPK4 as a tumor suppressor in skin epidermis, we carried out the two‐step cutaneous skin carcinogenesis assay (Fujiki et al, 1989). With a single treatment of DMBA (dimethylbenz[a]anthracene) followed by biweekly treatments of TPA (12‐O‐tetradecanoylphorbol‐13‐acetate), RIPK4 cKO mice displayed significantly enhanced tumorigenesis compared to their WT littermates (Fig 5C and D). The onset of tumors commenced as early as 8 weeks in cKO mice and 13 weeks in WT littermates. At week 11, 100% of cKO animals developed skin lesions, whereas more than 50% of WT animals remained tumor free even after 15 weeks. The average number of tumors in WT or cKO animals at different weeks is shown in Fig 5E. Loss of RIPK4 led to significantly more and larger tumors in cKO animals.
Histologically, the tumors in RIPK4 cKO animals displayed more advanced progression toward SCC. The characteristic squamous pearls were clearly visible by examining the H/E staining of tumors generated from RIPK4 cKO skin (Fig EV5). Islands of dysplastic squamous epithelial cells lying in the dermis distinctly indicated an invasive form of SCC. The necrotic keratinocytes were also visible in the center of the tumor in RIPK4 cKO animals. By contrast, tumors from the WT skin usually displayed intact basement membrane with hyperplasia of the overlying epidermis. This clearly characterized the benign nature of tumors in WT skin (Fig EV5). When stained for epidermal differentiation markers, loss of RIPK4 led to a significant decrease in the expression of Krt 10 and loricrin in skin tumors (Fig 5F and G), suggesting that RIPK4 regulates skin carcinogenesis through its role in epidermal differentiation.
Figure EV5. Loss of RIPK4 enhances skin tumorigenesis (related to Fig 5).
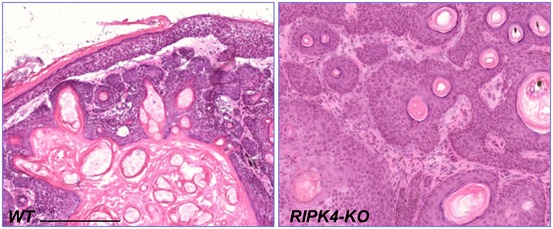
H/E staining of skin tumors generated from WT and RIPK4 cKO animals upon DMBA/TPA treatment. Scale bar = 200 μm.
RIPK4 mutations have been detected in human head and neck SCC, suggesting a critical function of RIPK4 in squamous differentiation and carcinogenesis (Stransky et al, 2011). In addition to mutations, expression of RIPK4 may be blunted when tumors initiate and progress to malignant cancers. Like Pkp1 (Fig 2G), RIPK4 expression is also blunted in TPCs of SCC, compared to normal skin epithelial stem and progenitor cells, where poorly differentiated, aggressively growing tumors express the lowest amount of Ripk4 (Fig 5H). Direct comparisons between Ripk4 and Pkp1 expression in these TPCs revealed a positive correlation (R 2 = 0.58, P = 0.0171) of these markers, with the highest expression in inter‐follicular progenitor cells (Fig 5I). Together, our data in normal skin and tumors suggest that the relative expression and activity of Ripk4 and Pkp1 could be explored as a prognostic measure for tumor differentiation and clinical outcome in future studies.
RIPK4 phosphorylation of Pkp1 plays an important role in skin stratification
Our results demonstrate a key role of RIPK4 in epidermal differentiation. To identify potential substrates of RIPK4 in a systematic manner, we again performed quantitative phosphoproteomics analysis with SILAC labeling of WT and RIPK4‐deficient epidermal progenitor cells (Fig EV6A). Both populations of cells were induced to differentiate in vitro upon calcium shift. From LC‐MS/MS analysis, we identified 2,487 phosphorylation sites, of which 2,163 phosphorylation sites were quantified. Among them, 167 phosphorylation sites were significantly down‐regulated (fold change > 2) in the RIPK4 KO cells and 92 phosphorylation sites were up‐regulated (Fig EV6A and B, and Dataset EV2). GO enrichment analysis of the results from WT and RIPK4‐deficient epidermal progenitor cells showed enrichment (P‐values < 0.05) of the same protein annotations found in our previous analysis with undifferentiated and differentiated epidermal progenitor cells, including cell junction (anchoring junctions, desmosome, adherence junctions, and focal adhesion) and skin development/differentiation annotations (epidermal cell differentiation and epidermis development) (Fig 6A). Interestingly, both phosphoproteomics analyses identified anchoring junction as the most enriched protein cluster annotation, which includes protein Pkp1.
Figure EV6. Phosphoproteomic changes upon loss of RIPK4 (related to Fig 6).
-
ADiagram of SILAC‐MS workflow. Differentiated RIPK4 KO cells (labeled with regular arginine and lysine) and differentiated WT cells (labeled with arginine and lysine containing heavy isotope) were mixed together and examined by LC‐MS/MS. Number of identified phosphorylation (Pi) peptide, protein, and sites are listed in the table below.
-
BThe distribution of normalized log2‐transformed ratios of phosphopeptides enriched in RIPK4 KO cells compared with WT epidermal stem cells.
-
CCell lysates were prepared from WT or RIPK4 KO keratinocytes and subjected to immunoblots with different antibodies as indicated.
-
DH/E staining of WT and RIPK4 KO skin grafts, as well as skin grafts generated from rescued cells. Scale bar = 100 μm.
-
ESkin sections from WT skin grafts were immunostained with different antibodies as indicated. Scale bar = 50 μm.
-
F, GThickness of Krt14 and Krt10 layers in different skin grafts were quantified and shown as box‐and‐whisker plots. The plots indicate the mean (solid diamond within the box), 25th percentile (bottom line of the box), median (middle line of the box), 75th percentile (top line of the box), 5th and 95th percentile (whiskers), 1st and 99th percentile (solid triangles) and minimum and maximum measurements (solid squares). Note significant rescue of RIPK4 KO phenotype upon expression of Pkp1 SE mutant transgene. One‐way ANOVA test was used for statistical analysis (data with different superscript letters P < 0.05), n > 15.
Figure 6. Phosphorylation of Pkp1 by RIPK4 regulates skin differentiation.
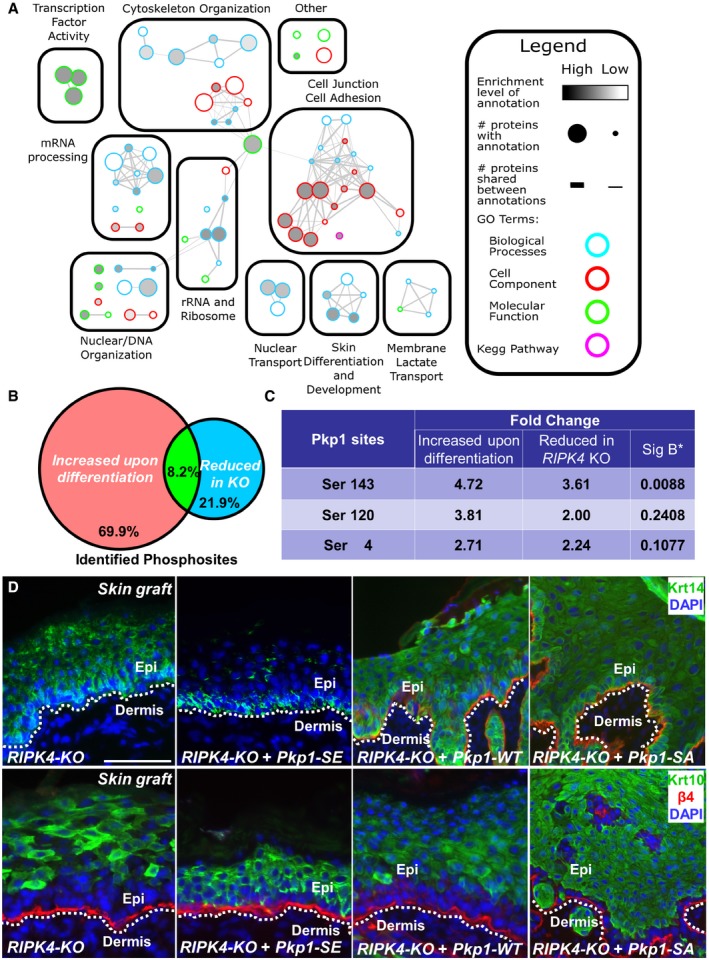
- Mapped enrichment results from proteomics analysis. Proteins of phosphosites with at least twofold of changes were analyzed for GO term and KEGG pathways enrichment using g:Profiler. Visualization of results was performed with Cytoscape and EnrichmentMapApp.
- The Venn diagram shows significant overlap of phosphosites that are decreased in RIPK4 KO cells with phosphosites that are increased upon epidermal differentiation.
- The three key phosphorylation sites at the head domain of Pkp1 show reduced phosphorylation upon deletion of RIPK4. The “Sig B*” column shows the P‐values for phosphorylation changes in RIPK4 KO as calculated by Signficance (Sig) B. Changes in Ser4 and Ser120 are more than twofold but not considered as statistically significant.
- RIPK4 KO cells were rescued with control or construct encoding Pkp1 or Pkp1 mutant. Rescued cells were grafted to nude mice, and skin stratification was determined by immunofluorescence staining with different antibodies as indicated. Scale bar = 100 μm.
By cross‐referencing with our original phosphoproteomics analysis, we obtained a list of 22 phosphorylation sites (18 unique proteins) that are significantly increased upon differentiation of WT progenitor cells, but lost in RIPK4 KO cells under the same condition (Fig 6B). Interestingly, 5 of the 22 phosphorylation sites corresponded to three desmosomal component proteins, Pkp1, Junctional plakoglobin, and desmoplakin. Modification of the three phosphorylation sites (serine 4, serine 120, and serine 143) at the Pkp1 head domain that were identified in our original search were all reduced in RIPK4 KO samples (Fig 6C), whereas overall protein level of Pkp1 was not significantly changed in RIPK4 KO skin (Fig EV6C). Although all three sites showed more than twofold decrease in phosphorylation, only the change in Ser 143 was statistically significant (P < 0.05) as determined by Sig B (Significance B) test.
Systematic examination of epidermal phosphoproteome upon loss of RIPK4 strongly suggests that phosphorylation of Pkp1 in the head domain could be critically involved in RIPK4‐mediated skin differentiation. To examine this hypothesis, we infected RIPK4 KO cells with control vector or lentiviral vector encoding WT Pkp1, the SA mutant of Pkp1, or the phosphomimetic mutant of Pkp1 (SE). When grafted to nude mice, exogenous expression of Pkp1 SE mutant can restore normal epidermal stratification, as shown by significantly thinner spinous layer (Krt‐10 staining) and restricted localization of Krt‐14‐positive cells at the basal layer (Figs 6D and EV6D–G). By contrast, control RIPK4 KO cells or KO cells rescued with WT Pkp1 or Pkp1 SA mutant gave rise to aberrant stratification of the epidermis, with thickened spinous layer and mislocalized basal cells, resembling RIPK4 KO skin in vivo (Figs 6D and EV6D–G). Together, our results strongly suggest that phosphorylation of Pkp1 head domain by RIPK4 plays a critical role in epidermal differentiation.
RIPK4 phosphorylation of Pkp1 promotes epidermal differentiation through MAP kinase signaling pathway
Accumulating evidence has indicated potential roles of desmosomal proteins in signal transduction. It has been reported that Pkp1 is present in both desmosome and nucleus (Schmidt & Jager, 2005). However, in stratified skin epidermis, endogenous Pkp1 exhibited cytoplasmic and cell junctional localization in basal and suprabasal keratinocytes, without significant localization in the cell nucleus (Fig 2B).
Recent study suggests that the desmosomal cadherin, DSG1 associates with Erbin (Erbb2 interacting protein), which can block ERK signaling by interacting with and disrupting the key Ras‐Raf scaffolding protein SHOC2 (leucine‐rich repeat protein SHOC2; Harmon et al, 2013). Ras/Raf and MAP kinase signaling pathway play a critical role in epidermal homeostasis (Dajee et al, 2002; Tarutani et al, 2003; Kern et al, 2011). The localization of DSG1 became more diffusive in Pkp1‐deficient skin (Figs 3B and EV2G). Consistently, localization of another key desmosomal protein, Dsp1 (desmoplakin 1) also became more diffusive in the cytoplasm upon deletion of Pkp1 (Fig 7A). Together, it suggests that Pkp1 could contribute to epidermal differentiation through a desmosome‐related signaling cascade.
Figure 7. Pkp1 regulates Dsg1 localization and MAP kinase signaling pathway.
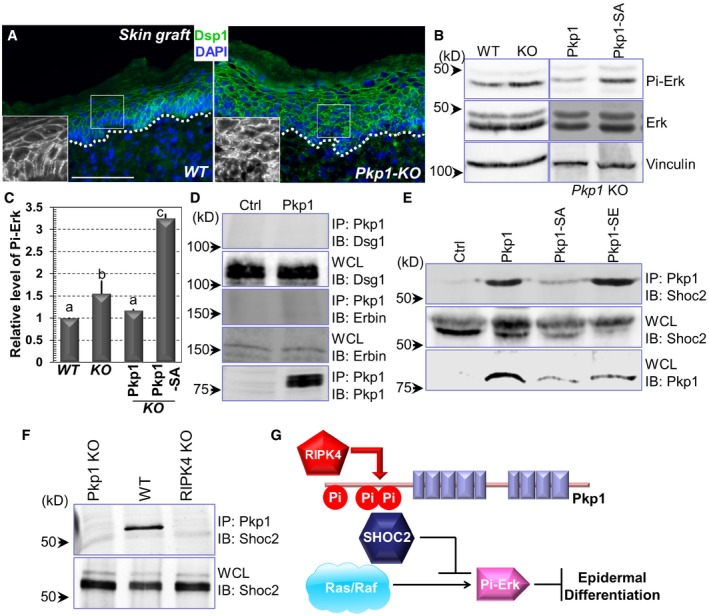
-
AWT and Pkp1 KO skins were collected and subjected to immunofluorescence staining with antibody against Dsp1 (desmoplakin 1) as indicated. Scale bar = 50 μm.
-
B, CErk phosphorylation level was determined by Western blot analysis using WT, Pkp1 KO, and rescued cells. Band intensity was determined by densitometry and quantified in (C). Error bars represent SD. Data with different superscript letters are significantly different, P < 0.05 (one‐way ANOVA), n > 3.
-
D, ECell lysates were collected from different cell lines as indicated and immuno‐precipitated with α‐Pkp1 antibody. Immunoprecipitates (IP) and aliquots of whole‐cell lysate (WCL) were immunoblotted with different antibodies as indicated.
-
FCell lysates were collected from Pkp1 KO, WT Pkp1, and RIPK4 KO cells. Lysates were immuno‐precipitated with Pkp1 antibody and immunoblotted with different antibodies as indicated.
-
GA working model of the regulation of epidermal differentiation by RIPK4 and Pkp1. Phosphorylation of Pkp1 by RIPK4 can suppress ERK signaling by association with SHOC2, which in turn enhances epidermal differentiation.
Interestingly, in Pkp1‐deficient cells, level of phosphorylated Erk was significantly elevated (Fig 7B, and quantification in Fig 7C). Re‐expression of WT Pkp1 in the KO cells can reduce Erk phosphorylation, whereas expression of Pkp1 SA mutant led to significantly higher level of phospho‐Erk in Pkp1 KO cells (Fig 7B, and quantification in Fig 7C), suggesting that phosphorylation of Pkp1 by RIPK4 can inhibit Erk signaling, and thus promote epidermal differentiation. To further examine the role of Pkp1 in epidermal differentiation, we tested the potential interaction of Pkp1 with DSG1, Erbin, and SHOC2. With biochemical pull‐down assay, our results indicated no physical interaction between Pkp1 and DSG1 or Erbin (Fig 7D). By contrast, WT Pkp1 and its phosphomimetic mutant (SE) but not the SA mutant of Pkp1 exhibited significant binding affinity with SHOC2 (Fig 7E). Consistent with this finding, interaction between WT Pkp1 and Shoc2 is significantly reduced in RIPK4 KO cells (Fig 7F). Taken together, our results suggest that RIPK4 phosphorylation of Pkp1 can promote SHOC2 binding with Pkp1 and enhance epidermal differentiation by functionally blocking Ras/MAP kinase signaling pathway (Fig 7G).
Discussion
The mammalian skin epidermis functions as an essential barrier against the environment damages (Blanpain & Fuchs, 2006; Fuchs, 2008). Skin keratinocytes elaborate the protective function by undergoing a complex and choreographed program of epidermal stratification or differentiation. Although morphological alterations during epidermal differentiation have been well documented, little is known about the signaling networks that govern this process. In this study, by employing a combinatory approach encompassing proteomics analysis with mouse genetics, molecular and cell biology studies, we present compelling evidence that phosphorylation of Pkp1, a desmosomal protein, by protein kinase RIPK4 plays an essential role in the differentiation of epidermal progenitor cells.
Desmosomes are intercellular adhesive junctions that associate with intermediate filaments and generate a three‐dimensional supracellular scaffolding providing epithelial tissue, such as skin epidermis with essential mechanical strength (Green & Gaudry, 2000; Getsios et al, 2004; Garrod & Chidgey, 2008). However, in addition to the mechanical role in mediating cell/cell junctions, emerging evidence has indicated that desmosomal proteins also participate in cellular signaling transductions (Green & Gaudry, 2000; Kitajima, 2014; Nitoiu et al, 2014; Broussard et al, 2015). Recent quantitative proteomics analysis revealed decreased expression of desmosomal proteins, such as DSG1 and DSG2, in both mouse and human skin SCCs (Zanivan et al, 2013), strongly suggesting a signaling role of desmosomal structural proteins in skin tissue homeostasis and carcinogenesis.
Desmosomes harbor multiple β‐catenin‐like armadillo family of nuclear and junctional proteins, such as plakophilins. Plakophilin family consists of three members (Pkp1 to 3) (Schmidt & Jager, 2005). While Pkp2 and 3 are present in almost all desmosome‐bearing cells, Pkp1 expression is restricted to the stratified epithelium, such as skin. In human, loss of function of Pkp1 leads to EDSF syndrome (McGrath et al, 1997; Sprecher et al, 2004), as exemplified by the development of trauma‐induced acantholytic blisters in skin, hyperkeratotic plaques on limbs and soles, dystrophic nail, and impaired hair follicle development. Pkp1‐deficient human skin epidermis is significantly thickened with increased intercellular spaces and poorly formed desmosomes. Overall, this suggests that Pkp1 is crucial for desmosomal stability and functional integrity of skin epidermis. In this report, we present compelling evidence that loss of Pkp1 leads to significant defects in epidermal differentiation, and its role in this process is dependent upon RIPK4‐mediated phosphorylation of its N‐terminal head domain. A recent study demonstrated that complete KO of Pkp1 in mice leads to growth retardation and perinatal lethality (Rietscher et al, 2016). The KO mice developed fragile skin with aberrant desmosome and disturbed tight junctions. Consistent with our findings, the Krt‐5 and Krt‐14 showed expanded expression toward suprabasal layers in the KO skin (Rietscher et al, 2016). Interestingly, our phosphoproteomics have also identified phosphorylation changes of other sites in Pkp1 and other desmosomal‐associated proteins, suggesting additional regulation of desmosomal proteins via other kinases during epidermal differentiation. The relevance of these changes in cell adhesion and epidermal homeostasis will be investigated in the future.
The potential role of Pkp1 in the signaling cascades that control skin stratification is not completely understood. It has been shown that Pkp1 head domain exhibits DNA binding affinity in vitro, and Pkp1 localization in the nucleus is dependent upon the N‐terminal head domain (Sobolik‐Delmaire et al, 2010). In addition, the head domain of Pkp1 also mediates binding with other desmosomal plaque proteins, including desmoplakin, desmocollin, and desmoglein (Schmidt & Jager, 2005). Our results suggest that Pkp1 can associate with SHOC2 after RIPK4‐mediated phosphorylation. This interaction will likely disrupt the Ras/Raf complex and inhibit ERK signaling, thus ultimately enhances epidermal differentiation (Fig 7G; Dajee et al, 2002; Tarutani et al, 2003; Kern et al, 2011; Harmon et al, 2013). It will be interesting and important to determine how the modification of Pkp1 head domain by RIPK4 may contribute to epidermal differentiation through Ras/Raf complex or potentially other signaling cascades. This issue merits further investigation in the future.
RIPK4 is a member of the receptor‐interacting protein serine/threonine kinase family, which functions as an integrator of stress signaling leading to the activation of NF‐κB, MAP kinases, cell death and inflammation (Green et al, 2011). Deficiency of RIPK4 in human leads to Bartsocas‐Papas syndrome, a complex congenital disorder characterized by multiple skin webs affecting the flexural surfaces accompanied by craniofacial anomalies (Kalay et al, 2012; Mitchell et al, 2012). RIPK4 KO in mice leads to perinatal lethality with severe defects in skin epidermal stratification (Holland et al, 2002). Despite its critical role in development, little is known about RIPK4's molecular functions. It has been recently demonstrated that RIPK4 can phosphorylate and activate IRF6 (interferon regulatory factor 6), a transcription factor involved in skin differentiation (Kwa et al, 2014, 2015; De Groote et al, 2015). In addition, RIPK4 has been shown to phosphorylate disheveled and participate in Wnt signal transduction (Huang et al, 2013). Our phosphoproteomics analysis has identified 167 phosphorylation sites that are specifically reduced in RIPK4 null keratinocytes, including many proteins involved in cell adhesion and junctional regulation, such as desmosomal proteins plakoglobin and desmoplakin. Although expression of Pkp1‐SE mutant can significantly reverse the skin stratification phenotype in RIPK4 cKO skin, the role of RIPK4 in skin differentiation could be diverse and future study will be essential to fully decipher the downstream signaling network.
Aberrant epidermal differentiation can make significant contribution to skin carcinogenesis. Interestingly, many genes that have been reported to mutate in SCCs are involved in differentiation, including IRF6, Notch, and P63 (Botti et al, 2011; Stransky et al, 2011; Nowell & Radtke, 2013; Missero & Antonini, 2017). Loss of RIPK4 or Pkp1 leads to dramatic expansion of the basal cell layer, suggesting that suppressed differentiation can greatly increase the “stem”‐like cells and make the tissue more vulnerable to carcinogenesis. The potential role of RIPK4 in tumorigenesis has been controversial. It has been shown that RIPK4 can enhance tumorigenesis by increasing Wnt signaling (Huang et al, 2013). Consistently, it has been shown that elevated expression of RIPK4 associates with the progression and poor prognosis of cervical SCC (Liu et al, 2015a). On the other hand, consistent with its role in tissue differentiation, RIPK4 loss of function has been identified in human head and neck SCC through exome sequencing (Stransky et al, 2011), and retroviral insertional mutagenesis also identified RIPK4 as a tumor suppressor in human heptocarcinogenesis (Heim et al, 2015). In this report, with mouse genetics, we present convincing evidence that RIPK4 acts as a tumor suppressor in skin epidermis. Loss of RIPK4 leads to aberrant skin development, tissue homeostasis, and greatly enhanced skin tumorigenesis in vivo. The controversy could result from different tumor types and/or different signal transduction mechanisms in different tissues. Additionally, we find that loss of Pkp1 leads to increased tumorigenesis in skin epidermis. This is congruent with the previous research that DNA methylation of Pkp1 gene was associated with esophagus adenocarcinoma (Kaz et al, 2012). It would be interesting to examine whether the RIPK4/Pkp1 signaling axis functions in tissue differentiation and other types of tumor, particularly in human cancers in the future.
Our results suggest that RIPK4/Pkp1 pathway regulates epidermal differentiation in both normal skin and skin tumors. Deletion of RIPK4 or Pkp1 leads to expansion of both basal cell layer and spinous (early differentiation) layer in the skin. Granular layer (late differentiation) is not significantly changed in the Pkp1 or RIPK4 KO skin. In skin tumors, interestingly, loss of RIPK4 or Pkp1 causes significant reduction of expression for both early and late differentiation markers. The difference could be due to the mutations or epigenetic changes in the skin tumor cells. For example, constitutively active Ras or MAP kinase pathway in the tumor cells can suppress keratinocyte differentiation by itself (Dajee et al, 2002; Tarutani et al, 2003; Khavari & Rinn, 2007). Loss of RIPK4 or Pkp1 in this background would likely result in more severe defects in epidermal differentiation.
Examination of skin phenotypes in vivo with traditional mouse genetics approach is technically challenging, lengthy and expensive. In this report, we took advantage of the well‐ established procedure for culturing skin epidermal progenitor cells and developed a new experimental platform by transplantation of skin organoids generated in vitro (Fig 2D). Transplanted WT epidermal cells can readily differentiate and generate a stratified epidermis indistinguishable from the recipient mouse skin, whereas deletion of RIPK4 or Pkp1 leads to aberrant epidermal differentiation, resembling genuine RIPK4 or Pkp1 KO skin (Holland et al, 2002; Rietscher et al, 2016). However, subtle difference does exist between our skin grafting model and traditional genetics model. For instance, deletion of RIPK4 results in expansion of both spinous and granular layers in the KO mice (Holland et al, 2002). By contrast, transplantation of RIPK4‐null epidermal cells leads to significant expansion of only the spinous layer. The difference could result from different strain background because skin transplantation was carried out with nude mice. Secondly, skin engraftment is preceded by an excisional wounding, which could lead to significant changes in the tissue microenvironment and cell signaling (Martin, 1997).
In closing, our findings provide critical insights into the molecular machineries of the intricate signaling network orchestrating the cross talk between cellular junctions and epidermal differentiation as well as skin tumorigenesis.
Materials and Methods
Antibodies, reagents, and plasmid DNA constructions
Guinea pig α‐Krt5, rabbit α‐Krt10, and loricrin antibodies were generous gifts from Dr. Elaine Fuchs at the Rockefeller University. Dsp antibody was a generous gift from Dr. Kathleen Green at the Northwestern University. Rabbit Krt 14 and Krt 10 antibodies were obtained from Covance (Princeton, NJ). Rat monoclonal β4‐integrin (CD104) was obtained from BD Pharmingen (Franklin lakes, NJ). RIPK4 antibody was obtained from Abnova (Taipei City, Taiwan). Ser10 phospho‐histone antibody was obtained from EMD Millipore (Billerica, MA). Mouse monoclonal antibody against vinculin, α‐Flag conjugated beads, and α‐Flag antibodies were obtained from Sigma (St. Louis, MO). Rabbit polyclonal antibodies against HA, Dsg1, Erk, phospho‐Erk, and Ki67 were obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Pkp1 antibody was obtained from Progen (GP‐PP1). SHOC2 antibody was obtained from Proteintech (Rosemont IL). Rabbit polycloncal antibody against RIPK4 was purchased from Abnova (Taiwan). Phos‐tag was obtained from Wako Chemicals (Japan). Other chemicals or reagents were obtained from Sigma, unless otherwise indicated.
Clustered regularly interspaced short palindromic repeats targeting vector against Pkp1 was generated with primers: CAC CGC TTG AGC GGA GAG TGG TTC A; AAA CTG AAC CAC TCT CCG CTC AAG. Pkp1 N‐terminal domain was cloned with primers: ATT GCG GCC GCT CAT GAA CCA CTC GCC GCT CAA GAC CG; GCA GAT CTT GGA GCT GGC CCT AGA GTG GCC AAA GGA C. Pkp1 rescue plasmids were constructed with primers: ATT GCG GCC GCT CAT GAA CCA CTC GCC GCT CAA GAC CG; CTA TGG AAT TCT TAG AAT CGG GAG GTG AAG TTC CTG AGG CTG TTG GC. Mutations in Pkp1 head domain (SA and SE) were introduced by synthesis of corresponding DNA gBlock (IDT).
SILAC‐MS and proteomic analysis
Undifferentiated and differentiated keratinocytes and differentiated RIPK4 KO cells were subjected to SILAC label. For heavy isotope medium, we used L‐lysine‐2HCl (4, 4, 5, 5‐D4) and L‐arginine‐HCl (μ‐13C6) (Cambridge Isotope Laboratories Inc, Andover, MA) to replace the regular lysine and arginine in the medium. Cells were lysed with a solution containing 8 M Urea, 100 mM NH4CO3, protease inhibitor cocktail (Roche), phosphatase inhibitors (Pierce). We mixed the same amount of heavy labeled proteins and light labeled proteins. The proteins were digested into peptides by trypsin. The peptides were desalted by C18 Sep‐pak column (Waters), followed by phosphopeptide enrichment using TiO2 beads. The phosphopeptide enrichment was based on a reported protocol (Mazanek et al, 2007). In brief, tryptic peptides were dissolved in loading buffer (6% TFA, 80% ACN, 1 M lactic acid), and then incubated with titanium dioxide beads (Titansphere, GL Sciences, Japan) for 30 min at room temperature. The titanium dioxide beads were then washed with loading buffer and wash buffer (0.5% TFA, 50% ACN). The phosphopeptides were eluted from the beads with 10% NH3H2O. The peptides were analyzed by Orbitrap Q Exactive (Thermo Scientific). The MS/MS spectra were searched with the MaxQuant software (version 1.4.1.2) against a composite target/decoy database to estimate false discovery rate (FDR).
SILAC‐MS heavy labeled and light labeled ratios were normalized and log2 converted. Proteins with phosphosites with more than twofold increase in phosphorylation were analyzed for GO terms and KEGG pathway analysis using g:Profiler (http://biit.cs.ut.ee/gprofiler/). Benjamini‐Hochberg FDR was used for significance threshold. Gene sets with < 0.05 P‐values and < 0.05 q‐values were visualized in enrichment maps using EnrichmentMapApp and Cytoscape.
Skin organotypic culture and grafting
Decellularized dermis (1 cm × 1 cm) was prepared from newborn CD1 mice. 1.5 × 106 cultured keratinocytes were seeded onto the dermis in cell culture insert. After overnight attachment, the skin culture was exposed to air/liquid interphase. To transplant to nude animals (2–3 months old), two 1 cm × 1 cm wounds were introduced to the back skin. After transplantation, the wound edge was sealed with surgical glue. The grafted animals were housed separately and the wound bandages were usually removed one week post‐surgery (Liu et al, 2015b; Yue et al, 2016). All the experiments had more than three biological replicates (independent skin grafts). For phenotypic analysis, at least three sections were taken from each graft for analysis and quantification by staining.
Generation of RIPK4 cKO mice
The RIPK4 conditionally targeted strain was kindly provided by EUCOMM (the European Conditional Mouse Mutagenesis Program). RIPK4 cKO animals were generated by breeding the original strain with Flp transgenic strain and K14‐Cre transgenic mice sequentially. To avoid potential maternal effect, we generally use male mice with K14‐Cre transgene during breeding. All mice used in this study were bred and maintained at the ARC (animal resource center) of the University of Chicago in accordance with institutional guidelines.
Histology and immunofluorescence
Skin or wound samples were embedded in OCT, sectioned, and fixed in 4% paraformaldehyde. Sections were subjected to hematoxylin and eosin staining or immunofluorescence staining as described (Guasch et al, 2007). Antibodies were diluted according to manufacturer's instruction unless indicated. Measurements of stratification markers were carried out using ImageJ.
Two‐step cutaneous skin carcinogenesis
Six‐ to eight‐week‐old RIPK4 cKO mice and wild‐type littermate control mice were treated on the dorsal skin with a single dose of DMBA (7,12‐dimethylbenz[a]anthracene) mutagen, which induces an irreversible and specific mutation in skin: an A‐to‐T transversion in codon 61 of Ha‐Ras. Then, the skin was treated twice a week with TPA (12‐O‐tetradecanoylphorbol‐13‐acetate) for 30 weeks.
Genomic meta‐analyses
Affymetrix gene expression data of tumor propagating cancer stem cells, epidermal progenitor cells and hair follicle stem cells (GSE29328) have been normalized and analyzed in Gene Pattern software as previously described (Schober & Fuchs, 2011). Expression values for Pkp1 and Ripk4 have been extracted from these data sets and their relative expression in a6b4hiCD34lo and a6b4hiCD34hi SCC cells from two independent controls, Tgfbr2‐deficient, Ptk2‐deficient, and Tgfbr2/Ptk2‐deficient SCCs was compared to a6b4hiCD34lo skin epithelial stem and progenitor cells.
Cell culture
Primary mouse keratinocytes were isolated from the epidermis of newborn mice using trypsin, after prior separation of the epidermis from the dermis by an overnight dispase treatment. Keratinocytes were plated on mitomycin C‐treated 3T3 fibroblast feeder cells until passage 3. Cells were cultured in E‐media supplemented with 15% serum with a final concentration of 0.05 mM Ca2+.
Protein biochemical analysis
Western blot was performed as described previously (Wu et al, 2004). Cell lysates were prepared with RIPA (radioimmunoprecipitation assay) buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 10% glycerol, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X‐100, 1% sodium deoxycholate, 0.1% SDS) containing protease inhibitors and phosphatase inhibitors. Equal amounts of the cell lysates were separated using SDS–polyacrylamide gel electrophoresis (PAGE) and electroblotted onto a NC membrane. The immunoblot was incubated with Odyssey blocking buffer (Li‐Cor) at room temperature for 1 h, followed by an overnight incubation with the primary antibody. Blots were washed three times with Tween‐20/Tris‐buffered saline (TBST) and incubated with a 1:10,000 dilution of secondary antibody for 1 h at room temperature. Blots were washed three times with TBST again. Visualization and quantification were carried out with the LI‐COR Odyssey scanner and software (LI‐COR Biosciences).
Protein phosphorylation was determined by Phos‐tag reagent (Wako pure Chemical Industries, Japan) according to manufacturer's instruction. The acrylamide‐pendant Phos‐tag ligand provided a phosphate affinity SDS–PAGE for mobility shift detection of protein phosphorylation. This methodology has now been well established and served as an excellent alternative approach for radioisotope‐free detection of protein phosphorylation (Matos et al, 2008; Ydenberg & Rose, 2009; Humke et al, 2010).
Screen with kinome cDNA library
Human kinase ORF (open reading frame) library was obtained from Addgene (library generated by the Center for Cancer Systems Biology, Dana Farber Cancer Institute), and supplemented with additional constructs generated by the lab (560 kinases in total). The kinase encoding ORFs were subcloned into mammalian expression vector (pEZYmyc‐His) through Gateway cloning. Expression of different kinase ORF in HEK293 cells was confirmed by Western blot.
To identify kinase that phosphorylates Pkp1, Pkp1 NT or NT mutant was co‐transfected with each individual kinase to HEK293 cells. Cells were labeled with [32P]‐orthophosphate. Pkp1 was isolated from cell lysate with HA conjugated beads, and phosphorylation of the protein was determined by liquid scintillation.
Statistical analysis
Statistical analysis was performed using Excel, OriginPro, or SciDAVis software. Box plots are used to describe the entire population without assumptions on the statistical distribution. Student's t‐test was used to assess the statistical significance (P‐value) of the difference for most experiments. For results in Figs 2E and 4E, Mann–Whitney U‐test was utilized to test the potential statistical significance. For results in Figs 3B–C and EV6E–F, one‐way ANOVA (analysis of variance) was used for statistical analysis.
Data availability
For this study, we will make our data available to the scientific community, which will avoid unintentional duplication of research. All the research data will be shared openly and timely in accordance with the most recent NIH guidelines (http://grants.nih.gov/grants/policy/data_sharing/).
Author contributions
PL, YZ, MS, MT, and XW designed the experiments. PL, SJ, YL, JY, and XG performed the experiments. PL, YL, S‐YC, YZ, MS, and MT analyzed the data. XW wrote the manuscript. All authors edited the manuscript.1
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Review Process File
Acknowledgements
We thank Dr. Elaine Fuchs at the Rockefeller University and Dr. Jerrold Turner at the University of Chicago for sharing reagents. We thank Drs. Kathleen Green and Spiro Getsios at the University of Chicago for sharing reagents and technical assistance. We thank Linda Degenstein from the University of Chicago transgenic core facility for her excellent technical assistance. The animal studies were carried out in the ALAAC‐accredited animal research facility at the University of Chicago. This work was supported by a grant R01‐AR063630 from the National Institutes of Health, the Research Scholar Grant (RSG‐13‐198‐01) from the American Cancer Society, and the V scholar award from V Foundation to XW. This work was supported by grants AA021434 and AA020265 from the National Institutes of Health to SC. This work was supported by the National Basic Research Program of China (973 Program) (No. 2014CBA02004) and the Shanghai Municipal Science and Technology Commission (No. 15410723100) to MT. PL is supported by the University of Chicago molecular and cellular biology training grant (T32, GM007183).
The EMBO Journal (2017) 36: 1963–1980
Note
Correction added on 3 July 2017, after first online publication: the Author contributions section has been updated.
References
- Bhr C, Rohwer A, Stempka L, Rincke G, Marks F, Gschwendt M (2000) DIK, a novel protein kinase that interacts with protein kinase Cdelta. Cloning, characterization, and gene analysis. J Biol Chem 275: 36350–36357 [DOI] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E (2006) Epidermal stem cells of the skin. Annu Rev Cell Dev Biol 22: 339–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botti E, Spallone G, Moretti F, Marinari B, Pinetti V, Galanti S, De Meo PD, De Nicola F, Ganci F, Castrignano T, Pesole G, Chimenti S, Guerrini L, Fanciulli M, Blandino G, Karin M, Costanzo A (2011) Developmental factor IRF6 exhibits tumor suppressor activity in squamous cell carcinomas. Proc Natl Acad Sci USA 108: 13710–13715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussard JA, Getsios S, Green KJ (2015) Desmosome regulation and signaling in disease. Cell Tissue Res 360: 501–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chahrour O, Cobice D, Malone J (2015) Stable isotope labelling methods in mass spectrometry‐based quantitative proteomics. J Pharm Biomed Anal 113: 2–20 [DOI] [PubMed] [Google Scholar]
- Chen L, Haider K, Ponda M, Cariappa A, Rowitch D, Pillai S (2001) Protein kinase C‐associated kinase (PKK), a novel membrane‐associated, ankyrin repeat‐containing protein kinase. J Biol Chem 276: 21737–21744 [DOI] [PubMed] [Google Scholar]
- Dajee M, Tarutani M, Deng H, Cai T, Khavari PA (2002) Epidermal Ras blockade demonstrates spatially localized Ras promotion of proliferation and inhibition of differentiation. Oncogene 21: 1527–1538 [DOI] [PubMed] [Google Scholar]
- De Groote P, Tran HT, Fransen M, Tanghe G, Urwyler C, De Craene B, Leurs K, Gilbert B, Van Imschoot G, De Rycke R, Guerin CJ, Holland P, Berx G, Vandenabeele P, Lippens S, Vleminckx K, Declercq W (2015) A novel RIPK4‐IRF6 connection is required to prevent epithelial fusions characteristic for popliteal pterygium syndromes. Cell Death Differ 22: 1012–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer EH (2013) Cellular regulation by protein phosphorylation. Biochem Biophys Res Commun 430: 865–867 [DOI] [PubMed] [Google Scholar]
- Fuchs E (2008) Skin stem cells: rising to the surface. J Cell Biol 180: 273–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiki H, Suganuma M, Yoshizawa S, Kanazawa H, Sugimura T, Manam S, Kahn SM, Jiang W, Hoshina S, Weinstein IB (1989) Codon 61 mutations in the c‐Harvey‐ras gene in mouse skin tumors induced by 7,12‐dimethylbenz[a]anthracene plus okadaic acid class tumor promoters. Mol Carcinog 2: 184–187 [DOI] [PubMed] [Google Scholar]
- Garrod D, Chidgey M (2008) Desmosome structure, composition and function. Biochem Biophys Acta 1778: 572–587 [DOI] [PubMed] [Google Scholar]
- Getsios S, Huen AC, Green KJ (2004) Working out the strength and flexibility of desmosomes. Nat Rev Mol Cell Biol 5: 271–281 [DOI] [PubMed] [Google Scholar]
- Green KJ, Gaudry CA (2000) Are desmosomes more than tethers for intermediate filaments? Nat Rev Mol Cell Biol 1: 208–216 [DOI] [PubMed] [Google Scholar]
- Green DR, Oberst A, Dillon CP, Weinlich R, Salvesen GS (2011) RIPK‐dependent necrosis and its regulation by caspases: a mystery in five acts. Mol Cell 44: 9–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasch G, Schober M, Pasolli HA, Conn EB, Polak L, Fuchs E (2007) Loss of TGFbeta signaling destabilizes homeostasis and promotes squamous cell carcinomas in stratified epithelia. Cancer Cell 12: 313–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon RM, Simpson CL, Johnson JL, Koetsier JL, Dubash AD, Najor NA, Sarig O, Sprecher E, Green KJ (2013) Desmoglein‐1/Erbin interaction suppresses ERK activation to support epidermal differentiation. J Clin Invest 123: 1556–1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heim D, Cornils K, Schulze K, Fehse B, Lohse AW, Brummendorf TH, Wege H (2015) Retroviral insertional mutagenesis in telomerase‐immortalized hepatocytes identifies RIPK4 as novel tumor suppressor in human hepatocarcinogenesis. Oncogene 34: 364–372 [DOI] [PubMed] [Google Scholar]
- Holland P, Willis C, Kanaly S, Glaccum M, Warren A, Charrier K, Murison J, Derry J, Virca G, Bird T, Peschon J (2002) RIP4 is an ankyrin repeat‐containing kinase essential for keratinocyte differentiation. Curr Biol 12: 1424–1428 [DOI] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F (2014) Development and applications of CRISPR‐Cas9 for genome engineering. Cell 157: 1262–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, McGann JC, Liu BY, Hannoush RN, Lill JR, Pham V, Newton K, Kakunda M, Liu J, Yu C, Hymowitz SG, Hongo JA, Wynshaw‐Boris A, Polakis P, Harland RM, Dixit VM (2013) Phosphorylation of Dishevelled by protein kinase RIPK4 regulates Wnt signaling. Science 339: 1441–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humke EW, Dorn KV, Milenkovic L, Scott MP, Rohatgi R (2010) The output of Hedgehog signaling is controlled by the dynamic association between Suppressor of Fused and the Gli proteins. Genes Dev 24: 670–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalay E, Sezgin O, Chellappa V, Mutlu M, Morsy H, Kayserili H, Kreiger E, Cansu A, Toraman B, Abdalla EM, Aslan Y, Pillai S, Akarsu NA (2012) Mutations in RIPK4 cause the autosomal‐recessive form of popliteal pterygium syndrome. Am J Hum Genet 90: 76–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaz AM, Luo Y, Dzieciatkowski S, Chak A, Willis JE, Upton MP, Leidner RS, Grady WM (2012) Aberrantly methylated PKP1 in the progression of Barrett's esophagus to esophageal adenocarcinoma. Genes Chromosom Cancer 51: 384–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern F, Niault T, Baccarini M (2011) Ras and Raf pathways in epidermis development and carcinogenesis. Br J Cancer 104: 229–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khavari TA, Rinn J (2007) Ras/Erk MAPK signaling in epidermal homeostasis and neoplasia. Cell Cycle 6: 2928–2931 [DOI] [PubMed] [Google Scholar]
- Kitajima Y (2014) 150(th) anniversary series: desmosomes and autoimmune disease, perspective of dynamic desmosome remodeling and its impairments in pemphigus. Cell Commun Adhes 21: 269–280 [DOI] [PubMed] [Google Scholar]
- Kwa MQ, Huynh J, Aw J, Zhang L, Nguyen T, Reynolds EC, Sweet MJ, Hamilton JA, Scholz GM (2014) Receptor‐interacting protein kinase 4 and interferon regulatory factor 6 function as a signaling axis to regulate keratinocyte differentiation. J Biol Chem 289: 31077–31087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwa MQ, Huynh J, Reynolds EC, Hamilton JA, Scholz GM (2015) Disease‐associated mutations in IRF6 and RIPK4 dysregulate their signalling functions. Cell Signal 27: 1509–1516 [DOI] [PubMed] [Google Scholar]
- Liu DQ, Li FF, Zhang JB, Zhou TJ, Xue WQ, Zheng XH, Chen YB, Liao XY, Zhang L, Zhang SD, Hu YZ, Jia WH (2015a) Increased RIPK4 expression is associated with progression and poor prognosis in cervical squamous cell carcinoma patients. Sci Rep 5: 11955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Yue J, Huang H, Gou X, Chen SY, Zhao Y, Wu X (2015b) Regulation of focal adhesion dynamics and cell motility by the EB2 and Hax1 protein complex. J Biol Chem 290: 30771–30782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez‐Pajares V, Yan K, Zarnegar BJ, Jameson KL, Khavari PA (2013) Genetic pathways in disorders of epidermal differentiation. Trends Genet 29: 31–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin P (1997) Wound healing–aiming for perfect skin regeneration. Science 276: 75–81 [DOI] [PubMed] [Google Scholar]
- Matos J, Lipp JJ, Bogdanova A, Guillot S, Okaz E, Junqueira M, Shevchenko A, Zachariae W (2008) Dbf4‐dependent CDC7 kinase links DNA replication to the segregation of homologous chromosomes in meiosis I. Cell 135: 662–678 [DOI] [PubMed] [Google Scholar]
- Mazanek M, Mituloviae G, Herzog F, Stingl C, Hutchins JR, Peters JM, Mechtler K (2007) Titanium dioxide as a chemo‐affinity solid phase in offline phosphopeptide chromatography prior to HPLC‐MS/MS analysis. Nat Protoc 2: 1059–1069 [DOI] [PubMed] [Google Scholar]
- McGrath JA, McMillan JR, Shemanko CS, Runswick SK, Leigh IM, Lane EB, Garrod DR, Eady RA (1997) Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndrome. Nat Genet 17: 240–244 [DOI] [PubMed] [Google Scholar]
- Missero C, Antonini D (2017) p63 in squamous cell carcinoma of the skin: more than a stem cell/progenitor marker. J Invest Dermatol 137: 280–281 [DOI] [PubMed] [Google Scholar]
- Mitchell K, O'Sullivan J, Missero C, Blair E, Richardson R, Anderson B, Antonini D, Murray JC, Shanske AL, Schutte BC, Romano RA, Sinha S, Bhaskar SS, Black GC, Dixon J, Dixon MJ (2012) Exome sequence identifies RIPK4 as the Bartsocas‐Papas syndrome locus. Am J Hum Genet 90: 69–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nekrasova O, Green KJ (2013) Desmosome assembly and dynamics. Trends Cell Biol 23: 537–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitoiu D, Etheridge SL, Kelsell DP (2014) Insights into desmosome biology from inherited human skin disease and cardiocutaneous syndromes. Cell Commun Adhes 21: 129–140 [DOI] [PubMed] [Google Scholar]
- Nowell C, Radtke F (2013) Cutaneous Notch signaling in health and disease. Cold Spring Harb Perspect Med 3: a017772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong SE, Foster LJ, Mann M (2003) Mass spectrometric‐based approaches in quantitative proteomics. Methods 29: 124–130 [DOI] [PubMed] [Google Scholar]
- Perdigoto CN, Valdes VJ, Bardot ES, Ezhkova E (2014) Epigenetic regulation of epidermal differentiation. Cold Spring Harb Perspect Med 4: a015263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prunieras M, Regnier M, Woodley D (1983) Methods for cultivation of keratinocytes with an air‐liquid interface. J Invest Dermatol 81: 28s–33s [DOI] [PubMed] [Google Scholar]
- Rietscher K, Wolf A, Hause G, Rother A, Keil R, Magin TM, Glass M, Niessen CM, Hatzfeld M (2016) Growth retardation, loss of desmosomal adhesion, and impaired tight junction function identify a unique role of plakophilin 1 in vivo . J Invest Dermatol 136: 1471–1478 [DOI] [PubMed] [Google Scholar]
- Schmidt A, Jager S (2005) Plakophilins–hard work in the desmosome, recreation in the nucleus? Eur J Cell Biol 84: 189–204 [DOI] [PubMed] [Google Scholar]
- Schober M, Fuchs E (2011) Tumor‐initiating stem cells of squamous cell carcinomas and their control by TGF‐beta and integrin/focal adhesion kinase (FAK) signaling. Proc Natl Acad Sci USA 108: 10544–10549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolik‐Delmaire T, Reddy R, Pashaj A, Roberts BJ, Wahl JK III (2010) Plakophilin‐1 localizes to the nucleus and interacts with single‐stranded DNA. J Invest Dermatol 130: 2638–2646 [DOI] [PubMed] [Google Scholar]
- Sprecher E, Molho‐Pessach V, Ingber A, Sagi E, Indelman M, Bergman R (2004) Homozygous splice site mutations in PKP1 result in loss of epidermal plakophilin 1 expression and underlie ectodermal dysplasia/skin fragility syndrome in two consanguineous families. J Invest Dermatol 122: 647–651 [DOI] [PubMed] [Google Scholar]
- Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, Carter SL, Voet D, Cortes ML, Auclair D, Berger MF, Saksena G, Guiducci C et al (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333: 1157–1160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarutani M, Cai T, Dajee M, Khavari PA (2003) Inducible activation of Ras and Raf in adult epidermis. Cancer Res 63: 319–323 [PubMed] [Google Scholar]
- Wu X, Suetsugu S, Cooper LA, Takenawa T, Guan JL (2004) Focal adhesion kinase regulation of N‐WASP subcellular localization and function. J Biol Chem 279: 9565–9576 [DOI] [PubMed] [Google Scholar]
- Wu X, Kodama A, Fuchs E (2008) ACF7 regulates cytoskeletal‐focal adhesion dynamics and migration and has ATPase activity. Cell 135: 137–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Boehm JS, Yang X, Salehi‐Ashtiani K, Hao T, Shen Y, Lubonja R, Thomas SR, Alkan O, Bhimdi T, Green TM, Johannessen CM, Silver SJ, Nguyen C, Murray RR, Hieronymus H, Balcha D, Fan C, Lin C, Ghamsari L et al (2011) A public genome‐scale lentiviral expression library of human ORFs. Nat Methods 8: 659–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ydenberg CA, Rose MD (2009) Antagonistic regulation of Fus2p nuclear localization by pheromone signaling and the cell cycle. J Cell Biol 184: 409–422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue J, Zhang Y, Liang WG, Gou X, Lee P, Liu H, Lyu W, Tang WJ, Chen SY, Yang F, Liang H, Wu X (2016) In vivo epidermal migration requires focal adhesion targeting of ACF7. Nat Commun 7: 11692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanivan S, Meves A, Behrendt K, Schoof EM, Neilson LJ, Cox J, Tang HR, Kalna G, van Ree JH, van Deursen JM, Trempus CS, Machesky LM, Linding R, Wickstrom SA, Fassler R, Mann M (2013) In vivo SILAC‐based proteomics reveals phosphoproteome changes during mouse skin carcinogenesis. Cell Rep 3: 552–566 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Review Process File
Data Availability Statement
For this study, we will make our data available to the scientific community, which will avoid unintentional duplication of research. All the research data will be shared openly and timely in accordance with the most recent NIH guidelines (http://grants.nih.gov/grants/policy/data_sharing/).