

Abstract
The recent elucidation of the structure of the carboxyl-terminal region of the hyperpolarization-activated cyclic nucleotide-modulated (HCN2) channel has prompted us to investigate a curious feature of this structure in HCN2 channels and in the related CNGA1 cyclic nucleotide-gated (CNG) channels. The crystallized fragment of the HCN2 channel contains both the cyclic nucleotide-binding domain (CNBD) and the C-linker region, which connects the CNBD to the pore. At the center of the fourfold-symmetric structure is a tunnel that runs perpendicular to the membrane. The narrowest part of the tunnel is ≈10 Å in diameter and is lined by a ring of negatively charged amino acids: D487, E488, and D489. Many ion channels have “charge rings” that focus permeant ions at the mouth of the pore and increase channel conductance. We used nonstationary fluctuation analysis and single-channel recording, coupled with site-directed mutagenesis and cysteine modification, to determine whether this part of HCN and CNG channels might be an extension of the permeation pathway. Our results indicate that modifying charge-ring amino acids affects gating but not ion permeation in HCN2 and CNG channels. Thus, this portion of the channel is not an obligatory part of the ion path but instead acts as a “gating ring.” The carboxyl-terminal region of these channels must hang below the pore much like the “hanging gondola” of voltage-gated potassium channels, but the permeation pathway must exit the protein before the level of the ring of charged amino acids.
Keywords: charge ring, fluctuation analysis, single-channel patch clamp, cyclic nucleotide-gated, hyperpolarization-activated cyclic nucleotide-modulated
Hyperpolarization-activated cyclic nucleotide-modulated (HCN) ion channels, also known as pacemaker channels, are critical components in the production of rhythmic activity in both cardiac and neural cells (1–6). Similar to other voltage-gated cation channels, HCN channels are tetramers. Each subunit has six transmembrane domains, denoted S1–S6, and a reentrant pore loop. The pore loops and the S6 transmembrane domains line the permeation path of the channel, and the S4 domains form the primary voltage sensor of HCN channels (7, 8). HCN channels also contain a cytoplasmic carboxyl-terminal domain that has sequence similarity with that of the cyclic nucleotide-gated (CNG) ion-channel family. The carboxyl-terminal region contains a cyclic nucleotide-binding domain (CNBD) that binds cytoplasmic cyclic nucleotide ligands and is connected to the pore by a C-linker region that is conserved among HCN and CNG channels (Fig. 1B) (9–11). HCN channels are weakly selective cation channels; they exclude divalent cations but allow both K+ and Na+ to permeate (K+ is favored) (9, 11, 12). Cyclic nucleotide binding to the CNBD of HCN channels modulates the open probability of the channel. Unlike most other voltage-gated channels, HCN channels are activated by membrane hyperpolarization. Binding of cyclic nucleotides to the CNBD shifts the voltage dependence of these channels to more depolarized potentials (5, 6, 13, 14).
Fig. 1.

Crystal structure of the HCN2 carboxyl-terminal region. (A) Electrostatic profile of the HCN2 carboxyl-terminal region calculated with grasp software. (Left) A side view of the structure, with one subunit removed, is shown. (Right) A view from below, looking from the cytoplasm toward the membrane. Negatively charged regions are shown as red, and positively charged regions are shown as blue. (B) Close-up view of the charge ring. D487 is shown as red, E488 is shown as green, and D489 is shown as blue. Below the ring is a sequence alignment showing the charge-ring region of HCN2 and CNGA1. Arrows identify D487, E488, and D489. (C) Possible permeation paths: ions might exit the channel before (yellow arrows) or after (green arrow) the charge ring.
CNG channels are critical components of sensory transduction pathways. They generate the primary electrical signals of vertebrate phototransduction, olfaction, and gustation (15–17). These channels are also found in other neural and nonneuronal tissues, in which their function is less clearly understood (18). CNG channels are nearly insensitive to membrane voltage despite having an S4 “voltage sensor” domain that is capable of supporting voltage sensitivity when transplanted into a voltage-sensitive channel (19, 20). The open probability of these channels is instead tightly regulated by binding of intracellular cyclic nucleotide ligands to the channel (16, 17, 21).
The x-ray crystallographic structure of the HCN2 carboxyl-terminal region has given us fresh perspective on the workings of cyclic nucleotide-modulated channels (22). The crystallized fragment contains both the C-linker and the CNBD. A striking feature of the HCN2 structure is a central tunnel that runs down the axis of the fourfold-symmetric channel, directly in line with the anticipated location of the channel pore (Fig. 1 A and B) (22). Because the crystallized fragment begins just at the end of the predicted S6 pore-forming domain, it is reasonable to hypothesize that the tunnel might constitute an exit pathway for the ions to move between the channel and the cytoplasm (Fig. 1C, green arrow). Along with its geometry and position, the central tunnel has another feature that suggests that it might be part of the permeation pathway. The narrowest part of the tunnel is ringed by a series of negatively charged amino acids: D487, E488, and D489 (Fig. 1 A and B). E488 points away from the aqueous center of the tunnel, facing the interior of the protein. D487 and D489, however, face the central axis and are directly accessible to the water-filled center of the tunnel. Negative charge rings are a common feature at the mouths of cation channels, where they help to focus the permeant cations and likely act as a “prefilter” by repelling intracellular anions from the immediate vicinity of the pore (23–30). Despite this architecture, it is possible that permeant ions leave the channel before reaching this tunnel (Fig. 1C, yellow arrows). If this is the case, and permeant ions need not flow through the tunnel, then the charge ring would likely have no critical role in ion focusing and permeation.
To determine whether the tunnel seen in the crystal structure is an obligatory part of the ion pathway in both HCN2 and CNGA1 channels, we altered the charge character of the charge ring and measured the effects on ion permeation and gating. We found that, although the carboxyl-terminal region modulates gating, it is not an obligatory part of the permeation pathway.
Materials and Methods
Mutagenesis. The wild-type HCN2 construct was kindly provided by Steven A. Siegelbaum (Columbia University, New York). The CNGA1cys-free constructs used in this study were a cysteineless version of CNGA1 in which all seven endogenous cysteine-encoding codons had been modified. Mutant constructs were made by standard site-directed mutagenesis methods (31). The PCR-amplified region of each mutant was sequenced completely by using the BigDye sequencing protocol (Applied Biosystems).
Oocyte Expression and Electrophysiology. cDNAs in the pGEMHE plasmid were linearized and then used as a template for RNA synthesis by using mMessage mMachine (Ambion, Austin, TX). RNA was injected into defolliculated Xenopus laevis oocytes as described (32). Recordings were made in the excised, inside-out patch-clamp configuration by using an Axopatch 200A patch-clamp amplifier (Axon Instruments, La Jolla, CA) and pulse acquisition software (HEKA Electronik, Lambrecht, Germany) (33). Patch pipettes for macroscopic recordings were pulled from glass micropipettes (VWR Scientific) and had resistances of 0.25–1 MΩ after fire polishing. Electrodes for single-channel recording had resistances of 1–3 MΩ and were coated with dental wax to reduce capacitance transients. Data were analyzed by using a combination of excel (Microsoft), igor pro (WaveMetrics, Lake Oswego, OR), and sigmaplot (Jandel, San Rafael, CA).
The base solution for both the extracellular (pipette) and intracellular (bath) solutions for HCN2-channel recordings consisted of 130 mM KCl, 0.2 mM EDTA, and 3 mM Hepes, pH 7.2. In addition, the intracellular solution contained 1 mM cAMP. The base solution for all CNGA1-channel recording solutions consisted of 130 mM NaCl, 0.2 mM EDTA, and 3 mM Hepes, pH 7.2. The extracellular pipette solution also contained 0.5 mM niflumic acid to block endogenous chloride conductances. Where indicated, intracellular solutions contained cyclic nucleotides that were diluted from 16 mM stock solutions to the desired concentration. For methanethiosulfonate ethyltrimethylammonium (MTSET) experiments, MTSET (Toronto Research Chemicals, New York) was dissolved in water to make a 100 mM stock solution that then was frozen in 100-μl aliquots and stored at –80°C. Just before MTSET application, a 100-μl aliquot of the stock solution was thawed and diluted to 2 mM in 2 mM cGMP-containing bath solution.
HCN2 Nonstationary Fluctuation Analysis. Fluctuation analysis data were collected by using a voltage-clamp protocol by which the membrane potential was held at 0 mV before stepping to –152 mV for 750 ms once every 3 s. Patches were selected for stability and allowed to equilibrate at least 10 min after excision before recording. Data were recorded with a sampling frequency of 20 kHz and filtered offline at 2 kHz. Neither sampling at higher frequency (40 kHz) nor changing the filter corner frequency (1–10 kHz) changed the single-channel conductance values obtained. Sweeps for each patch (85–180) were used to create a variance-versus-mean-current curve. The variance at zero open probability was subtracted from the mean current, and the curves then were fit with a parabolic equation:
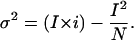 |
[1] |
σ2 is the variance, I is the mean macroscopic current, i is the single-channel current, and N is the number of channels in the patch. Ohm's law was used to transform the determined single-channel currents into single-channel conductance (γ) values (shown in Fig. 3B).
Fig. 3.

HCN2 unitary conductance is insensitive to charge-ring mutations. (A–C) Representative variance-versus-mean-current plots for HCN2, D487K, and D489R channels. Red lines are fits of a parabolic function (see Materials and Methods). The number of channels (N) and conductance (γ) from each curve is shown beneath the curves. (D) Box plot shows aggregate conductance values for each channel (HCN2, n = 4; D487K, n = 3; D489R, n = 6). Boxes indicate the 25–75% confidence intervals, and error bars show the 10–90% confidence intervals. The internal line is the median.
CNGA1cys-free Single-Channel Analysis. All CNGA1 experiments were begun 30 min after excision, after current run-up was complete (34). Macroscopic CNGA1 currents were corrected for leak by subtraction of the current recorded from the same patch in the absence of cyclic nucleotides. The voltage-clamp protocol for macroscopic CNGA1 records was always the same: 300-ms test pulses to voltages between +80 and –80 mV were applied in 20-mV increments at 1-s intervals from a holding potential of 0 mV. Measurements used for averaging and summary data figures were made early during the voltage step to +60 mV, after the capacitive transient had settled but before significant ion accumulation could occur (35). To measure CNGA1 single-channel currents, excised inside-out patches were voltage-clamped to a holding potential of 0 mV before stepping to test potentials between +20 and +100 mV in 20-mV increments for 1 s. Recordings were sampled at 10 kHz and filtered at 2 kHz for analysis.
CNGA1 Macroscopic Data Analysis. The CNGA1 macroscopic data at saturating cyclic nucleotide concentrations were fit with Eq. 2.
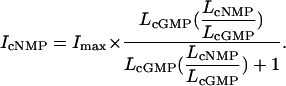 |
[2] |
IcNMP is IcGMP, IcAMP, or IcIMP, the experimentally determined macroscopic current with a saturating concentration of cyclic nucleotide (2.5 mM for cGMP, 16 mM for cAMP, and 16 mM for cIMP). Imax is the current if the channel open probability was 1. LcNMP is LcGMP, LcAMP, or LcIMP and represents the equilibrium constant for the channel-opening transition in the specified cyclic nucleotide. We assumed that the nucleotide selectivity and hence the ratios of LcAMP/LcGMP and LcIMP/LcGMP, are the same for all mutants tested. This assumption is reasonable, because none of these mutations are near the CNBD. Published ratios for LcIMP/LcGMP (0.06) and LcAMP/LcGMP (0.001) from CNGA1cys-free were used and held constant for fitting of L (36). Using these ratios and the measured currents for IcGMP, IcAMP, and IcIMP, we then fit current amplitudes with Eq. 2 by varying LcGMP and Imax by using a least-squares algorithm.
LcGMP values for the CNGA1 constructs were used to calculate the free energy (ΔG) of channel opening in cGMP (see Fig. 5D) by using Eq. 3,
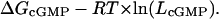 |
[3] |
R is the universal gas constant, and T is the temperature in degrees kelvin.
Fig. 5.

MTSET modification alters CNGA1cys-free channel gating. (A–C) Leak-subtracted current traces in response to steps from 0 to +60 or –60 mV in saturating cGMP (green), cIMP (blue), or cAMP (red) are superimposed. (A) Wild-type and D447K currents. (B and C) D447C and R449C constructs before and after exposure to 2 mM MTSET for 20 min. (D) Free energy of channel opening in cGMP calculated from fractional activation data such as those described for A. Boxes indicate the 25–75% confidence intervals, and error bars show the 10–90% confidence intervals. The internal line indicates the median value. (E) Change in the free energy of channel opening in response to MTSET modification. Error bars show the standard error of the mean.
The change in free energy of allosteric opening after addition of MTSET, ΔΔGMTSET (see Fig. 5C), was calculated from the experimentally determined LcGMP values by using Eq. 4.
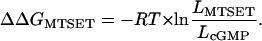 |
[4] |
LcGMP is measured before MTSET modification, and LMTSET is measured in the same manner as LcGMP but after exposing the patch to 2 mM MTSET for 20 min.
Statistics. All errors presented in the text and figures are presented as the standard error of the mean. Statistical significance was accorded to data with a P value of ≤0.05 as calculated by the Student t test.
Molecular Models. Molecular model representations in Fig. 1 were created with viewerpro 4.2 (Accelrys, San Diego) and grasp (37) software.
Results
To investigate the role of the HCN2 charge ring in ion permeation, we focused on two residues in the HCN2 carboxyl-terminal structure that project directly into the tunnel, D487 and D489. If these negatively charged side chains project into a cation-selective permeation pore, reversing their charge would be expected to alter the ability of cations to pass. D487 was mutated to lysine, and D489 was mutated to arginine, the identity of the analogous residue in CNGA1. The constructs were expressed in X. laevis oocytes and studied by the patch-clamp method in the excised inside-out patch configuration in the presence of 1 mM cAMP.
HCN2 Conductance Is Independent of the Charge Ring. Macroscopic recordings of HCN2 and the charge-reversal mutants are shown in Fig. 2. The mutant channels produced robust macroscopic currents with voltage dependences that were not significantly different than that of the HCN2 channel (HCN2 V1/2 =–107 ± 4.1 mV; D487K V1/2 = –114 ± 1.5 mV; and D489R V1/2 = –107 ± 3.0 mV).
Fig. 2.

Charge-ring mutations do not change HCN2 voltage-dependence. (A–C) Representative current records from inside-out patches from X. laevis oocytes expressing wild-type and mutant HCN2 channels. Membrane potential was held at 0 mV and then stepped to test potentials between –70 and –150 mV for 5 s before stepping to –40 mV for 1 s. Currents recorded as described for A were used to construct the conductance–voltage curves shown in D. Curves were made by taking the peak tail current amplitude and plotting versus the preceding test potential (black circles, HCN2; red triangles, D487K; blue squares, D489R). For comparison, curves were normalized to the asymptote of a fit of the Boltzmann equation to the data. The smooth curve is the fit to the HCN2 data. The Boltzmann equation was of the form: I/Imax = 1/[1 + e(V – V1/2)/kv]. I is the peak tail current at the test potential (V), Imax is the maximal tail current, V1/2 is the potential of half-maximal activation, and kv is the slope factor.
The large currents produced by D487K and D489R indicated that the permeation pathway was not totally obstructed by these mutations. Nonetheless it remained unclear whether the channel conductance might have been affected more subtly. The single-channel conductance of HCN channels is too small for direct observation of single-channel currents (14). Therefore, to test whether D487K and D489R mutations had altered ion conduction, we performed nonstationary fluctuation analysis (Fig. 3). Fluctuation analysis makes use of the variance or “noise” in macroscopic current recordings that arises from single-channel events to infer the properties of the single-channel currents (38). The variance-versus-mean-current curves were plotted from current records with steps to –152 mV for each construct and fit with a parabolic equation to determine the number of channels in the patch, as well as the single-channel conductance (Fig. 3 A–C). Fig. 3D shows compiled single-channel conductance data from multiple patches for the three HCN2-channel constructs. All constructs were determined to have single-channel conductances (γ) near 2.5 pS (HCN2 γ = 2.5 ± 0.33 pS; D487K γ = 2.3 ± 0.21 pS; D489R γ = 2.2 ± 0.09 pS). Neither D487K nor D489R had conductances significantly different from HCN2 (P > 0.05). Fluctuation analysis measurements can underestimate the conductance of single channels because of missed events. Nonetheless, the measurements for each construct were made in an identical manner; thus, any error should be systematic and therefore should not affect the comparison of conductances between constructs. Because charge-reversal mutations in the charge ring of HCN2 did not affect the single-channel conductance, it is highly unlikely that the tunnel seen in the HCN2 carboxyl-terminal structure is an obligate part of the permeation pathway in these channels.
CNGA1cys-free Single-Channel Conductance Is Independent of the Charge Ring. The sequence of CNG channels, similar to that of HCN2, has a string of charged residues analogous to those that surround the central tunnel of the HCN2 fragment of known structure (Fig. 1B). In CNGA1, however, the third charge is a positively charged arginine residue. Thus, the DED charge sequence in HCN2 is DER in CNGA1.
CNGA1 channels have several features that lead to technical advantages over HCN channels for examining the role of the carboxyl-terminal region in ion permeation and gating. First, CNGA1 channels have a much larger single-channel conductance, ≈20 pS (39–41), which allows recording of single-channel currents and direct observation of conductance differences that might arise from mutations in the charge ring. Second, cyclic nucleotide binding to the CNBD has a much larger effect on channel-gating energetics in CNG channels than HCN channels, increasing the ability to measure gating effects of the charge-ring residues. Third, a cysteine-free version of CNGA1 channels (CNGA1cys-free) is available (42). This cysteineless channel enables site-specific cysteine modification experiments that are not yet feasible in HCN2. For these reasons, we chose also to test the role of the charge-ring residues in the permeation and gating of CNGA1 channels.
We first made a charge-reversal mutation in the CNGA1cys-free channel. The D447K mutant in CNGA1 is equivalent to the D487K mutation in HCN2 (Fig. 1). D447K CNGA1cys-free channels produce robust currents in excised patches from X. laevis oocytes and are gated by cyclic nucleotides with the normal agonist selectivity (cGMP > cIMP > cAMP; see below). We recorded single-channel currents from excised inside-out patches of Xenopus oocytes that expressed either CNGA1cys-free or D447K channels. Saturating concentrations of cGMP (2 mM), cIMP (16 mM), or cAMP (16 mM) were used to induce channel opening. As shown previously for CNGA1cys-free channels, the single-channel conductance of D447K was similar for all three cyclic nucleotides (data not shown) (39, 43). Fig. 4A shows representative single-channel records for CNGA1cys-free channels in cIMP and D447K channels in cAMP. These conditions produced a similar open probability in the channels in which the single-channel conductance could be compared easily and measured accurately. Fig. 4B shows representative amplitude histograms from patches containing a single channel, and Fig. 4C shows single-channel current–voltage relations averaged from multiple patches for each mutant. The mean currents for CNGA1cys-free and D447K channels were not significantly different at any voltages between +20 and +100 mV (P > 0.05). Therefore, even at the single-channel level, mutating residue 447 from the negatively charged aspartic acid to a positively charged lysine has no significant effect on ion permeation.
Fig. 4.

D447K does not alter CNGA1cys-free unitary conductance. (A) Representative single-channel currents at +60 mV for CNGA1cys-free channels in 2 mM cGMP and D447K channels in 16 mM cIMP. Red dashed lines indicate open and closed current levels and are scaled identically for both constructs. (B) Amplitude histograms for 200 ms of single-channel record for a CNGA1cys-free or D447K channel. Smooth red curves are double Gaussian fits. (C) Mean single-channel current–voltage relations for CNGA1cys-free (filled circles, n = 3) or D447K (open circles, n = 4).
Charge-Ring Mutations Affect CNGA1 Channel Gating. In CNGA1cys-free channels, cAMP is a poor agonist, with saturating concentrations eliciting only 8 ± 3% of the maximal macroscopic current evoked by saturating levels of cGMP (Fig. 5A). cIMP is a stronger partial agonist, eliciting 67 ± 8% of the maximal current. D447K channels exhibited a higher fractional activation by partial agonists (Fig. 5A). In D447K channels, cAMP elicited 36 ± 5% and cIMP elicits 95 ± 1% of the maximal cGMP current. In saturating concentrations of cyclic nucleotide, the channels are liganded fully. Thus, the open probability is determined entirely by the ability of the ligand to stabilize the open state versus the closed state. In saturating cGMP, the channels are essentially open all of the time; therefore, the current level in saturating cGMP can be taken as the maximal current level. An increase in the current produced by cAMP and cIMP, and therefore the current relative to the maximal current evoked by cGMP, indicates that the mutation increased the relative stability of the open state of the channel, which is the case for the D447K mutant.
MTSET Modification Changes CNGA1 Gating, Not Conductance. To examine further the change in channel gating caused by disrupting the charge ring, we mutated either D447 or R449 to cysteine and measured the fractional activation by cyclic nucleotides before and after modification with the positively charged cysteine-modifying reagent MTSET (Fig. 5). Substituting a cysteine into the channel had a similar effect whether the cysteine is inserted at position 447, in place of a negatively charged aspartate (Fig. 5B), or at position 449 in place of a positively charged arginine (Fig. 5C). In both cases, the channel is potentiated relative to the CNGA1cys-free channel. As with the D447K mutation, D447C and R449C are activated more easily by the partial agonists cIMP and cAMP, much like the potentiation in D447K. Changes in the fractional activation by cGMP, cIMP, and cAMP were used to calculate the free energy of channel opening (ΔG) in cGMP (Fig. 5D). Both mutant channels were potentiated by ≈1 kcal/mol (1 kcal = 4.18 kJ) relative to CNGA1cys-free.
Comparing fractional activation levels before and after MTSET modification allowed us to quantify the change in free energy of channel opening induced by MTSET (ΔΔGMTSET; Fig. 5E). The CNGA1cys-free channel was unaffected by application of MTSET (Fig. 5E). Modification of D447C or R449C, however, caused changes in fractional activation by cIMP and cAMP. Addition of a positively charged group to cysteine D447C resulted in more potentiation, further increasing the fractional activation. This channel behaved similar to the D447K charge-reversal mutant (D447K ΔGcGMP = 491 ± 113, D447C+MTSET ΔGcGMP = 735 ± 225). MTSET modification of R449C resulted in inhibition, decreasing fractional activation by cIMP and cAMP, and caused R449C to behave like the CNGA1cys-free channel (CNGA1cys-free ΔGcGMP = 58 ± 14; R449C+MTSET ΔGcGMP = 52 ± 10). Thus, for both positions, removing the charge (be it positive or negative) potentiates the channel. For both channels, addition of the positive charge of MTSET results in a channel with behavior that mimics that of a channel with a positively charged amino acid at that position.
The effect of MTSET on D447C and R449C CNGA1 channels is significant (both functionally and statistically; P < 0.05). MTSET modulation of the mutant-channel currents indicates that the target cysteine was modified by the reagent and that the positively charged group was added to the channel. The gating effects of MTSET modification indicate that the normal charge conformation of the charge-ring structure is required for wild-type channel gating. If the unitary conductance of the channel had been affected by the charged group, the macroscopic current for the full agonist cGMP would necessarily be reduced as well. However, neither inward nor outward maximum currents were reduced by MTSET modification (Fig. 5 B and C), which indicates that although the charge residues affect channel gating, the tunnel seen in the HCN2 structure is not an obligate part of the permeation pathway in CNGA1 channels.
Discussion
The carboxyl-terminal region of CNG channels contains the ligand-binding domain, and the C-linker and is an important regulator of channel opening. Many studies have shown that mutagenic or chemical modification of this region have effects on channel gating (31, 44–51). The central tunnel seen in the crystal structure of the carboxyl-terminal region of HCN2 suggests that it might also be a part of the channel ion-permeation pathway. The crystallized fragment of the channel begins just after the predicted end of the S6 transmembrane domain that lines the inner pore of the channel. Thus, the crystallized region and the pore are adjacent, and there are no other apertures in the crystal structure that seem capable of allowing ion permeation. Nonetheless, we found that inserting a positive charge into the narrowest constriction of the tunnel in both HCN2 and CNGA1 channels had no effect on the ability of permeant ions to pass through the channel.
If permeant ions need not pass through the tunnel visualized in the HCN2 crystal structure, how do they escape from the channel pore? The most likely explanation is that ions leave the channel before reaching the charge ring (Fig. 1C, yellow arrows). If this is the case, there must be a path that allows ions access to the cytoplasm. Because of the fourfold symmetry of the channel, there most likely would be four such windows, which leads to a picture reminiscent of the T1 domain of Shaker and Kv K+ channels. The T1 domain hangs below the channel pore, held by four proteinaceous columns separating permeation windows in a “hanging-gondola” or “fenestrated-rotunda” conformation (52–54). In HCN and CNG channels, however, the gondola is formed by the carboxyl-terminal region, not the amino-terminal region as with the T1 domain. The crystallized fragment of HCN2 begins shortly after the S6 pore-forming regions and contains no apparent permeation windows before the tunnel examined here. New work examining the salt bridges seen in the crystal structure of the HCN2 carboxyl-terminal region indicates that the C-linkers may have been captured in the closed state despite the presence of a bound cAMP ligand (55). It is possible that the permeation windows that allow ions to leave the channel before the tunnel seen in the structure (Fig. 1C, yellow arrows) exist only in an open state of the channel that is not seen in the crystal structure.
Regardless of how the permeant ions escape the channel pore, our data indicate that they need not traverse the tunnel through the center of the carboxyl-terminal region. The fact that MTSET modification of cysteines at positions 447 and 449 of the CNGA1 changes the energetics of channel opening suggests that these residues likely move during channel gating. Thus, the C-linker and CNBD of HCN2 and CNGA1 seem to form a gating ring that modulates channel opening but is not itself an obligatory part of the ion-permeation pathway.
Acknowledgments
We thank Kevin Black, Shellee Cunnington, Gay Sheridan, and Heidi Utsugi for expert technical assistance; Kimberley Craven, Noah Shuart, and Justin Taraska for comments on the manuscript; Tinatin I. Brelidze, Galen E. Flynn, Leon Islas, Matthew C. Trudeau, and Jie Zheng for insightful discussions; and Nelson Olivier for help with the grasp image. This work was supported by National Eye Institute Grant EY10329 and the Howard Hughes Medical Institute.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HCN, hyperpolarization-activated cyclic nucleotide-modulated; CNG, cyclic nucleotide-gated; CNBD, cyclic-nucleotide binding domain; MTSET, methanethiosulfonate ethyltrimethylammonium.
References
- 1.Pape, H. C. & McCormick, D. A. (1989) Nature 340, 715–718. [DOI] [PubMed] [Google Scholar]
- 2.McCormick, D. A. & Pape, H. C. (1990) J. Physiol. (London) 431, 291–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brown, H. F., DiFrancesco, D. & Noble, S. J. (1979) Nature 280, 235–236. [DOI] [PubMed] [Google Scholar]
- 4.DiFrancesco, D. & Tortora, P. (1991) Nature 351, 145–147. [DOI] [PubMed] [Google Scholar]
- 5.Rosenbaum, T. & Gordon, S. E. (2004) Neuron 42, 193–196. [DOI] [PubMed] [Google Scholar]
- 6.Robinson, R. B. & Siegelbaum, S. A. (2003) Annu. Rev. Physiol. 65, 453–480. [DOI] [PubMed] [Google Scholar]
- 7.Chen, J., Mitcheson, J. S., Lin, M. & Sanguinetti, M. C. (2000) J. Biol. Chem. 275, 36465–36471. [DOI] [PubMed] [Google Scholar]
- 8.Vaca, L., Stieber, J., Zong, X., Ludwig, A., Hofmann, F. & Biel, M. (2000) FEBS Lett. 479, 35–40. [DOI] [PubMed] [Google Scholar]
- 9.Santoro, B., Liu, D. T., Yao, H., Bartsch, D., Kandel, E. R., Siegelbaum, S. A. & Tibbs, G. R. (1998) Cell 93, 717–729. [DOI] [PubMed] [Google Scholar]
- 10.Seifert, R., Scholten, A., Gauss, R., Mincheva, A., Lichter, P. & Kaupp, U. B. (1999) Proc. Natl. Acad. Sci. USA 96, 9391–9396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ludwig, A., Zong, X., Jeglitsch, M., Hofmann, F. & Biel, M. (1998) Nature 393, 587–591. [DOI] [PubMed] [Google Scholar]
- 12.Gauss, R., Seifert, R. & Kaupp, U. B. (1998) Nature 393, 583–587. [DOI] [PubMed] [Google Scholar]
- 13.Baruscotti, M. & Difrancesco, D. (2004) Ann. N.Y. Acad. Sci. 1015, 111–121. [DOI] [PubMed] [Google Scholar]
- 14.Accili, E. A., Proenza, C., Baruscotti, M. & DiFrancesco, D. (2002) News Physiol. Sci. 17, 32–37. [DOI] [PubMed] [Google Scholar]
- 15.Misaka, T., Kusakabe, Y., Emori, Y., Gonoi, T., Arai, S. & Abe, K. (1997) J. Biol. Chem. 272, 22623–22629. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura, T. & Gold, G. H. (1987) Nature 325, 442–444. [DOI] [PubMed] [Google Scholar]
- 17.Fesenko, E. E., Kolesnikov, S. S. & Lyubarsky, A. L. (1985) Nature 313, 310–313. [DOI] [PubMed] [Google Scholar]
- 18.Matulef, K. & Zagotta, W. N. (2003) Annu. Rev. Cell Dev. Biol. 19, 23–44. [DOI] [PubMed] [Google Scholar]
- 19.Tang, C. Y. & Papazian, D. M. (1997) J. Gen. Physiol. 109, 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karpen, J. W., Zimmerman, A. L., Stryer, L. & Baylor, D. A. (1988) Proc. Natl. Acad. Sci. USA 85, 1287–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yau, K. W. & Baylor, D. A. (1989) Annu. Rev. Neurosci. 12, 289–327. [DOI] [PubMed] [Google Scholar]
- 22.Zagotta, W. N., Olivier, N. B., Black, K. D., Young, E. C., Olson, R. & Gouaux, E. (2003) Nature 425, 200–205. [DOI] [PubMed] [Google Scholar]
- 23.Brelidze, T. I., Niu, X. & Magleby, K. L. (2003) Proc. Natl. Acad. Sci. USA 100, 9017–9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eismann, E., Muller, F., Heinemann, S. H. & Kaupp, U. B. (1994) Proc. Natl. Acad. Sci. USA 91, 1109–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuo, A., Gulbis, J. M., Antcliff, J. F., Rahman, T., Lowe, E. D., Zimmer, J., Cuthbertson, J., Ashcroft, F. M., Ezaki, T. & Doyle, D. A. (2003) Science 300, 1922–1926. [DOI] [PubMed] [Google Scholar]
- 26.Moorhouse, A. J., Keramidas, A., Zaykin, A., Schofield, P. R. & Barry, P. H. (2002) J. Gen. Physiol. 119, 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nimigean, C. M., Chappie, J. S. & Miller, C. (2003) Biochemistry 42, 9263–9268. [DOI] [PubMed] [Google Scholar]
- 28.Nishida, M. & MacKinnon, R. (2002) Cell 111, 957–965. [DOI] [PubMed] [Google Scholar]
- 29.Imoto, K., Busch, C., Sakmann, B., Mishina, M., Konno, T., Nakai, J., Bujo, H., Mori, Y., Fukuda, K. & Numa, S. (1988) Nature 335, 645–648. [DOI] [PubMed] [Google Scholar]
- 30.Haug, T., Sigg, D., Ciani, S., Toro, L., Stefani, E. & Olcese, R. (2004) J. Gen. Physiol. 124, 173–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gordon, S. E. & Zagotta, W. N. (1995) Neuron 14, 177–183. [DOI] [PubMed] [Google Scholar]
- 32.Zagotta, W. N., Hoshi, T. & Aldrich, R. W. (1989) Proc. Natl. Acad. Sci. USA 86, 7243–7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hamill, O. P., Marty, A., Neher, E., Sakmann, B. & Sigworth, F. J. (1981) Pflügers Arch. 391, 85–100. [DOI] [PubMed] [Google Scholar]
- 34.Molokanova, E., Trivedi, B., Savchenko, A. & Kramer, R. H. (1997) J. Neurosci. 17, 9068–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zimmerman, A. L., Karpen, J. W. & Baylor, D. A. (1988) Biophys. J. 54, 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johnson, J. P., Jr., & Zagotta, W. N. (2001) Nature 412, 917–921. [DOI] [PubMed] [Google Scholar]
- 37.Nicholls, A., Sharp, K. A. & Honig, B. (1991) Proteins 11, 281–296. [DOI] [PubMed] [Google Scholar]
- 38.Sigworth, F. J. (1980) J. Physiol. (London) 307, 131–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sunderman, E. R. & Zagotta, W. N. (1999) J. Gen. Physiol. 113, 601–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haynes, L. W., Kay, A. R. & Yau, K. W. (1986) Nature 321, 66–70. [DOI] [PubMed] [Google Scholar]
- 41.Zimmerman, A. L. & Baylor, D. A. (1986) Nature 321, 70–72. [DOI] [PubMed] [Google Scholar]
- 42.Matulef, K. & Zagotta, W. (2002) Neuron 36, 93–103. [DOI] [PubMed] [Google Scholar]
- 43.Sunderman, E. R. & Zagotta, W. N. (1999) J. Gen. Physiol. 113, 621–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gordon, S. E. & Zagotta, W. N. (1995) Proc. Natl. Acad. Sci. USA 92, 10222–10226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gordon, S. E., Oakley, J. C., Varnum, M. D. & Zagotta, W. N. (1996) Biochemistry 35, 3994–4001. [DOI] [PubMed] [Google Scholar]
- 46.Brown, R. L., Snow, S. D. & Haley, T. L. (1998) Biophys. J. 75, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Broillet, M. C. (2000) J. Biol. Chem. 275, 15135–15141. [DOI] [PubMed] [Google Scholar]
- 48.Zong, X., Zucker, H., Hofmann, F. & Biel, M. (1998) EMBO J. 17, 353–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paoletti, P., Young, E. C. & Siegelbaum, S. A. (1999) J. Gen. Physiol. 113, 17–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rosenbaum, T. & Gordon, S. E. (2002) Neuron 33, 703–713. [DOI] [PubMed] [Google Scholar]
- 51.Mottig, H., Kusch, J., Zimmer, T., Scholle, A. & Benndorf, K. (2001) J. Gen. Physiol. 118, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim, L. A., Furst, J., Gutierrez, D., Butler, M. H., Xu, S., Goldstein, S. A. & Grigorieff, N. (2004) Neuron 41, 513–519. [DOI] [PubMed] [Google Scholar]
- 53.Kobertz, W. R., Williams, C. & Miller, C. (2000) Biochemistry 39, 10347–10352. [DOI] [PubMed] [Google Scholar]
- 54.Sokolova, O., Kolmakova-Partensky, L. & Grigorieff, N. (2001) Structure (Cambridge) 9, 215–220. [DOI] [PubMed] [Google Scholar]
- 55.Craven, K. B. & Zagotta, W. N. (2004) J. Gen. Physiol. 124, 663–677. [DOI] [PMC free article] [PubMed] [Google Scholar]