

Abstract
Lafora disease (LD) is a fatal progressive epilepsy essentially caused by loss‐of‐function mutations in the glycogen phosphatase laforin or the ubiquitin E3 ligase malin. Glycogen in LD is hyperphosphorylated and poorly hydrosoluble. It precipitates and accumulates into neurotoxic Lafora bodies (LBs). The leading LD hypothesis that hyperphosphorylation causes the insolubility was recently challenged by the observation that phosphatase‐inactive laforin rescues the laforin‐deficient LD mouse model, apparently through correction of a general autophagy impairment. We were for the first time able to quantify brain glycogen phosphate. We also measured glycogen content and chain lengths, LBs, and autophagy markers in several laforin‐ or malin‐deficient mouse lines expressing phosphatase‐inactive laforin. We find that: (i) in laforin‐deficient mice, phosphatase‐inactive laforin corrects glycogen chain lengths, and not hyperphosphorylation, which leads to correction of glycogen amounts and prevention of LBs; (ii) in malin‐deficient mice, phosphatase‐inactive laforin confers no correction; (iii) general impairment of autophagy is not necessary in LD. We conclude that laforin's principle function is to control glycogen chain lengths, in a malin‐dependent fashion, and that loss of this control underlies LD.
Keywords: glycogen chain length, glycogen phosphorylation, Lafora disease, laforin, malin
Subject Categories: Genetics, Gene Therapy & Genetic Disease; Metabolism; Neuroscience
Introduction
Lafora disease (LD) is a recessively inherited teenage‐onset severe and fatal progressive myoclonus epilepsy. Its etiopathological hallmark is the Lafora body (LB), present throughout the brain in profuse ever‐increasing numbers and sizes (Lafora & Glueck, 1911; Minassian, 2001). LBs are overwhelmingly composed of an abnormally formed glycogen (Sakai et al, 1970) and likely result from gradual precipitation, aggregation and accumulation of abnormal glycogen.
Normally glycogen consists of highly and regularly branched roughly sphere‐shaped molecules, each composed of up to 55,000 glucose units. It is mainly synthesized by coordinated actions of glycogen synthase (GS), which elongates glucan chains forming α1‐4 interglucosidic linkages, and glycogen branching enzyme, which detaches a part of each newly formed chain and reattaches it to another glucan chain through an α1‐6 linkage. Glycogen is essentially degraded by glycogen phosphorylase and glycogen debranching enzyme. Glucan chains of glycogen have a wide chain length distribution with an average of ~13 units (Roach et al, 2012; Nitschke et al, 2013). High frequency of branching points and relatively short chains are critical for glycogen solubility. They prevent neighbouring chains from forming double helices, which otherwise exclude water, reduce the water solubility of the interacting chains and finally lead the entire molecule to precipitate. Incidentally, the main component of plant starch amylopectin is insoluble precisely because it is composed of longer chains that through clustered arrangement of branch points form crystalline areas of double helices (Roach et al, 2012; Cenci et al, 2014). Glycogen and amylopectin possess phosphate esters bound to a very small number of glucose units, in glycogen's case at carbon positions C2, C3 and C6 (Tagliabracci et al, 2011; Nitschke et al, 2013). The kinase(s) responsible for glycogen phosphorylation are not known, except that GS itself has been reported to be involved in C3 phosphorylation through what so far has been considered an undesirable side reaction of the enzyme (Tagliabracci et al, 2011; Contreras et al, 2016).
The abnormal glycogen constituting LBs has three characteristics: (i) its chain length distribution contains longer chains than normal glycogen, (ii) its phosphate levels are elevated, and (iii) its quantity is high, the latter reflecting the profuse deposition as LBs (Tagliabracci et al, 2008; Nitschke et al, 2013). LD is mainly caused by loss‐of‐function mutations in either the EPM2A (laforin) or EPM2B (malin) gene, and knockout of either in mouse recapitulates the disease (Minassian et al, 1998; Ganesh et al, 2002; Chan et al, 2003; Turnbull et al, 2010). Laforin is a glycogen phosphatase, its absence resulting in progressive glycogen hyperphosphorylation (Worby et al, 2006; Tagliabracci et al, 2008). Malin is a ubiquitin E3 ligase (Gentry et al, 2005), which in cell culture experiments, in a laforin‐dependent fashion, diminishes both GS and a protein involved in GS activation, R5/PTG, through proteasomal degradation (Vilchez et al, 2007; Worby et al, 2008). Malin, again in a laforin‐dependent manner, also diminishes a second protein that participates in GS activation, R6, in this case through autophagic degradation (Rubio‐Villena et al, 2013). Laforin and malin form a functional complex, the former (but not the latter) possessing a carbohydrate binding domain through which the complex is targeted to glycogen (Gentry et al, 2005; Lohi et al, 2005).
From the above results, two main hypotheses were advanced to explain the abnormal glycogen formation in LD: (i) absence of laforin or malin leads to increased GS activity, which outpacing branching leads to the formation of glycogen with long chains promoting water insolubility (Vilchez et al, 2007); and (ii) laforin deficiency results in non‐removal of erratically introduced phosphate and thus hyperphosphorylation of glycogen, which via an unknown mechanism alters its structure promoting glycogen precipitation (Tagliabracci et al, 2008; Roach, 2015). Both hypotheses, however, were challenged by more recent findings. (i) Tissues from laforin and malin knockout mice were shown to have unaltered GS activity (Tagliabracci et al, 2008; DePaoli‐Roach et al, 2010). (ii) When phosphatase‐inactive laforin was overexpressed in laforin knockout mice, LBs no longer formed and the mice were rescued from LD (Gayarre et al, 2014). The latter result was particularly provocative. It suggested that laforin's chief and only established physiological function (glycogen dephosphorylation) and the major and distinguishing pathological feature of LD glycogen (progressive hyperphosphorylation) were irrelevant to LB formation and LD.
Gayarre et al, however, left a number of critical questions unaddressed before the field could make a decided paradigm shift away from the centrality of glycogen phosphate in LB formation and LD towards exploring alternative disease mechanisms. They did not measure glycogen phosphate. It can, therefore, not be excluded that the overexpressed in vitro‐inactive laforin had retained some phosphatase activity in vivo or that there was a small amount of wild‐type (WT) laforin somehow still expressed in the transgenic mice. Laforin's phosphatase activity is catalysed by a cysteine residue (C265 in mouse, C266 in human), which Gayarre and colleagues had mutated to serine to inactivate the enzyme. Recently, crystal structures of laforin were determined (Raththagala et al, 2015; Sankhala et al, 2015) suggesting the presence of a putative second phosphatase site in laforin (C168 in mouse, C169 in human). The C168 site could be active in vivo, especially when overexpressed, and explain the rescue by laforin mutated only at C265.
The other two features specific to LD glycogen, chain length distribution and quantity, were also outside the scope of the above study and had not been analysed. As such, conclusions about the mechanism conferring rescue from LBs remained tentative. It was possible, for example, that the rescued mice did accumulate abnormal glycogen, which, coated with a large amount of hydrophilic protein (overexpressed laforin), remained soluble and did not coalesce into pathologically observable LBs.
In this work, we address all these points by characterizing glycogen in both laforin‐ and malin‐deficient mice as affected by overexpressed WT and phosphatase‐inactive laforin. Our results combine with previous studies to allow the proposal of a unifying hypothesis of LB pathogenesis in both laforin‐ and malin‐deficient LD.
Results
Amounts and molecular parameters of muscle glycogen in Epm2a −/− mice overexpressing phosphatase‐inactive laforin
We quantified total glycogen from skeletal muscle from the actual mice used in the Gayarre et al study (Gayarre et al, 2014). In Epm2a −/− mice, muscle glycogen content was fivefold increased, as in previous studies (Tagliabracci et al, 2008). In Epm2a −/− mice overexpressing WT laforin (Epm2a −/−.Laf), muscle glycogen content was normal. In Epm2a −/− mice overexpressing phosphatase‐inactive laforin (Epm2a −/−.C265SLaf), muscle glycogen content was also normal (Fig 1A), indicating that the prevention of LBs (which are aggregates of insoluble glycogen) by phosphatase‐inactive laforin (Gayarre et al, 2014) is indeed due to the prevention of generating insoluble glycogen, as opposed to merely maintaining the excess abnormal glycogen in a soluble state.
Figure 1. Phosphatase‐inactive laforin does not correct LD muscle glycogen hyperphosphorylation, but does correct its chain length distribution and prevents abnormal glycogen accumulation.
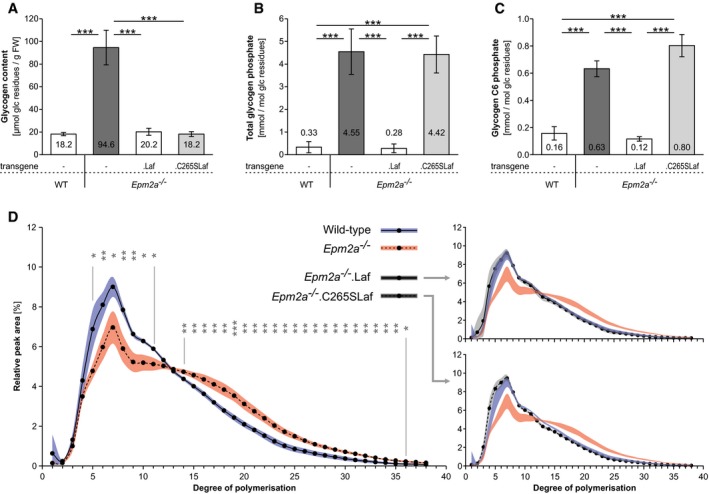
- Glycogen content.
- Glycogen total phosphate content.
- Glycogen carbon C6 phosphate content.
- Chain length distribution analyses.
We measured total glycogen phosphate in the above tissues, which in Epm2a −/− mice was 14‐fold increased, as in previous studies (Tagliabracci et al, 2008). Levels were normal in Epm2a −/−.Laf mice, but in Epm2a −/−.C265SLaf mice glycogen phosphate was just as high as in Epm2a −/− mice (Fig 1B). This indicates that phosphatase‐inactive laforin indeed has no phosphatase activity in vivo and does not normalize the hyperphosphorylation characteristic of LD glycogen. This in turn indicates that glycogen hyperphosphorylation does not cause the generation of insoluble glycogen.
Measurement of total glycogen phosphate requires relatively large amounts of purified glycogen (see Materials and Methods). Of the three glucosyl carbons phosphorylated in glycogen, C6 phosphorylation is the only one for which a sensitive method has been developed that allows site‐specific quantification. Moreover, C6 phosphate can be measured in very small amounts of glycogen, and as all three phosphorylation sites are proportionally high in LD (DePaoli‐Roach et al, 2015; Roach, 2015), it can be used as a measure of total glycogen phosphorylation. While sufficient amounts of glycogen can be purified from skeletal muscle for total glycogen phosphate measurement, this is currently unfeasible for brain glycogen, where C6 phosphorylation only would be quantified as an indicator of the total. Leading up to the brain studies (next sections), we measured C6 glycogen phosphate in skeletal muscle and found that it is, as expected, like total glycogen phosphate, increased in both the Epm2a −/− and Epm2a −/−.C265SLaf mice (Fig 1C).
Besides glycogen amount and phosphate content, the third feature that distinguishes LD from WT glycogen is chain length distribution. We measured skeletal muscle glycogen chain lengths in the above genotypes and found that the chain length distribution in Epm2a −/− mice is abnormal with a significantly increased proportion of longer chains, while in Epm2a −/−.C265SLaf mice (and of course Epm2a −/−.Laf mice) the chain length distribution is normal, indicating that phosphatase‐inactive laforin rescues the glycogen chain length abnormality characteristic of LD glycogen (Fig 1D).
To summarize, phosphatase‐inactive laforin does not correct the hyperphosphorylation of LD muscle glycogen, but does normalize the glycogen's chain length distribution preventing abnormal glycogen accumulation and LB formation.
Brain glycogen in Epm2a −/− and Epm2b −/− mouse lines overexpressing phosphatase‐inactive laforin
Generation of new mouse line
The implications of the Gayarre et al (2014) study and our above results are major: Laforin's phosphatase function is dispensable and glycogen hyperphosphorylation is not pathogenic. These results run counter to a leading LD hypothesis, in which glycogen phosphorylation is considered a damaging side product of GS activity that needs to be cleared by laforin (Tagliabracci et al, 2011; Raththagala et al, 2015; Roach, 2015; Contreras et al, 2016; Turnbull et al, 2016). To rule out the possibility of an artefact in the Gayarre et al mice, we generated a whole new set of Epm2a −/−, and this time also Epm2b −/− mice overexpressing phosphatase‐inactive laforin. Phosphatase‐inactive human laforin (C266SLaf) was used, in contrast to the phosphatase‐inactive murine laforin (C265SLaf) used by Gayarre et al (2014). We did so by crossing WT mice overexpressing C266SLaf available in our laboratory (Chan et al, 2004) with Epm2a −/− (Ganesh et al, 2002) and Epm2b −/− (Turnbull et al, 2010) mice. The resultant transgenic animals are termed Epm2a −/−.C266SLaf and Epm2b −/−.C266SLaf, respectively.
We simultaneously analysed glycogen and LBs from this new line and from the Gayarre et al Epm2a −/−.C265SLaf mice, focusing on brain, the disease‐relevant organ. Consistency of findings in Gayarre et al and our mice would confirm phosphatase‐inactive laforin's effect on glycogen and obviate results arising from any particularities of each mouse line studied. The differences between the mouse lines are transgene species (as mentioned above), presence or absence of a tag on the transgene protein, expression level, and mouse background (Chan et al, 2004; Gayarre et al, 2014). We had previously generated the C266SLaf mice (Chan et al, 2004) in an attempt to create an LD mouse model through a dominant‐negative approach. The phosphatase‐inactive laforin (C266SLaf) was supposed to outcompete WT laforin's phosphatase activity and lead to an effective loss of the endogenous laforin function. In fact, the resultant mice did have a few LBs and we reported this mouse as a mild, only‐pathological, model of LD, with no neurological phenotype (Chan et al, 2004). Compared to the laforin (and malin) knockout mice (Epm2a −/− and Epm2b −/− , respectively) (Ganesh et al, 2002; Turnbull et al, 2010), the number of LBs occurring in this model is orders of magnitude lower (Fig 2), and negligible for purposes of the present experiments.
Figure 2. Phosphatase‐inactive laforin prevents Epm2a −/− but not Epm2b −/− from LB formation.
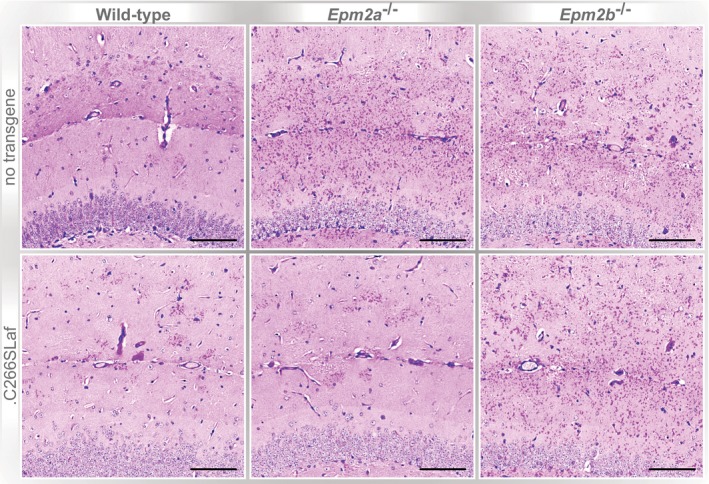
Representative PASD‐stained sections of hippocampi from WT, Epm2a −/− and Epm2b −/− mice in the absence or presence of the human C266S mutated laforin transgene (.C266SLaf). Scale bar equals 100 μm.Source data are available online for this figure.
Glycogen and LB analyses
Brain glycogen content was threefold to fourfold increased in both Epm2a −/− mouse lines and in Epm2b −/− mice, as previously published (Tagliabracci et al, 2008; DePaoli‐Roach et al, 2010; Turnbull et al, 2010). Brain glycogen content was normal in Epm2a −/−.C265SLaf and Epm2a −/−.C266SLaf mice, but remained elevated in Epm2b −/−.C266SLaf mice (Fig 3A and B). Thus, overexpressed phosphatase‐inactive laforin corrects the abnormal glycogen accumulation in laforin‐deficient LD but not in malin‐deficient LD. The latter indicates that the rescue in the former is not through some mechanism related to overexpressed protein (somehow physically solubilizing insoluble glycogen) as otherwise the same correction would be expected in the malin‐deficient background.
Figure 3. Phosphatase‐inactive laforin rescues Epm2a −/−, but not Epm2b −/− abnormal glycogen accumulation in the brain.
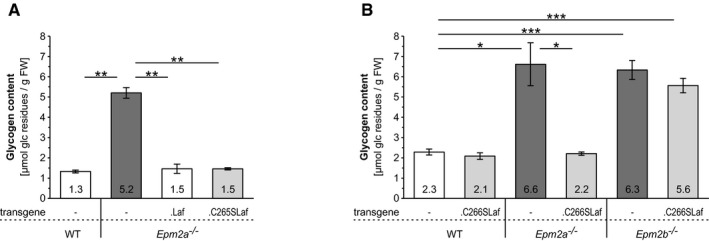
- Brain glycogen levels in the presence or absence of WT (.Laf) or C265S mutated (.C265Laf) murine laforin transgene (tissues from the Gayarre et al, 2014 study).
- Brain glycogen levels of WT, Epm2a −/− and Epm2b −/− mice in the presence or absence of human C266S mutated laforin (.C266SLaf) (tissues from the new mice generated in the present study).
Consistent with the rescue of the abnormal brain glycogen accumulation in the Epm2a −/−.C265SLaf and Epm2a −/−.C266SLaf mice, there were no LBs in the brains of these mice. LBs were, however, still present in the Epm2b −/−.C266SLaf mice, and to the same extent as in their Epm2b −/− controls (Fig 2), again indicating that phosphatase‐inactive laforin prevents LB formation in laforin‐deficient but not malin‐deficient LD.
We next measured brain glycogen phosphate (via C6 phosphate quantification). Due to particularities of brain tissue, this (and the below glycogen chain length analyses) required extensive new method development, detailed in the Materials and Methods section, and the results represent the first time these analyses have been possible in brain. Brain glycogen phosphate was threefold and twofold elevated in Epm2a −/− and Epm2b −/− mice, respectively, similar to muscle glycogen phosphate (Tagliabracci et al, 2008; DePaoli‐Roach et al, 2010; Turnbull et al, 2010). This increase was not corrected in any of the three phosphatase‐inactive laforin expressing mouse genotypes (Epm2a −/−.C265SLaf, Epm2a −/−.C266SLaf and Epm2b −/−.C266SLaf) (Fig 4). Taken together, the results in this section clearly indicate that hyperphosphorylation of glycogen is not pathogenic nor is it causative of abnormal glycogen accumulation and LB formation.
Figure 4. Phosphatase‐inactive laforin does not rescue glycogen hyperphosphorylation in Epm2a −/− or Epm2b −/− brain.
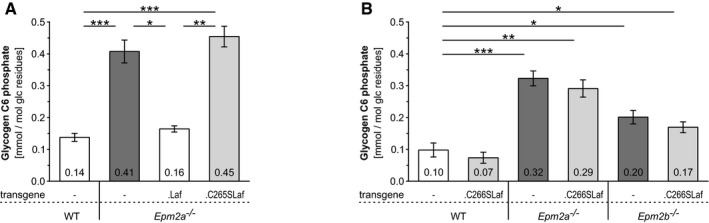
- Brain glycogen carbon C6 phosphate levels in the presence or absence of WT (.Laf) or C265S mutated (.C265Laf) murine laforin transgene.
- Brain glycogen C6 phosphate levels of WT, Epm2a −/− and Epm2b −/− mice in the presence or absence of human C266S mutated laforin (.C266SLaf).
Similar to previous findings in muscle glycogen (Fig 1D; see also Nitschke et al, 2013), but even more pronounced, brain glycogen chain length distributions were abnormal, significantly shifted towards longer chains, in Epm2a −/− and Epm2b −/− mice. They were normalized in Epm2a −/−.C265SLaf and Epm2a −/−.C266SLaf mice (indistinguishable from WT) and remained abnormal in Epm2b −/−.C266SLaf (Fig 5). This demonstrates that phosphatase‐inactive laforin can correct the brain glycogen chain length abnormality in laforin‐deficient LD, but not in malin‐deficient LD. These results tease apart, for the first time, the three characteristics of abnormal LD glycogen (glycogen accumulation, hyperphosphorylation, abnormal chain length distribution) and indicate that the altered glycogen chain length distribution, and not hyperphosphorylation, correlates with abnormal glycogen accumulation and LB formation. Moreover, phosphatase‐inactive laforin's ability to rescue the chain length abnormality (and the abnormal glycogen accumulation/LBs) depends on the presence of malin, as this correction does not occur in the malin‐deficient mice (Epm2b −/−.C266SLaf). This implies that laforin (irrespective of its phosphatase domain) and malin impact glycogen chain length distribution and prevent LB formation and LD concertedly. It directly links both proteins to glycogen structure in vivo and corroborates the close laforin–malin partnership previously suggested by cell culture overexpression experiments (Gentry et al, 2005; Lohi et al, 2005; Vilchez et al, 2007).
Figure 5. Phosphatase‐inactive laforin rescues brain glycogen chain length distribution in Epm2a −/− but not Epm2b −/− LD.
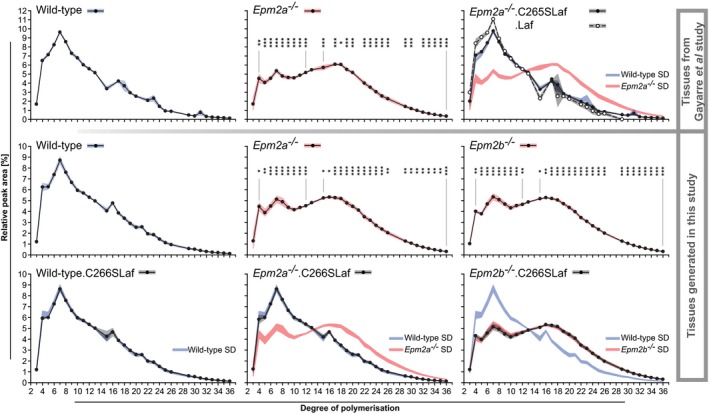
Top row from tissues from the Gayarre et al (2014) study; bottom two rows from mice generated in the present work. Chain lengths (x‐axes) are given as degrees of polymerization (DP), that is, numbers of glucose units per chain. Laf indicates WT laforin transgene expressed in indicated LD genotype. C265SLaf and C266SLaf indicate phosphatase‐inactive murine or human laforin, respectively, expressed in indicated LD genotype. Data are presented as means of relative peak areas for each DP with shades representing standard deviation (SD; blue, WT; red, Epm2a −/− or Epm2b −/−; grey, indicated transgenic mice; n > 5, for Epm2a −/−.Laf samples were pooled). In all panels relating to transgenic mice, in addition to the chain length distribution in these mice, SD shades of WT or respective knockout mutants (Epm2a −/− or Epm2b −/−) are shown to allow direct comparisons of the chain length distribution curves. Asterisks, statistical significance by ANOVA and post hoc analyses with regard to DP abundances in respective WT glycogen (*P < 0.05, **P < 0.01, ***P < 0.001; Appendix Table S4).
Effect of phosphatase‐inactive laforin on LD‐associated general defect in autophagy
LC3 becomes lipidated (i.e. LC3‐I converts to LC3‐II) as it joins proliferating autophagosomes during autophagy, and therefore decreased LC3‐II indicates decreased autophagy. p62 directs ubiquitinated proteins to autophagy, and thus, accumulation of p62 indicates failing autophagy. LD mouse brains were shown to have reduced LC3‐II and increased p62, suggesting that LD mice have impaired autophagy and that this defect underlies LB accumulation (Criado et al, 2012). Gayarre and colleagues reported normalization of LC3‐II levels in their Epm2a −/−.C265Laf mice, suggesting that phosphatase‐inactive laforin rescues laforin knockout mice by correcting the autophagic defect (Gayarre et al, 2014). We tested brain LC3‐II and p62 in the new mice generated for the present study and found no differences in either protein between WT, Epm2a −/−, Epm2b −/−, Epm2a −/−.C266Laf, and Epm2b −/−.C266Laf genotypes (Fig 6). These results imply that impaired autophagy may not be a common feature to all LD models and, therefore, it is not a strict requirement for LD.
Figure 6. C266S laforin's effect on LD‐associated general defect in autophagy.
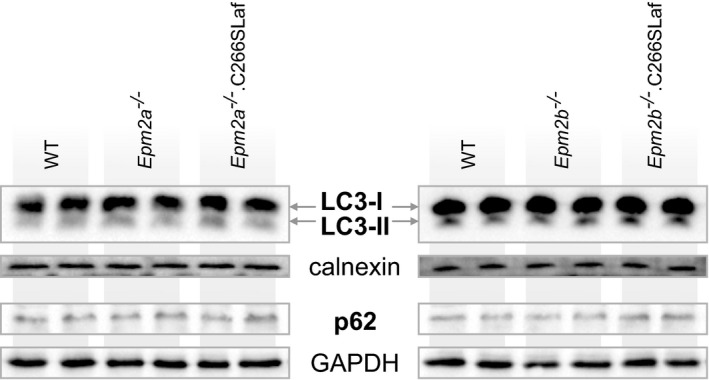
Representative Western blots of autophagy markers LC3 and p62 in protein extracts from laforin‐ and malin‐deficient mouse brains and their respective WT tissues. Loading controls were calnexin for LC3 detection in an autophagosome‐enriched membrane fraction and GAPDH for soluble p62.
Discussion
The report that phosphatase‐inactive laforin rescues laforin‐deficient LD (Gayarre et al, 2014) runs contrary to the widely accepted view that the disease is caused by lack of laforin‐mediated clearance of erratic glycogen phosphorylation (Raththagala et al, 2015; Roach, 2015; Contreras et al, 2016; Turnbull et al, 2016). In that publication, however, several important issues had remained unaddressed. First, possibly the rescue could be effected by the mere overabundance of (inactive) laforin solubilizing the malstructured (long‐chained) glycogen. Second, the rescue could have been due to the persistence of residual laforin phosphatase activity, either in the mice considered full laforin knockouts or in the overexpressed laforin transgene. Based on recently published structural information, laforin might possess a heretofore unrecognized second phosphatase site that would have been unaffected by the C265S transgene mutation (Sankhala et al, 2015). We now rule all these possibilities out. We show that the same inactive laforin overexpression does not correct the abnormal glycogen accumulation/LBs in malin‐deficient LD and that the correction in laforin‐deficient LD includes normalization of both glycogen chain distribution and content, unequivocally indicating that the rescue has a metabolic, glycogen‐related basis. We also show that the rescue occurs with no reduction in glycogen hyperphosphorylation, ruling out that it is due to any residual laforin phosphatase activity.
Our results show that glycogen hyperphosphorylation does not correlate with LB formation. Hyperphosphorylation is shared by Epm2a −/−, Epm2b −/−, Epm2a −/−.C265SLaf (rescued), Epm2a −/−.C266SLaf (rescued), and Epm2b −/−.C266SLaf (not rescued) genotypes and can therefore not be the cause of LB formation (Fig 7). The glycogen chain length abnormality, on the other hand, is shared by the Epm2a −/−, Epm2b −/−, and Epm2b −/−.C266SLaf (not rescued) mice, strictly correlating with LB formation and glycogen accumulation (Fig 7). This together with the well‐established facts that longer glucan chains are less water soluble as they form stable double helices and that isolated LBs consist of poorly branched glucans (Sakai et al, 1970; Gidley & Bulpin, 1987; Hejazi et al, 2009) strongly supports the notion that chain length abnormality is the actual cause of glycogen precipitation and, over time, accumulation into LBs.
Figure 7. Summary of phosphatase‐inactive laforin's effect in LD mouse model brains.
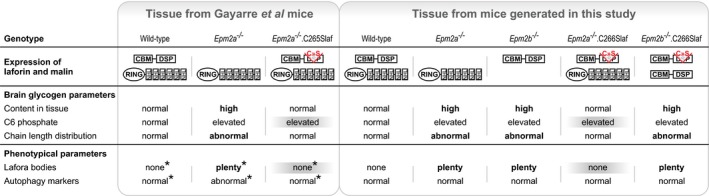
LD‐associated phenotypical parameters such as hippocampal LBs and autophagy impairment as well as brain glycogen content are compared with respect to the presence of laforin, malin, and the phosphatase‐inactive laforin transgene. Asterisks mark previously published results (Gayarre et al, 2014). Grey shadows emphasize the dissociation between hyperphosphorylation and LB occurrence. Bold letters indicate the association between abnormal chain length distribution and LBs.
Phosphatase‐inactive laforin rescues glycogen precipitation and accumulation in the Epm2a −/−.C265SLaf and Epm2a −/−.C266SLaf but not Epm2b −/−.C266SLaf mice. As the relevant difference between these genotypes is that the latter lacks malin and has an abnormal glycogen chain length pattern, it is safe to conclude that laforin confers its effect strictly together with malin. Previous publications afford insights into the mechanisms through which malin could prevent the generation of glycogen molecules with abnormally long chains. As mentioned, in cell culture experiments malin has consistently been shown to ubiquitinate, in a laforin‐dependent fashion, GS and proteins involved in GS activation (PTG/R5 and R6), targeting them to proteasomal or autophagic degradation (Vilchez et al, 2007; Worby et al, 2008; Rubio‐Villena et al, 2013). On the surface, this mechanism conflicted with the lack of overall elevated GS activity in LD mouse tissues (Tagliabracci et al, 2008; DePaoli‐Roach et al, 2010). These results can, however, be reconciled if the action of the laforin–malin complex is restricted to a small subset of glycogen molecules. Glycogen is heterogeneous, in both size (Wanson & Drochmans, 1968; Marchand et al, 2002; Nitschke et al, 2013) and chain length distribution (i.e., some glycogen molecules contain longer chains) (Palmer et al, 1983; Nitschke et al, 2013). The laforin–malin system would be required only for those molecules/molecule regions in which glucan chains approach thresholds to insolubility. Consistent with this is the observation that laforin's carbohydrate binding domain has greater affinity to longer rather than shorter glucan chains (Chan et al, 2004; Lohi et al, 2005; Dias et al, 2016). A reduced chain elongation would prevent progression towards precipitation, and since the activity would be localized, its absence would not result in detectably increased global GS activity.
Aguado et al (2010) reported that their murine Epm2a −/− LD model has an impairment of autophagy, and Criado et al (2012) that this impairment presents prior to the appearance of detectable glycogen accumulations/LBs. Gayarre et al (2014) showed that phosphatase‐inactive laforin restores autophagy as it rescues LD and proposed that laforin–malin prevents LB formation via autophagy. In contrast and in line with recent publications (Irimia et al, 2015; Wang et al, 2016), we do not detect presence of an autophagic impairment in the Epm2a −/− and Epm2b −/− animals used to prepare the new transgenic mice generated here and can, therefore, not confirm autophagy correction by laforin–malin. Additionally, other recent work showed that disturbances of autophagy markers present in LD are correctable by genetic reduction of GS activity (Duran et al, 2014), suggesting that the autophagy impairment may be secondary to the glycogenosis. Notwithstanding, it is entirely possible that laforin–malin acts locally to eliminate, through autophagy, individual glycogen molecules that are “at risk” of precipitation. In fact, malin was recently shown to be able to mediate K63‐linked ubiquitination, which can result in p62‐mediated substrate targeting to autophagy (Sanchez‐Martin et al, 2015). Since laforin preferentially binds glycogen molecules with longer chains (see above), it is conceivable that malin could ubiquitinate laforin through K63 ubiquitination and target laforin‐bound “at‐risk” glycogen molecules to autophagy.
As our results show that phosphorylation is not causative of glycogen precipitation and accumulation, what, if any, are the functions of glycogen phosphates? In starch metabolism, one established function of monophosphate esters of amylopectin is, through their high charges and hydrophilia, to separate amylopectin chains from each other, rendering them fully hydrated, and thus accessible to degrading enzymes (Edner et al, 2007; Hejazi et al, 2008; Kötting et al, 2009; Zeeman et al, 2010). As glycogen and amylopectin have a shared chemical basis (both consist of branched alpha‐glucan chains), effects of covalent modifications, such as phosphorylation, are expected to be similar. Phosphorylation should therefore enhance glycogen solubility and may represent a third mechanism to protect against precipitation of long‐chained “at‐risk” molecules. It is well established that neither starch‐degrading exo‐amylases nor glycogen phosphorylase can proceed beyond a phosphorylated glucosyl residue (Lomako et al, 1994; Kötting et al, 2009; Cenci et al, 2014; Emanuelle et al, 2016). Laforin's phosphatase role may be, similar to starch phosphatases (Gentry et al, 2007; Kötting et al, 2009), to remove the phosphates to allow reshortening of chains in “at‐risk” molecules and during glycogen digestion. Consistent with this is the recent demonstration that glycogen dephosphorylation by laforin occurs principally during glycogen degradation (Irimia et al, 2015). In the absence of laforin's phosphatase activity, cycles of partial glycogen digestion and resynthesis would lead to the time‐dependent phosphate accumulation that characterizes LD glycogen.
In summary, our results combine with recent work (Gayarre et al, 2014) to suggest that laforin's principle LD relevant function is mediated through malin and directed to preventing glycogen molecules with hyperextended chains. In the absence of either protein, some glycogen molecules at a time precipitate and gradually over time aggregate and amass into LBs, which, reaching a certain threshold profusion (at ~14 years of age in humans), initiate, and then drive the progressive myoclonus epilepsy.
Materials and Methods
Mouse lines, tissue collection, and histological stain for LBs
Tissues from Epm2a −/−.C265SLaf mice and their controls (see Results) were the same as utilized in Gayarre et al (2014; C57BL/6J). These mice were 9–12 months old and of both sexes. Epm2a −/−.C266SLaf and Epm2b −/−.C266SLaf mice and controls (see Results) were obtained following the crossing of both Epm2a −/− (Ganesh et al, 2002; C57BL/6J) and Epm2b −/− (Turnbull et al, 2010; C57BL/6J) mice with the C266SLaf mice described previously (Chan et al, 2004; 129SvJ). Mice were housed with environmental enrichment in ventilated cages at 20–22°C, fed a high‐quality, commercially available diet, water being automatically delivered. Brains of male and female mice were harvested after cervical dislocation of at least six 9‐ to 12‐month‐old mice from each genotype, split along the sagittal plane, one hemisphere immediately frozen in liquid nitrogen, the other fixed in 10% [w/v] formalin. The latter was embedded in paraffin, sectioned and stained using the periodic acid–Schiff diastase method (PASD; Chan et al, 2004). All animal procedures were approved by The Centre for Phenogenomics Animal Care Committee and are in compliance with the CCAC Guidelines and the OMAFRA Animals for Research Act.
Glycogen determination
Glycogen content was determined essentially as described previously (Suzuki et al, 2001). Briefly, frozen tissue was ground in liquid nitrogen, boiled in 30% [w/v] KOH, and precipitated thrice in 67% [v/v] ethanol and 15 mM LiCl (at least 1 h at −30°C). After redissolving in water, aliquots were digested with amyloglucosidase (Megazyme) in 80 mM sodium acetate buffer pH 4.5. Liberated glucose was determined enzymatically (Lowry & Passonneau, 1972).
Glycogen purification from muscle for phosphate determination
Quantification of total glycogen‐bound phosphate requires high glycogen purity, because its relative amounts are very low and non‐glycogen derived phosphoesters, for example, in nucleic acids, can severely affect the determination. Extracted muscle glycogen was thus further purified as previously published (Tagliabracci et al, 2007). Subsequently, glycogen was repeatedly precipitated in 67% ethanol and 15 mM LiCl (at least 1 h at −30°C) until the absence of nucleic acids could be confirmed by UV–vis spectra of glycogen solutions at concentrations of ~100 mM glucose equivalents.
Total glycogen phosphate quantification in muscle glycogen
Total glycogen‐bound phosphate was quantified as orthophosphate following enzymatic hydrolysis of all phospho‐monoesters. To facilitate complete orthophosphate release, highly purified muscle glycogen was incubated with phosphatase together with two glycogen hydrolysing enzymes. In a final volume of 65 μl, the reaction mixture contained 100 or 200 μg glycogen, 2.5 U Antarctic phosphatase (NEB), 0.1 U α‐amylase (Roche), 2 U amyloglucosidase (Megazyme), 50 mM bis–Tris–propane pH 6.0, 1 mM MgCl2, and 0.1 mM ZnCl2. Following overnight incubation at 37°C, 40 μl of this reaction mixture, and orthophosphate standards for calibration, was used to quantify ortho‐phosphate enzymatically as described previously (Nelson & Kaufman, 1987). Following the addition of equal volume of a buffer pH 7 (100 mM imidazole–HCl, 10 mM MgCl2, 5 mM EDTA), in a total volume of 80 μl, orthophosphate, via G1P, is converted to G6P using 0.02 mg rabbit phosphorylase a (Sigma), 67 μg purified rabbit muscle glycogen, and 1 U phosphoglucomutase (Sigma). Fluorescence (ex340/em470) was read until stable, first, after the addition of 150 μl 20 mM Tris pH 8, 60 μM NADP, and again, after the addition of 0.4 U G6PDH (Roche) in 5 μl 20 mM Tris pH 8. The G6PDH‐dependent increase in fluorescence was used to calculate the orthophosphate amounts in samples originally containing 100 or 200 μg glycogen. Glucose‐based phosphate amounts of the latter two measurements for each sample were averaged and deviated < 5% indicating that the enzymatic orthophosphate release was complete.
Glycogen purification from brain
Initial attempts to quantify C6 phosphate in brain glycogen were unsuccessful when following an isolation procedure similar to that used for muscle glycogen. Unexpected high values of apparent glucosyl C6 phosphate residues in hydrolysates of brain glycogen indicated the presence of a contaminant that greatly affected the measurements. The ratio of contaminant to glycogen was much higher in samples with low glycogen content, such as WT, suggesting that the contaminant is unrelated to glycogen but derives from other constituents of brain (whose glycogen content appears to be low as compared to other organs). Further experiments confirmed that the contaminant is very likely glucose 6‐phosphate released during acid hydrolysis from a glycan‐containing compound other than glycogen. This contamination is relevant only for tissues with low glycogen content, such as brain. Systematic experiments were conducted to remove the contaminant while retaining the little glycogen using spare WT mouse brains. After the extraction of glycogen from 100 to 400 mg ground frozen brain tissue in 30% KOH and three consecutive precipitations in 67% ethanol and 15 mM LiCl (see extraction of muscle glycogen for glycogen determination), the following procedure was established for all brain glycogen samples analysed in this study. The resulting pellet, containing glycogen, most likely proteins, and the contaminant, was resuspended in 100 μl water and subsequently extracted with 900 μl 2:1 methanol:chloroform [v/v] by vigorous mixing, incubation for 25 min at room temperature (RT) on a rotator, and 10 min at 85°C. After cooling to RT, the suspension was centrifuged for 20 min at 20°C and 16,000 × g, the supernatant discarded and the pellet dried briefly at 85°C. The extraction procedure was repeated twice, followed by resuspending the dry pellet in 300 μl water. After 20‐min incubation at 95°C with intermittent agitation, the solubilized glycogen was recovered in the supernatant (S1) obtained by centrifugation at RT (16,000 × g for 20 min). The pellet was extracted with 100 μl 0.5 M KOH at 95°C for 45 min with intermittent agitation. Subsequently, the extract, cooled to RT, was centrifuged (20 min, RT, 16,000 × g), and the resultant supernatant (S2) was combined with the solubilized glycogen (S1). The combined supernatants (S1 + S2) were subjected to several rounds of repeated ethanol precipitation (1,400 μl 67% ethanol, 15 mM LiCl, 70 min at −30°C), 20 min centrifugation at 4°C and 16,000 × g, carefully discarding the supernatant, and resolubilization of the pellet in water followed by 10‐min heat treatment (95°C, intermittent agitation). The ethanolic supernatant harbours the contaminant, the amount of which tends to be smaller with each round of precipitation. Glycogen pellets were considered to be contaminant free when the apparent G6P amount in the last ethanolic supernatant was below the detection limit. Using the enzymatic cycling method, the contaminant (apparent G6P) was quantified after drying the ethanolic supernatants (SpeedVac), dissolving the dry contaminant pellets in water, and subsequent acid hydrolysis (see below). Glycogen concentration was determined after dissolving the purified glycogen pellet as described above. As the number of sequential precipitations was adjusted to the persistence of the contaminant in the ethanolic supernatant, glycogen recovery varied but in most cases was between 50 and 80%.
Glycogen carbon C6 phosphate determination
Aliquots of purified muscle or brain glycogen preparations (see above) were subjected to acid hydrolysis (3 h, 95°C, 0.7 M HCl) and subsequently neutralized using 5 M KOH. G6P was determined alongside G6P standards using the enzymatic cycling assay previously described (Nitschke et al, 2013). G6P amounts were based on glucose amounts that were liberated during acid hydrolysis and determined enzymatically (Lowry & Passonneau, 1972).
Determination of glycogen chain length distributions
Purified muscle or brain glycogen (10–20 μg dry weight) was completely debranched by overnight incubation with 200 U isoamylase (Sigma) in 110 μl 10 mM sodium acetate at pH 5 and 37°C. Following heat inactivation (10 min at 95°C) and centrifugation (10 min at 20,000 × g), 90 μl of the supernatant was applied to high‐performance anion exchange chromatography with pulsed amperometric detection (HPAEC‐PAD; ThermoFisher, ICS5000). Separation of the oligoglucan chains was achieved on a CarboPac PA100 column and guard combination (ThermoFisher) at a constant flow rate of 1 ml/min with an elution profile of the two eluents A (150 mM NaOH) and B (150 mM NaOH, 500 mM sodium acetate) as follows: injection after 15 min 95% [v/v] A, 5% [v/v] B equilibration; 5 min 95% A, 5% B; 9 min linear gradient to 70% A, 30% B; 9 min linear gradient to 55% A, 45% B; 37 min linear gradient to 33% A, 67% B; 10 min 100% B. Chromatograms were analysed with Dionex Chromeleon software (version 7.2). For each chain length (degree of polymerization), relative peak areas were determined, which were then averaged among at least five biological replicates allowing the calculation of standard deviations (SD). Note that purified glycogen from all available brain samples of the genotype Epm2a −/−.Laf was pooled to obtain sufficiently large amounts of glycogen for the chain length distribution analysis. SD at each chain length as a measure of biological variance for this genotype was estimated by multiplying the average SD of the other genotypes (WT, Epm2a −/−, Epm2a −/−.C265SLaf) by two. As variances in relative peak areas between those genotypes are mostly equal, the estimated biological variance should include 95% of samples of the Epm2a −/−.Laf genotype.
Analysis of autophagy markers
For the detection of LC3 in brain tissue, a membrane fraction enriched in autophagosomes was prepared as described previously (Chu et al, 2009), and for p62 after extracting soluble protein as in Wang et al (2007); 15 μg total protein was separated on 14% SDS–PAGE using a Laemmli buffer system. Proteins of interest were detected after transfer to PVDF membranes using primary antibodies NB100‐2220 (LC3, Novus Biologicals), P0067 (p62, Sigma), sc‐25778, and sc‐11397 (GAPDH and calnexin, respectively, Santa Cruz), all at a 1:1,000 dilution.
Statistical analyses
Except where stated, all biochemical analyses were conducted in at least five biological replicates. Standard deviations (SD) or standard error of mean (SEM) was calculated as indicated. Averages of multiple groups of biological replicates were compared using one‐way analysis of variance (ANOVA) followed by post hoc analysis using homoscedastic Student's t‐ or heteroscedastic Welch's tests, both unpaired, two‐tailed, and with Holm–Bonferroni correction. The adequate post hoc test was selected based on equal or unequal variances of groups tested with F‐tests. Significance levels are indicated by asterisks (*P < 0.05, **P < 0.01, ***P < 0.001). For glycogen chain length distribution analyses, significance between genotypes was tested for each individual degree of polymerization.
Author contributions
BAM, FN, and MS conceived the study in collaboration with SRC. Experiments were conducted by FN, PW, and XZ with help from AMP, LI, LJL, and PB. Data were analysed by FN, EEC, and PW. The manuscript was written by FN, BAM, and MS with critical review from MAS, EEC, and SRC.
Conflict of interest
The authors declare that they have no conflict of interest.
The paper explained.
Problem
The fatal childhood‐onset epilepsy Lafora disease results from neuronal carbohydrate accumulations (Lafora bodies). Recent partly antagonistic hypotheses suggest that these accumulations originate from glycogen hyperphosphorylation, occurring when the glycogen phosphatase laforin is insufficiently functioning, or through deficiency in general autophagy.
Results
Through methodological advances, we are now able to measure brain glycogen structural parameters and show that glycogen chain lengths, not hyperphosphorylation nor deficiency in general autophagy, are critical for the formation of Lafora bodies. The Lafora disease proteins laforin and malin cooperate to regulate glycogen chain length and prevent glycogen malformation and precipitation into neurotoxic Lafora bodies.
Impact
The findings in this paper provide further insight into basic principles of glycogen metabolism, especially regarding the role of glycogen phosphate and its chain length distribution. Moreover, current hypotheses on the pathogenesis of Lafora disease are corrected while a view is presented that integrates previously contradicting results, sharpening the understanding of this horrible disease.
Supporting information
Appendix
Review Process File
Source Data for Figure 2
Acknowledgements
This work was supported by families and friends of the Chelsea's Hope Lafora Disease Research Fund, Associazione Italiana Lafora (AILA), France‐Lafora, the Milana and Tatjana Gajic Lafora Disease Foundation, Genome Canada, the Ontario Brain Institute (OBI) and The National Institute of Neurological Disorders and Stroke of the National Institutes of Health (NIH) under award number P01 NS097197. Mitchell A. Sullivan was supported by an NHMRC CJ Martin Fellowship (GNT1092451). BA Minassian holds the University of Toronto Michael Bahen Chair in Epilepsy Research and the University of Texas Southwestern UTSW Jimmy Elizabeth Westcott Distinguished Chair in Pediatric Neurology. We wish to thank Drs. Gino Cingolani (Thomas Jefferson University, Philadelphia, USA), and Michael Emes and Ian Tetlow (University of Guelph, Canada) for insightful discussions and the latter two for facilitating the use of the HPAEC‐PAD equipment.
EMBO Mol Med (2017) 9: 906–917
References
- Aguado C, Sarkar S, Korolchuk VI, Criado O, Vernia S, Boya P, Sanz P, de Cordoba SR, Knecht E, Rubinsztein DC (2010) Laforin, the most common protein mutated in Lafora disease, regulates autophagy. Hum Mol Genet 19: 2867–2876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cenci U, Nitschke F, Steup M, Minassian BA, Colleoni C, Ball SG (2014) Transition from glycogen to starch metabolism in Archaeplastida . Trends Plant Sci 19: 18–28 [DOI] [PubMed] [Google Scholar]
- Chan EM, Bulman DE, Paterson AD, Turnbull J, Andermann E, Andermann F, Rouleau GA, Delgado‐Escueta AV, Scherer SW, Minassian BA (2003) Genetic mapping of a new Lafora progressive myoclonus epilepsy locus (EPM2B) on 6p22. J Med Genet 40: 671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EM, Ackerley CA, Lohi H, Ianzano L, Cortez MA, Shannon P, Scherer SW, Minassian BA (2004) Laforin preferentially binds the neurotoxic starch‐like polyglucosans, which form in its absence in progressive myoclonus epilepsy. Hum Mol Genet 13: 1117–1129 [DOI] [PubMed] [Google Scholar]
- Chu CT, Plowey ED, Dagda RK, Hickey RW, Cherra IS, Clark RSB (2009) Chapter 11 autophagy in neurite injury and neurodegeneration: in vitro and in vivo models In Methods in Enzymol, Klionsky DJ. (ed.), Vol. 453, pp 217–249. San Diego, CA: Academic Press; [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras CJ, Segvich DM, Mahalingan K, Chikwana VM, Kirley TL, Hurley TD, DePaoli‐Roach AA, Roach PJ (2016) Incorporation of phosphate into glycogen by glycogen synthase. Arch Biochem Biophys 597: 21–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criado O, Aguado C, Gayarre J, Duran‐Trio L, Garcia‐Cabrero AM, Vernia S, San Millan B, Heredia M, Roma‐Mateo C, Mouron S et al (2012) Lafora bodies and neurological defects in malin‐deficient mice correlate with impaired autophagy. Hum Mol Genet 21: 1521–1533 [DOI] [PubMed] [Google Scholar]
- DePaoli‐Roach AA, Tagliabracci VS, Segvich DM, Meyer CM, Irimia JM, Roach PJ (2010) Genetic depletion of the malin E3 ubiquitin ligase in mice leads to lafora bodies and the accumulation of insoluble laforin. J Biol Chem 285: 25372–25381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePaoli‐Roach AA, Contreras CJ, Segvich DM, Heiss C, Ishihara M, Azadi P, Roach PJ (2015) Glycogen phosphomonoester distribution in mouse models of the progressive myoclonic epilepsy, Lafora disease. J Biol Chem 290: 841–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias DM, Furtado J, Wasielewski E, Cruz R, Costello B, Cole L, Faria TQ, Baaske P, Brito RMM, Ciulli A et al (2016) Biophysical characterization of laforin‐carbohydrate interaction. Biochem J 473: 335–345 [DOI] [PubMed] [Google Scholar]
- Duran J, Gruart A, Garcia‐Rocha M, Delgado‐Garcia JM, Guinovart JJ (2014) Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum Mol Genet 23: 3147–3156 [DOI] [PubMed] [Google Scholar]
- Edner C, Li J, Albrecht T, Mahlow S, Hejazi M, Hussain H, Kaplan F, Guy C, Smith SM, Steup M et al (2007) Glucan, water dikinase activity stimulates breakdown of starch granules by plastidial beta‐amylases. Plant Physiol 145: 17–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelle S, Brewer MK, Meekins DA, Gentry MS (2016) Unique carbohydrate binding platforms employed by the glucan phosphatases. Cell Mol Life Sci 73: 2765–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesh S, Delgado‐Escueta AV, Sakamoto T, Avila MR, Machado‐Salas J, Hoshii Y, Akagi T, Gomi H, Suzuki T, Amano K et al (2002) Targeted disruption of the Epm2a gene causes formation of Lafora inclusion bodies, neurodegeneration, ataxia, myoclonus epilepsy and impaired behavioral response in mice. Hum Mol Genet 11: 1251–1262 [DOI] [PubMed] [Google Scholar]
- Gayarre J, Duran‐Trio L, Criado Garcia O, Aguado C, Juana‐Lopez L, Crespo I, Knecht E, Bovolenta P, Rodriguez de Cordoba S (2014) The phosphatase activity of laforin is dispensable to rescue Epm2a‐/‐ mice from Lafora disease. Brain 137: 806–818 [DOI] [PubMed] [Google Scholar]
- Gentry MS, Worby CA, Dixon JE (2005) Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc Natl Acad Sci USA 102: 8501–8506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry MS, Dowen RH, Worby CA, Mattoo S, Ecker JR, Dixon JE (2007) The phosphatase laforin crosses evolutionary boundaries and links carbohydrate metabolism to neuronal disease. J Cell Biol 178: 477–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gidley MJ, Bulpin PV (1987) Crystallisation of malto‐oligosaccharides as models of the crystalline forms of starch: minimum chain‐length requirement for the formation of double helices. Carbohydr Res 161: 291–300 [Google Scholar]
- Hejazi M, Fettke J, Haebel S, Edner C, Paris O, Frohberg C, Steup M, Ritte G (2008) Glucan, water dikinase phosphorylates crystalline maltodextrins and thereby initiates solubilization. Plant J 55: 323–334 [DOI] [PubMed] [Google Scholar]
- Hejazi M, Fettke J, Paris O, Steup M (2009) The two plastidial starch‐related dikinases sequentially phosphorylate glucosyl residues at the surface of both the A‐ and B‐allomorph of crystallized maltodextrins but the mode of action differs. Plant Physiol 150: 962–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia JM, Tagliabracci VS, Meyer CM, Segvich DM, DePaoli‐Roach AA, Roach PJ (2015) Muscle glycogen remodeling and glycogen phosphate metabolism following exhaustive exercise of wild type and laforin knockout mice. J Biol Chem 290: 22686–22698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kötting O, Santelia D, Edner C, Eicke S, Marthaler T, Gentry MS, Comparot‐Moss S, Chen J, Smith AM, Steup M et al (2009) STARCH‐EXCESS4 is a laforin‐like Phosphoglucan phosphatase required for starch degradation in Arabidopsis thaliana . Plant Cell 21: 334–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafora GR, Glueck B (1911) Beitrag zur histopathologie der myoklonischen epilepsie. Z Gesamte Neurol Psychiatr 6: 1–14 [Google Scholar]
- Lohi H, Ianzano L, Zhao X‐C, Chan EM, Turnbull J, Scherer SW, Ackerley CA, Minassian BA (2005) Novel glycogen synthase kinase 3 and ubiquitination pathways in progressive myoclonus epilepsy. Hum Mol Genet 14: 2727–2736 [DOI] [PubMed] [Google Scholar]
- Lomako J, Lomako WM, Kirkman BR, Whelan WJ (1994) The role of phosphate in muscle glycogen. BioFactors 4: 167–171 [PubMed] [Google Scholar]
- Lowry OH, Passonneau JV. (eds) (1972) A flexible system of enzymatic analysis. New York: Academic Press; [Google Scholar]
- Marchand I, Chorneyko K, Tarnopolsky M, Hamilton S, Shearer J, Potvin J, Graham TE (2002) Quantification of subcellular glycogen in resting human muscle: granule size, number, and location. J Appl Physiol 93: 1598–1607 [DOI] [PubMed] [Google Scholar]
- Minassian BA, Lee JR, Herbrick JA, Huizenga J, Soder S, Mungall AJ, Dunham I, Gardner R, Fong CY, Carpenter S et al (1998) Mutations in a gene encoding a novel protein tyrosine phosphatase cause progressive myoclonus epilepsy. Nat Genet 20: 171–174 [DOI] [PubMed] [Google Scholar]
- Minassian BA (2001) Lafora's disease: towards a clinical, pathologic, and molecular synthesis. Pediatr Neurol 25: 21–29 [DOI] [PubMed] [Google Scholar]
- Nelson TJ, Kaufman S (1987) Two enzymatic methods for determination of the phosphate content of phosphoproteins. Anal Biochem 161: 352–357 [DOI] [PubMed] [Google Scholar]
- Nitschke F, Wang P, Schmieder P, Girard J‐M, Awrey DE, Wang T, Israelian J, Zhao X, Turnbull J, Heydenreich M et al (2013) Hyperphosphorylation of glucosyl C6 carbons and altered structure of glycogen in the neurodegenerative epilepsy Lafora disease. Cell Metab 17: 756–767 [DOI] [PubMed] [Google Scholar]
- Palmer TN, Macaskie LE, Grewal KK (1983) Spatial distribution of unit chains in glycogen. Carbohydr Res 115: 139–150 [Google Scholar]
- Raththagala M, Brewer MK, Parker MW, Sherwood AR, Wong BK, Hsu S, Bridges TM, Paasch BC, Hellman LM, Husodo S et al (2015) Structural mechanism of laforin function in glycogen dephosphorylation and Lafora disease. Mol Cell 57: 261–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach PJ, Depaoli‐Roach AA, Hurley TD, Tagliabracci VS (2012) Glycogen and its metabolism: some new developments and old themes. Biochem J 441: 763–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roach PJ (2015) Glycogen phosphorylation and Lafora disease. Mol Aspects Med 46: 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio‐Villena C, Garcia‐Gimeno MA, Sanz P (2013) Glycogenic activity of R6, a protein phosphatase 1 regulatory subunit, is modulated by the laforin‐malin complex. Int J Biochem Cell Biol 45: 1479–1488 [DOI] [PubMed] [Google Scholar]
- Sakai M, Austin J, Witmer F, Trueb L (1970) Studies in myoclonus epilepsy (Lafora body form): II. Polyglucosans in the systemic deposits of myoclonus epilepsy and in corpora amylacea. Neurology 20: 160 [DOI] [PubMed] [Google Scholar]
- Sanchez‐Martin P, Roma‐Mateo C, Viana R, Sanz P (2015) Ubiquitin conjugating enzyme E2‐N and sequestosome‐1 (p62) are components of the ubiquitination process mediated by the malin‐laforin E3‐ubiquitin ligase complex. Int J Biochem Cell Biol 69: 204–214 [DOI] [PubMed] [Google Scholar]
- Sankhala RS, Koksal AC, Ho L, Nitschke F, Minassian BA, Cingolani G (2015) Dimeric quaternary structure of human laforin. J Biol Chem 290: 4552–4559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y, Lanner C, Kim J‐H, Vilardo PG, Zhang H, Yang J, Cooper LD, Steele M, Kennedy A, Bock CB et al (2001) Insulin control of glycogen metabolism in knockout mice lacking the muscle‐specific protein phosphatase PP1G/R(GL). Mol Cell Biol 21: 2683–2694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Turnbull J, Wang W, Girard JM, Zhao X, Skurat AV, Delgado‐Escueta AV, Minassian BA, Depaoli‐Roach AA, Roach PJ (2007) Laforin is a glycogen phosphatase, deficiency of which leads to elevated phosphorylation of glycogen in vivo . Proc Natl Acad Sci USA 104: 19262–19266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Girard JM, Segvich D, Meyer C, Turnbull J, Zhao X, Minassian BA, DePaoli‐Roach AA, Roach PJ (2008) Abnormal metabolism of glycogen phosphate as a cause for Lafora disease. J Biol Chem 283: 33816–33825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagliabracci VS, Heiss C, Karthik C, Contreras CJ, Glushka J, Ishihara M, Azadi P, Hurley TD, Depaoli‐Roach AA, Roach PJ (2011) Phosphate incorporation during glycogen synthesis and Lafora disease. Cell Metab 13: 274–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull J, Wang P, Girard JM, Ruggieri A, Wang TJ, Draginov AG, Kameka AP, Pencea N, Zhao X, Ackerley CA et al (2010) Glycogen hyperphosphorylation underlies lafora body formation. Ann Neurol 68: 925–933 [DOI] [PubMed] [Google Scholar]
- Turnbull J, Tiberia E, Striano P, Genton P, Carpenter S, Ackerley CA, Minassian BA (2016) Lafora disease. Epileptic Disord 18: 38–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilchez D, Ros S, Cifuentes D, Pujadas L, Valles J, Garcia‐Fojeda B, Criado‐Garcia O, Fernandez‐Sanchez E, Medrano‐Fernandez I, Dominguez J et al (2007) Mechanism suppressing glycogen synthesis in neurons and its demise in progressive myoclonus epilepsy. Nat Neurosci 10: 1407–1413 [DOI] [PubMed] [Google Scholar]
- Wang W, Lohi H, Skurat AV, DePaoli‐Roach AA, Minassian BA, Roach PJ (2007) Glycogen metabolism in tissues from a mouse model of Lafora disease. Arch Biochem Biophys 457: 264–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Israelian L, Xue Y, Song S, Attisano L, Minassian B (2016) SGK1 (glucose transport), dishevelled2 (wnt signaling), LC3/p62 (autophagy) and p53 (apoptosis) proteins are unaltered in Lafora disease. All Res J Biol 7: 28–33 [PMC free article] [PubMed] [Google Scholar]
- Wanson J‐C, Drochmans P (1968) Rabbit skeletal muscle glycogen: a morphological and biochemical study of glycogen β‐particles isolated by the precipitation‐centrifugation method. J Cell Biol 38: 130–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby CA, Gentry MS, Dixon JE (2006) Laforin, a dual specificity phosphatase that dephosphorylates complex carbohydrates. J Biol Chem 281: 30412–30418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worby CA, Gentry MS, Dixon JE (2008) Malin decreases glycogen accumulation by promoting the degradation of protein targeting to glycogen (PTG). J Biol Chem 283: 4069–4076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeman SC, Kossmann J, Smith AM (2010) Starch: its metabolism, evolution, and biotechnological modification in plants. Annu Rev Plant Biol 61: 209–234 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File
Source Data for Figure 2