

Abstract
The impact of the mitochondrial permeability transition (MPT) on cellular physiology is well characterized. In contrast, the composition and mode of action of the permeability transition pore complex (PTPC), the supramolecular entity that initiates MPT, remain to be elucidated. Specifically, the precise contribution of the mitochondrial F1 FO ATP synthase (or subunits thereof) to MPT is a matter of debate. We demonstrate that F1 FO ATP synthase dimers dissociate as the PTPC opens upon MPT induction. Stabilizing F1 FO ATP synthase dimers by genetic approaches inhibits PTPC opening and MPT. Specific mutations in the F1 FO ATP synthase c subunit that alter C‐ring conformation sensitize cells to MPT induction, which can be reverted by stabilizing F1 FO ATP synthase dimers. Destabilizing F1 FO ATP synthase dimers fails to trigger PTPC opening in the presence of mutants of the c subunit that inhibit MPT. The current study does not provide direct evidence that the C‐ring is the long‐sought pore‐forming subunit of the PTPC, but reveals that PTPC opening requires the dissociation of F1 FO ATP synthase dimers and involves the C‐ring.
Keywords: ATP synthasome, ATP5G1, cyclosporine A, CYPD, regulated necrosis
Subject Categories: Autophagy & Cell Death, Membrane & Intracellular Transport
Introduction
The term “mitochondrial permeability transition” refers to a sudden and irreversible increase in the permeability of the inner mitochondrial membrane (IMM) to small solutes, leading to mitochondrial transmembrane potential (Δψm) dissipation, ATP synthesis arrest, unregulated entry of water into the mitochondrial matrix, and osmotic breakdown of the organelle 1. Thus, the MPT ultimately initiates a form of regulated cell death (RCD) that often manifests with necrotic morphological features 1, 2, 3.
The MPT can be triggered by the accumulation of Ca2+ ions in the cytosol or by intense oxidative stress, two conditions that characterize acute ischemic episodes 4, 5. Several attempts have been launched to pharmacologically inhibit MPT for neuro‐, cardio‐, or nephroprotection 6. However, results have been dismal, at least in part owing to our limited knowledge on the structure and mode of action of the supramolecular entity that induces MPT, the “permeability transition pore complex” (PTPC) 4.
One of the most debated points about the PTPC is the identity of its pore‐forming subunit. Various candidates have been ruled out by relatively robust genetic approaches 7, 8. Moreover, while peptidylprolyl isomerase F (PPIF; best known as CYPD) mediates a non‐redundant function in MPT 9, 10, there is a general consensus around the notion that CYPD regulates the PTPC but does not constitute its pore‐forming component 4.
Lately, the mitochondrial F1FO ATP synthase has attracted considerable attention as a potential pore‐forming component of the PTPC, for multiple reasons. First, it interacts with previously identified (yet dispensable) PTPc subunits including adenine nucleotide translocase (ANT) isoforms and solute carrier family 25 member 3 (SLC25A3, best known as PiC) in the context of the so‐called “ATP synthasome” 11. Second, multiple pharmacological regulators of the PTPC are substrates, products, or biochemical correlates of the activity of the ATP synthasome (e.g. ATP, ADP, inorganic phosphate, matrix pH) 1. Third, CYPD physically and functionally interacts with the peripheral stalk of the F1FO ATP synthase 12.
Two partially reconcilable models have been put forward to explain the contribution of the F1FO ATP synthase to MPT in molecular terms. On the one hand, it has been proposed that the F1FO ATP synthase would dimerize in the course of MPT and that such dimers would allow for the generation of a poorly selective channel with PTPC‐like activity 13. On the other hand, it has been suggested not only that the c subunit of the FO complex has a key role in the regulation of the MPT, but also that C‐rings (homo‐oligomeric, preassembled IMM channels composed of c subunits) de facto constitute the long‐sought PTPC pore‐forming subunit 14, 15, 16.
Both these models can be challenged. On the one hand, F1FO ATP synthase dimers exhibit no obvious pore‐forming features, dimerization has been reproducibly associated with improved bioenergetic metabolism and cell survival (rather than cell death) 17, and ρ0 cells (in which the F1FO ATP synthase is mostly monomeric) respond normally to MPT induction 18. On the other hand, it seems unlikely for C‐rings to lose their lipid plugs in relatively physiological conditions 19, and so far, a direct interaction between C‐rings and CYPD has not been identified. Thus, additional investigation is required to obtain precise structural and functional insights into the molecular machinery that mediate MPT.
The data presented here suggest that MPT involves the dissociation (not the association) of F1FO ATP synthase dimers in combination with the specific participation of C‐rings.
Results and Discussion
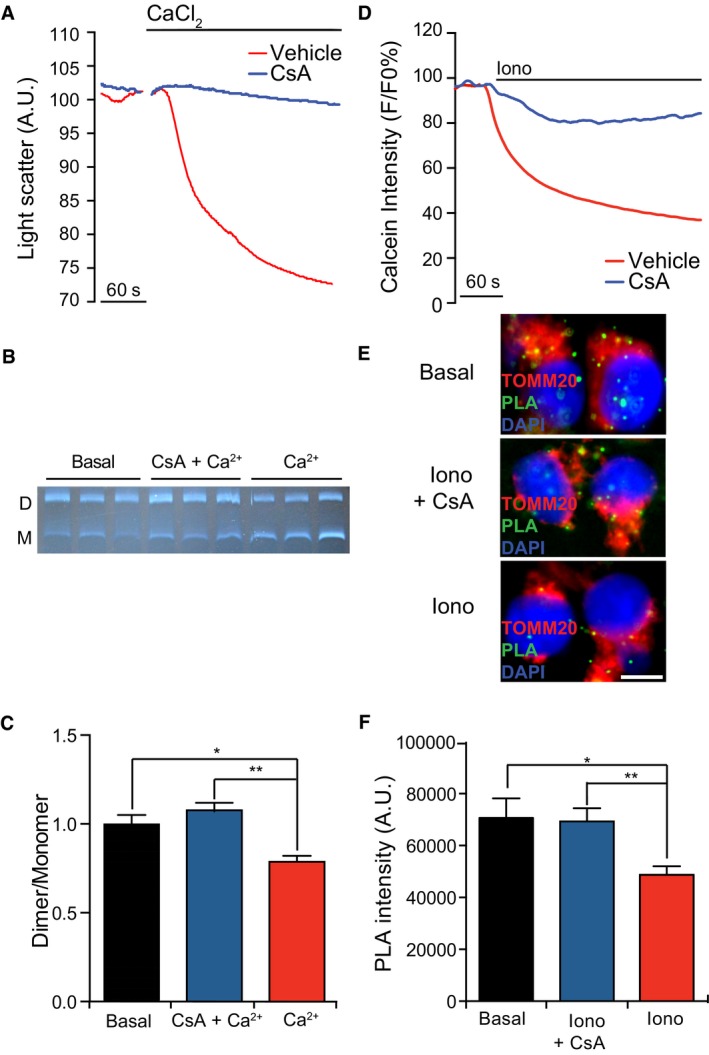
Experimental conditions that promote F1FO ATP synthase dimerization are often associated with a reduced propensity for PTPC opening and consequent MPT 17, 18. To obtain further insights into this issue, we purified rat liver mitochondria, placed them in energization buffer, and exposed them to control conditions or CsA prior to triggering MPT with 100 μM Ca2+. In setting, MPT‐associated mitochondrial swelling was monitored as a decrease in light diffraction of the preparation (Fig 1A). Mitochondria were then assessed for the relative abundance of F1FO ATP synthase dimers by blue‐native (BN) electrophoresis and in‐gel activity assay (Fig 1B). MPT induction by Ca2+ ions correlated with a partial accumulation of F1FO ATP synthase monomers over dimers, and this could be completely prevented by CsA (Fig 1A–C). As positive and negative controls for F1FO ATP synthase dimerization, we used rat heart mitochondria treated with digitonin (DIG) or n‐dodecyl β‐d‐maltoside (DM), respectively (Fig EV1A), as per established protocols 20, 21.
Figure 1. MPT is associated with dissociation of F1FO ATP synthase dimers.
-
A–CRepresentative traces of mouse liver mitochondria undergoing MPT in response to 100 μM CaCl2 alone or in the presence of 1 μM cyclosporine A (CsA) (A). Representative in‐gel activity assay after blue‐native PAGE (B) and quantification (C) of F1FO ATP synthase dimers (D) and monomers (M) in mouse liver mitochondria in basal conditions or after MPT induction as in panel (F). *P = 0.0001, **P = 0.0011 (ANOVA plus unpaired Student's t‐test). Results are representative of five independent experiments.
-
D–FRepresentative calcein/Co2+ quenching traces in HEK293T cells treated with 1 μM ionomycin (Iono), alone or in the presence of 1.6 μM CsA (D). Representative images (E) and quantification (F; n = 3) of PLA assays for F1FO ATP synthase dimers in HEK293T cells maintained in basal conditions or driven in MPT as in panel (D). *P = 0.0106, **P = 0.0137 (ANOVA plus unpaired Student's t‐test). Results are representative of three independent experiments.
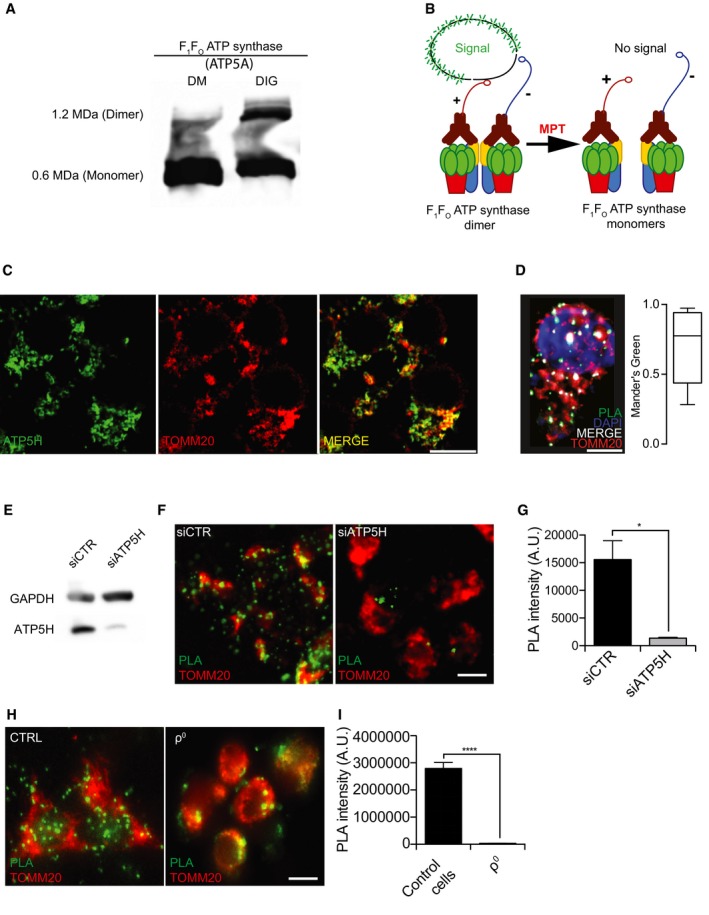
Figure EV1. Validation of F1FO ATP synthase dimerization status.
-
AHigh‐resolution image of F1FO ATP synthase dimers and monomers isolated from rat heart mitochondria (HM) by the use of n‐dodecyl β‐D‐maltoside (DM) or digitonin (DIG), upon separation by blue‐native PAGE and immunostaining for ATP5A.
-
BSchematic representation of PLA assay setting in physiological conditions (left) or in the course of MPT (right).
-
CImmunoreactivity of the ATP5H‐specific antibody used in the PLA assay as compared to an antibody specific for the mitochondrial marker TOMM20.
-
DMaximum intensity projection of z‐stack from a representative HEK293T cell stained for the (immuno)fluorescence microscopy‐based detection of DNA (DAPI, blue), mitochondria (TOMM20, red), and F1FO ATP synthase dimers (PLA, green). Quantification of the PLA signal colocalizing with TOMM20 is reported (whiskers: max and min; box: 10th and 90th percentile; line: median).
-
EAssessment of transfection efficiency in HEK293T cells transfected with a control siRNA (siCTR) or a siRNA targeting ATP5H (siATP5H) for 96 h. GAPDH levels were monitored to ensure equal lane loading.
-
F, GRepresentative images (F) and quantification (G) of PLA assays for F1FO ATP synthase dimers in HEK293T cells transfected with siCTR or siATP5H for 96 h. The results are representative of three independent experiments. *P = 0.0001 (unpaired Student's t‐test).
-
H, IRepresentative images (H) and quantification (I) of PLA assays for F1FO ATP synthase dimers in ρ0 cells and their wt counterpart HPA11. The results are representative of three independent experiments. ****P = 0.0001 (unpaired Student's t‐test).
Next, we set up a procedure to verify F1FO ATP synthase dimerization in cellula based on a dedicated proximity ligation assay (PLA) 22. In this setting, antibodies specific for ATP synthase, H+ transporting, mitochondrial Fo complex subunit D (ATP5H) were conjugated with either + or − PLA probes (Fig EV1B), and then used as a cocktail to stain HEK293T cells. This approach was expected to limit the analysis to transfected cells and hence avoid the underestimation expected by BN electrophoresis. HEK293T cells displayed a considerable amount of dotted staining colocalizing with the mitochondrial protein translocase of outer mitochondrial membrane 20 (TOMM20) (Fig EV1C and D). As a negative control, ATP5H was depleted by specific siRNAs, causing reduction in the PLA signal (Fig EV1E–G). Additionally, ρ0 cells displayed a reduced PLA signal as compared to their wild‐type (WT) counterparts (Fig EV1H and I). We concluded that our PLA assay was adequate to measure dimeric/oligomeric versus monomeric F1FO ATP synthase levels.
We next tested F1FO ATP synthase dimerization status in HEK293T cells responding to 1 μM ionomycin plus 1 mM Ca2+, alone or in the presence of CsA. Efficient and CsA‐sensitive MPT induction was verified by the calcein/Co2+ quenching assay (Fig 1D), whereas the F1FO ATP synthase dimerization status was conservatively evaluated by the PLA (Fig 1E). Irrespective of the underestimation of the relative abundance of F1FO ATP synthase dimers intrinsically associated with this approach (only heterologous proximity between + and − probes could be detected), MPT was accompanied by a significant decrease in PLA signal that did not occur in the presence of CsA (Fig 1F). These findings suggest that PTPC opening and consequent MPT correlate with the destabilization of F1FO ATP synthase dimers and that such a dissociation can be blocked by the pharmacological MPT inhibition.
Since CYPD binds to the F1FO ATP synthase in a CsA‐sensitive manner 12, we speculated that such an interaction provokes a structural rearrangement that destabilizes F1FO ATP synthase dimers. We reasoned that if PTPC opening involves the dissociation of F1FO ATP synthase dimers, artificially stabilizing dimers would inhibit MPT. We tested this prediction by transfecting HEK293T cells with a small‐interfering RNA (siRNA) against ATPase inhibitory factor 1 (ATPIF1), a subunit of the F1FO ATP synthase known to promote dimerization 23. Robust ATPIF1 depletion (> 95% reduction in ATPIF1 levels) sensitized HEK293T cells to PTPC opening by ionomycin plus Ca2+ (Fig 2A–C).
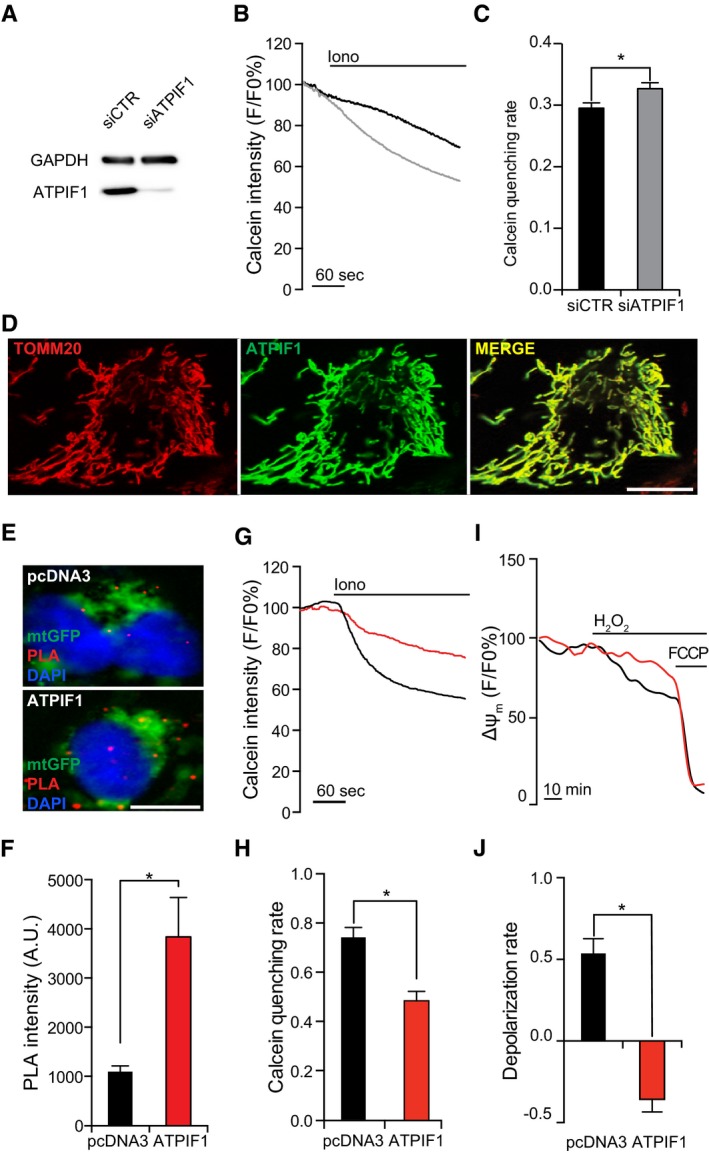
Figure 2. Stabilization of F1FO ATP synthase dimers by ATPIF1 inhibits MPT.
-
AAssessment of transfection efficiency in HEK293T cells transfected with a control siRNA (siCTR) or a siRNA targeting ATPIF1 (siATPIF1) for 96 h. GAPDH levels were monitored to ensure equal lane loading.
-
B, CRepresentative calcein/Co2+ quenching traces (B) and quenching rate quantification (C) in HEK293T cells transfected with siCTR or siATPIF1 for 96 h, and then treated with 1 μM ionomycin (Iono). Results are representative of three independent experiments. *P = 0.0135 (unpaired Student's t‐test).
-
DRepresentative colocalization of ATPIF1 with TOMM20 in HEK293T cells transfected with a plasmid for ATPIF1 overexpression.
-
E, FRepresentative images (E) and quantification (F) of PLA assays for F1FO ATP synthase dimers in HEK293T cells co‐transfected with a plasmid coding for mtGFP and with pcDNA3 or a plasmid for the overexpression of ATPIF1. Results are representative of five independent experiments. *P = 0.0001 (unpaired Student's t‐test).
-
G, HRepresentative calcein/Co2+ quenching traces (G) and quenching rate quantification (H) in HEK293T cells transfected as in (E, F) and treated with 1 μM Iono. Results are representative of five independent experiments. *P = 0.0001 (unpaired Student's t‐test).
-
I, JRepresentative mitochondrial transmembrane potential (Δψm) recordings (I) and depolarization rate quantification (J; n = 3) in HEK293T cells transiently as in (E, F) and exposed to 500 μM H2O2. 1 μM carbonyl cyanide p‐trifluoromethoxyphenylhydrazone (FCCP) was employed as control for depolarization. Results are representative of five independent experiments. *P = 0.0001 (unpaired Student's t‐test).
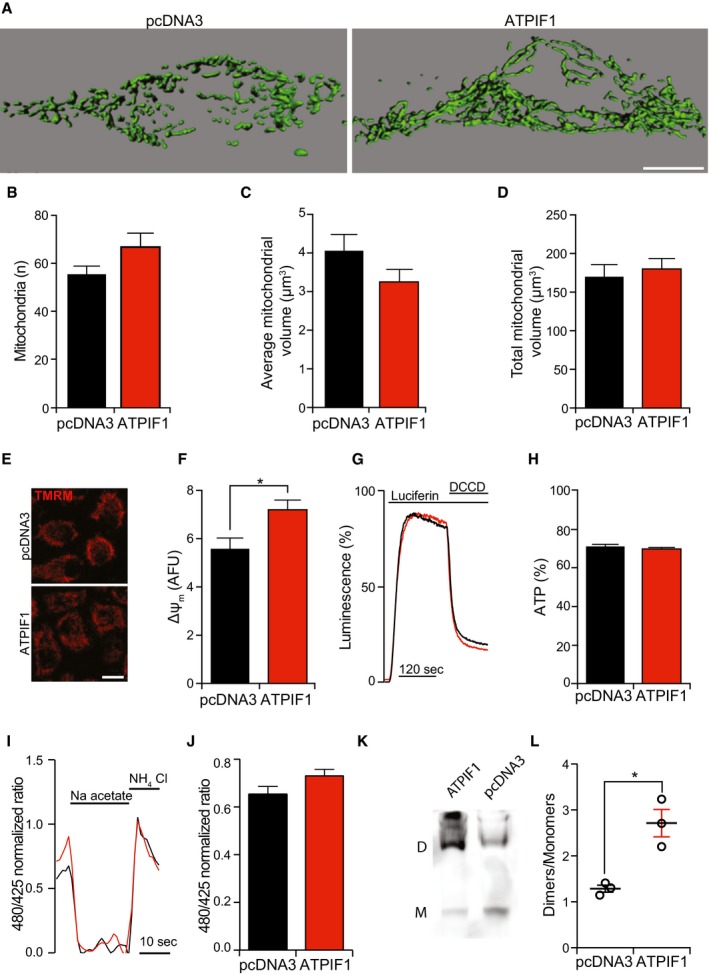
We next tested the consequences of ATPIF1 overexpression. Overexpressed ATPIF1 could be visualized by immunofluorescence microscopy and perfectly co‐localized with TOMM20 (Fig 2D). HEK293T cells overexpressing ATPIF1 did not exhibit morphological signs of mitochondrial deterioration 24, including fragmentation (Fig EV2A–C), total mass reduction (Fig EV2A and D), and depolarization (Fig EV2E and F) as compared to cells transfected with an empty vector. Co‐expressing ATPIF1 with a luciferase directed to the mitochondrial matrix demonstrated that ATPIF1 levels do not significantly affect mitochondrial ATP content, neither in basal conditions, nor upon inhibition of ATP synthesis with 75 μM N,N‐dicyclohexylcarbodiimide (DCCD) (Fig EV2G and H). Finally, the GFP‐based pH reporter mtAlpHi 25 revealed no differences in mitochondrial matrix pH (Fig EV2I and J).
Figure EV2. Mitochondrial parameters in ATPIF1‐overexpressing HEK293T cells.
-
A–DRepresentative rendering (A) and quantification of morphological parameters (count, B; average volume, C; total volume, D) in HEK293T cells transiently transfected with pcDNA3 or with a plasmid for the overexpression of ATPIF1. The results are representative of three independent experiments. Scale bar = 10 μm.
-
E, FRepresentative images (E) and quantification (F) of TMRM staining in HEK293T cells transiently transfected with pcDNA3 or with a plasmid for the overexpression of ATPIF1. The results are representative of five independent experiments. *P = 0.0001 (unpaired Student's t‐test). Scale bar = 10 μm.
-
G, HRepresentative traces (G) and quantification (H) of mitochondrial ATP‐dependent light emission in HEK293T cells transiently transfected with pcDNA3 or with a plasmid for the overexpression of ATPIF1. 75 μM N,N‐dicyclohexylcarbodiimide (DCCD) was employed to completely inhibit mitochondrial ATP synthesis. The results are representative of five independent experiments.
-
I, JRepresentative traces (I) and quantification (J) of mtAlphi/ECFP ratio in HEK293T cells transiently co‐transfected with pcDNA3 or with a plasmid for the overexpression of ATPIF1. 30 mM Na acetate or 30 mM NH4Cl was employed to detect the minimum and maximum ratio, respectively. The results are representative of four independent experiments.
-
K, LRepresentative immunoblotting for the detection of the ATP5A1 subunit after blue‐native PAGE (K) and quantification (L) of F1FO ATP synthase dimers (D) and monomers (M) in HEK293T cells transiently transfected with pcDNA3 or with a plasmid for the overexpression of ATPIF1. The results are representative of three independent experiments. *P = 0.0095 (unpaired Student's t‐test).
HEK293T cells overexpressing ATPIF1 manifested a drastic increase in the relative abundance of F1FO ATP synthase dimers as determined by PLA (quantification restricted to mtGFP+ objects) (Fig 2E and F) and BN electrophoresis (Fig EV2K and L), prompting us to test their sensitivity to MPT elicited by ionomycin plus Ca2+. ATPIF1‐overexpressing HEK293T cells display an increased resistance to MPT induction by ionomycin plus Ca2+ as compared to their control counterparts (Fig 2G and H). Since one of the major consequences of the MPT is mitochondrial depolarization, we performed long‐term confocal imaging on control and ATPIF1‐overexpressing HEK293T cells treated with the MPT‐inducer hydrogen peroxide (H2O2), upon staining with the Δψm‐sensitive probe tetramethylrhodamine methyl ester (TMRM). Control cells (transfected with an empty pcDNA3‐based vector) treated with 500 μM H2O2 experienced relatively slow mitochondrial depolarization, which could be virtually prevented by ATPIF1 overexpression (Fig 2I and J).
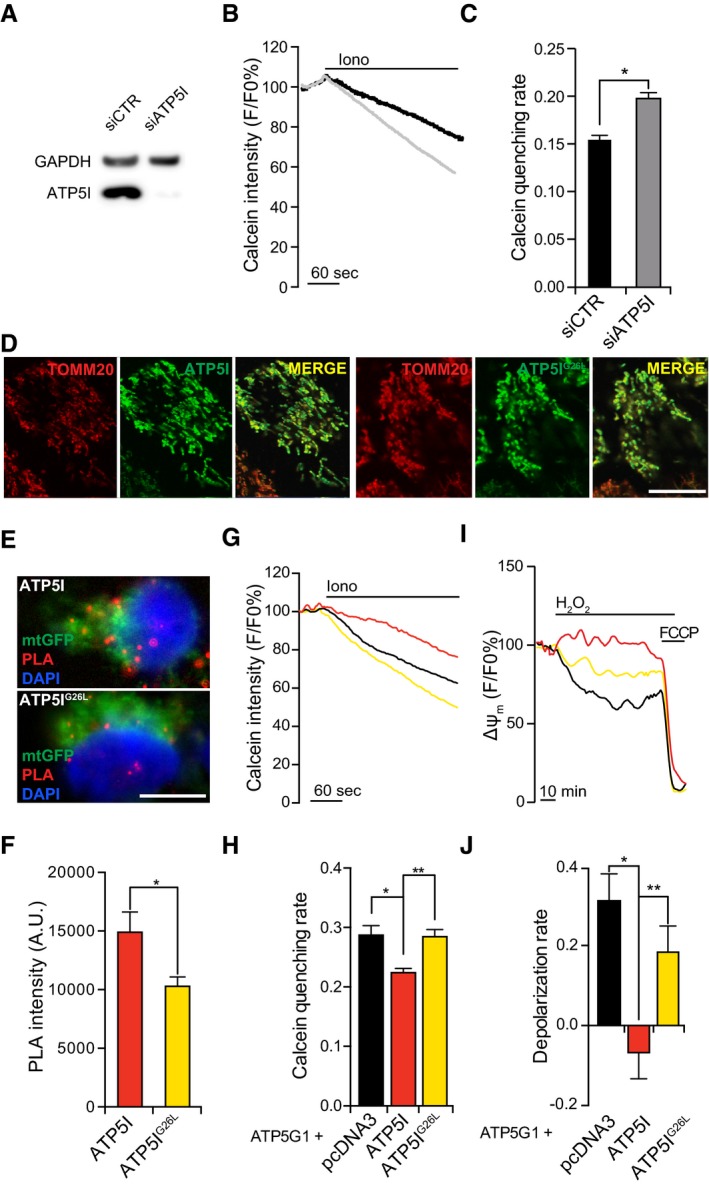
To corroborate these observations, we evaluated MPT sensitivity after manipulating the expression of ATP synthase, H+ transporting, mitochondrial FO complex subunit E (ATP5I), which is critical for dimerization 26. Per se, robust ATP5I depletion (> 97% reduction in ATP5I levels after 96 h) considerably sensitized HEK293T cells to MPT induction by ionomycin plus Ca2+ (Fig 3A–C).
Figure 3. Modulation of F1FO ATP synthase dimers via ATP5I impacts on MPT.
-
AAssessment of transfection efficiency in HEK293T cells transfected with a control siRNA (siCTR) or a siRNA targeting ATP5I (siATP5I) for 96 h. GAPDH levels were monitored to ensure equal lane loading.
-
B, CRepresentative calcein/Co2+ quenching traces (B) and quenching rate quantification (C) in HEK293T cells transfected with siCTR or siATP5I for 96 h, and then treated with 1 μM ionomycin (Iono). Results are representative of five independent experiments. *P = 0.0001 (unpaired Student's t‐test).
-
DRepresentative colocalization of ATP5I or ATP5IG26L with TOMM20 in HEK293T cells transfected with constructs for ATP5I or ATP5IG26L overexpression, respectively.
-
E, FRepresentative images (E) and quantification (F) of PLA assays for F1FO ATP synthase dimers in HEK293T cells co‐transfected with a plasmid coding for mtGFP and with constructs for the overexpression of ATP5I or ATP5IG26L. Results are representative of three independent experiments. *P = 0.0001 (unpaired Student's t‐test).
-
G, HRepresentative calcein/Co2+ quenching traces (G) and quenching rate quantification (H) in HEK293T cells co‐transfected with an ATP5G1‐encoding construct and pcDNA or with plasmids for the overexpression of ATP5I or ATP5IG26L and treated with 1 μM Iono. Results are representative of five independent experiments. *P = 0.0007, **P = 0.0007 (ANOVA plus unpaired Student's t‐test).
-
I, JRepresentative mitochondrial transmembrane potential (Δψm) recordings (I) and depolarization rate quantification (J) in HEK293T cells transfected as in (G, H) and exposed to 500 μM H2O2. 1 μM carbonyl cyanide p‐trifluoromethoxyphenylhydrazone (FCCP) was added as control for depolarization. Results are representative of 3–5 independent experiments. *P = 0.0001, **P = 0.0191 (ANOVA plus unpaired Student's t‐test).
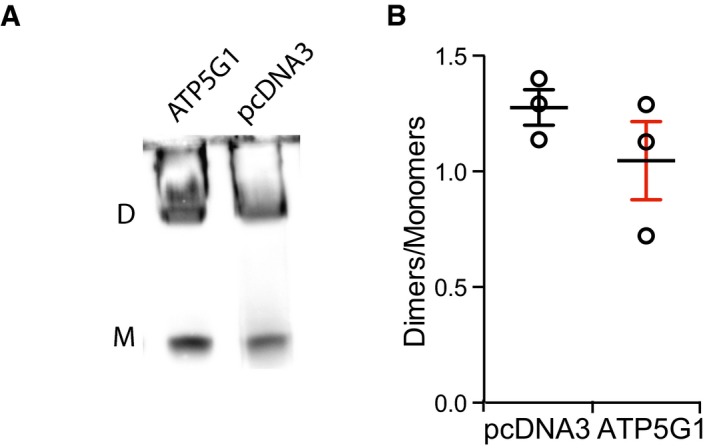
Next, we tested whether overexpressing ATP5I would have any effect on PTPC opening in conditions that sensitize cells to MPT. To this aim, we took advantage of the fact that overexpressing the c subunit of the F1FO ATP synthase not only promotes (some extent of) spontaneous MPT, but also potently sensitizes cells to MPT induction by ionomycin plus Ca2+ 16, irrespective of alterations of dimer/monomer ratio (as confirmed by BN electrophoresis) (Fig EV3A and B). A critical residue required for ATP5I to stabilize F1FO ATP synthase dimers was characterized in yeast 26. We transposed this mutation to human ATP5I and generated a glycine‐to‐leucine substitution (ATP5IG26L) mutant. Next, we co‐transfected HEK293T cells with cDNAs for the overexpression of ATP5G1 together with either WT ATP5I or ATP5IG26L, and monitored the relative abundance of F1FO ATP synthase dimers, MPT sensitivity, and mitochondrial depolarization rate.
Figure EV3. Dimerization status during ATP5I overexpression.
-
A, BRepresentative immunoblotting for the detection of the ATP5A1 subunit after blue‐native PAGE (A) and quantification (B) of F1FO ATP synthase dimers (D) and monomers (M) in HEK293T cells transiently transfected with pcDNA3 or with a plasmid for the overexpression of ATP5G1. The results are representative of three independent experiments and expressed as mean ± SEM.
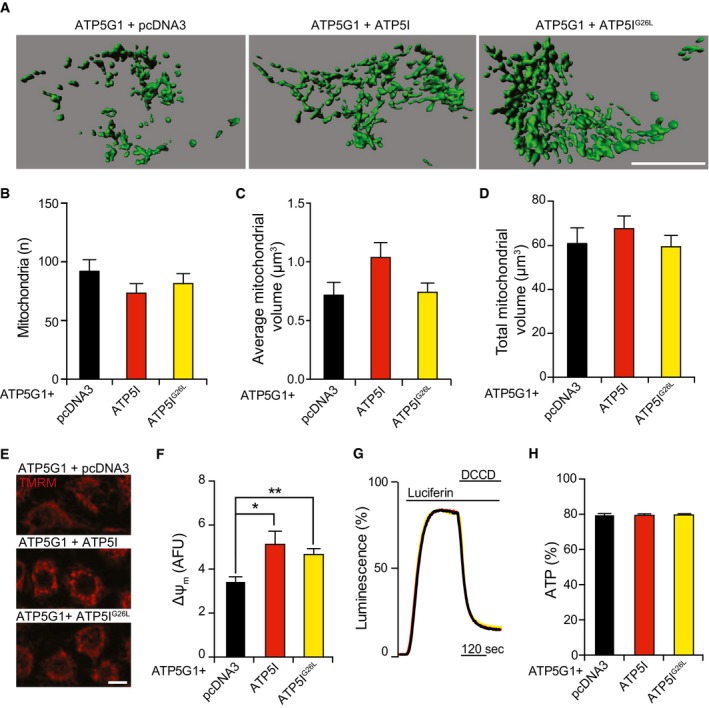
We detected high levels of ATP5G1 and ATP5I or ATP5IG26L, which properly co‐localized at mitochondria (Fig 3D). The overexpression of either ATP5I or ATP5IG26L tended to revert the moderate morphological alterations imposed by ATP5G1 overexpression on the mitochondrial network, although in a statistically subsignificant manner (Fig EV4A–D). Conversely, ATP5I (and partially ATP5IG26L) remarkably increased Δψm in HEK293T cells co‐overexpressing ATP5G1 (Fig EV4E and F) an effect previously attributed to improved F1FO ATP synthase dimerization (in yeast) 27, although this had no impact on mitochondrial ATP content (Fig EV4G and H). Importantly, the overexpression of ATP5I (but not ATP5IG26L) increased the relative abundance of F1FO ATP synthase dimers in ATP5G1‐overexpressing HEK293T cells (Fig 3E and F), while limiting their sensitivity to MPT induction by ionomycin plus Ca2+ (Fig 3G and H). Along similar lines, ATP5I (but less so ATP5IG26L) prevented mitochondrial depolarization in HEK293T cells exposed to H2O2 (Fig 3I and J). ATP5IG26L‐expressing cells displayed partial protection from MPT triggered by H2O2, possibly reflecting the incomplete inactivation of the dimerization domain. These data indicate that stabilizing F1FO ATP synthase dimers (by two genetically distinct approaches) limits PTPC opening and MPT and that the dissociation of F1FO ATP synthase dimers is a cause, not a consequence, of MPT.
Figure EV4. Mitochondrial parameters in ATP5G1‐ and ATP5I‐co‐overexpressing HEK293T cells.
-
A–DRepresentative rendering (A) and quantification of morphological parameters (count, B; average volume, C; total volume, D) in HEK293T cells transiently transfected with a construct for the overexpression of ATP5G1 plus pcDNA3 or a plasmid for the overexpression of ATP5I or ATP5IG26L. The results are representative of three independent experiments. Scale bar = 10 μm.
-
E, FRepresentative images (E) and quantification (F) of TMRM staining in HEK293T cells transiently transfected with a construct for the overexpression of ATP5G1 plus pcDNA3 or a plasmid for the overexpression of ATP5I or ATP5IG26L. The results are representative of three independent experiments. *P = 0.0060, **P = 0.0345 (Kruskal–Wallis test with Dunn's correction for multiple comparison). Scale bar = 10 μm.
-
G, HRepresentative traces (G) and quantification (H) of mitochondrial ATP‐dependent light emission in HEK293T cells transiently transfected with a construct for the overexpression of ATP5G1 plus pcDNA3 or a plasmid for the overexpression of ATP5I or ATP5IG26L. 75 μM DCCD was employed to completely inhibit mitochondrial ATP synthesis at the end of the assay. The results are representative of three independent experiments.
Since MPT apparently involves the disassembly of F1FO ATP synthase dimers, we hypothesized that the c subunit might contribute to PTPC opening in a subsequent step of the process. We previously demonstrated that depleting the c subunit with specific sRNAs is sufficient to inhibit MPT 16. Along similar lines, it was reported that substitution of a residue in the highly conserved glycine zipper domain of the c subunit results in a deformation of C‐rings that is paralleled by increased MPT sensitivity 14. Such domain displays extraordinary conservation across species, possibly because it allows for the tight disposition of c subunits within C‐rings 28. Indeed, substituting glycine residues with alanines increased the distance between c subunits in C‐rings from I. tartaricus, and substituting G25 with serine dramatically reduced the stability of the complex 29.
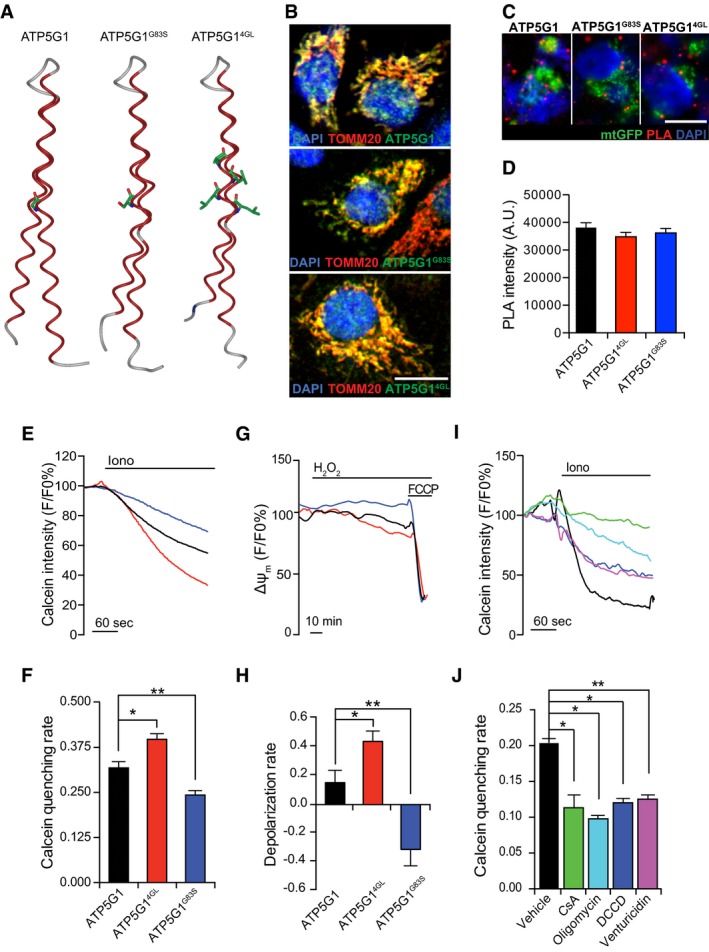
To test whether C‐rings are determinant in PTPC opening, we transposed the G25S mutation identified by Pogoryelov et al to human ATP5G1, generating the ATP5G1G83S mutant. In addition, we created an ATP5G1 variant in which all key glycines were substituted with leucines (ATP5G14GL), as per the findings from Alavian and colleagues 14. Bioinformatic studies based on the RaptorX web server 30 predicted that both ATP5G1G83S and ATP5G14GL would exhibit a structural deformation at the level of the first helix (Fig 4A). Transfecting HEK293T cells with cDNAs encoding WT ATP5G1, ATP5G1G83S, or ATP5G14GL resulted in robust mitochondrial expression of WT or mutant ATP5G1 (Fig 4B) and did not affect F1FO ATP synthase dimerization, as assessed by PLA (quantification restricted to mtGFP+ objects) (Fig 4C and D). ATP5G14GL‐overexpressing HEK293T cells were more sensitive to MPT induction than HEK293T cells overexpressing WT ATP5G1. Conversely, overexpression of ATP5G1G83S increased the ability of HEK293T cells to resist MPT induction by ionomycin and Ca2+ (Fig 4E and F). Along similar lines, ATP5G14GL accelerated, while ATP5G1G83S retarded, mitochondrial depolarization driven by H2O2 (Fig 4G and H). These results suggest that the glycine zipper domain of the c subunit plays a central role in PTPC opening.
Figure 4. C‐ring conformation affects PTPC opening and consequent MPT.
-
ABioinformatic prediction of the alterations imposed by the G83S and the 4GL mutations on ATP5G1 structure.
-
BRepresentative colocalization of ATP5G1, ATP5G1G83S or ATP5G14GL with TOMM20 in HEK293T cells transfected with constructs for ATP5G1, ATP5G1G83S or ATP5G14GL overexpression, respectively. Scale bar = 10 μm.
-
C, DRepresentative images (C) and quantification (D) of PLA assays for F1FO ATP synthase dimers in HEK293T cells co‐transfected with a plasmid coding for mtGFP and constructs for the overexpression of ATP5G1, ATP5G1G83S or ATP5G14GL. Results are representative of three independent experiments. Scale bar = 10 μm.
-
E, FRepresentative calcein/Co2+ quenching recordings (E) and quenching rate quantification (F) in HEK293T cells transfected as in (C, D) and then treated with 1 μM ionomycin (Iono). Results are representative of three independent experiments. *P = 0.0352, **P = 0.0453 (ANOVA plus unpaired Student's t‐test).
-
G, HRepresentative mitochondrial transmembrane potential (Δψm) readings (G) and depolarization rate quantification (H) in HEK293T cells transfected as in (C, D) and then exposed to 500 μM H2O2. 1 μM carbonyl cyanide p‐trifluoromethoxyphenylhydrazone (FCCP) was employed at the end of the assay as a positive control for depolarization. Results are representative of five independent experiments. *P = 0.0006, **P = 0.0270 (ANOVA plus unpaired Student's t‐test).
-
I, JRepresentative calcein/Co2+ quenching traces (I) and quenching rate quantification (J) in HEK293T cells pre‐treated with 1.6 μM cyclosporine A (CsA), 10 μM oligomycin, 15 μM N,N‐dicyclohexylcarbodiimide (DCCD) or 10 μM venturicidin, and then exposed to 1 μM Iono. The results are representative of three independent experiments. *P = 0.0001, **P = 0.0033 (ANOVA plus unpaired Student's t‐test).
To validate this notion, we harnessed the fact that some of the most potent inhibitors of the F1FO ATP synthase (e.g. oligomycin, venturicidin, and DCCD) bind to a unique pocket of the C‐ring that is partially formed by the c subunit glycine domain 31. In particular, we tested whether these agents would affect the sensitivity of HEK293T cells to MPT induction by ionomycin plus Ca2+ in calcein/Co2+ quenching assays. Indeed, all the compounds reduced calcein quenching rate as efficiently as CsA (Fig 4I and J).
To determine whether the dissociation of F1FO ATP synthase dimers or the rearrangement of C‐rings drives MPT, we co‐transfected HEK293T cells with cDNAs coding for ATPIF1 and ATP5G1 or ATP5G14GL, and tested their sensitivity to MPT induction by ionomycin plus Ca2+ in calcein/Co2+ quenching assays. Surprisingly, the overexpression of ATPIF1 (which stabilizes F1FO ATP synthase dimers) limited the sensitization of HEK293T cells to MPT imposed by the overexpression of ATP5G1 or ATP5G14GL (Fig 5A and B) suggesting that the dissociation of F1FO ATP synthase dimers is required for C‐rings to assume a conformation compatible with PTPC opening. Along similar lines, depletion of ATP5I (which destabilizes F1FO ATP synthase dimers) retained the ability to facilitate MPT induction by ionomycin plus Ca2+ even when ATP5G1 was overexpressed, an effect that was lost in ATP5G1G83S‐overexpressing cells (Fig 5C and D). Altogether, these data suggest that a proper C‐ring conformation is required for MPT induction once F1FO ATP synthase dimers have dissociated, underscoring the importance of the C‐ring in PTPC opening. These findings do not imply that the C‐ring constitutes the pore‐forming component of the PTPC, as lipids are expected to occupy its lumen even during MPT, which is inconsistent with the PTPC channel properties 32. Nonetheless, our findings do not exclude that F1FO ATP synthase dimers prevent C‐rings to interact with other partners for the generation of a hitherto obscure pore‐generating structure. Indeed, the c subunit has recently been shown to generate complexes with inorganic phosphate and polyhydroxybutyrate supporting the formation of PTPC‐like currents 33.
Figure 5. Critical role of C‐rings in MPT in cellula and ex vivo .
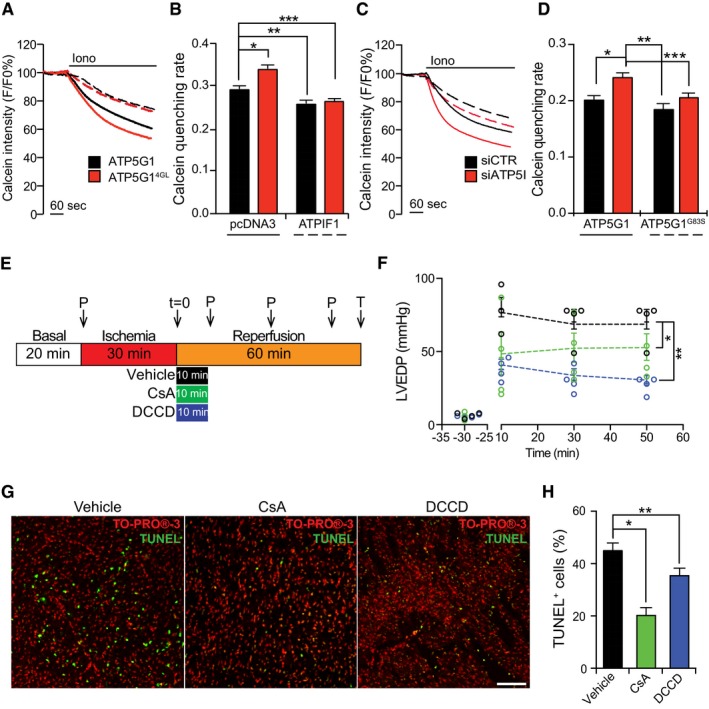
-
A, BRepresentative calcein/Co2+ quenching recordings (A) and quenching rate quantification (B) in HEK293T cells transfected with an empty plasmid or with a construct for the overexpression of ATPIF1, combined with an ATP5G1‐ or an ATP5G14GL‐encoding plasmid, and exposed to 1 μM ionomycin (Iono). Results are representative of three independent experiments. *P = 0.0030, **P = 0.0035, ***P = 0.0078 (ANOVA plus unpaired Student's t‐test).
-
C, DRepresentative calcein/Co2+ quenching recordings (C) and quenching rate quantification (D) in HEK293T cells transfected with a control siRNA (siCTR) or with a siRNA specific for ATP5I (siATP5I), combined with an ATP5G1‐ or an ATP5G1G83S‐encoding plasmid, and then treated with 1 μM Iono. Results are representative of three independent experiments. *P = 0.0001, **P = 0.00001, ***P = 0.0235 (ANOVA plus unpaired Student's t‐test).
-
EDesign of the experimental approach to study cardiac ischemia/reperfusion damage ex vixo. P, pressure readings. T, TUNEL assay.
-
FQuantification of LVEDP in rat hearts subjected to ischemia ex vivo and then reperfused with vehicle only (black), or with vehicle supplemented with 0.2 μM cyclosporine A (CsA; green) or 0.2 μM N,N‐dicyclohexylcarbodiimide (DCCD; blue), as detailed in panel (E). Results are representative of four independent experiments and expressed as individual readings. *P = 0.0001, **P = 0.0032 (ANOVA plus unpaired Student's t‐test).
-
G, HRepresentative images (G) and quantification (H) of TUNEL assays in rat hearts subjected to ischemia ex vivo and then reperfused with vehicle only, or with vehicle supplemented with 0.2 μM CsA or 0.2 μM DCCD, as detailed in panel (E). TO‐PRO®‐3 was employed as a nuclear counterstain. Results are representative of three independent experiments. *P = 0.0054, **P = 0.0006 (ANOVA plus unpaired Student's t‐test).
Considering the dramatic effects of C‐ring‐targeting agents on PTPC opening in vitro, we determined whether these drugs also limit MPT ex vivo, in a model of cardiac reperfusion injury. We isolated beating hearts from adult male Wistar rats and placed them in a Langendorff system, continuously perfused with Krebs–Henseleit buffer (KHB) bubbled with oxygen at 37°C. The hearts were allowed to stabilize for 20 min, and then, retrograde perfusion was progressively stopped to generate 30 min of global ischemia, followed by 1 h of reperfusion (Fig 5E). Upon reperfusion with KHB only, left ventricular end diastolic pressure (LVEDP) never returned to baseline levels, suggesting irreversible tissue damage (Fig 5F). Conversely, when 0.2 μM CsA was administered throughout the first 10 min of reperfusion, LVEDP values were significantly closer to baseline, confirming cardioprotection. Importantly, 0.2 μM DCCD was even more potent than CsA in limiting the changes in LVEDP imposed by ischemia/reperfusion ex vivo (Fig 5F). At the end of the procedure, hearts were analyzed for cell death incidence by terminal deoxynucleotidyl transferase dUTP nick‐end labeling (TUNEL) assays. In control conditions, as many as 45% of cardiomyocytes were TUNEL+, a percentage that was significantly reduced in the presence of CsA or DCCD (Fig 5G and H). These findings confirm the ability of C‐ring‐targeting agents to inhibit PTPC opening ex vivo as efficiently as the prototypic MPT inhibitor CsA.
Taken together, our findings suggest that PTPC opening is a multistep process that involves disassembly of F1FO ATP synthase dimers and rearrangement of C‐rings and that the c subunit of the F1FO ATP synthase may constitute a promising target for the development of novel cardio‐, neuro‐, or nephroprotective agents.
Materials and Methods
Chemicals, cell cultures, and transfections
Unless otherwise noted, chemicals were purchased from Sigma‐Aldrich, cell culture media and supplements from Thermo Fischer Scientific, and plasticware from Corning Life Sciences. Human embryonic kidney HEK293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin G sodium and 100 μg/ml streptomycin sulfate. For RNA interference, HEK293T cells were transfected with a commercial control siRNA (AllStars Negative Control siRNA) or with a set of siRNAs specific for ATP5I (siATP5I) (SI04272905; SI04299232; SI00308112; SI04155228), all from Qiagen, by means of the HiPerfect® transfection reagent (Qiagen), as per manufacturer's instructions 34. For transient overexpression experiments, a pCMV6‐entry‐based plasmid coding for MYC‐tagged ATP5G1 (RC200292) or ATP5I (RC215565) under the control of the CMV immediate early promoter was obtained from OriGene. The gene encoding ATP5G1G83S and ATP5G14GL cloned into pCMV6‐Entry was synthesized by Blue Heron Biotechnology, Inc. The gene encoding mutant ATP5IG26L cloned into pCMV6‐Entry was synthesized by Bio‐Fab Research. Plasmid encoding for human ATPIF1 was kindly gifted by Dr. Michelangelo Campanella. Plasmid encoding for mtAlphI and mtCFP were kindly gifted from Prof. Tullio Pozzan. For the quantification of mitochondrial ATP levels, a VR1012‐based construct encoding a mitochondrially targeted variant of the Photinus pyralis luciferase under the control of the CMV immediate early promoter was employed 35. A pcDNA3‐based plasmid coding for a mitochondrially targeted variant of GFP (mtGFP) under the control of the CMV immediate early promoter was employed for the evaluation of mitochondrial ultrastructure 36. Plasmid co‐transfections were performed via the standard Ca2+‐phosphate technique 37.
Antibodies
For immunofluorescence, immunoblotting, and PLA, the following primary antibodies were used: rabbit anti‐GAPDH [#2118] (1:5,000) from Cell Signaling; mouse anti‐ATP sintase inhibitory factor 1 [ab‐110277] (1:100 for immunofluorescence, 1:1,000 for Western blotting), rabbit anti‐ATP5I [ab‐122241] (1:100 for immunofluorescence, 1:1,000 for Western blotting), rabbit anti‐ATP5G1 [ab‐181243] (1:100), mouse anti‐ATP5H [ab‐110275] (1:100) from Abcam; and rabbit anti‐TOM20 [sc‐11415] (1:100) from Santa Cruz Biotechnology.
Animal models
Liver or heart mitochondria were obtained from 3‐month‐old female C57BL/6J mice. Heart for ex vivo reperfusion injury was obtained from 6‐week‐old male Wistar rats. All animals were kept in polysulfone‐ventilated cages with wire bar lids from stainless steel, under controlled conditions: 12‐h dark/light cycle; 50–60% humidity; temperature 21–23°C; water and food (standard diet) ad libitum. It was also guaranteed appropriate enrichments for nest building and to improve social conditions.
All animal procedures were performed in accordance with (i) the Institutional Animal Care and Use Committee (IACUC), (ii) the guidelines established in the Principles of Laboratory Animal Care (directive 86/609/EEC), (iii) the guidelines approved by Local (Warsaw) Ethical Committee (188/2015/IBD and 188W/2015/IBD) based on Poland national laws, and (iv) approved by the Italian Ministry of Health.
Proximity ligation assays
Cell were fixed in 4% PFA for 10 min at 37°C, washed in PBS, placed in 1 mM EDTA buffer (pH 8.0) for 20 min at 100°C (to improve the epitope‐antibody binding) and then at room temperature for an additional 10 min. Thereafter, cells were permeabilized with 0.05% Triton X‐100 for 10 min at 37°C, and unspecific binding sites were blocked by incubating cells in 0.05% Triton X‐100 supplemented with 2% bovine serum albumin (BSA) for 45 min at 37°C. Upon overnight incubation with ATP5H‐specific antibodies previously conjugated to + or − PLA oligonucleotide probes (antibody dilution 1:100; 4°C), as per the instructions of Duolink In Situ ® Probemaker kits (Sigma‐Aldrich), detection was performed as follows. A ligation‐ligase solution was added to each sample for 30 min at 37°C, washed twice for 2 min with Duolink In Situ ® Wash Buffer A, an amplification‐polymerase solution was added for 100 min at 37°C and washed twice for 10 min with 1× Duolink In Situ ® Wash Buffer A. Eventually, slides were stained with Duolink In Situ ® Detection Reagent Green or Red, mounted using DAPI‐containing Duolink In Situ ® Mounting Medium, and protein proximity was evaluated on a Axiovert 200M fluorescence microscope equipped with a 40× water immersion objective (N.A. 1.2, from Carl Zeiss Microscopy, LLC) as a function of global signal intensity (quantification limited to GFP+ objects).
Calcein/Co2+ quenching assays
PTPC opening was assayed as previously described 38. Briefly, HEK293T cells were loaded with 1 mM calcein acetoxymethyl ester and Co2+ as instructed by the Image‐IT® LIVE Mitochondrial Transition Pore Assay Kit (Thermo Fischer Scientific). Cells were then imaged based on 490 ± 20 nm excitation and 525‐nm longpass emission filters on a Axiovert 200M fluorescence microscope equipped with a 40× water immersion objective (N.A. 1.2, from Carl Zeiss Microscopy, LLC). Finally, images were analyzed with MetaMorph® (Molecular Devices), and quenching rate was calculated as the slope of the fluorescence trace over a period of 60 s after stimulation.
Mitochondrial ATP
HEK293T cells expressing a mitochondrially targeted variant of the Photinus pyralis luciferase were perfused with a modified Krebs–Ringer buffer containing 125 mM NaCl, 5 mM KCl, 1 mM Na3PO4, 1 mM MgSO4, 1 mM CaCl2, 20 μM luciferin, and 20 mM HEPES buffer (pH 7.4) at 37°C, and luciferin‐dependent luminescence was monitored with a customized luminometer (Elettrofor), as previously described 35.
Mitochondrial swelling
Mitochondria were isolated from mouse livers according to conventional procedures and placed in energized buffer containing respiratory substrates 39. Thereafter, 100 μM CaCl2 alone or upon pre‐treatment with 1 μM CsA was used to promote PTPC opening, which was followed spectrometrically by measuring 90° light scattering at 540 nm.
Mitochondrial pH indicator
Cells were co‐transfected with plasmids encoding mtAlpHi and mtECFP for 48 h, and then imaged on a LSM 510 confocal microscope (Carl Zeiss Microscopy, LLC). For calibrating mtAlpHi signal on mitochondrial pH, 30 mM sodium acetate and 30 mM NH4Cl were employed. All the kinetics were normalized on the minimum and maximal mtAlpHi/mtECFP signal ratio detected (in the presence of sodium acetate and NH4Cl, respectively). Basal normalized ratios were used as readout of mitochondrial matrix pH.
Mitochondrial morphology
Mitochondrial morphology was assayed as previously described 38. Briefly, HEK293T cells expressing a mitochondrially targeted variant of GFP 40 were imaged with an IX‐81 automated epifluorescence microscope (Olympus) equipped with a 60× oil immersion objective (N.A. 1.35, from Olympus) and an ORCA‐R2 CCD camera (Hamamatsu Photonics K.K.). Selected cells were followed over time, and Z‐stacks were subjected to digital deconvolution by means of a Wiener deconvolution filter and a theoretical point‐spread function provided by the Xcellence software package (Olympus). GFP+ objects were quantified with the “3D object counter” plug‐in of the open‐source Fiji software (freely available at http://fiji.sc/), whereas 3D representations were obtained with the “3D Viewer” plug‐in.
Blue‐native SDS–PAGE
Cell or liver mitochondrial pellets were lysed in 50 mM Bis‐Tris buffer supplemented with in 1 M aminocaproic acid (pH 7.0). Upon protein content quantification by means of the Protein Assay Kit (Bio‐Rad), digitonin (3 mg/mg total protein) was added to the samples, and lysates were incubated on ice for 30 min, and then centrifuged at 14,000 g for 20 min to remove insolubilized material. Thereafter, native lysates were combined with 5% Serva Blue G and 60 μg of proteins was loaded and separated on a freshly prepared 5–12% polyacrylamide gel. As an internal standard, 40 μg of proteins from mouse liver mitochondria was used. At the end of the run, the gel was equilibrated in 25 mM Tris buffer supplemented with 192 mM glycine and 0.1% SDS (pH 8.3) for 30 min, followed by conventional transfer on polyvinyldifluoride (PVDF) membranes (Bio‐Rad) and immunoblotting with antibodies specific for ATP5A1 and secondary donkey anti‐mouse antibodies labeled with IRdye (Li‐Cor, Biosciences) (1:5,000 in TBS supplemented with 0.1% Tween and 5% BSA). Detection was performed with the Odyssey infrared Imaging System (Li‐Cor Biosciences). Signal intensity was analyzed by ImageJ (https://fiji.sc/#). As an alternative, gels were stained with a Pb(NO3)2 solution in the presence of ATP, and directly visualized according to previously published procedures 41.
Immunoblotting
Immunoblotting was performed as previously described, with minor modifications 42. In brief, cells were washed, harvested, and lysed in RIPA buffer (50 mM Tris–HCl pH 7.8, 150 mM NaCl, 1% IGEPAL CA‐630, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM dithiothreitol) supplemented with 1 mM phenylmethylsulfonyl fluoride and Complete Protease Inhibitor Cocktail® (Roche Diagnostics Corp.). Thereafter, protein extracts (30 μg/lane) were separated on precast 4–12% SDS–PAGE gels (Thermo Fischer Scientific), electrotransfered onto PVDF membranes (Bio‐Rad), and probed with antibodies specific for ATP5I, ATPIF1, or GAPDH. Finally, membranes were incubated with appropriate HRP‐labeled secondary antibodies (Thermo Fischer Scientific). A conventional chemiluminescent substrate (#34080, from Thermo Fischer Scientific) and the ImageQuant LAS 4010 (GE Healthcare) were employed for detection.
Immunofluorescence microscopy
Immunofluorescence microscopy was performed according to standard procedures 43. Briefly, cells were fixed in 4% PFA for 20 min at room temperature, washed three times in PBS, and permeabilized with 0.1% Triton X‐100 for 5 min at room temperature. Thereafter, unspecific binding sites were blocked by incubating cells in PBS supplemented with 2% BSA (blocking buffer) for 1 h at room temperature. Cells were then incubated overnight at 4°C with primary antibodies specific for TOMM20, ATP5I, ATPIF1, or ATPG51. Finally, primary antibodies were revealed by means of appropriate AlexaFluor 488® or AlexaFluor 594® conjugates (Thermo Fischer Scientific). Images were acquired with a LSM 510 confocal microscope (Carl Zeiss Microscopy, LLC) through a Plan‐Apochromat 63×/1.4 oil objective (Carl Zeiss Microscopy, LLC).
Ex vivo ischemia/reperfusion injury
Ischemia/reperfusion was studied ex vivo according to the Langendorff model, with minor modifications 44, 45. In brief, upon euthanasia, the hearts of 6‐week‐old male Wistar rats were rapidly excised, immediately arrested in ice‐cold Krebs–Henseleit buffer (KHB; pH 7.4; 4°C), cannulated, and retrogradely perfused through the aorta with warm KHB (37°C) bubbled with 95% O2 and 5% CO2. The number of animals needed for the study was determined by the GPower software (http://www.gpower.hhu.de/). We used a multivariate analysis of variance (MANOVA) with “a priori” power analysis considering an α err prob = 0.05; an effect size = 0.2 and a statistical power = 0.8. Upon removal of the left atrial appendage, to obtain an isovolumetrically beating preparation, a latex fluid‐filled balloon was inserted into the LV cavity via the atrium and connected by fluid‐filled polyethylene catheter to a pressure transducer (1290 A OPT 002, Hewlett Packard). At the start of each experiment, the fluid in the balloon was increased stepwise to achieve a constant diastolic pressure between 0.5 and 1 mmHg. Before each experiment, the balloon fluid was adjusted to obtain a diastolic pressure of 1.0 mmHg. After a 20‐min stabilization period, hearts were subjected to global normothermic ischemia by halting perfusion for 30 min, followed by a 60‐min period of reperfusion. LV pressures (mmHg) were recorded immediately prior to ischemia as well as 10, 30, and 50 min after reperfusion using a programmable acquisition system (BM IDAS, Biomedica Mangoni). At the end of the procedure, hearts were frozen, cut into 16 μm thick sections by a conventional cryostat system, fixed in 4% PFA, and stained with the DeadEnd™ Fluorometric TUNEL kit (Promega), as per the manufacturer's instructions. Upon staining with TO‐PRO®‐3 (Thermo Fischer Scientific), sections were imaged on a LSM 510 confocal microscope (Carl Zeiss Microscopy, LLC), and nuclei from dead cells (in green) and all cells (in red) were counted.
Statistical procedures
Unless otherwise indicated, assays were performed in triplicate independent instances, yielding comparable results. Data, which are presented as means ± SEM, were analyzed with Microsoft Excel (Microsoft Co.). Statistical significance was determined by t‐test (two groups) of one‐way or two‐way ANOVA (three or more groups) with Bonferroni correction. P‐values < 0.05 were considered statistically significant. Normal distribution of data was assessed by the D'Agostino & Pearson omnibus normality test. F‐test was used to compare variances between groups. A P‐value < 0.05 was considered significant. All data are reported as mean ± SEM. Exact P‐values are indicated in the figure legends.
Author contributions
MB and PP conceived the study. MB, CM, GM, GP, GA, and ML‐A performed experiments and analyzed results. MB, CG, PR, GC, RF, GK, MRW, LG, and PP provided guidance and/or senior supervision over the study or part thereof. MB, LG, and PP wrote the manuscript. MB and CM prepared the figures under supervision from LG and PP. All authors provided input and corrections on the preparation of the manuscript and figures.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
PP is grateful to Camilla degli Scrovegni for continuous support. PP is supported by the Italian Ministry of Education, University and Research (COFIN no. 20129JLHSY_002, FIRB no. RBAP11FXBC_002, and Futuro in Ricerca no. RBFR10EGVP_001), the Italian Cystic Fibrosis Research Foundation (19/2014), and Telethon (GGP15219/B). PP and CG are supported by local funds from the University of Ferrara and the Italian Association for Cancer Research (IG‐18624 and MFAG‐13521). MRW and ML‐A are supported by the National Science Centre, Poland (grant 2014/15/B/NZ1/00490), grant W100/HFSC/2011, and HFSP grant RGP0027/2011. GK is supported by the French Ligue contre le Cancer (équipe labellisée); the French Agence National de la Recherche (ANR)—Projets Blancs; the ANR under the frame of E‐Rare‐2 (the ERA‐Net for Research on Rare Diseases); the French Association pour la recherche sur le cancer (ARC); Cancéropôle Ile‐de‐France; the French Institut National du Cancer (INCa); the Fondation Bettencourt Schueller; the Fondation de France; the French Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC, #333); the LabEx Immuno‐Oncology; the SIRIC (sites de recherche intégrée sur le cancer) Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE); the SIRIC Cancer Research and Personalized Medicine (CARPEM); the Swiss Bridge Foundation; the Swiss Institute for Experimental Cancer Research (ISREC); and the Paris Alliance of Cancer Research Institutes (PACRI). LG is supported by a start‐up grant from the Department of Radiation Oncology, Weill Cornell Medical College (New York, NY, US) and Sotio a.c. (Prague, Czech Republic).
EMBO Reports (2017) 18: 1077–1089
See also: C Chinopoulos (July 2017)
Contributor Information
Mariusz R Wieckowski, Email: m.wieckowski@nencki.gov.pl.
Lorenzo Galluzzi, Email: deadoc@vodafone.it.
Paolo Pinton, Email: paolo.pinton@unife.it.
References
- 1. Bonora M, Wieckowski MR, Chinopoulos C, Kepp O, Kroemer G, Galluzzi L, Pinton P (2015) Molecular mechanisms of cell death: central implication of ATP synthase in mitochondrial permeability transition. Oncogene 34: 1475–1486 [DOI] [PubMed] [Google Scholar]
- 2. Galluzzi L, Bravo‐San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, Alnemri ES, Altucci L, Andrews D, Annicchiarico‐Petruzzelli M et al (2015) Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 22: 58–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sharpe JC, Arnoult D, Youle RJ (2004) Control of mitochondrial permeability by Bcl‐2 family members. Biochim Biophys Acta 1644: 107–113 [DOI] [PubMed] [Google Scholar]
- 4. Izzo V, Bravo‐San Pedro JM, Sica V, Kroemer G, Galluzzi L (2016) Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol 26: 655–667 [DOI] [PubMed] [Google Scholar]
- 5. Morciano G, Giorgi C, Bonora M, Punzetti S, Pavasini R, Wieckowski MR, Campo G, Pinton P (2015) Molecular identity of the mitochondrial permeability transition pore and its role in ischemia‐reperfusion injury. J Mol Cell Cardiol 78: 142–153 [DOI] [PubMed] [Google Scholar]
- 6. Conrad M, Angeli JP, Vandenabeele P, Stockwell BR (2016) Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov 15: 348–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallace DC (2004) The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427: 461–465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD (2007) Voltage‐dependent anion channels are dispensable for mitochondrial‐dependent cell death. Nat Cell Biol 9: 550–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW et al (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434: 658–662 [DOI] [PubMed] [Google Scholar]
- 10. Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ (2005) Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA 102: 12005–12010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Green DR, Galluzzi L, Kroemer G (2014) Cell biology. Metabolic control of cell death. Science 345: 1250256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Giorgio V, Bisetto E, Soriano ME, Dabbeni‐Sala F, Basso E, Petronilli V, Forte MA, Bernardi P, Lippe G (2009) Cyclophilin D modulates mitochondrial F0F1‐ATP synthase by interacting with the lateral stalk of the complex. J Biol Chem 284: 33982–33988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, Glick GD, Petronilli V, Zoratti M, Szabo I et al (2013) Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci USA 110: 5887–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, Li H, Nabili P, Hockensmith K, Graham M et al (2014) An uncoupling channel within the c‐subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci USA 111: 10580–10585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Azarashvili T, Odinokova I, Bakunts A, Ternovsky V, Krestinina O, Tyynela J, Saris NE (2014) Potential role of subunit c of F0F1‐ATPase and subunit c of storage body in the mitochondrial permeability transition. Effect of the phosphorylation status of subunit c on pore opening. Cell Calcium 55: 69–77 [DOI] [PubMed] [Google Scholar]
- 16. Bonora M, Bononi A, De Marchi E, Giorgi C, Lebiedzinska M, Marchi S, Patergnani S, Rimessi A, Suski JM, Wojtala A et al (2013) Role of the c subunit of the FO ATP synthase in mitochondrial permeability transition. Cell Cycle 12: 674–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Daum B, Walter A, Horst A, Osiewacz HD, Kuhlbrandt W (2013) Age‐dependent dissociation of ATP synthase dimers and loss of inner‐membrane cristae in mitochondria. Proc Natl Acad Sci USA 110: 15301–15306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Masgras I, Rasola A, Bernardi P (2012) Induction of the permeability transition pore in cells depleted of mitochondrial DNA. Biochim Biophys Acta 1817: 1860–1866 [DOI] [PubMed] [Google Scholar]
- 19. Gerle C (2016) On the structural possibility of pore‐forming mitochondrial FoF1 ATP synthase. Biochim Biophys Acta 1857: 1191–1196 [DOI] [PubMed] [Google Scholar]
- 20. Minauro‐Sanmiguel F, Wilkens S, Garcia JJ (2005) Structure of dimeric mitochondrial ATP synthase: novel F0 bridging features and the structural basis of mitochondrial cristae biogenesis. Proc Natl Acad Sci USA 102: 12356–12358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wittig I, Schagger H (2008) Structural organization of mitochondrial ATP synthase. Biochim Biophys Acta 1777: 592–598 [DOI] [PubMed] [Google Scholar]
- 22. Fredriksson S, Gullberg M, Jarvius J, Olsson C, Pietras K, Gustafsdottir SM, Ostman A, Landegren U (2002) Protein detection using proximity‐dependent DNA ligation assays. Nat Biotechnol 20: 473–477 [DOI] [PubMed] [Google Scholar]
- 23. Garcia JJ, Morales‐Rios E, Cortes‐Hernandez P, Rodriguez‐Zavala JS (2006) The inhibitor protein (IF1) promotes dimerization of the mitochondrial F1F0‐ATP synthase. Biochemistry 45: 12695–12703 [DOI] [PubMed] [Google Scholar]
- 24. Scott SV, Cassidy‐Stone A, Meeusen SL, Nunnari J (2003) Staying in aerobic shape: how the structural integrity of mitochondria and mitochondrial DNA is maintained. Curr Opin Cell Biol 15: 482–488 [DOI] [PubMed] [Google Scholar]
- 25. Abad MF, Di Benedetto G, Magalhaes PJ, Filippin L, Pozzan T (2004) Mitochondrial pH monitored by a new engineered green fluorescent protein mutant. J Biol Chem 279: 11521–11529 [DOI] [PubMed] [Google Scholar]
- 26. Arselin G, Giraud MF, Dautant A, Vaillier J, Brethes D, Coulary‐Salin B, Schaeffer J, Velours J (2003) The GxxxG motif of the transmembrane domain of subunit e is involved in the dimerization/oligomerization of the yeast ATP synthase complex in the mitochondrial membrane. Eur J Biochem 270: 1875–1884 [DOI] [PubMed] [Google Scholar]
- 27. Bornhovd C, Vogel F, Neupert W, Reichert AS (2006) Mitochondrial membrane potential is dependent on the oligomeric state of F1F0‐ATP synthase supracomplexes. J Biol Chem 281: 13990–13998 [DOI] [PubMed] [Google Scholar]
- 28. Pandini A, Kleinjung J, Taylor WR, Junge W, Khan S (2015) The phylogenetic signature underlying ATP synthase c‐ring compliance. Biophys J 109: 975–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pogoryelov D, Klyszejko AL, Krasnoselska GO, Heller EM, Leone V, Langer JD, Vonck J, Muller DJ, Faraldo‐Gomez JD, Meier T (2012) Engineering rotor ring stoichiometries in the ATP synthase. Proc Natl Acad Sci USA 109: E1599–E1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kallberg M, Wang H, Wang S, Peng J, Wang Z, Lu H, Xu J (2012) Template‐based protein structure modeling using the RaptorX web server. Nat Protoc 7: 1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Symersky J, Osowski D, Walters DE, Mueller DM (2012) Oligomycin frames a common drug‐binding site in the ATP synthase. Proc Natl Acad Sci USA 109: 13961–13965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou W, Marinelli F, Nief C, Faraldo‐Gomez JD (2017) Atomistic simulations indicate the c‐subunit ring of the F1Fo ATP synthase is not the mitochondrial permeability transition pore. Elife 6: e23781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Elustondo PA, Nichols M, Negoda A, Thirumaran A, Zakharian E, Robertson GS, Pavlov EV (2016) Mitochondrial permeability transition pore induction is linked to formation of the complex of ATPase C‐subunit, polyhydroxybutyrate and inorganic polyphosphate. Cell Death Discov 2: 16070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. de La Motte Rouge T, Galluzzi L, Olaussen KA, Zermati Y, Tasdemir E, Robert T, Ripoche H, Lazar V, Dessen P, Harper F et al (2007) A novel epidermal growth factor receptor inhibitor promotes apoptosis in non‐small cell lung cancer cells resistant to erlotinib. Cancer Res 67: 6253–6262 [DOI] [PubMed] [Google Scholar]
- 35. Jouaville LS, Pinton P, Bastianutto C, Rutter GA, Rizzuto R (1999) Regulation of mitochondrial ATP synthesis by calcium: evidence for a long‐term metabolic priming. Proc Natl Acad Sci USA 96: 13807–13812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T (1998) Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 280: 1763–1766 [DOI] [PubMed] [Google Scholar]
- 37. Pinton P, Ferrari D, Magalhaes P, Schulze‐Osthoff K, Di Virgilio F, Pozzan T, Rizzuto R (2000) Reduced loading of intracellular Ca2+ stores and downregulation of capacitative Ca2+ influx in Bcl‐2‐overexpressing cells. J Cell Biol 148: 857–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bonora M, Morganti C, Morciano G, Giorgi C, Wieckowski MR, Pinton P (2016) Comprehensive analysis of mitochondrial permeability transition pore activity in living cells using fluorescence‐imaging‐based techniques. Nat Protoc 11: 1067–1080 [DOI] [PubMed] [Google Scholar]
- 39. Crouser ED, Gadd ME, Julian MW, Huff JE, Broekemeier KM, Robbins KA, Pfeiffer DR (2003) Quantitation of cytochrome c release from rat liver mitochondria. Anal Biochem 317: 67–75 [DOI] [PubMed] [Google Scholar]
- 40. De Giorgi F, Ahmed Z, Bastianutto C, Brini M, Jouaville LS, Marsault R, Murgia M, Pinton P, Pozzan T (1999) Targeting GFP to organelles. Methods Cell Biol 58: 75–85 [DOI] [PubMed] [Google Scholar]
- 41. Nijtmans LG, Henderson NS, Holt IJ (2002) Blue Native electrophoresis to study mitochondrial and other protein complexes. Methods 26: 327–334 [DOI] [PubMed] [Google Scholar]
- 42. Vitale I, Senovilla L, Jemaa M, Michaud M, Galluzzi L, Kepp O, Nanty L, Criollo A, Rello‐Varona S, Manic G et al (2010) Multipolar mitosis of tetraploid cells: inhibition by p53 and dependency on Mos. EMBO J 29: 1272–1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hoffmann J, Vitale I, Buchmann B, Galluzzi L, Schwede W, Senovilla L, Skuballa W, Vivet S, Lichtner RB, Vicencio JM et al (2008) Improved cellular pharmacokinetics and pharmacodynamics underlie the wide anticancer activity of sagopilone. Cancer Res 68: 5301–5308 [DOI] [PubMed] [Google Scholar]
- 44. Bell RM, Mocanu MM, Yellon DM (2011) Retrograde heart perfusion: the Langendorff technique of isolated heart perfusion. J Mol Cell Cardiol 50: 940–950 [DOI] [PubMed] [Google Scholar]
- 45. Ceconi C, Cargnoni A, Francolini G, Parinello G, Ferrari R (2009) Heart rate reduction with ivabradine improves energy metabolism and mechanical function of isolated ischaemic rabbit heart. Cardiovasc Res 84: 72–82 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3