

Abstract
Sequence variations in the triggering receptor expressed on myeloid cells 2 (TREM2) have been linked to an increased risk for neurodegenerative disorders such as Alzheimer's disease and frontotemporal lobar degeneration. In the brain, TREM2 is predominantly expressed in microglia. Several disease‐associated TREM2 variants result in a loss of function by reducing microglial phagocytosis, impairing lipid sensing, preventing binding of lipoproteins and affecting shielding of amyloid plaques. We here investigate the consequences of TREM2 loss of function on the microglia transcriptome. Among the differentially expressed messenger RNAs in wild‐type and Trem2−/− microglia, gene clusters are identified which represent gene functions in chemotaxis, migration and mobility. Functional analyses confirm that loss of TREM2 impairs appropriate microglial responses to injury and signals that normally evoke chemotaxis on multiple levels. In an ex vivo organotypic brain slice assay, absence of TREM2 reduces the distance migrated by microglia. Moreover, migration towards defined chemo‐attractants is reduced upon ablation of TREM2 and can be rescued by TREM2 re‐expression. In vivo, microglia lacking TREM2 migrate less towards injected apoptotic neurons, and outgrowth of microglial processes towards sites of laser‐induced focal CNS damage in the somatosensory cortex is slowed. The apparent lack of chemotactic stimulation upon depletion of TREM2 is consistent with a stable expression profile of genes characterizing the homoeostatic signature of microglia.
Keywords: Alzheimer's disease, chemotaxis, microglia, neurodegeneration, TREM2
Subject Categories: Immunology, Molecular Biology of Disease, Neuroscience
Introduction
Neurodegenerative diseases share common pathological and mechanistic principles. The most obvious common denominator are insoluble deposits formed by misfolded proteins, which are usually highly expressed in affected cells or in affected areas of the central nervous system (CNS) 1. Furthermore, for several neurodegenerative diseases, evidence exists that soluble oligomers, which may be rather stable intermediates of the aggregation process of amyloidogenic proteins, are the executers of neurotoxicity 2. Onset and progression of individual neurodegenerative diseases are also driven by shared seeding and spreading mechanisms, which were originally described and thought to be specific to prion disorders 3. However, the most commonly shared pathological principle of basically all neurodegenerative diseases and many other neurological disorders is gliosis associated with an inflammatory response 4. The critical role of the inflammatory response in neurodegeneration is strongly supported by the genetic linkage of sequence variants in the triggering receptor expressed on myeloid cells 2 (TREM2), which are associated with an increased risk for several neurodegenerative disorders including Alzheimer's disease (AD), frontotemporal lobar degeneration (FTLD), Parkinson's disease, FTLD‐like syndrome and Nasu–Hakola disease 5, 6, 7, 8, 9, 10, 11, 12, 13. Within the nervous system, TREM2 is predominantly expressed in microglia 14, 15, the resident immune cells of the brain. Microglia are known to rapidly respond to neuronal insults and pathological protein deposits in the brain by increased chemotactic migration, cytokine release, phagocytosis and proliferation (for review see 16). TREM2 is a type‐1 trans‐membrane protein, which is shed on the cell surface 17, 18. Certain disease‐associated sequence variants, which are located in the immunoglobulin‐like domain of TREM2, affect correct folding and lead to the retention of the misfolded protein within the endoplasmic reticulum 17 and consequently a reduction of its cellular functions such as phagocytosis, proliferation, pro‐survival signalling, lipid sensing, ApoE binding and release of cytokines 17, 19, 20, 21, 22, 23. Lack of cell surface TREM2 can be assessed in humans by quantitative analysis of soluble TREM2 (sTREM2) within biological fluids 17, 24, 25, 26. Indeed, patients with a homozygous TREM2 p.T66M mutation have no detectable sTREM2 in blood and cerebrospinal fluid (CSF) 17, 25. Moreover, increased levels of CSF sTREM, as it is observed in early stages of AD, may reflect a protective activation response of microglia 24, 27. Neuropathological consequences of a loss of TREM2 function have been detected in mouse models for AD‐like plaque pathology. In such models, reduced Trem2 function consistently led to reduced clustering of microglia around amyloid plaques 28, 29, 30, 31. As a consequence, a diffuse type of amyloid plaques accumulates, which may be due to reduced phagocytic clearance of the plaque halo 29. Alternatively, reduced barrier function may lead to enhanced amyloid plaque growth and as a consequence increased disease progression 30. Indeed, such diffuse plaques, which accumulate in mice deficient of Trem2, are associated with severe axonal dystrophy 29, 30. Together, these findings suggest that disease‐associated TREM2 mutations affect disease onset due to a loss of function. We therefore compared the transcriptome of microglia derived from wild‐type (WT) or Trem2 knockout (Trem2−/−) mice with the aim to identify gene clusters pinpointing to a pivotal TREM2‐dependent function.
Results
Trem2 deficiency affects expression profiles of genes involved in chemotaxis
To address the impact of a Trem2 loss of function on microglial gene expression, we isolated microglia from brains of adult WT and Trem2−/− mice by fluorescence‐activated cell sorting (FACS) using anti‐FCRLS and anti‐CD11b antibodies 15. Microglia were analysed for changes in their transcriptional profile using NanoString‐based chips containing 482 microglia‐enriched genes 15. Gene expression levels in each sample were normalized against the geometric mean of six housekeeping genes including Cltc, Gapdh, Gusb, Hprt1, Pgk1 and Tubb5. Within the set of 482 genes analysed, more than half them were unchanged, 88 were upregulated and 34 were downregulated (Dataset EV1). A scatter plot of the normalized and averaged gene expression levels from Trem2−/− mice against WT mice demonstrates that none of the genes show a fold change greater than two but several genes indicate a fold change of < 0.5 (Fig 1A). We then performed a gene ontology pathway analysis. This revealed clusters of dysregulated genes (Fig 1B) that were associated with chemotactic motility, wounding and cytokine response (Table 1 and Dataset EV3). Literature searches pointed out that specifically, the downregulated genes Ccl2, Il1b, Tnf and Spp1 of the category cell motility (Table 1) are putative direct targets of Trem2 20, 32, 33. Furthermore, application of pathway enrichment analysis separately to significantly up‐ and downregulated genes from the NanoString‐based screen revealed that in particular, downregulated genes contribute to chemotaxis and migration (compare the top significant functional categories of Datasets EV1 and EV2). Significantly downregulated genes (e.g. Il1b, Vegfa) involved in chemotaxis and migration are highlighted in Fig EV1.
Figure 1. Disturbed expression of gene clusters involved in chemotaxis in the absence of Trem2.
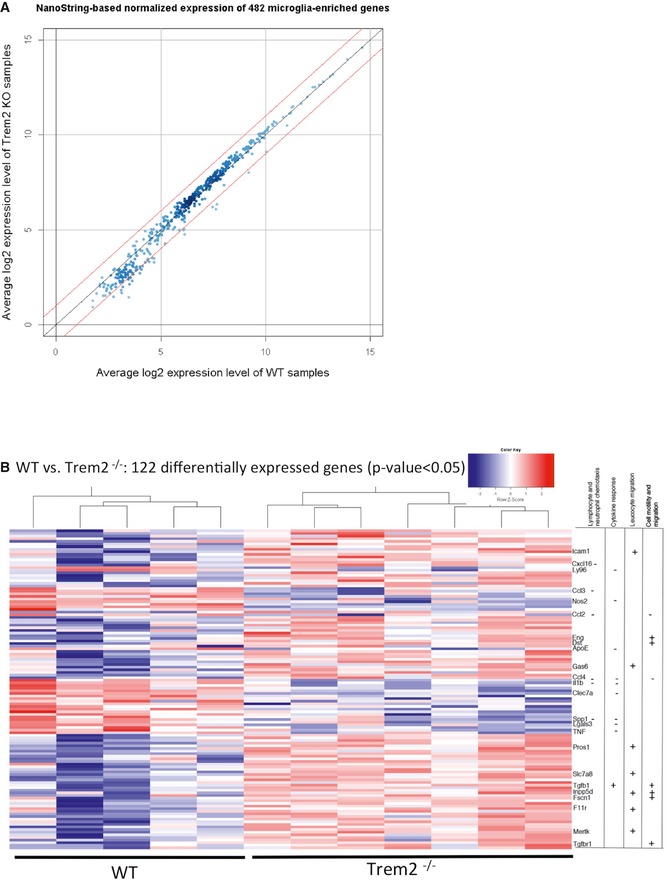
- Scatter plot of the normalized and averaged log2 gene expression level of Trem2−/− mice (KO) against wild‐type (WT) samples. The upper red line corresponds to a fold change of 2 while the lower red line indicates a fold change of 0.5.
- Gene expression profile of the normalized log2 transformed levels (for genes with P < 0.05 after Student's t‐test and FDR < 0.158) from the NanoString‐based chips represented by a heat map. For generation of the heat map, the heatmap.2 function within the gplots package of R statistical software was used. Hierarchical clustering by the hclust function (method = “complete”) was applied to group experimental conditions (columns) as well as intensities of genes (rows) for the heat map. Rows are scaled and represented as z‐score. A dendrogram is shown only for the columns. For the assignment of genes to functional categories at the right site of the heat map, the enrichment analysis of the GeneRanker was used but also a pathway analysis with help of the Pathway Studio software (Copyright 2014, Elsevier) as well as manual literature searches. WT N = 5, Trem2−/− N = 7.
Source data are available online for this figure.
Table 1.
Disturbed regulation of gene clusters involved in chemotaxis and migration
| GO term | Upregulated | Downregulated |
|---|---|---|
| Locomotion | INPP5d, CSF3R, DST, P2RY6, PROS1, LIMK1, ABI3, GAB1, SLC7A8, TGFBR1, GAS6, TGFBR2, MERTK, ICAM1, TNFRSF11A, TGFB1, RGMB, SEMA4D, FSCN1 | TREM2, CCL2, IL1B, ITGAX, PTPRM, VEGFA, CNTN1, SPP1, CXCL16, APOE, TNF, CSF1, PGRMC1 |
| Cellular component movement | INPP5d, CSF3R, DST, P2RY6, PROS1, ABI1, LIMK1, ABI3, GAB1, SLC7A8, TGFBR1, GAS6, TGFBR2, MERTK, ICAM1, TNFRSF11A, TGFB1, RGMB, SEMA4D, FSCN1 | TREM2, CCL2, IL1B, ITGAX, PTPRM, VEGFA, CNTN1, SPP1, CXCL16, APOE, TNF, CSF1, PGRMC1 |
| Cell migration | INPP5d, CSF3R, P2RY6, PROS1, ABI1, LIMK1, ABI3, GAB1, SLC7A8, TGFBR1, GAS6, TGFBR2, MERTK, ICAM1, TNFRSF11A, TGFB1, SEMA4D, FSCN1 | CCL2, IL1B, ITGAX, PTPRM, VEGFA, CNTN1, SPP1, CXCL16, APOE, TNF, CSF1 |
| Cell motility | INPP5d, CSF3R, PROS1, SLC7A8, GAS6, MERTK, ICAM1, TNFRSF11A, DST | CCL2, IL1B, ITGAX, VEGFA, SPP1, CXCL16, TNF |
| Leucocyte migration | INPP5D, CSF3R, PROS1, SLC7A8, GAS6, MERTK, ICAM1, TNFRSF11A | CCL2, IL1B, ITGAX, VEGFA, SPP1, CXCL16, TNF |
| Response to wounding | INPP5d, FGD2, STAB 1, CSF3R, DST, BASP1, ABI1, TIMP2, LIMK1, INPP4B, TANC2, GAB1, MEF2C, BIN1, NUAK1, TJP1, TGFBR1, GAS6, TGFBR2, MERTK, ICAM1, SPARC, CKB, SALL3, TNFRSF11A, FADS1, TGFB1, USP2, RGMB, SEMA4D | TREM2, CCL2, IL1B, ITGAX, PTPRM, ATF3, VEGFA, CNTN1, SPP1, APOE, TNF, CD83, CSF1, PGRMC1 |
| Neutorophil and lymphocyte chemotaxis | CSF3R | SPP1, IL1B, CXCL16, CCL2 |
Differentially regulated pathways in Trem2−/− microglia isolated from adult mice. Enrichment of 122 differentially regulated genes in the Gene Ontology (GO) category biological process by the GeneRanker (Genomatix). Significantly overrepresented GO terms with an adjusted P‐value < 0.05 were deduced. An adjusted P‐value was estimated from the result of 1,000 simulated null hypothesis queries according to Berriz et al 67.
Figure EV1. Heat map of significantly expressed genes.
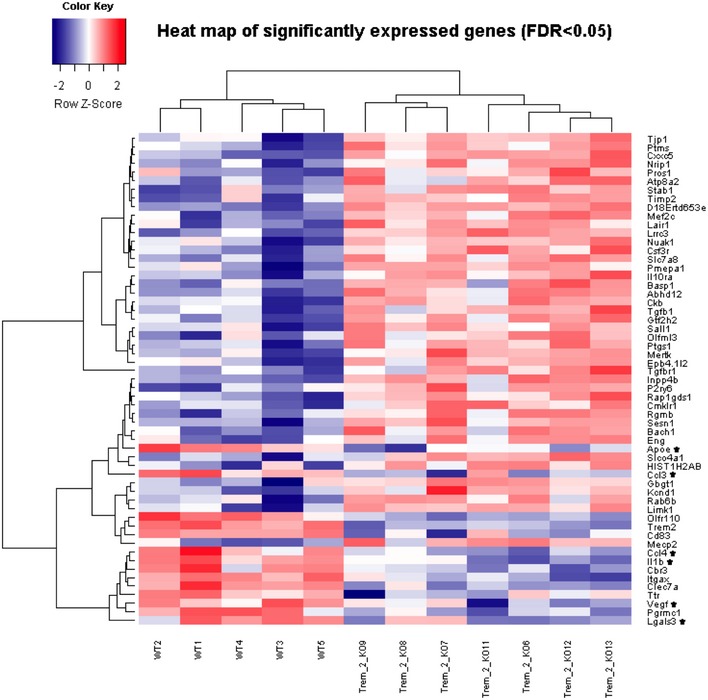
The heat map is based on normalized log2 expression levels of 57 significantly changed genes (FDR < 0.05) from the NanoString‐based chips. For generation of the heat map, the heatmap.2 function within the gplots package of R statistical software was used. Hierarchical clustering by the hclust function (method = “complete”) was applied to group experimental conditions (columns) as well as intensities of genes (rows). Rows were scaled and represented as z‐score. A dendrogram is shown for the samples (columns) as well as for gene expression levels (rows). Genes which show decreased expression levels in Trem2−/− mice (KO) compared to wild‐type mice (WT) and known to be involved in the functional categories chemotaxis and migration are highlighted by an asterisk.
To validate a potential impact of Trem2 loss of function on chemotactic migration, we used independent in vitro, ex vivo and in vivo approaches.
Trem2 deficiency impairs chemotaxis in organotypic slice cultures
First, we utilized our recently established ex vivo assay 34 of co‐culturing of young (P5) WT and old (12–14 months) APPPS1 35 organotypic brain slices. This assay allows the quantitative investigation of chemotactic migration of young microglia towards injured and dying tissue derived from APPPS1 mice (Fig 2A). Microglia were visualized using Cx3cr1‐GFP 36 as a microglia reporter as well as staining of CD68 (Fig 2A) and analysed after 7 days in vitro (DIV). Baseline migration of CD68‐positive cells was observed, when slices from young WT mice were cultured alone, and was significantly reduced when brain slices were prepared from young Trem2−/− mice (Fig 2B; for quantification see Fig 2D). Upon co‐culturing of young WT slices with brain slices derived from old APPPS1 mice, the distance migrated by CD68‐positive cells increased significantly (Fig 2C; for quantification see Fig 2D). This is in striking contrast to CD68‐positive cells devoid of Trem2. Here, the distance migrated was significantly less and no different to that observed when young tissue was cultured alone (Fig 2C and D). This suggests that the old dying tissue may provide chemotactic signals, which attract WT‐ but not Trem2‐deficient microglia and indicate a defect in sensing of certain stimuli and/or a reduced capacity for migration upon a loss of Trem2 function.
Figure 2. Migration deficits of Trem2−/− microglia in an ex vivo assay.
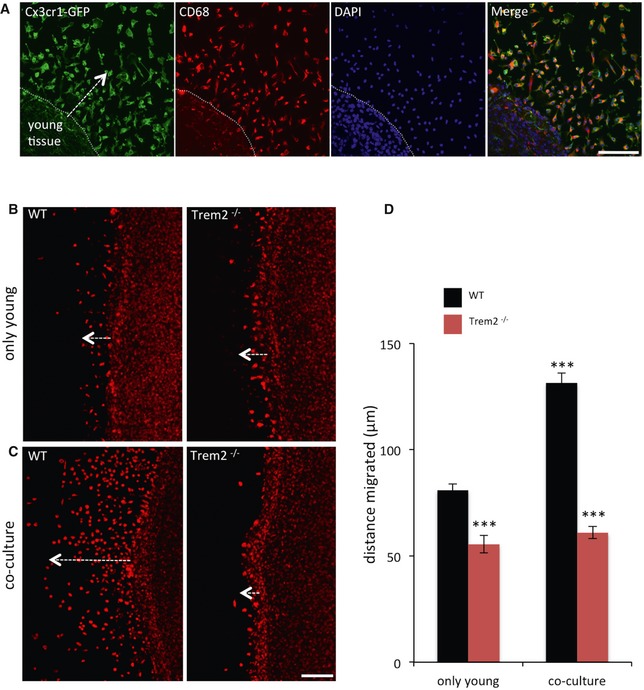
- Cx3cr1‐GFP‐ (green) and CD68 (red)‐positive young microglial cells migrate towards old tissue in co‐cultures of old APP/PS1 and young Cx3cr1+/GFP brain slices (7 DIV). DAPI (blue) was used to counterstain the nuclei.
- CD68‐positive cells migrate out of WT and Trem2−/− brain slices cultured alone (7 DIV). Trem2−/− microglia migrate much shorter distances compared to WT.
- Enhanced migration of CD68‐positive microglia in co‐cultures of old APP/PS1 and young WT brain slices is abolished in the absence of Trem2.
- Quantitative analysis of the distance migrated by WT or Trem2−/− CD68‐positive cells in (B) and (C). N = 4 WT and 4 Trem2−/− mice, from each mouse, two slices were prepared, and on average, 230–550 cells per genotype were analysed. Data are presented as mean ± s.e.m., ***P < 0.001 by using a one‐way analysis of variance with Tukey post hoc comparison. P‐values: WT young only versus Trem2−/− young only: ***P = 0.001; WT young only versus WT co‐culture: ***P = 0.001; Trem2−/− young only versus Trem2−/− co‐culture: P = 0.8999; WT co‐culture versus Trem2−/− co‐culture: ***P = 0.001.
Trem2 deficiency decreases migration towards defined chemo‐attractants
To specifically investigate the migration capacity towards defined chemo‐attractants, we took advantage of a transwell assays 37. Trem2 is known to act via phosphorylation and activation of Syk 38. We therefore tested whether inhibition of Syk activation in the N9 microglia cell line 39 might affect their response to chemokines in a transwell assay. Indeed, migration was almost entirely blocked when N9 microglia were pre‐incubated with the general tyrosine kinase inhibitor genistein or the Syk‐selective inhibitor piceatannol (Fig 3A). Next, we used a CRISPR/CAS9‐modified N9 line, which completely lacks Trem2 expression 40 to address whether Trem2 loss of function affects the response to chemotactic stimuli. Using either CCL2 or C5a as chemotactic stimulus, we observed a significantly decreased migration of Trem2‐deficient N9 microglia towards both chemotactic stimuli (Fig 3B and C). Importantly, re‐expression of mouse Trem2 in the Trem2‐deficient cell line fully rescued the chemotactic response towards C5a (Fig 3D and E).
Figure 3. Migration of N9 microglia cells towards chemotactic stimuli in a transwell assay.
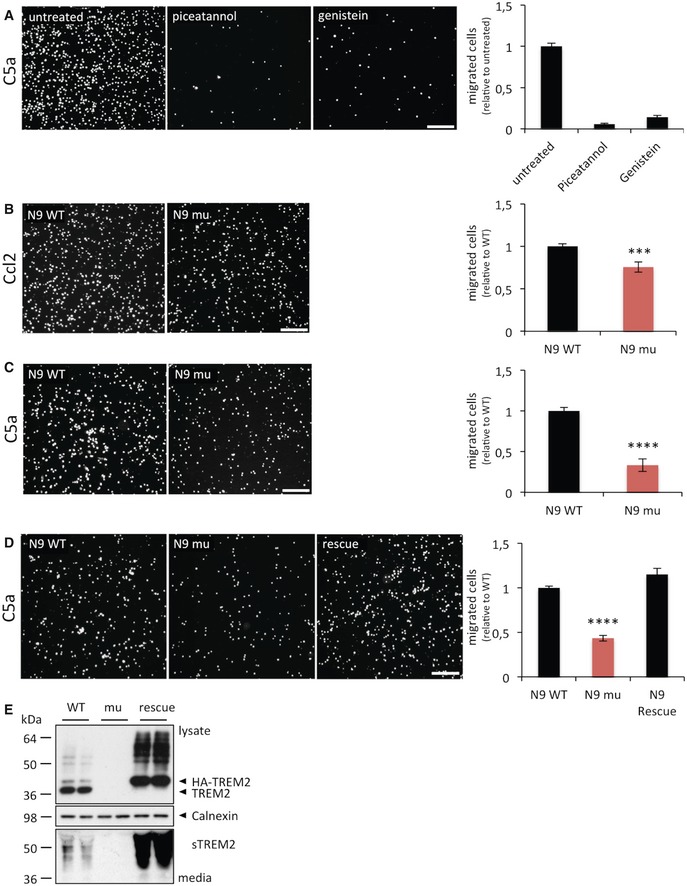
- N9 cell migration in the presence and absence of the general tyrosine kinase inhibitor genistein and the Syk‐selective inhibitor piceatannol. P = 0.00001, N = 12 for each condition.
- Migration capacity of wild‐type N9 microglia (WT) or Trem2‐deficient N9 microglia (N9 mu) towards 100 ng/ml recombinant mouse Ccl2. P = 0.00041, N = 18 for WT and N = 18 for Trem2−/−.
- Migration capacity of wild‐type N9 microglia (WT) or Trem2‐deficient N9 microglia (N9 mu) towards 25 ng/ml recombinant mouse C5a. P = 0.00001, N = 19 for WT and N = 17 for Trem2−/−.
- The migration deficit of Trem2‐deficient N9 cells is fully restored upon expression of mouse Trem2. P = 0.00001, N = 6.
- Western blot analysis of lysates (upper panel) and media (lower panel) from WT, mutant and N9 cells re‐expressing TREM2 using the anti‐murine TREM2 antibody 5F4 raised against the murine TREM2 extracellular domain. sTREM2, soluble TREM2. Calnexin (middle panel), loading control.
Trem2 deficiency impairs chemotactic migration towards apoptotic neurons in vivo
As the innate immune cells of the brain, microglia are the first cells to respond to neuronal damage 16, 41. To test whether the chemotactic deficit that we observed in vitro and ex vivo can be recapitulated in vivo, we measured the ability of microglia to migrate towards apoptotic neurons injected into the brain. Apoptotic neurons are an important hallmark of neurodegeneration, and they are known to release “find‐me” signals to attract phagocytes 42, 43. We stereotactically injected Alexa fluor‐488‐labelled apoptotic neurons in the cortex of adult WT and Trem2−/− mice. Brains were fixed 6 h postinjection and stained with the microglia‐specific antibody anti‐P2ry12 15. WT microglia were attracted to and accumulated around the injected dead neurons as early as 6 h postinjection (Fig 4A). In contrast, we observed a significant reduction in the number of clustered microglia around apoptotic neurons in the absence of Trem2 (Fig 4B; for quantification see Fig 4C). This confirms a critical role of Trem2 in the attraction of microglia towards sites of neuronal injury in vivo.
Figure 4. Reduced migration of Trem2‐deficient microglia towards apoptotic neurons in vivo .
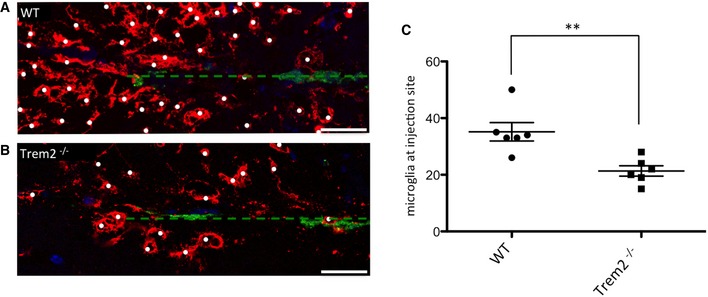
-
A, BMigration of microglia towards injected Alexa 488‐labelled apoptotic neurons (green) in WT (A) and Trem2−/− (B) mouse brains. Microglial cells are visualized with the anti‐P2ry12 antibody (red) and nuclei with DAPI (blue). Dashed line indicates injection channel. Scale bar, 25 μm.
-
CQuantification of the number of microglia (labelled with white dots) clustered around the injection site. N = 6 individual mice per genotype. **P = 0.004 according to a two‐tailed Student's t‐test. The scatter plot shows data points from individual experiments. Horizontal lines represent mean values, error bars indicate standard deviations.
Source data are available online for this figure.
Trem2 deficiency affects outgrowth of microglia processes
Finally, we investigated whether microglia lacking Trem2 are still capable to respond to CNS tissue injury via rapid extension of their processes. To allow in vivo imaging of process extension, we crossed WT and Trem2−/− mice with mice expressing green fluorescent protein (GFP) under the control of the Iba1 promoter 44. In the WT‐ and Trem2‐deficient offspring, we induced a focal CNS damage in the somatosensory cortex using a focussed laser beam 45, 46, 47 and acquired time‐lapse images of the microglial response using in vivo two‐photon microscopy (Fig 5A). We then determined the speed of the microglial process extension towards the lesion site (Fig 5A–C). This revealed a significant delay in the response of Trem2−/− microglia close to the injury (immediate area) but not further away (intermediate and distant areas; Fig 5B; for quantification Fig 5C) confirming that the ability of Trem2‐deficient microglia to react to nervous system damage is reduced in vivo.
Figure 5. Impaired wound response in Trem2‐deficient microglia in vivo .
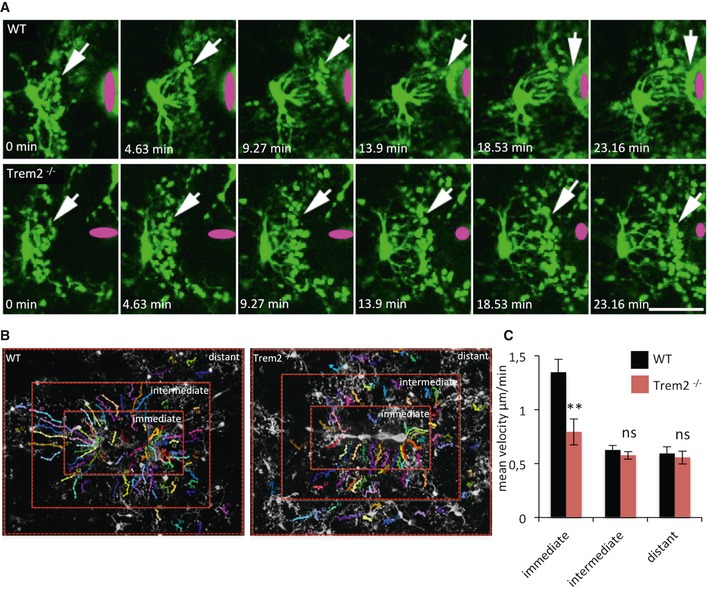
- Early response of Iba1‐GFP WT or Trem2−/− microglia towards a laser lesion (indicated in magenta). White arrows point to the microglial processes. Time between each frame is 278 seconds. Scale bar, 25 μm.
- Areas used to quantitate process outgrowth speeds.
- Quantification of the speed of process outgrowth. Data are presented as mean ± s.e.m., **P = 0.0016 according to Mann–Whitney U‐test (N = number of injuries: WT = 13 injuries in five mice; Trem2−/− = 8 injuries in three mice).
Source data are available online for this figure.
Trem2‐deficient microglia display a homoeostatic mRNA signature
Systems biological studies revealed a homoeostatic physiological mRNA signature 15, which is distinct from mRNA profiles of microglia in neurodegenerative diseases 48, 49. In mouse models for AD and ALS, many genes typically expressed under physiological conditions such as Csf1r, Cx3cr1, Tmem119, Tgfbr1, P2ry12 or Il10ra are selectively downregulated 49, 50. Since apparently microglia lacking TREM2 are unable to respond to various types of chemo‐attractants and neuronal injury, we asked whether in Trem2−/− microglia the homoeostatic mRNA signature is preserved. Microglia were analysed for changes in the expression of selected homoeostatic genes using the above‐described NanoString‐based chips 15 (Fig 6A). This revealed that expression of homoeostatic genes was not suppressed but rather fully maintained and even slightly but significantly increased (Fig 6B).
Figure 6. Sustained expression of microglial homoeostatic genes in the absence of Trem2.
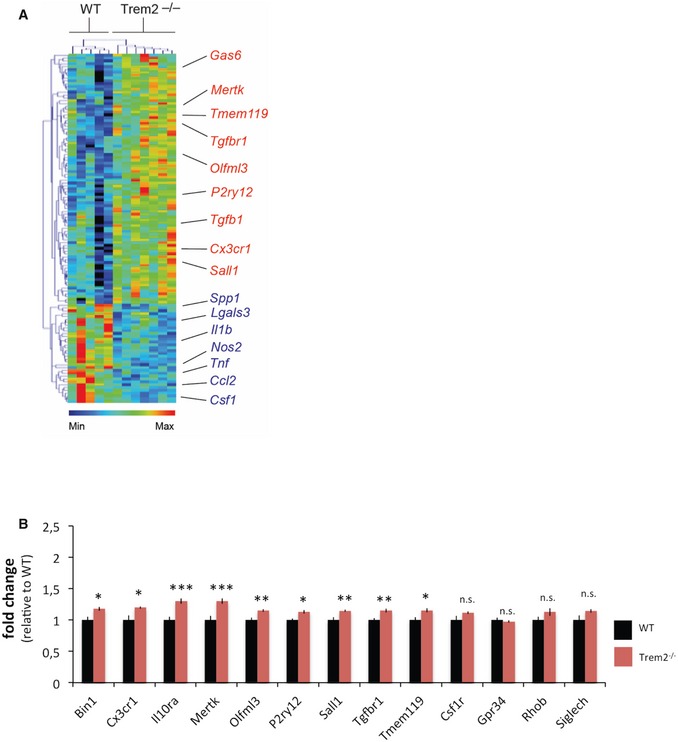
- Heat map demonstrating altered expression levels of microglial genes in FACS‐sorted microglia from WT and Trem2−/− mice as determined by NanoString analysis. Heat map and hierarchical clustering of microglia were analysed with the MG400 chip. Results were log‐transformed, normalized and centred, and populations and genes were clustered by Pearson correlation using MeV v4.6.
- Expression levels of homoeostatic microglial genes in FACS‐sorted microglia derived from WT and Trem2−/− mice as determined by NanoString analysis. Bars show levels of mRNA transcripts in the respective models normalized to six independent housekeeping genes. Values indicate mean ± s.e.m. per 100 ng of total RNA. *P < 0.05, **P < 0.01, ***P < 0.001 by two‐tailed Student's t‐test. P‐values: Trem2−/− versus WT, Bin1 *P = 0.012, Cx3cr1 *P = 0.012, Il10ra ***P = 0.001, Mertk ***P = 0.002, Olfml3 **P = 0.005, P2ry12 *P = 0.011, Sall1 **P = 0.005, Tgfbr1 **P = 0.006, Tmem119 *P = 0.02, Csf1r P = 0.081, Gpr34 P = 0.505, Rhob P = 0.164, Siglech P = 0.062. Data were generated from the same samples as in Fig 1; WT N = 5, Trem2−/− N = 7.
Source data are available online for this figure.
Discussion
Microglia are the resident macrophages of the central nervous system and play a pivotal role in synaptic pruning and apoptotic cell clearance during development of the nervous system 51, 52, 53, 54, 55, 56. In the adult brain, microglia are of central importance for brain homoeostasis. Furthermore, in almost all neurodegenerative disorders, microgliosis is a major pathological hallmark although it is not entirely clear whether activated microglia drive or slow disease progression. Increasing evidence for a pathological role of microglia in onset and progression of neurodegenerative diseases is derived from genomewide association studies, which led to the identification of TREM2 and other microglia‐related risk genes 7, 11. Certain mutations in TREM2 inhibit its maturation and transport to the cell surface and result in reduced phagocytosis, lipid sensing and ApoE binding, thus clearly indicating a loss of function 17, 19, 20, 21, 22, 23. A loss of function of TREM2 in disease is further supported by null mutations in TREM2 and its binding partner DAP12, which are causative for Nasu–Hakola disease 57. However, the molecular and cellular mechanisms responsible for dysfunctional microglia in the absence of TREM2 are not well understood. Understanding such consequences will help to unravel the physiological and pathological function of TREM2 and may open new avenues for therapeutic modulation of neurodegenerative disorders. Moreover, systems analyses may shed light on the important question whether microglia prevent or promote disease progression. To obtain insights into Trem2‐associated functional pathways, we isolated microglia from brains of WT or Trem2−/− mice using previously characterized highly selective antibodies 15 and compared their homoeostatic mRNA signature using the NanoString technology. We found that in the absence of Trem2, the homoeostatic signature was not suppressed, since many of the characteristic genes found to be expressed under resting conditions were expressed at even slightly higher levels. Interestingly, one of the genes whose expression is even slightly upregulated is Sall1. Sall1 acts to maintain microglial identity in vivo, and its inactivation leads to the conversion of microglia from resting into an inflammatory state 58. Thus, failure of Sall1 suppression in the absence of Trem2 may further stabilize a resting state of microglia. Moreover, microglia deficient for Trem2 showed a significant increase in TGFbr1. TGFb1 signalling is required for maintaining the unique molecular signature that is characteristic for adult resting microglia in vivo 15. Furthermore, expression of TGFbr1 is downregulated in a mouse model for familial amyotrophic lateral sclerosis (ALS) as well as in human sporadic and familial ALS 48. Based on these findings, we predict that a loss of function of Trem2 may lock microglia in a resting state and prevents them from being activated. Since several loss‐of‐function variants of TREM2 are associated with neurodegenerative conditions in humans, this also suggests that a normal function of TREM2 may be a protective response, which could be involved in slowing disease progression. This is in line with the finding that sTREM2 increases during healthy ageing, which may reflect an increase of TREM2 function via enhanced TREM2 levels on the cell surface 24, 27, 59. Moreover, sTREM2 was even further enhanced in asymptomatic stages of patients of the DIAN cohort 27 and in patients with very mild cognitive impairment in a sporadic AD cohort 24. The increase of sTREM2 was also associated with increased CSF levels of total tau and phosphorylated tau suggesting that TREM2 expression and function may be stimulated by injured neurons and that the failure to appropriately respond to such injuries may be associated with accelerated disease progression. Moreover, failure of Trem2−/− microglia to appropriately respond to chemotactic stimuli, which we confirmed by a wide variety of independent assays, is also in line with a protective function of WT TREM2. This is also consistent with the observation that microglia fail to cluster around amyloid plaques in the absence of Trem2 31, 60, 61. Moreover, treatment of microglia with TGFb1 attenuates microglial migration towards aggregated amyloid β‐peptide by dysregulating chemotactic genes 62. This finding is consistent with the upregulation of Tgfbr1 in the absence of Trem2 and our prediction that microglia are locked in a resting state when Trem2 is absent or dysfunctional due to disease‐associated sequence variants. However, we performed all phenotypic analyses in the complete absence of Trem2. It therefore remains to be shown if single AD‐associated point mutations expressed at the endogenous level would also result in similar loss‐of‐function phenotypes.
Materials and Methods
Animal experiments and licence
The Cx3cr1GFP/GFP reporter line 36 was obtained from the Jackson Laboratory and bred with C57BL/6J WT to obtain CX3CR1+/GFP mice. Iba1‐GFP mice are described in 44.
Hemizygous APPPS1 mice overexpressing human APPKM670/671NL and PS1L166P under the control of the Thy‐1 promoter (line 21) were kindly provided by Mathias Jucker (Hertie‐Institute for Clinical Brain Research, University of Tübingen and DZNE‐Tübingen) 35 and bred in a C57BL/6J background. Trem2 KO mice were kindly provided by Marco Colonna (Washington University, School of Medicine) 63.
All animal experiments were performed in accordance with local animal handling laws.
Microglia isolation
For microglia isolation, adult male mice (3.5–4 months) were sacrificed by CO2 followed by a cervical dislocation. Brains were homogenized in HBSS by mechanical dissociation; cell suspensions were filtered through a 100‐μm cell strainer and separated in a 40–70% Percoll gradient (GE Healthcare). Mononuclear cells were collected from the interface and washed once with blocking buffer (0.2% bovine serum albumin in HBSS). These cells were then stained sequentially with monoclonal FCRLS 15 and CD11b antibodies (BD Biosciences) and sorted using a Becton Dickinson FACSARIA II cell sorter. Total RNA was isolated and gene expression profiling was performed using the NanoString technology as described 15, 48.
Data normalization and analysis
Gene expression levels in each sample were normalized against the geometric mean of six housekeeping genes including Cltc, Gapdh, Gusb, Hprt1, Pgk1 and Tubb5. Based on the normalized gene expression levels of NanoString‐based chips, a two‐tailed Student's t‐test assuming equal variance was applied to each gene to compare the difference between the Trem2−/− group consisting of seven mice and the WT group consisting of five mice. A false discovery rate (FDR) for each P‐value was calculated according to Benjamini and Hochberg 64 by using p.adjust (method = “BH”) in R statistical software (www.r-project.org). Entrez Gene IDs of genes which show P < 0.05 were then imported into the GeneRanker program from Genomatix (Munich, Germany) by using the database versions ElDorado 12‐2013 and Genomatix Literature Mining 11‐2013 to perform an enrichment analysis on the Gene Ontology (GO) functional categories.
Organotypic slice cultures and ex vivo migration assay
Organotypic slice cultures were performed and immunostained as described by Daria et al 34. In the co‐culture paradigm, two slices of young P5 tissue (WT, Trem2−/− or Cx3cr1+/GFP) were plated together with two slices of old (10–12 months) APPPS1 tissue. The migration distance was measured 7 days after the culturing ex vivo. The migration distance was measured using Fiji 65 by drawing a line between the migrated cell and the slice culture border. In total, up to 550 (WT) and 400 (Trem2−/−) cells were analysed from eight slices (four mice per genotype).
Cell migration in transwell assays
N9 cells 39 and the CRISPR/CAS9‐generated TREM2 mutant cell line were described before 40. Re‐expression of WT TREM2 in TREM2 knockout cells was achieved by transfecting N‐terminal tagged full‐length murine Trem2 in a pcDNA3.1 vector. Twenty‐four hours after transfection, cells were selected with 200 μg/ml Zeocin for 2 weeks. Single‐cell clones were cultured in media containing 100 μg/ml Zeocin medium, followed by Western blot analysis.
N9 cell migration towards 100 ng/ml of recombinant murine JE/MCP‐1 (CCL2); Peprotech) or 20 ng/ml of recombinant murine complement component C5a (R&D Systems) was tested in transwell assays (COSTAR 24‐well plate with inserts, 8‐μm pore, Corning, NY). For Syk inhibition, cells were incubated overnight in 50 μM genistein (Sigma) or piceatannol (Tocris).
For the transwell assays, 600 μl media (DMEM complemented with GlutaMAX™ (Life Technologies)) together with the attractant were added to the bottom well and 5 × 105 cells were cultured in 100 μl media in the top well. Migration was assayed for 2 h at 37°C and 5% CO2. Subsequently, inserts were removed and washed three times with PBS. Cells were then fixed with 4% PFA for 15 min, followed by permeabilization with PBS containing 0.2% Triton X‐100. Stationary cells were removed from the top, and cells that migrated through the filter to the lower side of the transwell filter were stained with DAPI and images were taken. Two fields for each well were selected for imaging, and the migrated cells were quantified using Fiji software.
Stereotactic injection of apoptotic neurons
To induce apoptosis, neurons were carefully detached from the plate surface by repeated washes with PBS. Neurons were irradiated with UV light (302 nm) with an intensity of 6 × 15 W for 15 min. Cells were harvested by centrifugation, and the pellet processed for downstream applications. Neurons were incubated for 15 min at 37°C with 2 μl of the labelling dye (Alexa488 5‐SDP Ester or Alexa405 5‐SDP, Life Technologies). To block and capture residual dye, cells were resuspended in 1 ml FBS and washed twice with PBS. Total apoptotic cell number was determined using trypan blue staining and resuspended at a density of ~100,000 cells per 2 μl for stereotactic injection. Mice were anesthetized by i.p. injection of a mixture of ketamine (100 mg/kg), xylazine (10 mg/kg) and acepromazine (3 mg/kg); 2 μl of Alexa488‐labelled apoptotic neurons or saline solution was distributed intrahippocampal and intracortical bilaterally (Y: ± 1.5 mm; X: −2 mm; Z: −2 mm and −1 mm) using stereotaxic equipment (Harvard Apparatus). After recovery from surgery, animals were returned to their home cages. Postsurgery (16 h), mice were euthanized by CO2 inhalation and perfused for subsequent experiments.
For histological analyses of microglia migration towards Alexa 488‐labelled and injected apoptotic neurons, brains were perfused with ice‐cold HBSS, fixed in 4% buffered formalin and subsequently embedded in paraffin. Stepwise sections were taken and paraffin sections (3 μm) were collected when the injection channel was clearly visible. After dewaxing, antigen retrieval was performed by boiling the sections for 30 min in 10 mM citrate buffer, pH 6.0. Sections were washed, permeabilized with 0.2% Triton X‐100 (Roche) in TBS and blocked for 1 h (Protein‐Free T20 (TBS) Blocking buffer #37071 Thermo Fischer). The microglia‐specific antibody P2ry12 15 was incubated in blocking buffer overnight at 4°C with constant shaking, followed by intensive washing and incubation with an Alexa‐555‐coupled anti‐rabbit secondary antibody for 1 h. Sections were washed again and mounted with DAPI‐Fluoromount‐G (SouthernBiotech). Data acquisition was performed using a Leica Sp5 confocal microscope and Leica application suite software (LAS‐AF‐lite).
In vivo imaging of process outgrowth
To image the microglia process outgrowth upon laser lesion in controlled conditions, cranial windows were implanted over the somato‐sensory cortex of Iba1‐GFP WT and Iba1‐GFP/Trem2−/− mice 14 days prior to imaging and laser lesion induction (for details, see 66). On the day of imaging, animals were anaesthetized using a mixture of medetomidine, midazolom and fentanyl (MMF; 150–200 μl/kg) 30 min prior to the start of the experiment. Laser lesions of ~150 μm length were performed 30 μm below the cortical surface, and the microglia response was recorded every 5 min for 90 min over an area of 250,000 μm2 from the cortical surface to 70 μm depth.
The extension of individual processes was tracked using the mTrackJ plug‐in of the Fiji software 65 package. Briefly, cells located in a field of 20,000, 60,000 or 120,000 μm2 (immediate, intermediate and distant areas, respectively) around the injury were selected for manual tracking. The data were exported to Excel for analysis, and the mean velocity of the extension was calculated for each microglia process. More than 200 outgrowing tracks from a minimum of three independent experiments were analysed for each experimental condition manner. Data analysis was confirmed by independent co‐workers in a blinded manner.
MG400 microglia nCounter chip design
The MG400 chip was designed using the quantitative NanoString nCounter platform. Selection of genes is based on analyses that identified genes and proteins, which are specifically or highly expressed in adult mouse microglia 15.
Author contributions
CH and FM conceived the study and analysed the results. CH wrote the manuscript with help from all co‐authors. FM carried out the transwell assays, isolated microglia, prepared RNA samples, performed the ex vivo, in vitro and in vivo data analyses. GK helped with the microglia isolation and interpretation of the results. AD and ST conducted the ex vivo culturing and immunofluorescence analysis. GW generated the genetically modified N9 cells. NS, TM and MK conceived and performed the two‐photon imaging experiments. DT and WW performed the pathway analysis. AC helped with the transwell assays and generation of the N9 mutant cell line. BB provided technical assistance. SB assisted with cell sorting. OB, CM and SK conducted the NanoString analysis and injection of apoptotic neurons.
Conflict of interest
C.H. is an advisor of F. Hoffmann‐La Roche. All other authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) within the framework of the Munich Cluster for Systems Neurology (EXC 1010 SyNergy), the European Research Council under the European Union's Seventh Framework Program (FP7/2007–2013)/ERC Grant Agreement No. 321366‐Amyloid, the general legacy of Mrs. Ammer, the MetLife award and the Cure Alzheimer's Fund. We thank Kristin Hartmann for technical assistance. T.M was further supported by the Center for Integrated Protein Science Munich (CIPSM, EXC 114), SFB 870 and Research Grants Mi 694/4‐1(SPP1710)/7‐1/8‐1. Further support came from the European Research Council under the European Union's Seventh Framework Program (FP/2007‐2013; ERC Grant Agreement n. 616791) and the Hans‐and‐Ilse‐Breuer Foundation. This work was supported by the German Science Foundation Collaborative Research Centre (CRC) 870. In addition, this work was funded in part by the Helmholtz Portfolio Theme “Supercomputing and Modeling for the Human Brain” (SMHB). O.B. is supported by NIH‐NINDS (1R01NS088137), NIA (1R01AG051812), National Multiple Sclerosis Society (5092A1), Amyotrophic Lateral Sclerosis Association (ALSA2087) and the Nancy Davis Foundation. We thank Jochen Herms for providing Iba1‐GFP mice.
EMBO Reports (2017) 18: 1186–1198
References
- 1. Aguzzi A, Haass C (2003) Games played by rogue proteins in prion disorders and Alzheimer's disease. Science 302: 814–818 [DOI] [PubMed] [Google Scholar]
- 2. Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid beta‐peptide. Nat Rev Mol Cell Biol 8: 101–112 [DOI] [PubMed] [Google Scholar]
- 3. Jucker M, Walker LC (2013) Self‐propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501: 45–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss‐Coray T, Vitorica J, Ransohoff RM et al (2015) Neuroinflammation in Alzheimer's disease. Lancet Neurol 14: 388–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Borroni B, Ferrari F, Galimberti D, Nacmias B, Barone C, Bagnoli S, Fenoglio C, Piaceri I, Archetti S, Bonvicini C et al (2014) Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging 35: 934.e7–934.e10 [DOI] [PubMed] [Google Scholar]
- 6. Cady J, Koval ED, Benitez BA, Zaidman C, Jockel‐Balsarotti J, Allred P, Baloh RH, Ravits J, Simpson E, Appel SH et al (2014) TREM2 variant p. R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol 71: 449–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guerreiro R, Bilgic B, Guven G, Bras J, Rohrer J, Lohmann E, Hanagasi H, Gurvit H, Emre M (2013) Novel compound heterozygous mutation in TREM2 found in a Turkish frontotemporal dementia‐like family. Neurobiol Aging 34: 2890.e1–2890.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guerreiro R, Hardy J (2013) TREM2 and neurodegenerative disease. N Engl J Med 369: 1569–1570 [DOI] [PubMed] [Google Scholar]
- 9. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S et al (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368: 117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Guerreiro RJ, Lohmann E, Bras JM, Gibbs JR, Rohrer JD, Gurunlian N, Dursun B, Bilgic B, Hanagasi H, Gurvit H et al (2013) Using exome sequencing to reveal mutations in TREM2 presenting as a frontotemporal dementia‐like syndrome without bone involvement. JAMA Neurol 70: 78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ et al (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368: 107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, Hatanpaa KJ, Lomen‐Hoerth C, Kertesz A, Bigio EH et al (2013) TREM2 in neurodegeneration: evidence for association of the p. R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener 8: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klunemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW, De Bleecker JL, Rossor MN, Marienhagen J, Klein HE et al (2005) The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology 64: 1502–1507 [DOI] [PubMed] [Google Scholar]
- 14. Colonna M, Wang Y (2016) TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci 17: 201–207 [DOI] [PubMed] [Google Scholar]
- 15. Butovsky O, Jedrychowski MP, Moore CS, Cialic R, Lanser AJ, Gabriely G, Koeglsperger T, Dake B, Wu PM, Doykan CE et al (2014) Identification of a unique TGF‐beta‐dependent molecular and functional signature in microglia. Nat Neurosci 17: 131–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ransohoff RM (2016) A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19: 987–991 [DOI] [PubMed] [Google Scholar]
- 17. Kleinberger G, Yamanishi Y, Suarez‐Calvet M, Czirr E, Lohmann E, Cuyvers E, Struyfs H, Pettkus N, Wenninger‐Weinzierl A, Mazaheri F et al (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6: 243ra86 [DOI] [PubMed] [Google Scholar]
- 18. Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, Walter J (2013) Sequential proteolytic processing of the triggering receptor expressed on myeloid cells‐2 (TREM2) protein by ectodomain shedding and gamma‐secretase‐dependent intramembranous cleavage. J Biol Chem 288: 33027–33036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Colonna M (2003) TREMs in the immune system and beyond. Nat Rev Immunol 3: 445–453 [DOI] [PubMed] [Google Scholar]
- 20. Sieber MW, Jaenisch N, Brehm M, Guenther M, Linnartz‐Gerlach B, Neumann H, Witte OW, Frahm C (2013) Attenuated inflammatory response in triggering receptor expressed on myeloid cells 2 (TREM2) knock‐out mice following stroke. PLoS ONE 8: e52982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Atagi Y, Liu CC, Painter MM, Chen XF, Verbeeck C, Zheng H, Li X, Rademakers R, Kang SS, Xu H et al (2015) Apolipoprotein E is a ligand for triggering receptor expressed on myeloid cells 2 (TREM2). J Biol Chem 290: 26043–26050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yeh Felix L, Wang Y, Tom I, Gonzalez Lino C, Sheng M (2016) TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid‐beta by microglia. Neuron 91: 328–340 [DOI] [PubMed] [Google Scholar]
- 23. Bailey CC, DeVaux LB, Farzan M (2015) The triggering receptor expressed on myeloid cells 2 binds apolipoprotein E. J Biol Chem 290: 26033–26042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Suarez‐Calvet M, Kleinberger G, Araque Caballero MA, Brendel M, Rominger A, Alcolea D, Fortea J, Lleo A, Blesa R, Gispert JD et al (2016) sTREM2 cerebrospinal fluid levels are a potential biomarker for microglia activity in early‐stage Alzheimer's disease and associate with neuronal injury markers. EMBO Mol Med 8: 466–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Piccio L, Deming Y, Del‐Aguila JL, Ghezzi L, Holtzman DM, Fagan AM, Fenoglio C, Galimberti D, Borroni B, Cruchaga C (2016) Cerebrospinal fluid soluble TREM2 is higher in Alzheimer disease and associated with mutation status. Acta Neuropathol 131: 925–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Heslegrave A, Heywood W, Paterson R, Magdalinou N, Svensson J, Johansson P, Ohrfelt A, Blennow K, Hardy J, Schott J et al (2016) Increased cerebrospinal fluid soluble TREM2 concentration in Alzheimer's disease. Mol Neurodegener 11: 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Suarez‐Calvet M, Araque Caballero MA, Kleinberger G, Bateman RJ, Fagan AM, Morris JC, Levin J, Danek A, Ewers M, Haass C et al (2016) Early changes in CSF sTREM2 in dominantly inherited Alzheimer's disease occur after amyloid deposition and neuronal injury. Sci Transl Med 8: 369ra178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, Holtzman DM (2014) Altered microglial response to Abeta plaques in APPPS1‐21 mice heterozygous for TREM2. Mol Neurodegener 9: 20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S et al (2016) TREM2‐mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med 213: 667–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yuan P, Condello C, Keene CD, Wang Y, Bird TD, Paul SM, Luo W, Colonna M, Baddeley D, Grutzendler J (2016) TREM2 haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron 90: 724–739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, Xu G, Margevicius D, Karlo JC, Sousa GL et al (2015) TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer's disease mouse models. J Exp Med 212: 287–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ma J, Zhou Y, Xu J, Liu X, Wang Y, Deng Y, Wang G, Xu W, Ren R, Liu X et al (2014) Association study of TREM2 polymorphism rs75932628 with late‐onset Alzheimer's disease in Chinese Han population. Neurol Res 36: 894–896 [DOI] [PubMed] [Google Scholar]
- 33. Zhang WQ, Huang SH, Huang X, Li JH, Ye P, Xu J, Zheng PZ, Shen HY, Huang JR (2016) Regulation of human mesenchymal stem cell differentiation by TREM‐2. Hum Immunol 77: 476–482 [DOI] [PubMed] [Google Scholar]
- 34. Daria A, Colombo A, Llovera G, Hampel H, Willem M, Liesz A, Haass C, Tahirovic S (2016) Young microglia restore amyloid plaque clearance of aged microglia. EMBO J 36: 583–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Radde R, Bolmont T, Kaeser SA, Coomaraswamy J, Lindau D, Stoltze L, Calhoun ME, Jaggi F, Wolburg H, Gengler S et al (2006) Abeta42‐driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep 7: 940–946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20: 4106–4114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Badie B, Schartner J, Klaver J, Vorpahl J (1999) In vitro modulation of microglia motility by glioma cells is mediated by hepatocyte growth factor/scatter factor. Neurosurgery 44: 1077–1082; discussion 1082‐3 [DOI] [PubMed] [Google Scholar]
- 38. Paradowska‐Gorycka A, Jurkowska M (2013) Structure, expression pattern and biological activity of molecular complex TREM‐2/DAP12. Hum Immunol 74: 730–737 [DOI] [PubMed] [Google Scholar]
- 39. Ferrari D, Villalba M, Chiozzi P, Falzoni S, Ricciardi‐Castagnoli P, Di Virgilio F (1996) Mouse microglial cells express a plasma membrane pore gated by extracellular ATP. J Immunol 156: 1531–1539 [PubMed] [Google Scholar]
- 40. Xiang X, Werner G, Bohrmann B, Liesz A, Mazaheri F, Capell A, Feederle R, Knuesel I, Kleinberger G, Haass C (2016) TREM2 deficiency reduces the efficacy of immunotherapeutic amyloid clearance. EMBO Mol Med 8: 992–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kettenmann H, Hanisch UK, Noda M, Verkhratsky A (2011) Physiology of microglia. Physiol Rev 91: 461–553 [DOI] [PubMed] [Google Scholar]
- 42. Medina CB, Ravichandran KS (2016) Do not let death do us part: “find‐me” signals in communication between dying cells and the phagocytes. Cell Death Differ 23: 979–989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ravichandran KS, Lorenz U (2007) Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol 7: 964–974 [DOI] [PubMed] [Google Scholar]
- 44. Hirasawa T, Ohsawa K, Imai Y, Ondo Y, Akazawa C, Uchino S, Kohsaka S (2005) Visualization of microglia in living tissues using Iba1‐EGFP transgenic mice. J Neurosci Res 81: 357–362 [DOI] [PubMed] [Google Scholar]
- 45. Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo . Science 308: 1314–1318 [DOI] [PubMed] [Google Scholar]
- 46. Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S, Littman DR, Dustin ML, Gan WB (2005) ATP mediates rapid microglial response to local brain injury in vivo . Nat Neurosci 8: 752–758 [DOI] [PubMed] [Google Scholar]
- 47. Fourgeaud L, Traves PG, Tufail Y, Leal‐Bailey H, Lew ED, Burrola PG, Callaway P, Zagorska A, Rothlin CV, Nimmerjahn A et al (2016) TAM receptors regulate multiple features of microglial physiology. Nature 532: 240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Butovsky O, Jedrychowski MP, Cialic R, Krasemann S, Murugaiyan G, Fanek Z, Greco DJ, Wu PM, Doykan CE, Kiner O et al (2015) Targeting miR‐155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol 77: 75–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Holtman IR, Raj DD, Miller JA, Schaafsma W, Yin Z, Brouwer N, Wes PD, Moller T, Orre M, Kamphuis W et al (2015) Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: a co‐expression meta‐analysis. Acta Neuropathol Commun 3: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chiu IM, Morimoto ET, Goodarzi H, Liao JT, O'Keeffe S, Phatnani HP, Muratet M, Carroll MC, Levy S, Tavazoie S et al (2013) A neurodegeneration‐specific gene‐expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep 4: 385–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet‐Wadiche LS, Tsirka SE, Maletic‐Savatic M (2010) Microglia shape adult hippocampal neurogenesis through apoptosis‐coupled phagocytosis. Cell Stem Cell 7: 483–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Schafer Dorothy P, Lehrman Emily K, Kautzman Amanda G, Koyama R, Mardinly Alan R, Yamasaki R, Ransohoff Richard M, Greenberg Michael E, Barres Ben A, Stevens B (2012) Microglia sculpt postnatal neural circuits in an activity and complement‐dependent manner. Neuron 74: 691–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schafer DP, Lehrman EK, Stevens B (2013) The “quad‐partite” synapse: microglia‐synapse interactions in the developing and mature CNS. Glia 61: 24–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B et al (2007) The classical complement cascade mediates CNS synapse elimination. Cell 131: 1164–1178 [DOI] [PubMed] [Google Scholar]
- 55. Mazaheri F, Breus O, Durdu S, Haas P, Wittbrodt J, Gilmour D, Peri F (2014) Distinct roles for BAI1 and TIM‐4 in the engulfment of dying neurons by microglia. Nat Commun 5: 4046 [DOI] [PubMed] [Google Scholar]
- 56. Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L et al (2011) Synaptic pruning by microglia is necessary for normal brain development. Science 333: 1456–1458 [DOI] [PubMed] [Google Scholar]
- 57. Paloneva J, Kestila M, Wu J, Salminen A, Bohling T, Ruotsalainen V, Hakola P, Bakker AB, Phillips JH, Pekkarinen P et al (2000) Loss‐of‐function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet 25: 357–361 [DOI] [PubMed] [Google Scholar]
- 58. Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, Nishinakamura R, Becher B, Greter M (2016) Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol 17: 1397–1406 [DOI] [PubMed] [Google Scholar]
- 59. Henjum K, Almdahl IS, Arskog V, Minthon L, Hansson O, Fladby T, Nilsson LN (2016) Cerebrospinal fluid soluble TREM2 in aging and Alzheimer's disease. Alzheimers Res Ther 8: 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ulrich JD, Holtzman DM (2016) TREM2 function in Alzheimer's Disease and neurodegeneration. ACS Chem Neurosci 7: 420–427 [DOI] [PubMed] [Google Scholar]
- 61. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH et al (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160: 1061–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Huang WC, Yen FC, Shie F‐S, Pan C‐M, Shiao Y‐J, Yang C‐N, Huang F‐L, Sung Y‐J, Tsay H‐J (2010) TGF‐β1 blockade of microglial chemotaxis toward Aβ aggregates involves SMAD signaling and down‐regulation of CCL5. J Neuroinflammation 7: 11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Turnbull IR, Gilfillan S, Cella M, Aoshi T, Miller M, Piccio L, Hernandez M, Colonna M (2006) Cutting edge: TREM‐2 attenuates macrophage activation. J Immunol 177: 3520–3524 [DOI] [PubMed] [Google Scholar]
- 64. Benjamini YHY (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B 57: 289–300 [Google Scholar]
- 65. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Holtmaat A, Bonhoeffer T, Chow DK, Chuckowree J, De Paola V, Hofer SB, Hubener M, Keck T, Knott G, Lee WC et al (2009) Long‐term, high‐resolution imaging in the mouse neocortex through a chronic cranial window. Nat Protoc 4: 1128–1144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Berriz GF, King OD, Bryant B, Sander C, Roth FP (2003) Characterizing gene sets with FuncAssociate. Bioinformatics 19: 2502–2504 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Dataset EV2
Dataset EV3
Review Process File
Source Data for Figure 1
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6