

Abstract
The association between nicotinic acetylcholine receptor (nAChR) dysfunction and cognitive decline in Alzheimer's disease (AD) has been widely exploited for its therapeutic potential. The effects of chronic nicotine exposure on Aβ accumulation have been studied in both humans and animal models, but its therapeutic efficacy for AD neuropathology is still unresolved. To date, no in vivo studies have addressed the consequences of activating nAChRs on tau pathology. To determine the effects of chronic nicotine administration on Aβ and tau pathology, we chronically administrated nicotine to a transgenic model of AD (3xTg-AD) in their drinking water. Here, we show that chronic nicotine intake causes an up-regulation of nicotinic receptors, which correlated with a marked increase in the aggregation and phosphorylation state of tau. These data show that nicotine exacerbates tau pathology in vivo. The increase in tau phosphorylation appears to be due to the activation of p38-mitogen-activated protein kinase, which is known to phosphorylate tau in vivo and in vitro. We also show that the 3xTg-AD mice have an age-dependent reduction of α7nAChRs compared with age-matched nontransgenic mice in specific brain regions. The reduction of α7nAChRs is first apparent at 6 months of age and is restricted to brain regions that show intraneuronal Aβ42 accumulation. Finally, this study highlights the importance of testing compounds designed to ameliorate AD pathology in a model with both neuropathological lesions because of the differential effects it can have on either Aβ or tau.
Keywords: Aβ, tangles, plaques, β-amyloid-3xTg-AD
The loss of cholinergic neurons is a critical event in the pathogenesis of Alzheimer's disease (AD) (1, 2). Acetylcholine (ACh) is a key neuromodulator in the synaptic mechanisms involved with learning and memory (3) and acts through two major receptor subtypes: nicotinic acetylcholine receptors (nAChRs) and muscarinic acetylcholine receptors (mAChRs). Whereas mAChRs are metabotropic, nAChRs are ligand-gated ion channels formed by a combination of five subunits (α, β, γ, δ, and ε), each encoded by a member of a gene superfamily (4). The α7 and α4β2 are two of the major nicotinic receptors subtypes present in the brain (4, 5). For the past several years, a mainstay of AD therapy has been aimed at inhibiting acetylcholinesterase, the enzyme responsible for degrading ACh in the synaptic cleft, and thereby increasing ACh levels in the brain (6). These compounds slow the phenotypical memory impairments; however, their effectiveness is diminished over time.
The AD brain is characterized by two pathological hallmarks: amyloid plaques, which are mainly composed of the Aβ peptide, and neurofibrillary tangles (NFTs), which consist of hyperphosphorylated tau protein. Several studies have shown that nAChRs are selectively reduced in the AD brain, particularly in regions harboring plaques and neurofibrillary tangles, suggesting a potential relationship between nicotinic receptors and AD neuropathology (5, 7–9). Notably, chronic nicotine treatment has also been shown to reduce the plaque burden in amyloid-β precursor protein (APP) transgenic mice, although the steady-state levels of soluble Aβ1–40 and Aβ1–42 remained unaltered (10, 11). These results suggest that nicotine could be used as a possible anti-AD agent. However, the animal model used in this study does not develop tau pathology, and the effects of nicotine administration on this critical neuropathological hallmark could not be addressed. In fact, the effects of nicotine on tau pathology have not been studied in vivo, and to date only two in vitro studies have investigated the relationship between nicotinic receptors and tau pathology. Both studies found that nicotine increases tau phosphorylation in SH-SY5Y and SK-N-MC neuroblastoma cells and in hippocampal synaptosomes (12, 13). In particular, Wang et al. (13) showed that application of nicotine or Aβ1–42 are equally effective at increasing tau phosphorylation in systems enriched in α7nAChRs, such as hippocampal synaptosomes. However, application of nicotine or Aβ1–42 to systems containing low levels of α7nAChRs failed to increase tau phosphorylation, leading the authors to conclude that α7nAChRs mediate Aβ-induced tau pathology (13).
To determine the role of nAChRs in the pathogenesis of AD, we monitored steady-state levels of this receptor subtype in the CNS of 3xTg-AD mice, in which both plaques and tangles develop in a regional and age-dependent manner (14, 15). Here, we report a selective loss of nAChRs in brain regions containing plaques and tangles. We also examined the effect of chronic nicotine administration on the onset of Aβ and tau pathology in young, prepathologic 3xTg-AD mice. We report that SDS-soluble Aβ levels were unaffected by this treatment, consistent with prior findings (10, 11). However, this treatment significantly modulated the onset of the tau pathology, increasing tau hyperphosphorylation and aggregation compared with age- and gender-matched control mice. The mechanism underlying the nicotine-dependent increase in tau phosphorylation is likely mediated by an increase in p38 activity, a Ca2+-dependent kinase capable of phosphorylating tau in vivo (16, 17). These data show that nicotine exposure exacerbates tau pathology in vivo. Although the use of nicotinic receptor agonists may have a positive effect on reducing Aβ plaque burden (11), the potential adverse effects on tau suggests that the use of nicotine as a possible therapeutic drug for AD should be carefully reevaluated.
Materials and Methods
Mice. The 3xTg-AD mice were derived by comicroinjecting two independent transgenes encoding human APPSwe and the human tauP301L (both under control of the mouse Thy1.2 regulatory element) into single-cell embryos harvested from homozygous mutant PS1M146V knockin (PS1-KI) mice (15). For these studies, 1-month-old hemizygous 3xTg-AD and age-matched nontransgenic (NonTg) mice were chronically exposed with either nicotine [-]-1-Methyl-2-[3-pyridyl]pyrrolidine, (Sigma) or vehicle alone (2% sucrose) in their drinking water for 5 months. All mice were given ad libitum access to food and water.
Antibodies. The antibodies used were as follows: anti-Aβ 6E10 (Signet Laboratories, Dedham, MA), anti-Aβ 1560 (Chemicon), both raised against amino acids 1–17 of Aβ, anti-Tau HT7 (raised against amino acids 159–163), AT8 (recognizes phosphorylated Ser-202 and Thr-205), AT180 (recognizes phosphorylated Ser-231) (Innogenetics, Ghent, Belgium), AT270 (recognizes phosphorylated Thr-181), anti-GSK3β-pY216 (BioSource International, Camarillo, CA), anti-β-actin (Sigma), anti-p38, which recognizes phosphorylated Thr-180 and Tyr-182 (Cell Signaling Technology, Beverly, MA), and anti-CDK5, which recognizes amino acids 268–283 (Calbiochem).
Protein Extraction and Western Blot. Brains were homogenized in a solution of 2% SDS in H2O containing 0.7 mg/ml Pepstatin A supplemented with complete Mini protease inhibitor tablets (Roche, Gipf-Oberfrick, Switzerland) and phosphatase inhibitors 1:100 (Calbiochem). The homogenized mixes were sonicated to sheer the DNA and centrifuged at 4°C for 1 h at 100,000 × g. The supernatant was stored as soluble fraction. The pellet was rehomogenized in 70% formic acid and centrifuged as above. The supernatant was stored as the insoluble fraction.
Proteins from the soluble fraction were resolved by SDS/PAGE {10% [bis(2-hydroxyethyl)amino]tris(hydroxymethyl)methane from Invitrogen} under reducing conditions and transferred to a nitrocellulose membrane. The membrane was incubated in a 5% solution of nonfat milk for 1 h at 20°C. After overnight incubation at 4°C with primary antibody, the blots were washed in Tween 20-TBS (T-TBS) (0.02% Tween 20/100 mM Tris, pH 7.5/150 nM NaCl) for 20 min and incubated at 20°C with secondary antibody for 1 h. The blots were washed in T-TBS for 20 min and incubated for 5 min with SuperSignal (Pierce), washed, and exposed.
ELISA. Aβ1–40 and Aβ1–42 levels were measured by using a sensitive sandwich ELISA system. Proteins from the soluble fraction (see above) were loaded directly onto ELISA plates, and formic acid fractions were diluted 1:20 in neutralization buffer (1 M Tris base/0.5 M NaH2PO dibasic) before loading. Maxisorp immunoplates (Nalge Nunc) were coated with monoclonal antibody 20.1, a specific antibody against Aβ1–16 (from William E. Van Nostrand, Stony Brook University, Stony Brook, NY) in coating buffer (0.1 M NaCO3, pH 9.6), and blocked with 3% BSA. Synthetic Aβ standards (Bachem) were defibrillated by dissolving in hexafluoroisopropanol (HFIP) at 1 mg/ml, and the HFIP evaporated with a stream of N2. The defibrillated Aβ was dissolved in DMSO at 1 mg/ml. Standards of both Aβ1–40 and Aβ1–42 were made in antigen capture buffer (20 mM NAH2PO4/2 mM EDTA/0.4 M NaCl/0.5 g of CHAPS/1% BSA, pH 7.0) and loaded onto ELISA plates in duplicate. Samples were loaded in duplicate and incubated overnight at 4°C. Plates were washed and probed with either horseradish peroxidase-conjugated anti-Aβ 35–40 (MM32–13.1.1 for Aβ40) or anti-Aβ 35–42 (MM40–21.3.4 for Aβ42) overnight at 4°C. 3,3′,5,5′-tetramethylbenzidine was used as the chromagen, and the reaction was stopped with the addition of 30% O-phosphoric acid and read at 450 nm on a plate reader (Labsystems, Sunnyvale, CA).
Binding Studies. Mice were killed by CO2 asphyxiation, and brains were extracted and frozen in isopentane at -20°C. Coronal sections (20 μm thick) through selected brain regions were prepared on a cryostat at -20°C and labeled with [125I]epibatidine for measurement of α4β2 nAChR binding sites (18) or [125I]α-bungarotoxin for measurement of α7 nAChRs (19). For [125I]epibatidine binding, slides were preincubated at room temperature for 10 min in buffer (50 mM Tris·HCl/120 mM NaCl/5mMKCl/2.5 mM CaCl2/1mM MgCl2, pH 7.4) and incubated with 0.08 nM radioligand at room temperature for 90 min in the absence and presence of nicotine (300 μM) to define nonspecific binding. Slides were rinsed twice for 10 min in ice-cold buffer, dipped briefly in ice-cold water, and blown dry. For [125I]αbungarotoxin binding, a similar method was used except that the buffer consisted of 50 nM Tris (pH 7.4) with 120 nM NaCl. Slides were incubated at room temperature for 2 h with [125I]αbungarotoxin (5 nM) in the absence and presence of α-cobratoxin (10 μM) to define nonspecific binding. Slides were apposed to Kodak Biomax MR film for 24 h, and the resulting autoradiograms were developed and fixed.
Autoradiograms were quantified with a computer-based image analysis system (mcid, Imaging Research, St. Catherine's, ON, Canada) by using calibrated standards of ref. 19. A calibration curve of optical density against radioligand concentration (fmol/mg of tissue) was constructed by using values of known radioactivity. Optical densities in discrete regions of the autoradiographic images were measured, and corresponding values of radioactivity were determined by interpolation from the calibration curve. Specific binding values in each region were determined by subtracting residual binding in the presence of excess inhibitor from total binding values. Although [125I]epibatidine labels heterogeneous sites within rodent brain, previous pharmacological analysis has shown selective binding of this radioligand to α4β2 nAChRs in the regions chosen for analysis in this study (18).
Immunohistochemistry. Mice were killed by CO2 asphyxiation, and the brains were fixed for 48 h in 4% paraformaldehyde in TBS. Free-floating sections (50 μm thick) were obtained by using a vibratome slicing system. Endogenous peroxidase activity was quenched for 30 min in H2O2, and sections were subsequently incubated in 90% formic acid for 7 min to expose the epitope. The appropriate primary antibody was applied, and sections were incubated overnight at 4°C. Sections were subsequently washed in TBS to remove excess primary antibody. Sections were incubated in the appropriate secondary antibody for 1 h at 20°C. After a final wash of 20 min, slides were developed with diaminobenzidine substrate by using the avidin-biotin horseradish peroxidase system (Vector Laboratories). The primary antibodies were applied at the following dilutions: 1:1,000 for 6E10, 1:3,000 for 1560, 1:1,000 for HT7, 1:200 for AT8, and 1:500 for AT100 and AT270.
Densitometric Analysis. Photomicrographs were taken by using a Zeiss digital camera, imported into the scion image system (National Institutes of Health), and converted to black and white images. Threshold intensity was manually set and kept constant, and the number of pixels were determined for both Aβ and tau immunostained sections. The data were subsequently analyzed by ANOVA or t test comparison by using prism software (GraphPad, San Diego).
Results
Contradictory results have emerged from epidemiological studies of cigarette smoking and the risk of developing AD (20–22). To investigate the in vivo effects of chronic nicotine exposure on the onset of Aβ and tau pathology, prepathologic 1-month-old hemizygous 3xTg-AD and NonTg mice were exposed ad libitum to drinking water spiked with nicotine (also supplemented with 2% sucrose) (Table 1). As a control, age- and genotyped-matched mice were treated with drinking water supplemented with 2% sucrose alone.
Table 1. Nicotine dosing paradigm.
| Day | Concentration, μg/ml |
|---|---|
| 1 | 25 |
| 2–3 | 50 |
| 4–6 | 100 |
| 7–22 | 200 |
| 23–26 | 400 |
| 27 to the end of treatment | 600 |
One-month-old hemizygous 3xTg-AD and NonTg mice (n = 5 per group) were exposed ad libitum to drinking water spiked with nicotine (supplemented with 2% sucrose). The concentration of nicotine was gradually increased during the first month of treatment. As a control, age- and genotype-matched mice were treated with drinking water supplemented with 2% sucrose alone.
α7nAChRs Receptor Levels Are Reduced in the 3xTg-AD Mice. Loss of nAChRs is an established feature of AD (23–26). To determine whether these receptors are reduced in the 3xTg-AD mice, we performed a double-blind receptor-binding study to measure the steady-state levels of α7 and α4β2 nAChRs. No difference in the steady-state levels of the α4β2-nAChRs was found between 3xTg-AD and age-matched NonTg mice across various brain regions (Fig. 1A, compare 3xTg-AD untreated and NonTg untreated). However, we found a significant decrease in the levels of α7nAChRs in the hippocampus, retrosplenial and parietal cortices, and thalamus of 6-month-old 3xTg-AD mice compared with age-matched NonTg mice (Fig. 1B, compare 3xTg-AD untreated and NonTg untreated). At this age, the only apparent neuropathological change in the hippocampus is the intraneuronal accumulation of Aβ42 (Fig. 1 C–H). It is important to note that, in the auditory cortex, a brain region that does not show any intracellular Aβ immunoreactivity at this age (Fig. 1G), the levels of α7nAChRs were not significantly different between 3xTg-AD and NonTg mice (Fig. 1B). Thus, the loss of nAChRs in selective brain regions is another critical aspect of AD neuropathology that is mimicked by the 3xTg-AD and appears to be restricted to brain regions that show intraneuronal Aβ immunoreactivity.
Fig. 1.

Reduction in α7nAChRs levels in selective brain regions correlates with intraneuronal Aβ. (A) Steady-state levels of α4β2nAChRs were not significantly altered between untreated NonTg and 3xTg-AD mice in all brain regions analyzed, except the thalamus, where we found significantly more receptors in the 3xTg-AD mice (P = 0.011). Chronic nicotine administration significantly increased α4β2 levels in all brain regions in the 3xTg-AD and NonTg mice. (B) Steady-state levels of α7nAChRs were significantly lower in untreated 6-month-old 3xTg-AD compared with age-matched untreated NonTg mice. This effect was evident only in brain regions that accumulate intraneuronal Aβ, such as hippocampus, retrosplenial and parietal cortices, and thalamus (P < 0.05). No statistically significant alteration was apparent in the auditory cortex, which does not show any intraneuronal Aβ accumulation at this age (P > 0.05). Chronic nicotine administration did not alter the steady-state levels of α7nAChRs because the levels between treated and untreated mice were not statistically significant. (C–H) Immunohistochemical analysis by using an anti-Aβ42 specific antibody shows the buildup of intraneuronal Aβ in the hippocampus (C), the retrosplenial and parietal cortices (D–F) and the thalamus (G). Intraneuronal Aβ accumulation is not apparent in the auditory cortex at this age (H). E and F show a higher magnification of the retrosplenial and parietal cortex, respectively. Hi, hippocampus; Th, thalamus; Re, retrosplenial cortex; Pa, parietal cortex; Au, auditory cortex; rf, rhinal fissure.
We next used autoradiography binding studies to determine whether the levels of nAChRs were altered as a consequence of nicotine administration. We found that the steady-state levels of α4β2 receptors significantly increased after nicotine exposure in both NonTg and 3xTg-AD mice compared with age-matched untreated mice (Fig. 1A). In contrast, the steady-state levels of α7nAChRs remained unchanged in both NonTg and 3xTg-AD mice (Fig. 1B). These results are consistent with other studies showing up-regulation of α4β2nAChRs after chronic nicotine administration (27).
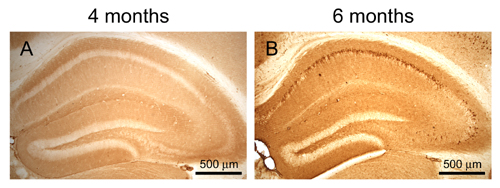
To determine whether there is a temporal relationship between the reduction in the steady-state levels of α7nAChRs and intraneuronal Aβ accumulation, and to exclude the possibility that this reduction is due to an overexpression artifact, we investigated the α7nAChR levels in the hippocampus of 2- and 4-month-old hemizygous 3xTg-AD mice. We focused on the hippocampus because this brain region develops both plaque and tangle pathology, and it is a major structure impacted in AD. We found that, at both 2 and 4 months of age, the 3xTg-AD mice do not show a significant reduction in hippocampal α7nAChR levels. In contrast, by 6 months of age, there is a significant reduction in the α7nAChR levels between 3xTg-AD and age-matched NonTg mice (Fig. 2A). Therefore, we conclude that α7nAChRs are reduced in an age-dependent manner in the 3xTg-AD mice, and the reduction correlates with the buildup of intraneuronal Aβ (Fig. 2 B and C and Fig. 6, which is supporting information on the PNAS web site).
Fig. 2.

Age-dependent reduction in α7nAChRs levels in the hippocampus of 3xTg-AD mice. (A) Quantitative analysis of α-bungarotoxin binding reveals an age-related decrease in α7nAChR steady-state levels in the hippocampus of 3xTg-AD mice beginning at 6 months of age (P < 0.001). At 2 and 4 months of age, no difference in the α7nAChRs steady-state levels is apparent between 3xTg-AD and NonTg mice, indicating that the 3xTg-AD mice are not born with this deficit (P = 0.208 and 0.087 for 2 and 4 month olds, respectively). The reduction in α7nAChRs correlates with intraneuronal Aβ accumulation because 2- and 4-month-old 3xTg-AD mice do not show any immunoreactivity with an anti-Aβ antibody. (B) A representative microphotograph of the hippocampus of a 4-month-old hemizygous 3xTg-AD mouse stained with an Aβ42 specific-antibody. Note the lack of immunostaining in the CA1 pyramidal neurons defined by the arrows. (C) By 6 months of age, hemizygous 3xTg-AD mice show prominent intraneuronal Aβ42 buildup in CA1 pyramidal neurons after staining with an anti-Aβ specific antibody.
Nicotine Treatment Does Not Alter APP Processing. To determine whether APP processing was modulated by chronic nicotine administration, we compared steady-state levels of APP and its derivatives in treated and untreated 3xTg-AD mice. We found no significant difference in the steady-state levels of full-length APP by quantitative Western blot analysis between untreated and treated 3xTg-AD mice (Fig. 3 A and B). Likewise, no statistically significant difference was apparent in C99 steady-state levels (Fig. 3 A and B). Therefore, these results suggest that chronic nicotine exposure does not alter the β-secretase cleavage of APP.
Fig. 3.

APP processing and Aβ deposition are not altered after chronic nicotine administration. (A) Immunoblot shows that APP and C99 levels are not significantly different between treated and untreated 3xTg-AD mice. (B) Quantitative analysis of blots in A after normalizing to β-actin shows that nicotine administration did not significantly alter the steady-state levels of APP or C99. (C and D) Immunohistochemical analysis by using an anti-Aβ42 specific antibody shows that Aβ deposition was not altered after chronic nicotine administration. (E) Densitometric analysis of C and D did not reveal any significant change in the Aβ load in the hippocampus of treated versus untreated 3xTg-AD mice. (F) Sandwich ELISA revealed that Aβ40 and Aβ42 steady-state levels were unaltered after chronic nicotine administration (n = 5 per group). Although there appears to be reduced Aβ40 levels in the treated mice, it did not achieve significance (P = 0.332 and 0.676 for Aβ40 and Aβ42, respectively). (G) The ratio of Aβ42/Aβ40 was also unchanged by the nicotine administration (P = 0.198).
We next determined whether the onset of Aβ accumulation in the brain of the 3xTg-AD mice was altered by nicotine administration. No difference in the extent of intracellular Aβ immunoreactivity in the hippocampus was apparent between treated and untreated 3xTg-AD mice, as determined by immunohistochemical analysis with 6E10- and Aβ42-specific antibodies (compare Fig. 3 C and D and Fig. 7, which is published as supporting information on the PNAS web site). Quantitative assessment confirmed no significant difference in the Aβ load between treated and untreated mice (Fig. 3E). Note that, at this age, the 3xTg-AD mice do not show any extracellular Aβ deposits and nicotine administration did not affect the onset of plaque development.
To determine whether nicotine exposure modulates Aβ levels, we measured Aβ40 and Aβ42 levels by sandwich ELISA. This analysis indicated that the levels of SDS-soluble Aβ40 and Aβ42 were not significantly different between treated and untreated 3xTg-AD mice (Fig. 3F). The ratio of Aβ42/Aβ40 was also unaffected by nicotine administration (Fig. 3G). Levels of SDS-insoluble Aβ40 and Aβ42 were below detection levels at this age. Based on these results, we conclude that chronic nicotine exposure does not modulate APP processing or affect the onset of plaque deposition in this model. These data are in agreement with other in vivo studies showing that nicotine administration does not change SDS-soluble Aβ levels (10, 11).
Tau Phosphorylation Is Enhanced After Nicotine Administration. To determine the effect of chronic nicotine administration on tau pathology, we compared the phosphorylation and aggregation state of tau between treated and untreated 3xTg-AD mice. We focused on the hippocampus because this brain region is the first region to develop tau pathology in the 3xTg-AD mice. At 6 months, the first notable change we typically observe is the occurrence of tau immunoreactivity within the somatodendritic compartment of CA1 pyramidal neurons. In 6-month-old hemizygous mice, even though the CA1 pyramidal neurons show somatodendritic labeling of tau, they remain immunonegative for MC1, AT8, AT180, or PHF-1; it is not until the hemizygous mice are >1 year old that prominent immunostaining for these conformational and phospho-specific epitopes is apparent (14, 15).
We found that administering nicotine for 5 months markedly increased somatodendritic labeling of tau in CA1 pyramidal neurons of 6-month-old 3xTg-AD mice compared with age-matched untreated mice (compare Fig. 4 A and B). Note that, in the treated mice, virtually all of the CA1 neurons show robust tau somatodendritic immunostaining, indicating tau mislocalization from the axonal compartment, an early event in tangle formation (Fig. 4B). Moreover, we found a significant increase in tau phosphorylation at Thr-181 (as detected by phospho-specific anti-tau antibody AT270) in the hippocampus of treated mice (compare Fig. 4 C and D).
Fig. 4.

Phosphorylation and aggregation of tau are enhanced after chronic nicotine administration. Immunohistochemical analysis uses the human-specific anti-tau antibody, HT7 (A and B) and AT270, which recognizes tau phosphorylated at Thr-181 (C and D). Chronic nicotine administration exacerbates tau pathology in the hippocampus of the 3xTg-AD mice because both HT7 and AT270 immunoreactivity is increased. Note the increase in the number of HT7- and AT270-positive neurons in the CA1 subfield in B and D compared with A and C. (E) Immunoblot analysis by using the phospho-specific tau antibody AT270 shows increased tau phosphorylation in the brains of treated versus untreated 3xTg-AD mice. The arrow in E points to the 64-kDa band, which indicates a pathological aggregated form of tau. Note the shift from faster migrating tau species to the slower 64-kDa band, consistent with an increase in the tau aggregation state induced by chronic nicotine administration. (F) Quantitative analysis indicates a significant increase in the 64-kDa band in the brains of treated versus untreated mice (P < 0.005). (G and H) The increase in tau phosphorylation was confirmed by using another phospho-specific tau antibody, AT8, which also shows a significant increase in tau phosphorylation at Ser-202 (P < 0.05).
To better quantify the increase in tau phosphorylation between treated and untreated mice, we measured the levels of the 64-kDa isoform of tau. This species is characteristic of tau found in neurofibrillary tangle in AD, frontotemporal dementia, and parkinsonism linked to chromosome (FTDP-17) brains and indicates the presence of tau aggregates (28). Chronic nicotine administration caused a 2-fold increase in the 64-kDa tau isoform (Fig. 4 E and F). Notably, there is a shift in the electrophoretic mobility of tau that is induced by chronic nicotine treatment from a lower molecular mass band to the 64-kDa species (arrow in Fig. 4E). The increase in tau phosphorylation was also evident by quantitative Western blot analysis by using the phospho-specific anti-tau antibody, AT8, which recognizes phosphorylated tau at Ser-202. The phosphorylation of tau at Ser-202 and Thr-181, induced by chronic nicotine exposure, modulates the kinetics of tau binding to microtubules leading to the destabilization of the neuronal cytoskeleton (29).
Nicotine Administration Increases the Levels of Activated p38-Mitogen-Activated Protein (MAP) Kinase. The finding that tau phosphorylation was increased by chronic nicotine administration raised the question as to the mechanism underlying this process. Several kinases are known to phosphorylate tau in vivo, including many Ca2+-dependent kinases. We used quantitative Western blot analysis to screen a panel of candidate molecules (with antibodies against the activated form of these kinases). We found that the steady-state levels of the active forms of CDK5, ERK1, ERK2 (data not shown), and GSK3β were not significantly different in the brains of treated and untreated 3xTg-AD (Fig. 5 A and B). However, we found a significant increase in the steady-state levels of activated p38-MAP kinase (Fig. 5 C and D). Therefore, based on these findings, we conclude that the increase in tau phosphorylation induced by chronic nicotine administration is likely due to the activation of p38-MAP kinase.
Fig. 5.

Chronic nicotine exposure selectively increases the steady-state levels of activated p38-MAP kinase. To determine which kinase is responsible for the increase in tau phosphorylation after nicotine administration, the levels of several candidate tau kinases were measured by quantitative Western blot analysis. A and B show that the steady-state levels of GSK3α and GSK3β are not significantly altered between treated and untreated 3xTg-AD mice. C and D show that the activated form of p38-MAP kinase is significantly increased in 3xTg-AD mice after chronic nicotine administration (P < 0.05).
Discussion
We explored the role of α7nAChRs in the pathogenesis of AD by analyzing the regional and age-dependent profile of this receptor subtype in a transgenic model of AD. We report that the 3xTg-AD mice show a significant age-dependent decrease in the steady-state levels of α7nAChRs in several brain regions. In the human AD brain, α7nAChRs are also diminished (8, 9, 30); thus, the 3xTg-AD mice mimic another important molecular phenotypic feature of AD neuropathology. In the transgenic brain, we find that the loss of α7nAChRs is preceded by intracellular Aβ accumulation and is restricted to brain regions that develop Aβ pathology, particularly the hippocampus and the cortex. Notably, we did not find a reduction in α7nAChRs in the auditory cortex, a brain region that is spared of any intracellular Aβ immunoreactivity. Therefore, as in the human condition, in the 3xTg-AD mice, the loss of α7nAChRs appears to be restricted to brain regions that develop Aβ pathology.
As a part of this study, we investigated the effects of chronic nicotine administration on the onset of Aβ and tau pathology in the 3xTg-AD mice. We report that APP processing and SDS-soluble Aβ levels were not significantly altered by this chronic treatment. Tau phosphorylation and aggregation, however, were significantly increased in CA1 pyramidal neurons of treated versus untreated age-matched mice. The mechanism underlying the nicotine-induced phosphorylation of tau appears to be selectively mediated by p38-MAP kinase because other putative tau kinases, including GSK3β, Erk1, Erk2, and CDK5, were unaffected by this treatment. Our results concur with prior in vitro studies showing that nicotine application leads to enhanced tau phosphorylation in SH-SY5Y and SK-N-MC neuroblastoma cells and in hippocampal synaptosomes (12, 13). These results are also consistent with an epidemiologic study showing a positive correlation between the amount of smoking and the neurofibrillary tangle load in brains of 301 patients with a known history of smoking (22). Therefore, it appears that nicotine can selectively exacerbate tau pathology.
Cholinergic dysfunction and Aβ accumulation are a central component in the pathogenesis of AD, although the mechanism linking these two features remains to be established. In vitro experiments have shown that Aβ strongly binds to α7nAChRs and subsequently can activate intracellular pathways, thereby altering neuronal function and implicating α7nAChRs as a potential link between Aβ pathology and cholinergic dysfunction (31, 32). Further evidence supporting a link between Aβ and α7nAChRs is the finding that, in the human brain, regions that are more susceptible to AD neuropathology, such as the hippocampus and cortex, express the highest α7nAChRs levels (5). After Aβ binds to α7nAChRs, internalization of the Aβ–nAChRs complex can occur and may lead to a buildup of intracellular Aβ, an event that has been shown to interfere with neuronal function, including proteasome activity (33–37). Aβ may also induce tau pathology by activating different kinases, leading to an increase in tau phosphorylation and microtubule destabilization (13, 38–40). In this regard, it is notable that Aβ binding to α7nAChRs increases intracellular Ca2+ concentration, which in turn can lead to the activation of different kinases (41).
In vitro studies have shown that nicotine administration can mimic some aspects of Aβ-induced pathology, such as the effect on tau phosphorylation, which is mediated by the elevation of intracellular Ca2+ concentration that accompanies activation of nAChRs (12, 13). Higher intracellular Ca2+ levels lead to the activation of Ca2+-dependent kinases, thereby altering the balance between tau phosphorylation and dephosphorylation, resulting in a net increase in the steady-state levels of phosphorylated tau. The results presented here are in vivo evidence showing that an nAChR agonist, such as nicotine, increases tau phosphorylation at Ser-202 and Thr-181. These two residues are known to modulate the kinetics of tau binding to microtubules, which can eventually lead to microtubule instability (29). These data also support the hypothesis that nAChRs may be a link between Aβ and tau pathology, although other mechanisms underlying Aβ-induced tau pathology certainly cannot be excluded, because it is likely that multiple pathways are responsible for Aβ-induced neurotoxicity.
Nicotine has been considered as a potential therapeutic agent in AD (42). Supporting this hypothesis, Nordberg et al. (11) showed that chronic nicotine administration lowered the plaque load in APP transgenic mice with extensive Aβ plaque pathology. Our data are not inconsistent with those findings because they also found that soluble Aβ levels were not altered by this treatment. Nevertheless, our findings suggest that the use of nicotine as a potential therapy for AD should be reevaluated because nicotine can significantly increase the phosphorylation and aggregation state of tau, a fundamental component in AD neuropathology. Therefore, the results presented here highlight the importance of testing compounds designed to ameliorate AD pathology in a model that develops plaques and tangles because of the differential effects that the treatment can have on either Aβ or tau.
Supplementary Material
Acknowledgments
This work was supported by National Institute on Aging Grant AG0212982 (to F. M. LaFerla), Alzheimer's Association Grant IIRG-02-3767 (to F. M. LaFerla), and National Institute on Drug Abuse Grant DA10612 (to F. M. Leslie).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: Ach, acetylcholine; AD, Alzheimer's disease; APP, amyloid-β precursor protein; MAP, mitogen-activated protein; nAChrR, nicotinic acetylcholine receptor; NonTg, nontransgenic.
References
- 1.Davies, P. & Maloney, A. J. (1976) Lancet 2, 1403. [DOI] [PubMed] [Google Scholar]
- 2.Muir, J. L. (1997) Pharmacol. Biochem. Behav. 56, 687-696. [DOI] [PubMed] [Google Scholar]
- 3.Woolf, N. J. (1996) Neurobiol. Learn. Mem. 66, 258-266. [DOI] [PubMed] [Google Scholar]
- 4.Wevers, A. & Schroder, H. (1999) J. Alzheimers Dis. 1, 207-219. [DOI] [PubMed] [Google Scholar]
- 5.Wevers, A., Monteggia, L., Nowacki, S., Bloch, W., Schutz, U., Lindstrom, J., Pereira, E. F., Eisenberg, H., Giacobini, E., de Vos, R. A., et al. (1999) Eur. J. Neurosci. 11, 2551-2565. [DOI] [PubMed] [Google Scholar]
- 6.Ibach, B. & Haen, E. (2004) Curr. Pharm. Des. 10, 231-251. [DOI] [PubMed] [Google Scholar]
- 7.Nordberg, A. (1994) Neurochem. Int. 25, 93-97. [DOI] [PubMed] [Google Scholar]
- 8.Burghaus, L., Schutz, U., Krempel, U., de Vos, R. A., Jansen Steur, E. N., Wevers, A., Lindstrom, J. & Schroder, H. (2000) Brain Res. Mol. Brain Res. 76, 385-388. [DOI] [PubMed] [Google Scholar]
- 9.Banerjee, C., Nyengaard, J. R., Wevers, A., de Vos, R. A., Jansen Steur, E. N., Lindstrom, J., Pilz, K., Nowacki, S., Bloch, W. & Schroder, H. (2000) Neurobiol. Dis. 7, 666-672. [DOI] [PubMed] [Google Scholar]
- 10.Hellstrom-Lindahl, E., Court, J., Keverne, J., Svedberg, M., Lee, M., Marutle, A., Thomas, A., Perry, E., Bednar, I. & Nordberg, A. (2004) Eur. J. Neurosci. 19, 2703-2710. [DOI] [PubMed] [Google Scholar]
- 11.Nordberg, A., Hellstrom-Lindahl, E., Lee, M., Johnson, M., Mousavi, M., Hall, R., Perry, E., Bednar, I. & Court, J. (2002) J. Neurochem. 81, 655-658. [DOI] [PubMed] [Google Scholar]
- 12.Hellstrom-Lindahl, E., Moore, H. & Nordberg, A. (2000) J. Neurochem. 74, 777-784. [DOI] [PubMed] [Google Scholar]
- 13.Wang, H. Y., Li, W., Benedetti, N. J. & Lee, D. H. (2003) J. Biol. Chem. 278, 31547-31553. [DOI] [PubMed] [Google Scholar]
- 14.Oddo, S., Caccamo, A., Kitazawa, M., Tseng, B. P. & LaFerla, F. M. (2003) Neurobiol. Aging 24, 1063-1070. [DOI] [PubMed] [Google Scholar]
- 15.Oddo, S., Caccamo, A., Shepherd, J. D., Murphy, M. P., Golde, T. E., Kayed, R., Metherate, R., Mattson, M. P., Akbari, Y. & LaFerla, F. M. (2003) Neuron 39, 409-421. [DOI] [PubMed] [Google Scholar]
- 16.Hensley, K., Floyd, R. A., Zheng, N. Y., Nael, R., Robinson, K. A., Nguyen, X., Pye, Q. N., Stewart, C. A., Geddes, J., Markesbery, W. R., et al. (1999) J. Neurochem. 72, 2053-2058. [DOI] [PubMed] [Google Scholar]
- 17.Dalrymple, S. A. (2002) J. Mol. Neurosci. 19, 295-299. [DOI] [PubMed] [Google Scholar]
- 18.Perry, D. C., Xiao, Y., Nguyen, H. N., Musachio, J. L., Davila-Garcia, M. I. & Kellar, K. J. (2002) J. Neurochem. 82, 468-481. [DOI] [PubMed] [Google Scholar]
- 19.Ospina, J. A., Broide, R. S., Acevedo, D., Robertson, R. T. & Leslie, F. M. (1998) J. Neurochem. 70, 1061-1068. [DOI] [PubMed] [Google Scholar]
- 20.Lee, P. N. (1994) Neuroepidemiology 13, 131-144. [DOI] [PubMed] [Google Scholar]
- 21.Merchant, C., Tang, M. X., Albert, S., Manly, J., Stern, Y. & Mayeux, R. (1999) Neurology 52, 1408-1412. [DOI] [PubMed] [Google Scholar]
- 22.Ulrich, J., Johannson-Locher, G., Seiler, W. O. & Stahelin, H. B. (1997) Acta Neuropathol. 94, 450-454. [DOI] [PubMed] [Google Scholar]
- 23.Sugaya, K., Giacobini, E. & Chiappinelli, V. A. (1990) J. Neurosci. Res. 27, 349-359. [DOI] [PubMed] [Google Scholar]
- 24.Whitehouse, P. J., Martino, A. M., Antuono, P. G., Lowenstein, P. R., Coyle, J. T., Price, D. L. & Kellar, K. J. (1986) Brain Res. 371, 146-151. [DOI] [PubMed] [Google Scholar]
- 25.Nordberg, A. & Winblad, B. (1986) Neurosci. Lett. 72, 115-119. [DOI] [PubMed] [Google Scholar]
- 26.Nordberg, A., Alafuzoff, I. & Winblad, B. (1992) J. Neurosci. Res. 31, 103-111. [DOI] [PubMed] [Google Scholar]
- 27.Flores, C. M., Rogers, S. W., Pabreza, L. A., Wolfe, B. B. & Kellar, K. J. (1992) Mol. Pharmacol. 41, 31-37. [PubMed] [Google Scholar]
- 28.Spillantini, M. G. & Goedert, M. (1998) Trends Neurosci. 21, 428-433. [DOI] [PubMed] [Google Scholar]
- 29.Xie, H., Litersky, J. M., Hartigan, J. A., Jope, R. S. & Johnson, G. V. (1998) Brain Res. 798, 173-183. [DOI] [PubMed] [Google Scholar]
- 30.Wevers, A., Burghaus, L., Moser, N., Witter, B., Steinlein, O. K., Schutz, U., Achnitz, B., Krempel, U., Nowacki, S., Pilz, K., et al. (2000) Behav. Brain Res. 113, 207-215. [DOI] [PubMed] [Google Scholar]
- 31.Wang, H. Y., Lee, D. H., D'Andrea, M. R., Peterson, P. A., Shank, R. P. & Reitz, A. B. (2000) J. Biol. Chem. 275, 5626-5632. [DOI] [PubMed] [Google Scholar]
- 32.Dineley, K. T., Westerman, M., Bui, D., Bell, K., Ashe, K. H. & Sweatt, J. D. (2001) J. Neurosci. 21, 4125-4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tseng, B. P., Kitazawa, M. & LaFerla, F. M. (2004) Curr. Alzheimer Res. 1, 231-239. [DOI] [PubMed] [Google Scholar]
- 34.LaFerla, F. M., Tinkle, B. T., Bieberich, C. J., Haudenschild, C. C. & Jay, G. (1995) Nat. Genet. 9, 21-30. [DOI] [PubMed] [Google Scholar]
- 35.D'Andrea, M. R., Nagele, R. G., Wang, H. Y., Peterson, P. A. & Lee, D. H. (2001) Histopathology 38, 120-134. [DOI] [PubMed] [Google Scholar]
- 36.Nagele, R. G., D'Andrea, M. R., Anderson, W. J. & Wang, H. Y. (2002) Neuroscience 110, 199-211. [DOI] [PubMed] [Google Scholar]
- 37.Oddo, S., Billings, L., Kesslak, J. P., Cribbs, D. H. & LaFerla, F. M. (2004) Neuron 43, 321-332. [DOI] [PubMed] [Google Scholar]
- 38.Alvarez, A., Toro, R., Caceres, A. & Maccioni, R. B. (1999) FEBS Lett 459, 421-426. [DOI] [PubMed] [Google Scholar]
- 39.Busciglio, J., Lorenzo, A., Yeh, J. & Yankner, B. A. (1995) Neuron 14, 879-888. [DOI] [PubMed] [Google Scholar]
- 40.Takashima, A., Noguchi, K., Sato, K., Hoshino, T. & Imahori, K. (1993) Proc. Natl. Acad. Sci. USA 90, 7789-7793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dineley, K. T., Bell, K. A., Bui, D. & Sweatt, J. D. (2002) J. Biol. Chem. 277, 25056-25061. [DOI] [PubMed] [Google Scholar]
- 42.Sabbagh, M. N., Lukas, R. J., Sparks, D. L. & Reid, R. T. (2002) J. Alzheimers Dis. 4, 317-325. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}