

Abstract
Although the burden of heart failure with preserved ejection fraction (HFpEF) is increasing, there is no therapy available that improves prognosis. Clinical trials using beta blockers and angiotensin converting enzyme inhibitors, cardiac-targeting drugs that reduce mortality in heart failure with reduced ejection fraction (HFrEF), have had disappointing results in HFpEF patients. A new “whole-systems” approach has been proposed for designing future HFpEF therapies, moving focus from the cardiomyocyte to the endothelium. Indeed, dysfunction of endothelial cells throughout the entire cardiovascular system is suggested as a central mechanism in HFpEF pathophysiology. The objective of this review is to provide an overview of current knowledge regarding endothelial dysfunction in HFpEF. We discuss the molecular and cellular mechanisms leading to endothelial dysfunction and the extent, presence, and prognostic importance of clinical endothelial dysfunction in different vascular beds. We also consider implications towards exercise training, a promising therapy targeting system-wide endothelial dysfunction in HFpEF.
1. Introduction
Heart failure (HF) is the most frequent cause of hospitalization in people over 65 years, and incidence is still increasing. Despite improved medical management, prognosis is grim, especially for heart failure with preserved ejection fraction (HFpEF) which has a 65% mortality rate at 5 years [1]. In contrast to heart failure with reduced ejection fraction (HFrEF), timely diagnosis of HFpEF remains a challenge and current standard therapy fails to improve prognosis [2]. Beta blockers and renin-angiotensin-aldosterone axis antagonists, drugs that mainly target the heart and have reduced mortality in HFrEF, had disappointing results in HFpEF trials [3–5]. As such, a “whole-systems” approach has been proposed, moving therapeutic focus in HFpEF away from the cardiomyocyte [6, 7].
Although HFpEF emerged as a distinct HF phenotype about three decades ago and about half of patients fall into this category, its pathogenesis remains incompletely understood. Beside advanced age, female sex, and sedentary lifestyle, HFpEF is associated with comorbidities such as arterial hypertension, diabetes, obesity, chronic obstructive pulmonary disease, and renal dysfunction [8]. Cardiac and extracardiac adjustments to these comorbidities can become maladaptive and lead to the HFpEF syndrome, with exercise intolerance as its main symptom. This maladaptation is characterized by structural changes such as myocardial hypertrophy and fibrosis, driven by a neurohormonal imbalance and systemic cytokine overexpression [9]. As a third mechanism, dysfunction of endothelial cells throughout the entire cardiovascular (CV) system has been put forward as the link between comorbidities and the pathophysiology of HFpEF. This builds on experimental evidence by Brutsaert et al. in the 1980s that the interaction between endothelial cells and cardiomyocytes directly influences diastolic function [10, 11].
Clinical endothelial dysfunction (ED) is recognized as a precursor to many CV diseases including HF [12]. Moreover, its prognostic value is proven in cohorts ranging from an unselected general population over patients at risk for CV disease (hypertension, chronic kidney disease) to patients with established CV disease [13]. Endothelial function is an independent predictor of survival in HF patients [14]. Exercise intolerance, the cardinal symptom in HFpEF, is objectively measured by peak pulmonary oxygen uptake (VO2peak) which is determined by the product of cardiac output and arteriovenous oxygen (O2) difference. Hence, both O2 delivery mechanisms (cardiac output, peripheral vascular function) as well as O2 utilizing factors (skeletal muscle) contribute to exercise intolerance [15]. Reduced endothelial-dependent vasodilation on exertion limits systemic O2 delivery, precipitating the switch to an anaerobic metabolism and thereby exacerbating fatigue and dyspnea [16]. ED also forms an attractive therapeutic target due to its reversibility at early stages [17]. This has shifted the search for an effective HFpEF therapy towards interventions correcting ED.
Exercise training is one of the most successful approaches to improve and even correct ED [18, 19]. Exercise-based cardiac rehabilitation programs have already earned their merit by improving symptoms and reducing mortality in various CV diseases, including HFrEF [20, 21]. The additional beneficial effects on other comorbidities and risk factors make exercise training conceptually a promising therapy for HFpEF [22].
In this review, we will focus on different aspects of ED in HFpEF. First, we briefly review the underlying molecular mechanisms leading to ED. We list the existing evidence on the presence of ED in distinct vascular beds and the clinical importance relative to HFpEF. Finally, the effects of exercise training on endothelial function are discussed, portending important implications for HFpEF treatment.
2. The Endothelium Is More than a Barrier
The endothelium was long considered a mere protective layer between the blood and different extravascular tissues. We now know that endothelial cells are dynamic, highly interacting cells regulating blood vessel function and homeostasis. The healthy endothelium prevents platelet and leukocyte adhesion and aggregation, inhibits smooth muscle proliferation, and regulates vascular tone through release of vasoactive substances, all of which are essential in organ perfusion [23]. Nitric oxide (NO) is the major effector molecule, formed from its precursor L-arginine by endothelial NO synthase (eNOS) in response to stimuli such as shear stress, cytokines, and platelet-derived factors. In endothelial cells, NO inhibits expression of leukocyte adhesion molecules, reducing vascular inflammation and atherosclerosis. By diffusing into platelets and vascular smooth muscle cells, NO stimulates the soluble guanylate cyclase—cyclic guanosine monophosphate—protein kinase G (sGC-cGMP-PKG) pathway, hereby inhibiting platelet aggregation and inducing vasorelaxation [23]. NO also diffuses to cardiomyocytes adjacent to coronary microvascular and endocardial endothelial cells, modulating cardiac function [24]. Finally, NO mobilizes stem cells and progenitor cells important for vascular homeostasis and repair [25].
In the setting of CV disease risk factors (smoking, aging, hypercholesterolemia, hypertension, hyperglycemia, and obesity), the endothelium loses these regulatory functions [26, 27]. Reactive oxygen species play an important role, reacting with NO to form toxic peroxynitrite, thereby reducing NO bioavailability. This disturbance of endothelial homeostasis can lead to a vasoconstrictory, proinflammatory, and prothrombotic phenotype at risk for CV disease [12]. The term “endothelial dysfunction” refers to these phenotypic alterations. Figure 1 summarizes the most important molecular influences on healthy and dysfunctional endothelium.
Figure 1.

Pathophysiology of endothelial dysfunction. Healthy endothelium maintains a balance between vasodilating, anti-inflammatory, and anti-thrombotic factors on one side and vasoconstricting, inflammatory, and thrombotic factors on the other. In endothelial dysfunction, increased oxidative stress caused by comorbidities tips the balance over to a vasoconstricting, inflammatory, and thrombotic profile. AT2=angiotensin 2, COX=cyclooxygenase, ET=endothelin, NO=nitric oxide, NOX=nicotinamide adenine dinucleotide phosphate oxidase, ONOO−=peroxynitrite, Ortho=orthosympathetic nerve activity, PGI2=prostacyclin, ROS=reactive oxygen species.
Repair of diseased endothelium does not solely depend on proliferation of existing endothelial cells. Bone marrow-derived endothelial progenitor cells can be mobilized to sites of endothelial injury or ischemia. They are able to proliferate, exert beneficial paracrine effects through secreting vascular growth factors, and finally integrate into the endothelial layer by differentiating into endothelial cells [28, 29].
3. Evaluation of Endothelial Function
ED is recognized as the first—but still reversible—step to overt atherosclerosis. As such, several diagnostic evaluation methods have been developed, with the goal to identify high-risk populations and start preventive therapy early. At the other end of the spectrum, presence and severity of ED is related to a negative outcome in established coronary ischemic heart disease and HFrEF [30].
Usually, endothelial function is measured as vasodilation in response to an endothelium-specific stimulus. This includes drugs, such as acetylcholine, but a short period of local ischemia also elicits endothelium-specific hyperemia. The amount of vasodilation can be assessed invasively (e.g., coronary angiography, intravascular flow wires), although noninvasive methods are more widely used nowadays. The percentage dilation of the brachial artery in response to forearm ischemia, measured by ultrasound, is called flow-mediated dilation (FMD) [31]. The more recent EndoPAT™ device (Itamar Medical, Israel) uses a fingertip probe to measure arterial tone. The response to ischemia is calculated automatically and is called reactive hyperemia index (RHI) [32]. Details and advantages of these and other techniques to measure endothelial function have been reviewed previously [26, 33]. Generally, FMD is considered a measure of the response to shear stress in conduit vessels (macrovascular), which is largely NO dependent, while RHI measures microvascular dilatation to shear stress, which involves other vascular mediators in addition to NO [34].
4. Endothelial Dysfunction in HFPEF: Cause or Consequence?
Impaired coronary endothelial-dependent vasodilation was found in nonischemic dilated cardiomyopathy, highlighting the implication of the endothelium in HFrEF regardless of the presence of atherosclerosis [35]. Moreover, ED is not only limited to the coronary arteries, but is equally present in other vascular beds, indicating the systemic nature of ED in HFrEF.
In 2013, Paulus and Tschöpe hypothesized that ED plays a causal role in the development of HFpEF [10]. They postulate that the comorbid illnesses seen in HFpEF are the primary impellent of a systemic inflammatory state, leading to coronary microvascular ED. Indeed, elevated levels of inflammatory cytokines are seen in HFpEF patients [36]. In asymptomatic patients, biomarkers of inflammation predict the onset of HFpEF but not HFrEF [37]. Circulating inflammatory cytokines activate and inflame the endothelium throughout the vascular system, including the coronary microvasculature. This coronary microvascular endothelial inflammation is seen in animal models of HFpEF and in human cardiac biopsies [38, 39]. Reduced endothelium-dependent vasodilation is seen in animal models as well [40].
Reduced NO signaling from dysfunctional endothelium then influences adjacent cardiomyocytes and cardiac fibroblasts through the sGC-cGMP-PKG pathway [24]. Lower myocardial PKG content eventually leads to functional and structural cardiac changes associated with HFpEF [41]. These include delayed myocardial relaxation, increased cardiomyocyte stiffness, cardiac hypertrophy, and interstitial fibrosis [10]. Cardiac-endothelial interaction is reviewed in more detail in 6.2.
However, a two-way interaction between HFpEF and ED exists. Once HF develops, the syndrome maintains a vicious circle, further impairing endothelial function. HFpEF itself causes a systemic inflammatory state with high levels of circulating proinflammatory cytokines, increasing production of reactive oxygen species and exerting direct deleterious effects on eNOS expression [42, 43]. Neurohormonal activation in HFpEF increases oxidative stress and activates collagen synthesis [44]. Thus, HFpEF worsens system-wide ED, causing a downward spiral eventually leading to progressive HF.
5. Clinical Importance: Endothelial Dysfunction as Prognostic Marker in HFPEF
As there is no universally accepted cutoff for defining ED, the actual prevalence of ED in HFpEF is unknown. In community studies, endothelial function declines with age and presence of CV risk factors [45, 46]. Understandably, FMD and RHI values are lower in populations with established CV disease, including HF patients [14, 47]. In one of the first studies proving reduced RHI in HFpEF, Borlaug et al. estimated the prevalence of ED in HFpEF patients at 42% [48]. Of note, the cutoff to define ED in this study was arbitrarily chosen as RHI <2.0, which is substantially higher than the original reference value defined in coronary artery disease patients (RHI <1.67) [49]. The prevalence of ED found in the Borlaug study could as such be overestimated.
In the largest study to date, measuring endothelial function in 321 Japanese HFpEF patients, Akiyama et al. found that a RHI below the median predicted CV events [47]. For each decrease of 1.0 in RHI, CV risk increased 20%. The prognostic significance of ED in HFpEF patients was independent of clinical, echocardiographic, and neurohormonal factors. This was later confirmed in a smaller study by Matsue et al. [50]. Of note, both Japanese studies propose a prognostic cutoff value for RHI <1.63, close to the original reference value of RHI <1.67. Applied to the large Akiyama study, this implies an ED prevalence of 50% in HFpEF patients. Full details of studies measuring peripheral ED in HFpEF can be found in Table 1.
Table 1.
Studies assessing peripheral endothelial function in HFpEF patients compared to a control population.
| Reference | Technique | Outcome variable | Study design |
Number of patients | Number of HFpEF patients |
Control groups | Result | P value |
|---|---|---|---|---|---|---|---|---|
| Studies assessing macrovascular endothelial function | ||||||||
| Hundley et al., 2006 [150] |
FMD (magnetic resonance) |
% change in femoral artery area |
Case-control | 30 | 9 | Healthy, matched for age | FMD comparable in HFpEF and healthy,(12 ± 1 versus 14 ± 2%), no difference in shear rate |
ns |
| Haykowsky et al., 2013 [61] | FMD (ultrasound) |
% dilatation brachial artery |
Case-control | 111 | 60 | Young healthy group, matched for gender Old healthy group, matched for age and gender |
FMD better in young healthy (6.13 ± 0.53%) FMD comparable in HFpEF and old healthy (3.64 ± 0.28 versus 4.00 ± 0.38%), |
<0.001 versus young ns versus old |
| Farrero et al., 2014 [94] |
FMD (ultrasound) |
% dilatation brachial artery |
Case-control | 70 | 28 | Hypertensive, matched for age | FMD significantly lower in HFpEF + PHT (1.95 [−0.81–4.92] versus 5.02 [3.90–10.12] %), no difference in shear rate (p = 0.47) |
0.002 |
| Kishimoto et al., 2017 [151] |
FMD (ultrasound) |
% dilatation brachial artery |
Case-control | 206 | 41 | Subjects without heart failure, unmatched |
FMD significantly lower in HFpEF (2.9 ± 2.1 versus 4.6 ± 2.7%) |
0.0002 |
| Studies assessing microvascular endothelial function | ||||||||
| Balmain et al., 2007 [65] |
Laser Doppler; venous occlusion plethysmography |
Perfusion units; mL/100mL blood |
Case-control | 36 | 12 | Coronary heart disease patients, unmatched |
Cutaneous blood flow lower in HFpEF patients, venous capacitance was not different versus control∗ |
<0.001 |
| Borlaug et al., 2010 [48] |
RHI (PAT) | Ln (PAT ratio 60–120 sec) | Case-control | 50 | 21 | Hypertensive group; Healthy group; both matched for age and gender |
RHI significantly lower in HFpEF versus healthy (0.85 ± 0.42 versus 1.33 ± 0.34), but not in HFpEF versus hypertensive (0.85 ± 0.42 versus 0.92 ± 0.38) |
<0.05∗ ns |
| Akiyama et al., 2012 [47] |
RHI (PAT) | Ln (PAT ratio 90–150 sec) | Prospective cohort |
494 | 321 | Healthy, matched for age, gender, and presence of hypertension and diabetes mellitus |
RHI significantly lower in HFpEF (0.53 ± 0.20 versus 0.64 ± 0.20) |
<0.001 |
| Vitiello et al., 2014 [152] |
Venous occlusion plethysmography |
mL/100mL blood | Case-control | 32 | 18 | Healthy, unmatched | Venous capacitance was not different versus healthy |
ns |
| Yamamoto et al., 2015 [64] |
RHI (PAT) | Not reported | Case-control | 128 | 64 | Healthy, matched for age, gender and comorbidities |
RHI significantly lower in HFpEF (1.70 [1.55 – 1.88] versus 2.01 [1.64 – 2.42]) |
<0.001 |
| Studies assessing both macro- and microvascular endothelial function | ||||||||
| Maréchaux et al., 2016 [62] |
FMD (ultrasound) | % dilatation brachial artery | Case-control | 90 | 45 | Hypertensive, matched for age, sex, and presence of diabetes mellitus | FMD significantly lower in HFpEF patients (3.6 [0.4 – 7.4] versus 7.2 [3.2 – 12.7] %) |
0.001 |
| RHI (Laser Doppler) |
Perfusion units | Cutaneous blood flow lower in HFpEF patients (135 [104 – 206] versus 177 [139 – 216] units) |
0.03 | |||||
| Lee et al., 2016 [63] |
FMD (ultrasound) | % dilatation brachial artery |
Case-control | 48 | 24 | Healthy, matched for age, sex, and brachial artery diameter |
FMD lower in HFpEF (3.06 ± 0.68 versus 5.06 ± 0.53), but no difference in FMD when corrected for shear rate |
ns |
| RHI (ultrasound) | Blood flow AUC after cuff release |
AUC lower in HFpEF (454 ± 35 versus 659 ± 63 mL/min) |
0.03 | |||||
AUC: area under the curve; FMD: flow mediated dilatation; HFpEF: heart failure with preserved ejection fraction; Ln: natural logarithm; ns: not significant; PAT: peripheral arterial tonometry; PHT: pulmonary hypertension; RHI: reactive hyperemia index; ∗exact numbers not reported.
Given the central role of ED in the development of HFpEF, this estimated prevalence of ED of 42–50% seems low. However, to be more precise, 42–50% of HFpEF patients have peripheral ED as defined by a given RHI cutoff. In our opinion, the other 50% fail to show decreased RHI because of the following reasons. First, it takes time before microvascular inflammation is translated to clinically measureable disturbances in vasoreactivity. Second, the cutoff of RHI <1.63 reflects a value useful for clinical prognosis, but has not been correlated with pathophysiological changes such as endothelial inflammation and reduced NO bioavailability. Third, RHI and FMD show poor agreement which suggests different mechanisms are measured [51]. Possibly, FMD more accurately reflects reduced NO signaling, but data on FMD in HFpEF is incomplete (no large or prognostic studies), and a cutoff defining ED is not available. Perhaps, it is more correct to state that the prevalence of ED in HFpEF is hard to estimate based on current data, but almost half of patients have reduced peripheral endothelial-dependent vasodilation compared to controls, which is linked to increased CV events.
Another clinical clue to the importance of ED is the relation with exercise intolerance, objectively measured by cardiopulmonary exercise testing and determination of VO2peak. This is related to adverse prognosis, since VO2peak is one of the strongest predictors of mortality in HFpEF [52]. The Fick principle (VO2 = cardiac output • arteriovenous O2 difference) states that VO2peak can be limited by either a central factor, cardiac output, or peripheral O2 extraction. The latter is influenced by oxygenation of the blood in the lungs, O2 carrying capacity of the blood, appropriate distribution of blood to the peripheral tissues, and adequate tissue O2 extraction from the blood. A key factor is the oxygen diffusion capacity (DO2), which can be a limiting factor in both pulmonary and skeletal muscle O2 kinetics. Applying Fick's law of diffusion (VO2 = DO2 • (capillary pO2 – intracellular pO2) with pO2 being partial oxygen pressure) in exercising muscle, where intracellular pO2 is very low, the capillary pO2 determines the O2 diffusion gradient. As such, capillary pO2 can limit VO2 during exercise [53]. Adequate endothelial function is necessary for an appropriate exercise-induced increase in blood flow to the muscles [54]. As capillary pO2 is determined by the instantaneous balance between VO2 and perfusion, ED can also limit capillary pO2 [53]. In theory, ED can thus limit VO2 both by reducing capillary blood flow and limiting O2 diffusion.
Reduced cardiac output on exertion was long considered the main mechanism behind exercise intolerance in HFpEF [55]. Chronotropic incompetence and reduced peak stroke volume have both been implicated as the most important factor limiting VO2peak [56]. More recently, a peripheral limitation to exercise capacity in HFpEF has been put forward. Borlaug et al. reported reduced systemic vascular resistance and lower RHI at peak exercise in HFpEF patients compared to hypertensive and healthy controls [48]. Haykowsky et al. even suggested that a failure to increase peripheral O2 extraction during exercise is the predominant factor limiting VO2peak [57]. The rest-to-peak change in peripheral O2 extraction was the strongest independent predictor of VO2peak in their study. This was later confirmed using exercise hemodynamics and exercise echocardiography [58, 59]. Although the dominant limiting factor to VO2peak remains controversial, clearly peripheral elements play a role in determining exercise capacity in HFpEF. We further elaborate this finding in the next section.
6. Various Vascular Beds Display Endothelial Dysfunction in HFPEF
Theoretically, many clinical findings related to the HFpEF syndrome could be explained by a system-wide ED, leading to alterations in several organ systems. In Figure 2, we postulate that systemic ED is the underlying pathophysiological mechanism by which HFpEF risk factors lead to exercise intolerance. Systemic inflammation induced by HFpEF risk factors creates oxidative stress at the level of the endothelium throughout the vasculature, reducing NO availability for adjacent cells pertaining to all organs implicated in exercise performance.
Figure 2.

Role of system-wide endothelial dysfunction in HFpEF pathophysiology. Comorbidities induce systemic inflammation, creating oxidative stress in endothelial cells system-wide. Reduced NO bioavailability through reduction of NO to ONOO− causes endothelial dysfunction. In different vascular beds, endothelial dysfunction has heterogeneous effects, which manifest as the cardinal HFpEF symptom of exercise intolerance. COPD=chronic obstructive pulmonary disease, CRP=C-reactive protein, ED=endothelial dysfunction, IL-6=interleukin-6, NO=nitric oxide, ONOO−=peroxynitrite, ROS=reactive oxygen species, RV=right ventricle, TNFα=tumor necrosis factor alpha.
In what follows, we review the evidence of the presence, extent, and underlying mechanisms of ED in different vascular beds and the corresponding organs.
6.1. Peripheral Vasculature and Skeletal Muscle
The peripheral circulation is the preferred organ system for measuring endothelial-dependent vasodilation, because of the easy, noninvasive measurement and the good correlation with “gold standard” invasive coronary vasodilation [60]. Studies evaluating peripheral endothelial function in HFpEF are summarized in Table 1.
Evidence regarding macrovascular ED in HFpEF is conflicting. The largest study to date reported no significant difference in FMD between HFpEF patients and healthy volunteers matched for age and gender [61]. In contrast, in almost all studies assessing microvascular peripheral endothelial function through RHI measurement, HFpEF patients have evidence of microvascular ED [48, 62–65]. Also, prognostic significance for ED in HFpEF has only been proven for microvascular dysfunction [47]. Of note, many studies have different methodologies even when using the same technique for measuring endothelial function. Control groups are often heterogeneous and unmatched, few studies using FMD adhere to the most recent guidelines that state shear stimulus must be reported, [66, 67] and different cutoffs for identifying ED are used. These disparities complicate the interpretation of study results.
Besides vasodilatory dysfunction of the afferent arteries to the working muscle, reduced peripheral O2 extraction in HFpEF can also result from skeletal muscle dysfunction. HFpEF patients indeed have abnormalities in skeletal muscle mass, composition, capillary density, and oxidative metabolism. In contrast to the high prevalence of obesity, HFpEF patients have reduced lean leg mass [68]. This could be related to adipose tissue infiltration in muscle, which shows a similar correlation with exercise capacity. A markedly lower VO2peak indexed to lean body mass in HFpEF patients further confirms that abnormalities in skeletal muscle perfusion and/or metabolism contribute to exercise intolerance [69]. Mitochondria are important regulators of skeletal muscle metabolism. Recently, reductions in muscle mitochondrial content, oxidative capacity, and expression of key mitochondrial proteins were found in muscle biopsies of HFpEF patients [70]. These changes were related to VO2peak, emphasizing muscle mitochondrial dysfunction is likely a limiting factor to exercise capacity. Other possible underlying molecular changes could be a switch from oxidative slow-twitch type I fibers to glycolytic fast-twitch type II fibers which reduces oxidative capacity, increased muscle fatigability, and a reduction in skeletal muscle capillary density [71].
The latter is especially intriguing, as it links these skeletal muscle abnormalities to vascular dysfunction. Kitzman et al. demonstrated a severely reduced capillary-to-fiber ratio in muscles of HFpEF patients, related to VO2peak [72]. A lower capillary density, and hence reduced capillary blood supply, may also underlie the muscle fiber atrophy seen in animal and human HFpEF studies [69, 71]. Also, as muscle blood flow assumes an important role in limiting VO2 kinetics, the authors suggest a decreased O2 diffusion to contracting muscle limits exercise capacity in HFpEF. As mentioned above, ED could play a role in this limitation of diffusive capacity by reducing the pO2 driving gradient.
When leg blood flow is measured by ultrasound Doppler, HFpEF patients indeed have a reduced muscle blood flow during exercise compared to healthy controls, even at relatively low workloads of 10-15 W [73]. Stroke volume and heart rate were similar in HFpEF and control patients in this study, again implying a vascular (and not cardiac) limitation of exercise capacity. Also, HFpEF patients fail to augment peripheral O2 extraction during exercise with a greater increase in blood pressure than controls [59]. This suggests that a reduced vasodilatory capacity prevents appropriate distribution of blood flow during exercise, leading to limitation of exercise capacity [55]. Possibly, microvascular ED contributes more than macrovascular ED at the level of the muscle vascular bed, as Haykowsky et al. found a peripheral limitation of exercise capacity but no decrease in FMD [61, 74].
In summary, there is evidence for microvascular ED in HFpEF, predictive of long-term CV morbidity. Reports on macrovascular dysfunction are conflicting, and all studies suffer from methodological disparities. Also, HFpEF patients suffer numerous changes in skeletal muscles which correlate with reduced VO2peak, including mitochondrial dysfunction, fiber atrophy, and reduced oxidative capacity. Possibly, skeletal muscle abnormalities are linked to vascular dysfunction through a reduction in muscle capillary density, which limits muscle blood flow and O2 diffusion during exercise.
6.2. Heart
Traditionally, coronary endothelial function is measured by intracoronary infusion of a vasodilating substance such as acetylcholine. Subsequently, microvascular function can be estimated by measuring coronary flow reserve (CFR), the ratio of coronary blood flow after the vasodilating stimulus over blood flow at rest. In HFrEF patients, CFR correlates with VO2peak, invasive and echocardiographic hemodynamics, and mortality [75–77]. Tschöpe et al. measured CFR in patients with diastolic dysfunction, showing a reduced vasodilatory response to intracoronary acetylcholine infusion even before onset of HF symptoms [78]. Furthermore, invasively measured CFR is reduced in HFpEF patients and CFR correlates with echocardiographic measures of diastolic function and LV hypertrophy [76, 78, 79]. Interestingly, two studies in HFrEF patients showed no relationship between CFR and peripheral endothelial function [75, 80]. As such, different pathophysiological mechanisms may lie at the origin of coronary and peripheral ED.
As mentioned above, reduced NO bioavailability leads to both structural and functional changes in HFpEF. Structurally, HFpEF hearts are characterized by interstitial fibrosis and both macroscopic and microscopic hypertrophy [10]. Hemodynamically, diastolic dysfunction is evident as slowed ventricular relaxation on one hand and decreased compliance due to myocardial stiffness on the other hand [9].
In the normal heart, endothelial NO bursts directly modulate relaxation in a beat-to-beat way [81]. High levels of peroxynitrite (ONOO−), however, increase diastolic calcium content and thus delay cardiomyocyte relaxation [82]. Through its effects on sGC, NO is also able to modify cardiomyocyte stiffness and hypertrophy. sGC increases cGMP production, which in turn increases cellular PKG content. PKG acutely reduces cardiomyocyte stiffness through phosphorylation of the giant protein titin, the most important regulator of passive myocardial stiffness. Also, PKG functions as a brake on several pathways implicated in left ventricular hypertrophy. The sGC-cGMP-PKG pathway and its targets are indeed downregulated in HFpEF animals [83, 84]. Low PKG content has also been found in myocardial biopsies from HFpEF patients [41].
Finally, NO exerts direct antifibrotic effects in the heart by counteracting endothelin-1, angiotensin II, and aldosterone. Reduced NO bioavailability leaves profibrotic actions of these molecules unopposed, promoting proliferation of fibroblasts and myofibroblasts [85].
In summary, microvascular cardiac endothelium modulates diastolic function and development of LV hypertrophy and fibrosis. Coronary microvascular function, as measured by CFR, is reduced in HFpEF but does not relate to peripheral ED.
6.3. Lungs
Pulmonary hypertension (PHT) at rest is highly prevalent in HFpEF patients, with up to 83% affected [86]. Patients often have an exaggerated elevation of pulmonary artery pressures during exercise [87, 88]. This increased afterload on the right ventricle (RV) and the presence of common risk factors explain the high prevalence of RV dysfunction in HFpEF, which is associated with increased morbidity and mortality [89].
Passive transition of elevated end-diastolic pressure explains only part of the elevated pulmonary artery pressures in HFpEF [86]. As in patients with HFrEF and pulmonary arterial hypertension, impaired NO-dependent pulmonary vasodilation has been described in HFpEF patients. The Mayo Clinic group has spearheaded research in this field, proving abnormal RV and pulmonary artery hemodynamics both at rest and on exertion [88]. Although initially an increased pulmonary vasodilatory capacity was suggested based on dobutamine infusion [90], recent invasive measurements showed reduced exercise-induced pulmonary vasodilation in HFpEF [88].
Pulmonary arterial endothelial function was disturbed, and pulmonary artery pressures were higher in an animal infarct model of HFpEF, while aortic endothelial function and intracardiac pressures remained unaltered [91]. This could mean that pulmonary vascular ED even precedes systemic ED in HFpEF. Indeed, as the pulmonary circulation is primarily flow-driven in contrast to the pressure-driven systemic circulation, it may be more susceptible to the influence of shear stress and ED [92]. More recently, a murine model of HFpEF with PHT was established by blocking vascular endothelial growth factor receptors in obese and hypertensive rats. Oral administration of nitrite, which acts as NO donor, prevented the development of PHT but could not reverse established PHT [93]. These findings are compatible with “reversible” pulmonary ED playing an early role in the establishment of PHT, while “fixed”vascular remodeling occurs in more advanced stages.
In a cohort of 28 HFpEF patients with PHT that had severe macrovascular ED (FMD median 1.95%), Farrero et al. found a significant inverse correlation between FMD and pulmonary vascular resistance. No correlation was found with capillary wedge pressure [94]. While this does not prove a causal relationship, it is plausible that more severe HFpEF is related with more severe ED in the systemic and pulmonary vasculature, ultimately leading to PHT. This would corroborate the concept of whole-body ED in HFpEF.
PHT is also induced through reactive pulmonary vasoconstriction and vascular remodeling [95]. This process is largely mediated by NO, as pulmonary vascular reactivity is maintained by continuous local NO production [95]. A systemic reduced NO bioavailability, as found in HFpEF, causes vascular smooth muscle dysfunction in the pulmonary vasculature, paving the way for PHT [96].
Pulmonary function itself is frequently disturbed in HFpEF patients, with 59% suffering airflow limitation on spirometry [97]. As pulmonary impairment increases with symptom severity, pulmonary edema is a likely explanation. But diaphragm dysfunction may also contribute by increasing work of breathing. The diaphragm exhibits similar changes as skeletal muscle in HFpEF, including fiber atrophy, decreased oxidative capacity, impaired mitochondrial function, and increased fatigability [71]. As ED possibly underlies several skeletal muscle alterations, ED could also be a pathophysiological factor in diaphragm dysfunction, forming the link between skeletal muscle and respiratory abnormalities in HFpEF.
Pulmonary gas exchange is impaired in up to 83% of HFpEF patients, showing true O2 diffusion limitation at rest in 59% [97]. At exercise, diffusion abnormalities are exacerbated in HFpEF patients compared to healthy individuals [98]. These findings provide further evidence that exercise capacity is limited by O2 diffusion in both the systemic and the pulmonary microcirculation.
In summary, PHT is a frequent and ominous finding in HFpEF patients. Vascular remodeling and reactive pulmonary vasoconstriction, caused by a reduced systemic NO bioavailability, play an important role in its development. Spirometry, diaphragm function, and pulmonary diffusion capacity are frequently impaired in HFpEF patients. Possibly, ED plays a role by impairing O2 diffusion in the pulmonary microcirculation and causing adverse changes in diaphragm muscle composition similar to those in skeletal muscle.
6.4. Kidneys
HFpEF can induce renal dysfunction, and vice versa. Chronic kidney disease is highly prevalent in HFpEF patients (30–34% in large outcome trials) [99, 100]. Moreover, HF mortality is increased by concurrent renal impairment [101].
Clinically, endothelial function is impaired in patients with even mild chronic kidney disease, whether measured by RHI or FMD [102, 103]. Furthermore, worse endothelial function correlates with worse diastolic function on echocardiography [104]. Studies on the impact of renal disease on progression of ED in HFpEF are currently still lacking, but it is certainly an interesting field for future research [105].
HFpEF can cause renal dysfunction in different ways [106]. First, hemodynamic factors impair glomerular blood flow. Renal congestion due to elevated central venous pressure increases efferent glomerular pressure [107]. Additionally, fixed stroke volume and chronotropic incompetence reduce cardiac output on exertion, which impairs afferent blood flow [108]. The net result is decreased glomerular blood flow, leading to renovascular and glomerular injury and activating sodium retention pathways [109]. Second, the systemic inflammation that accompanies HFpEF has deleterious effects on the kidneys. Leukocyte recruitment causes renal fibrosis through transforming growth factor ß-mediated fibroblast stimulation. Also, systemic inflammation reduces NO bioavailability as described above. Renal blood flow is dependent on systemic NO supply, which is reduced in HFpEF [110]. In a metabolic syndrome rat model of HFpEF, degradation of peritubular and glomerular microvasculature is linked with progressive glomerulosclerosis [111]. Interestingly, in this last study, microscopic renal damage was evident before onset of HFpEF.
On the other hand, renal disease can also lead to HFpEF. In long-term follow-up of >8500 chronic kidney disease patients, 34% was diagnosed with new-onset HFpEF [112]. Possible mechanisms include, again, worsening endothelial function and inducing systemic inflammation [105]. Several important feedback mechanisms, regulated by the kidney and disturbed in renal failure, induce ED: vitamin D deficiency, erythropoietin deficiency, elevated parathyroid hormone levels, and phosphorus excess [113–115]. Also, the endothelium is involved in sodium handling. Sodium retention could increase intracellular sodium, which disrupts endothelial homeostasis [116]. Asymmetric dimethyl arginine, a retention product found in kidney failure, is a competitive inhibitor of eNOS and increases endothelial oxidative stress [106].
In summary, HFpEF and chronic kidney disease are mutually influencing conditions. ED is an important risk factor for both diseases, and interesting pathophysiological links exist.
7. Exercise Training: The Silver Lining on the Cloud
Cardiac rehabilitation programs have been a mainstay of HFrEF treatment after it was discovered that training is safe and reduces hospitalizations [21]. The evidence in HFpEF, however, is still emerging. Several medium-sized single-center studies demonstrated substantial benefit of training in HFpEF patients [117–122]. Three recent meta-analyses concluded that exercise training in HFpEF increases VO2peak and physical function scores [123–125]. Diastolic function (measured by E/e' ratio and left atrial volume) also improved with exercise in the landmark Ex-DHF trial [117]. These results have led to a class I, level of evidence A recommendation for exercise training in HF patients regardless of their ejection fraction in recent European Society of Cardiology HF guidelines [2]. Although no recommendations are made towards the intensity of exercise training, existing evidence suggests diverging effects of standard moderate-intensity aerobic training (at 60–70% of VO2peak) and high intensity interval training (adding short intervals at 80–90% VO2peak). In a single-center trial, high intensity training in HFrEF patients led to superior increases in VO2peak and ejection fraction compared to moderate training [126]. Unfortunately, these findings could not be replicated in the large multicenter SmartEx trial [127]. Of note, the majority of patients exercised below the prescribed target in the high-intensity group and above target in the moderate group. A pilot study in 15 HFpEF patients showed superior effects of high intensity interval training on exercise capacity and diastolic function [128]. However, the lack of VO2peak improvement in patients training at moderate intensity contrasts with the earlier studies.
The ongoing OptimEx study aims to study optimal exercise dose in 180 HFpEF patients with regard to aerobic capacity [129]. Also, this trial will reevaluate the effect of exercise training on FMD in HFpEF patients and add much-needed information on microvascular function.
In the contemporary “whole-systems” approach towards HFpEF therapy, ED forms an attractive target due to its systemic nature and reversibility in early stages. Improving ED in HFpEF can be achieved through correction of comorbidities, increasing NO bioavailability, or antioxidative therapy. Sadly, none of these approaches alone has thus far been successful in decreasing HFpEF-related morbidity or mortality. Exercise training integrates all three mechanisms, forming a promising systemically oriented therapy [7].
Both peripheral endothelial function and muscle metabolism are beneficially influenced by exercise. Exercise increases NO production by upregulating and phosphorylating eNOS through increased shear stress and vascular endothelial growth factor 2 release [19]. Exercise training also reduces oxidative stress by downregulating angiotensin receptors and nicotinamide adenine dinucleotide phosphate oxidase [130]. In addition, the anti-inflammatory and permeability decreasing properties of exercise may contribute to improvement of endothelial function [22].
Circulating progenitor cells could add to these favorable changes [131]. Endothelium-repairing endothelial progenitor cells are mobilized from the bone marrow by stimuli such as ischemia and cytokine release, under control of circulating angiogenic T lymphocytes [132, 133]. Our group has shown that the number of circulating angiogenic T lymphocytes and their functional capacity increase with exercise training, both in healthy subjects and HF patients [134]. The acute exercise-induced changes in circulating angiogenic T lymphocyte function wane with exercise training, suggesting that repetitive exercise bouts progressively lead to endothelial repair [135]. Another group has recently shown increases in endothelial progenitor cell number and function in HF patients as well [136].
Molecular determinants of exercise-induced effects specific to HFpEF are still poorly investigated. In HFpEF rats, exercise training restored endothelial-dependent vasodilation measured ex vivo in organ baths [40]. Endothelial function correlated well with eNOS expression, which was reduced in HFpEF rats and recovered after exercise training. Matrix metalloproteinase activity, which is an indirect measure of extracellular matrix degradation and thus vessel wall modulation, was increased in HFpEF and blunted by exercise training while the endothelial cell layer remained intact. This suggests exercise-induced vascular changes extend beyond the endothelium.
In a secondary analysis of the Ex-DHF trial, circulating cytokines and hormones were analyzed in HFpEF patients before and after training [137]. Inflammatory cytokines (interleukins 1ß, 6, and 10 and tumor necrosis factor alpha) showed no change with exercise. Interestingly, levels of the growth hormone releasing peptide ghrelin, which inhibits cardiomyocyte and endothelial cell apoptosis in vitro, increased by exercise training. Clearly, molecular determinants underlying the exercise-induced benefits in HFpEF deserve further in-depth exploration.
Clinically, peripheral endothelial function shows improvement after exercise training in patients with CV risk factors, coronary atherosclerosis, and HFrEF [138–140]. Of note, when comparing high intensity interval training to moderate training in HFrEF, endothelial function (as measured by FMD) and mitochondrial function (determined from muscle biopsies) improved only by high intensity training [126]. In HFpEF patients, Haykowsky et al. found that exercise training can increase peripheral O2 extraction. The increase in VO2peak was almost entirely attributable to an improvement in peripheral function (i.e., improved vascular and/or skeletal muscle functions) [141]. In a study by Fu et al., aerobic interval training increased muscle perfusion and muscle O2 extraction in HFpEF patients. This increase in muscle vascular function was the only significant predictor of VO2peak. Interestingly, this phenomenon was not seen in HFrEF patients, for whom improved cardiac output was the only predictor of VO2peak [142].
Conversely, Kitzman et al. could not demonstrate an improvement of FMD after training HFpEF patients, despite an increase in VO2peak [74]. A possible confounder could be that FMD was measured in the postprandial state in the Kitzman study, while guidelines advise to assess FMD in a fasting state because of a significant influence of food ingestion [67, 143]. In addition, the intensity of the exercise training protocol in this study was rather moderate and therefore could have failed to induce changes in macrovascular endothelial function.
There is no data regarding the effects of training on coronary, pulmonary, or renal ED in HFpEF patients. However, studies in patients with other CV diseases suggest exercise training is indeed able to improve regional endothelial function. Coronary endothelial function is improved by cardiac rehabilitation in dilated cardiomyopathy and coronary atherosclerosis [138, 144]. In patients with chronic kidney disease, changes in several molecular markers (asymmetric dimethyl arginine, glutathione, and lipid peroxidation products) suggest increased NO bioavailability through exercise training [106]. Unfortunately, Van Craenenbroeck et al. found that exercise training did not improve FMD nor cellular markers of vascular function, despite an increase in VO2peak [102]. However, data on microvascular function is lacking.
8. Future Directions
Considering HFpEF as a multisystem syndrome rather than an isolated cardiac disease could lead us to alternative research approaches and eventually to successful therapies. The heterogeneity of the HFpEF patient population has frequently been cited as one of the reasons major trials have failed to prove a benefit for pharmacological treatment [145]. Efforts to subdivide HFpEF patients into different phenotypes have only started recently [146–148]. In the spectrum of HFpEF as a multisystem pathology, some patients seem younger and suffer less cardiac impairment, some have important metabolic disorders and more severe cardiac disease including RV and pulmonary vascular involvement, and others have a predominant renal dysfunction. Importantly, prognosis between phenotypes differs substantially [147]. The greatest challenges for future HFpEF research will be to correctly stratify patients into phenogroups and to design clinical trials accordingly. Whether endothelial function measurement could aid in identifying the correct HFpEF phenotype in patients is still unknown.
Also, a one-size-fits-all therapeutic approach is probably not the best strategy for the heterogeneous HFpEF population. A treatment algorithm based on presence of different comorbidities has recently been proposed [146]. Keeping in mind the important effects of even low-level exercise, matching or stratifying groups for physical activity seems reasonable when designing HFpEF trials, although maintaining statistical power will require a delicate balance. Rather, we support further subdividing of HFpEF based on large phenotyping studies to better characterize this heterogeneous population. Clinical trials could then be focused on a well-defined subgroup, eliminating confounding by other phenotypes.
Unravelling the beneficial effects of exercise training in HFpEF could lead to patient-specific new therapies. Such a tailored approach can be useful in patients who are unable to exercise, or as add-on to a training program. Pharmacological or nonpharmacological correction of comorbidities, increase of NO bioavailability, and antioxidative therapy are possible targets, some of which are being explored in clinical trials already [119, 149]. These can be combined with exercise training to compose a truly personalized treatment for each patient (Figure 3).
Figure 3.
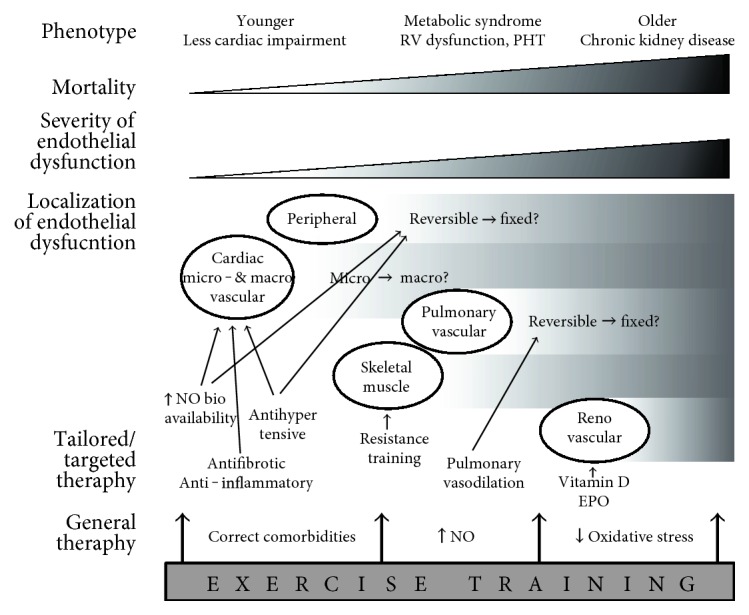
Possibilities for exercise training and targeted therapies depending on HFpEF phenotype. Cardiac ED is an early hallmark in all HFpEF patients. In older patients, pulmonary and renal vasculature are more frequently involved, and mortality is higher. HFpEF therapy could be tailored for each phenotype. Younger patients could still benefit from correction of comorbidities, preventing further systemic inflammation and ED. Increasing NO bioavailability, antifibrotic, or anti-inflammatory therapy could also be useful in early stages. Pulmonary vasodilation can only be effective when pulmonary vascular ED is manifested and still reversible. Exercise training has possible benefits at each stage, as it is able to correct comorbidities (weight loss, better glycemic control), increase NO bioavailability, and reduce systemic oxidative stress. EPO=erythropoietin, NO=nitric oxide, PHT=pulmonary hypertension, RV=right ventricle.
9. Conclusions
HFpEF is a multisystem pathology. Cardiac dysfunction is not the sole causative factor, but interacts with a heterogeneous range of organ dysfunctions, including pulmonary, renal, peripheral vascular, and skeletal muscle dysfunctions. Endothelial dysfunction could be a central mechanism in this system-wide CV maladaptation, as such it forms an attractive target for future HFpEF therapies. Exercise training is thus far the only therapy with proven beneficial effects in HFpEF. While exercise training does not improve macrovascular ED in HFpEF, evidence does suggest peripheral vascular and/or skeletal muscle function is enhanced. This warrants a shift in both fundamental and clinical research towards endothelial-targeted therapies, including exercise training, in the search for an effective therapeutic strategy for HFpEF.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
EMVC is supported by the fund for scientific research-Flanders (FWO) as Senior clinical investigator. Research Group Cardiovascular Diseases is part of the Infla-Med Research Consortium of Excellence.
References
- 1.Roger V. L. Epidemiology of heart failure. Circulation Research. 2013;113(6):646–660. doi: 10.1161/CIRCRESAHA.113.300268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ponikowski P., Voors A. A., Anker S. D., et al. 2016 ESC guidelines for the diagnosis and treatment of acute and chronic heart failure. European Heart Journal. 2016;37(27):2129–2200. doi: 10.1093/eurheartj/ehw128. [DOI] [PubMed] [Google Scholar]
- 3.Yusuf S., Pfeffer M. A., Swedberg K., et al. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-preserved trial. Lancet. 2003;362(9386):777–781. doi: 10.1016/S0140-6736(03)14285-7. [DOI] [PubMed] [Google Scholar]
- 4.Pitt B., Pfeffer M. A., Assmann S. F., et al. Spironolactone for heart failure with preserved ejection fraction. The New England Journal of Medicine. 2014;370(15):1383–1392. doi: 10.1056/NEJMoa1313731. [DOI] [PubMed] [Google Scholar]
- 5.Conraads V. M., Metra M., Kamp O., et al. Effects of the long-term administration of nebivolol on the clinical symptoms, exercise capacity, and left ventricular function of patients with diastolic dysfunction: results of the ELANDD study. European Journal of Heart Failure. 2012;14(2):219–225. doi: 10.1093/eurjhf/hfr161. [DOI] [PubMed] [Google Scholar]
- 6.Lim S. L., Lam C. S. P., Segers V. F. M., Brutsaert D. L., De Keulenaer G. W. Cardiac endothelium–myocyte interaction: clinical opportunities for new heart failure therapies regardless of ejection fraction. European Heart Journal. 2015;36(31):2050–2060. doi: 10.1093/eurheartj/ehv132. [DOI] [PubMed] [Google Scholar]
- 7.De Keulenaer G. W., Segers V. F. M., Zannad F., Brutsaert D. L. The future of pleiotropic therapy in heart failure. Lessons from the benefits of exercise training on endothelial function. European Journal of Heart Failure. 2017;19:603–614. doi: 10.1002/ejhf.735. [DOI] [PubMed] [Google Scholar]
- 8.Bhatia R. S., Tu J. V., Lee D. S., et al. Outcome of heart failure with preserved ejection fraction in a population-based study. The New England Journal of Medicine. 2006;355(3):260–269. doi: 10.1056/NEJMoa051530. [DOI] [PubMed] [Google Scholar]
- 9.Borlaug B. A. The pathophysiology of heart failure with preserved ejection fraction. Nature Reviews. Cardiology. 2014;11(10):507–515. doi: 10.1038/nrcardio.2014.83. [DOI] [PubMed] [Google Scholar]
- 10.Paulus W. J., Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction. Journal of the American College of Cardiology. 2013;62(4):263–271. doi: 10.1016/j.jacc.2013.02.092. [DOI] [PubMed] [Google Scholar]
- 11.Brutsaert D. L., Sys S. U. Relaxation and diastole of the heart. Physiological Reviews. 1989;69(4):1228–1315. doi: 10.1152/physrev.1989.69.4.1228. [DOI] [PubMed] [Google Scholar]
- 12.Widlansky M. E., Gokce N., Keaney J. F., Vita J. A. The clinical implications of endothelial dysfunction. Journal of the American College of Cardiology. 2003;42(7):1149–1160. doi: 10.1016/S0735-1097(03)00994-X. [DOI] [PubMed] [Google Scholar]
- 13.Xu Y., Arora R. C., Hiebert B. M., et al. Non-invasive endothelial function testing and the risk of adverse outcomes: a systematic review and meta-analysis. European Heart Journal. Cardiovascular Imaging. 2014;15(7):736–746. doi: 10.1093/ehjci/jet256. [DOI] [PubMed] [Google Scholar]
- 14.Katz S. D., Hryniewicz K., Hriljac I., et al. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure. Circulation. 2005;111(3):310–314. doi: 10.1161/01.CIR.0000153349.77489.CF. [DOI] [PubMed] [Google Scholar]
- 15.Haykowsky M. J., Tomczak C. R., Scott J. M., Paterson D. I., Kitzman D. W. Determinants of exercise intolerance in patients with heart failure and reduced or preserved ejection fraction. Journal of Applied Physiology. 2015;119(6):739–744. doi: 10.1152/japplphysiol.00049.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Upadhya B., Haykowsky M. J., Eggebeen J., Kitzman D. W. Exercise intolerance in heart failure with preserved ejection fraction: more than a heart problem. Journal of Geriatric Cardiology. 2015;12(3):294–304. doi: 10.11909/j.issn.1671-5411.2015.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Celermajer D. S. Endothelial dysfunction: does it matter? Is it reversible? Journal of the American College of Cardiology. 1997;30(2):325–333. doi: 10.1016/s0735-1097(97)00189-7. [DOI] [PubMed] [Google Scholar]
- 18.Hambrecht R., Gielen S., Linke A., et al. Effects of exercise training on left ventricular function and peripheral resistance in patients with chronic heart failure. Journal of the American Medical Association. 2000;283(23):3095–3101. doi: 10.1001/jama.283.23.3095. [DOI] [PubMed] [Google Scholar]
- 19.Hambrecht R., Fiehn E., Weigl C., et al. Regular physical exercise corrects endothelial dysfunction and improves exercise capacity in patients with chronic heart failure. Circulation. 1998;98(24):2709–2715. doi: 10.1161/01.cir.98.24.2709. [DOI] [PubMed] [Google Scholar]
- 20.Taylor R. S., Brown A., Ebrahim S., et al. Exercise-based rehabilitation for patients with coronary heart disease: systematic review and meta-analysis of randomized controlled trials. The American Journal of Medicine. 2004;116(10):682–692. doi: 10.1016/j.amjmed.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 21.O’Connor C., Whellan D., Lee K., et al. Efficacy and safety of exercise training in patients with chronic heart failure: HF-ACTION randomized controlled trial. Journal of the American Medical Association. 2009;301(14):1439–1450. doi: 10.1001/jama.2009.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gielen S., Schuler G., Adams V. Cardiovascular effects of exercise training: molecular mechanisms. Circulation. 2010;122(12):1221–1238. doi: 10.1161/CIRCULATIONAHA.110.939959. [DOI] [PubMed] [Google Scholar]
- 23.Pacher P., Beckman J. S., Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiological Reviews. 2007;87(1):315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brutsaert D. L. Cardiac endothelial-myocardial signaling: its role in cardiac growth, contractile performance, and rhythmicity. Physiological Reviews. 2003;83(1):59–115. doi: 10.1152/physrev.00017.2002. [DOI] [PubMed] [Google Scholar]
- 25.Özüyaman B., Ebner P., Niesler U., et al. Nitric oxide differentially regulates proliferation and mobilization of endothelial progenitor cells but not of hematopoietic stem cells. Thrombosis and Haemostasis. 2005;94(4):770–772. doi: 10.1160/TH05-01-0038. [DOI] [PubMed] [Google Scholar]
- 26.Bruyndonckx L., Hoymans V. Y., Van Craenenbroeck A. H., et al. Assessment of endothelial dysfunction in childhood obesity and clinical use. Oxidative Medicine and Cellular Longevity. 2013;2013:19. doi: 10.1155/2013/174782.174782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bruyndonckx L., Hoymans V. Y., Lemmens K., Ramet J., Vrints C. J. Childhood obesity-related endothelial dysfunction: an update on pathophysiological mechanisms and diagnostic advancements. Pediatric Research. 2016;79(6):831–837. doi: 10.1038/pr.2016.22. [DOI] [PubMed] [Google Scholar]
- 28.Van Craenenbroeck E. M., Conraads V. M. Endothelial progenitor cells in vascular health: focus on lifestyle. Microvascular Research. 2010;79(3):184–192. doi: 10.1016/j.mvr.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 29.Van Craenenbroeck E. M., Van Craenenbroeck A. H., van Ierssel S., et al. Quantification of circulating CD34+/KDR+/CD45dim endothelial progenitor cells: analytical considerations. International Journal of Cardiology. 2013;167(5):1688–1695. doi: 10.1016/j.ijcard.2012.10.047. [DOI] [PubMed] [Google Scholar]
- 30.Fischer D., Rossa S., Landmesser U., et al. Endothelial dysfunction in patients with chronic heart failure is independently associated with increased incidence of hospitalization, cardiac transplantation, or death. European Heart Journal. 2005;26(1):65–69. doi: 10.1093/eurheartj/ehi001. [DOI] [PubMed] [Google Scholar]
- 31.Moens A. L., Goovaerts I., Claeys M. J., Vrints C. J. Flow-mediated vasodilation: a diagnostic instrument, or an experimental tool? Chest. 2005;127(6):2254–2263. doi: 10.1378/chest.127.6.2254. [DOI] [PubMed] [Google Scholar]
- 32.Bruyndonckx L., Radtke T., Eser P., et al. Methodological considerations and practical recommendations for the application of peripheral arterial tonometry in children and adolescents. International Journal of Cardiology. 2013;168(4):3183–3190. doi: 10.1016/j.ijcard.2013.07.236. [DOI] [PubMed] [Google Scholar]
- 33.Arrebola-Moreno A. L., Laclaustra M., Kaski J. C. Noninvasive assessment of endothelial function in clinical practice. Revista Española de Cardiología (English Edition) 2012;65(1):80–90. doi: 10.1016/j.recesp.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 34.Hamburg N. M., Benjamin E. J. Assessment of endothelial function using digital pulse amplitude tonometry. Trends in Cardiovascular Medicine. 2009;19(1):6–11. doi: 10.1016/j.tcm.2009.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Treasure C. B., Vita J. A., Cox D. A., et al. Endothelium-dependent dilation of the coronary microvasculature is impaired in dilated cardiomyopathy. Circulation. 1990;81(3):772–779. doi: 10.1161/01.CIR.81.3.772. [DOI] [PubMed] [Google Scholar]
- 36.Collier P., Watson C. J., Voon V., et al. Can emerging biomarkers of myocardial remodelling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure? European Journal of Heart Failure. 2011;13(10):1087–1095. doi: 10.1093/eurjhf/hfr079. [DOI] [PubMed] [Google Scholar]
- 37.Kalogeropoulos A., Georgiopoulou V., Psaty B. M., et al. Inflammatory markers and incident heart failure risk in older adults. The health ABC (health, aging, and body composition) study. Journal of the American College of Cardiology. 2010;55(19):2129–2137. doi: 10.1016/j.jacc.2009.12.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Franssen C., Chen S., Unger A., et al. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC: Heart Failure. 2016;4(4):312–324. doi: 10.1016/j.jchf.2015.10.007. [DOI] [PubMed] [Google Scholar]
- 39.Westermann D., Lindner D., Kasner M., et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circulation: Heart Failure. 2011;4(1):44–52. doi: 10.1161/CIRCHEARTFAILURE.109.931451. [DOI] [PubMed] [Google Scholar]
- 40.Adams V., Alves M., Fischer T., et al. High-intensity interval training attenuates endothelial dysfunction in a dahl salt-sensitive rat model of heart failure with preserved ejection fraction. Journal of Applied Physiology. 2015;119(6):745–752. doi: 10.1152/japplphysiol.01123.2014. [DOI] [PubMed] [Google Scholar]
- 41.Van Heerebeek L., Hamdani N., Falcão-Pires I., et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012;126(7):830–839. doi: 10.1161/CIRCULATIONAHA.111.076075. [DOI] [PubMed] [Google Scholar]
- 42.Feldman A. M., Combes A., Wagner D., et al. The role of tumor necrosis factor in the pathophysiology of heart failure. Journal of the American College of Cardiology. 2000;35(3):537–544. doi: 10.1016/s0735-1097(99)00600-2. [DOI] [PubMed] [Google Scholar]
- 43.Yoshizumi M., Perrella M. A., Burnett J. C., Lee M. E. Tumor necrosis factor downregulates an endothelial nitric oxide synthase mRNA by shortening its half-life. Circulation Research. 1993;73(1):205–209. doi: 10.1161/01.RES.73.1.205. [DOI] [PubMed] [Google Scholar]
- 44.Bishu K., Deswal A., Chen H. H., et al. Biomarkers in acutely decompensated heart failure with preserved or reduced ejection fraction. American Heart Journal. 2012;164(5, article e3):763–770. doi: 10.1016/j.ahj.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamburg N. M., Keyes M. J., Larson M. G., et al. Cross-sectional relations of digital vascular function to cardiovascular risk factors in the Framingham heart study. Circulation. 2008;117(19):2467–2474. doi: 10.1161/CIRCULATIONAHA.107.748574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Benjamin E. J., Larson M. G., Keyes M. J., et al. Clinical correlates and heritability of flow-mediated dilation in the community: the Framingham heart study. Circulation. 2004;109(5):613–619. doi: 10.1161/01.CIR.0000112565.60887.1E. [DOI] [PubMed] [Google Scholar]
- 47.Akiyama E., Sugiyama S., Matsuzawa Y., et al. Incremental prognostic significance of peripheral endothelial dysfunction in patients with heart failure with normal left ventricular ejection fraction. Journal of the American College of Cardiology. 2012;60(18):1778–1786. doi: 10.1016/j.jacc.2012.07.036. [DOI] [PubMed] [Google Scholar]
- 48.Borlaug B. A., Olson T. P., Lam C. S. P., et al. Global cardiovascular reserve dysfunction in heart failure with preserved ejection fraction. Journal of the American College of Cardiology. 2010;56(11):845–854. doi: 10.1016/j.jacc.2010.03.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bonetti P. O., Pumper G. M., Higano S. T., Holmes D. R., Kuvin J. T., Lerman A. Noninvasive identification of patients with early coronary atherosclerosis by assessment of digital reactive hyperemia. Journal of the American College of Cardiology. 2004;44(11):2137–2141. doi: 10.1016/j.jacc.2004.08.062. [DOI] [PubMed] [Google Scholar]
- 50.Matsue Y., Suzuki M., Nagahori W., et al. Endothelial dysfunction measured by peripheral arterial tonometry predicts prognosis in patients with heart failure with preserved ejection fraction. International Journal of Cardiology. 2013;168(1):36–40. doi: 10.1016/j.ijcard.2012.09.021. [DOI] [PubMed] [Google Scholar]
- 51.Hamburg N. M., Palmisano J., Larson M. G., et al. Relation of brachial and digital measures of vascular function in the community: the framingham heart study. Hypertension. 2011;57(3):390–396. doi: 10.1161/HYPERTENSIONAHA.110.160812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shafiq A., Brawner C. A., Aldred H. A., et al. Prognostic value of cardiopulmonary exercise testing in heart failure with preserved ejection fraction. The Henry ford HospITal CardioPulmonary EXercise testing (FIT-CPX) project. American Heart Journal. 2016;174:167–172. doi: 10.1016/j.ahj.2015.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hirai D. M., Musch T. I., Poole D. C. Exercise training in chronic heart failure: improving skeletal muscle O2 transport and utilization. American Journal of Physiology. Heart and Circulatory Physiology. 2015;309(9):H1419–H1439. doi: 10.1152/ajpheart.00469.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sarelius I., Pohl U. Control of muscle blood flow during exercise: local factors and integrative mechanisms. Acta Physiologica. 2010;199(4):349–365. doi: 10.1111/j.1748-1716.2010.02129.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Little W. C., Borlaug B. A. Exercise intolerance in heart failure with preserved ejection fraction: what does the heart have to do with it? Circulation: Heart Failure. 2015;8(2):233–235. doi: 10.1161/CIRCHEARTFAILURE.114.001966. [DOI] [PubMed] [Google Scholar]
- 56.Santos M., Opotowsky A. R., Shah A. M., Tracy J., Waxman A. B., Systrom D. M. Central cardiac limit to aerobic capacity in patients with exertional pulmonary venous hypertension: implications for heart failure with preserved ejection fraction. Circulation: Heart Failure. 2014;8(2):278–285. doi: 10.1161/CIRCHEARTFAILURE.114.001551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haykowsky M. J., Brubaker P. H., John J. M., Stewart K. P., Morgan T. M., Kitzman D. W. Determinants of exercise intolerance in elderly heart failure patients with preserved ejection fraction. Journal of the American College of Cardiology. 2011;58(3):265–274. doi: 10.1016/j.jacc.2011.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shimiaie J., Sherez J., Aviram G., et al. Determinants of effort intolerance in patients with heart failure. JACC: Heart Failure. 2015;3(10):803–814. doi: 10.1016/j.jchf.2015.05.010. [DOI] [PubMed] [Google Scholar]
- 59.Dhakal B. P., Malhotra R., Murphy R. M., et al. Mechanisms of exercise intolerance in heart failure with preserved ejection fraction: the role of abnormal peripheral oxygen extraction. Circulation: Heart Failure. 2014;8(2):286–294. doi: 10.1161/CIRCHEARTFAILURE.114.001825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anderson T. J., Uehata A., Gerhard M. D., et al. Close relation of endothelial function in the human coronary and peripheral circulations. Journal of the American College of Cardiology. 1995;26(5):1235–1241. doi: 10.1016/0735-1097(95)00327-4. [DOI] [PubMed] [Google Scholar]
- 61.Haykowsky M. J., Herrington D. M., Brubaker P. H., Morgan T. M., Hundley W. G., Kitzman D. W. Relationship of flow-mediated arterial dilation and exercise capacity in older patients with heart failure and preserved ejection fraction. The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences. 2013;68(2):161–167. doi: 10.1093/gerona/gls099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maréchaux S., Samson R., Van Belle E., et al. Vascular and microvascular endothelial function in heart failure with preserved ejection fraction. Journal of Cardiac Failure. 2016;22(1):3–11. doi: 10.1016/j.cardfail.2015.09.003. [DOI] [PubMed] [Google Scholar]
- 63.Lee J. F., Barrett-O’Keefe Z., Garten R. S., et al. Evidence of microvascular dysfunction in heart failure with preserved ejection fraction. Heart. 2016;102(4):278–284. doi: 10.1136/heartjnl-2015-308403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yamamoto E., Hirata Y., Tokitsu T., et al. The pivotal role of eNOS uncoupling in vascular endothelial dysfunction in patients with heart failure with preserved ejection fraction. International Journal of Cardiology. 2015;190:335–337. doi: 10.1016/j.ijcard.2015.04.162. [DOI] [PubMed] [Google Scholar]
- 65.Balmain S., Padmanabhan N., Ferrell W. R., Morton J. J., McMurray J. J. V. Differences in arterial compliance, microvascular function and venous capacitance between patients with heart failure and either preserved or reduced left ventricular systolic function. European Journal of Heart Failure. 2007;9(9):865–871. doi: 10.1016/j.ejheart.2007.06.003. [DOI] [PubMed] [Google Scholar]
- 66.Pyke K. E., Tschakovsky M. E. The relationship between shear stress and flow-mediated dilatation: implications for the assessment of endothelial function. The Journal of Physiology. 2005;568(Part 2):357–369. doi: 10.1113/jphysiol.2005.089755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thijssen D. H., Black M. A., Pyke K. E., et al. Assessment of flow-mediated dilation in humans: a methodological and physiological guideline. American Journal of Physiology. Heart and Circulatory Physiology. 2011;300(1):H2–H12. doi: 10.1152/ajpheart.00471.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Haykowsky M. J., Brubaker P. H., Morgan T. M., Kritchevsky S., Eggebeen J., Kitzman D. W. Impaired aerobic capacity and physical functional performance in older heart failure patients with preserved ejection fraction: role of lean body mass. The Journals of Gerontology. Series A, Biological Sciences and Medical Sciences. 2013;68(8):968–975. doi: 10.1093/gerona/glt011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haykowsky M. J., Kouba E. J., Brubaker P. H., Nicklas B. J., Eggebeen J., Kitzman D. W. Skeletal muscle composition and its relation to exercise intolerance in older patients with heart failure and preserved ejection fraction. The American Journal of Cardiology. 2014;113(7):1211–1216. doi: 10.1016/j.amjcard.2013.12.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Molina A. J. A., Bharadwaj M. S., Van Horn C., et al. Skeletal muscle mitochondrial content, oxidative capacity, and Mfn2 expression are reduced in older patients with heart failure and preserved ejection fraction and are related to exercise intolerance. JACC: Heart Failure. 2016;4(8):0–9. doi: 10.1016/j.jchf.2016.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bowen T. S., Rolim N. P. L., Fischer T., et al. Heart failure with preserved ejection fraction induces molecular, mitochondrial, histological, and functional alterations in rat respiratory and limb skeletal muscle. European Journal of Heart Failure. 2015;17(3):263–272. doi: 10.1002/ejhf.239. [DOI] [PubMed] [Google Scholar]
- 72.Kitzman D. W., Nicklas B., Kraus W. E., et al. Skeletal muscle abnormalities and exercise intolerance in older patients with heart failure and preserved ejection fraction. American Journal of Physiology. Heart and Circulatory Physiology. 2014;306(9):H1364–H1370. doi: 10.1152/ajpheart.00004.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lee J. F., Barrett-O’Keefe Z., Nelson A. D., et al. Impaired skeletal muscle vasodilation during exercise in heart failure with preserved ejection fraction. International Journal of Cardiology. 2016;211:14–21. doi: 10.1016/j.ijcard.2016.02.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kitzman D. W., Brubaker P. H., Herrington D. M., et al. Effect of endurance exercise training on endothelial function and arterial stiffness in older patients with heart failure and preserved ejection fraction: a randomized, controlled, single-blind trial. Journal of the American College of Cardiology. 2013;62(7):584–592. doi: 10.1016/j.jacc.2013.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dini F. L., Ghiadoni L., Conti U., et al. Coronary flow Reserve in Idiopathic Dilated Cardiomyopathy: relation with left Ventricular Wall stress, natriuretic peptides, and endothelial dysfunction. Journal of the American Society of Echocardiography. 2009;22(4):354–360. doi: 10.1016/j.echo.2009.01.013. [DOI] [PubMed] [Google Scholar]
- 76.Snoer M., Monk-Hansen T., Olsen R. H., et al. Coronary flow reserve as a link between diastolic and systolic function and exercise capacity in heart failure. European Heart Journal. Cardiovascular Imaging. 2013;14(7):677–683. doi: 10.1093/ehjci/jes269. [DOI] [PubMed] [Google Scholar]
- 77.Anantharam B., Janardhanan R., Hayat S., et al. Coronary flow reserve assessed by myocardial contrast echocardiography predicts mortality in patients with heart failure. European Journal of Echocardiography. 2011;12(1):69–75. doi: 10.1093/ejechocard/jeq109. [DOI] [PubMed] [Google Scholar]
- 78.Tschöpe C., Bock C.-T., Kasner M., et al. High prevalence of cardiac parvovirus B19 infection in patients with isolated left ventricular diastolic dysfunction. Circulation. 2005;111(7):879–886. doi: 10.1161/01.CIR.0000155615.68924.B3. [DOI] [PubMed] [Google Scholar]
- 79.Teragaki M., Yanagi S., Toda I., et al. Coronary flow reserve correlates left ventricular diastolic dysfunction in patients with dilated cardiomyopathy. Catheterization and Cardiovascular Interventions. 2003;58(1):43–50. doi: 10.1002/ccd.10349. [DOI] [PubMed] [Google Scholar]
- 80.Snoer M., Monk-Hansen T., Olsen R., et al. Insulin resistance and exercise tolerance in heart failure patients: linkage to coronary flow reserve and peripheral vascular function. Cardiovascular Diabetology. 2012;11(1):p. 97. doi: 10.1186/1475-2840-11-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pinsky D. J., Patton S., Mesaros S., et al. Mechanical transduction of nitric oxide synthesis in the beating heart. Circulation Research. 1997;81(3):372–379. doi: 10.1161/01.RES.81.3.372. [DOI] [PubMed] [Google Scholar]
- 82.Kohr M. J., Davis J. P., Ziolo M. T. Peroxynitrite increases protein phosphatase activity and promotes the interaction of phospholamban with protein phosphatase 2a in the myocardium. Nitric Oxide. 2009;20(3):217–221. doi: 10.1016/j.niox.2009.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hamdani N., Franssen C., Lourenço A., et al. Myocardial titin hypophosphorylation importantly contributes to heart failure with preserved ejection fraction in a rat metabolic risk model. Circulation: Heart Failure. 2013;6(6):1239–1249. doi: 10.1161/CIRCHEARTFAILURE.113.000539. [DOI] [PubMed] [Google Scholar]
- 84.Takimoto E., Champion H. C., Li M., et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nature Medicine. 2005;11(2):214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 85.Hou J., Kato H., Cohen R. A., Chobanian A. V., Brecher P. Angiotensin II-induced cardiac fibrosis in the rat is increased by chronic inhibition of nitric oxide synthase. The Journal of Clinical Investigation. 1995;96(5):2469–2477. doi: 10.1172/JCI118305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lam C. S. P., Roger V. L., Rodeheffer R. J., Borlaug B., Enders F. T., Redfield M. M. Pulmonary hypertension in heart failure with preserved ejection fraction: a community-based study. Journal of the American College of Cardiology. 2009;53(13):1119–1126. doi: 10.1016/j.jacc.2008.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Borlaug B. A., Nishimura R. A., Sorajja P., Lam C. S. P., Redfield M. M. Exercise hemodynamics enhance diagnosis of early heart failure with preserved ejection fraction. Circulation: Heart Failure. 2010;3(5):588–595. doi: 10.1161/CIRCHEARTFAILURE.109.930701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Borlaug B. A., Kane G. C., Melenovsky V., Olson T. P. Abnormal right ventricular-pulmonary artery coupling with exercise in heart failure with preserved ejection fraction. European Heart Journal. 2016;37(43):3293–3302. doi: 10.1093/eurheartj/ehx184. ehw 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mohammed S. F., Hussain I., Abou Ezzeddine OF, et al. Right ventricular function in heart failure with preserved ejection fraction: a community-based study. Circulation. 2014;130(25):2310–2320. doi: 10.1161/CIRCULATIONAHA.113.008461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Andersen M. J., Hwang S. J., Kane G. C., et al. Enhanced pulmonary vasodilator reserve and abnormal right ventricular: pulmonary artery coupling in heart failure with preserved ejection fraction. Circulation: Heart Failure. 2015;8(3):542–550. doi: 10.1161/CIRCHEARTFAILURE.114.002114. [DOI] [PubMed] [Google Scholar]
- 91.Driss A B., Devaux C., Henrion D., et al. Hemodynamic stresses induce endothelial dysfunction and remodeling of pulmonary artery in experimental compensated heart failure. Circulation. 2000;101(23) doi: 10.1161/01.cir.101.23.2764. [DOI] [PubMed] [Google Scholar]
- 92.Lam C. S. P., Brutsaert D. L. Endothelial dysfunction: a pathophysiologic factor in heart failure with preserved ejection fraction. Journal of the American College of Cardiology. 2012;60(18):1787–1789. doi: 10.1016/j.jacc.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 93.Lai Y.-C., Tabima D. M., Dube J. J., et al. SIRT3-AMPK activation by nitrite and metformin improves hyperglycemia and normalizes pulmonary hypertension associated with heart failure with preserved ejection fraction (PH-HFpEF) Circulation. 2016;133(8):717–731. doi: 10.1161/CIRCULATIONAHA.115.018935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Farrero M., Blanco I., Batlle M., et al. Pulmonary hypertension is related to peripheral endothelial dysfunction in heart failure with preserved ejection fraction. Circulation: Heart Failure. 2014;7(5):791–798. doi: 10.1161/CIRCULATIONAHA.115.018935. [DOI] [PubMed] [Google Scholar]
- 95.Cooper C. J., Landzberg M. J., Anderson T. J., et al. Role of nitric oxide in the local regulation of pulmonary vascular resistance in humans. Circulation. 1996;93(2):266–271. doi: 10.1161/01.CIR.93.2.266. [DOI] [PubMed] [Google Scholar]
- 96.Segers V. F. M., Brutsaert D. L., De Keulenaer G. W. Pulmonary hypertension and right heart failure in heart failure with preserved left ventricular ejection fraction. Current Opinion in Cardiology. 2012;27(3):273–280. doi: 10.1097/HCO.0b013e3283512035. [DOI] [PubMed] [Google Scholar]
- 97.Andrea R., López-Giraldo A., Falces C., et al. Lung function abnormalities are highly frequent in patients with heart failure and preserved ejection fraction. Heart, Lung & Circulation. 2014;23(3):273–279. doi: 10.1016/j.hlc.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 98.Olson T. P., Johnson B. D., Borlaug B. A. Impaired pulmonary diffusion in heart failure with preserved ejection fraction. JACC: Heart Failure. 2016;4(6):490–498. doi: 10.1016/j.jchf.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Massie B. M., Carson P. E., McMurray J. J., et al. Irbesartan in patients with heart failure and preserved ejection fraction. The New England Journal of Medicine. 2008;359(23):2456–2467. doi: 10.1161/CIRCULATIONAHA.116.024593. [DOI] [PubMed] [Google Scholar]
- 100.Hillege H. L., Nitsch D., Pfeffer M. A., et al. Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation. 2006;113(5):671–678. doi: 10.1161/CIRCULATIONAHA.105.580506. [DOI] [PubMed] [Google Scholar]
- 101.Damman K., Valente M. A. E., Voors A. A., O’Connor C. M., van Veldhuisen D. J., Hillege H. L. Renal impairment, worsening renal function, and outcome in patients with heart failure: an updated meta-analysis. European Heart Journal. 2014;35(7):455–469. doi: 10.1093/eurheartj/eht386. [DOI] [PubMed] [Google Scholar]
- 102.Van Craenenbroeck A. H., Van Craenenbroeck E. M., Van Ackeren K., et al. Effect of moderate aerobic exercise training on endothelial function and arterial stiffness in CKD stages 3-4: a randomized controlled trial. American Journal of Kidney Diseases. 2015;66(2):285–296. doi: 10.1053/j.ajkd.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 103.Hirata Y., Sugiyama S., Yamamoto E., et al. Endothelial function and cardiovascular events in chronic kidney disease. International Journal of Cardiology. 2014;173(3):481–486. doi: 10.1016/j.ijcard.2014.03.085. [DOI] [PubMed] [Google Scholar]
- 104.Gevaert A., Van Craenenbroeck A. H., Shivalkar B., Lemmens K., Vrints C. J., Van Craenenbroeck E. M. Preclinical diastolic dysfunction is related to impaired endothelial function in chronic kidney disease patients (Abstract) European Heart Journal - Cardiovascular Imaging. 2015;16(Supplement 2):p. S4. doi: 10.1093/ehjci/jev260. [DOI] [Google Scholar]
- 105.ter Maaten J. M., Damman K., Verhaar M. C., et al. Connecting heart failure with preserved ejection fraction and renal dysfunction: the role of endothelial dysfunction and inflammation. European Journal of Heart Failure. 2016;18(6):588–598. doi: 10.1002/ejhf.497. [DOI] [PubMed] [Google Scholar]
- 106.Van Craenenbroeck A. H., Van Craenenbroeck E. M., Kouidi E., Vrints C. J., Couttenye M. M., Conraads V. M. Vascular effects of exercise training in CKD: current evidence and pathophysiological mechanisms. Clinical Journal of the American Society of Nephrology. 2014;9(7):1305–1318. doi: 10.2215/CJN.13031213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mullens W., Abrahams Z., Francis G. S., et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. Journal of the American College of Cardiology. 2009;53(7):589–596. doi: 10.1016/j.jacc.2008.05.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Klein D. A., Katz D. H., Beussink-Nelson L., Sanchez C. L., Strzelczyk T. A., Shah S. J. Association of Chronic Kidney Disease with Chronotropic Incompetence in heart failure with preserved ejection fraction. The American Journal of Cardiology. 2015;116(7):1093–1100. doi: 10.1016/j.amjcard.2015.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tang W. H. W., Mullens W. Cardiorenal syndrome in decompensated heart failure. Heart. 2010;96(4):255–260. doi: 10.1136/hrt.2009.166256. [DOI] [PubMed] [Google Scholar]
- 110.Damkjær M., Vafaee M., Møller M. L., et al. Renal cortical and medullary blood flow responses to altered NO availability in humans. American Journal of Physiology. Regulatory, Integrative and Comparative Physiology. 2010;299(6):R1449–R1455. doi: 10.1152/ajpregu.00440.2010. [DOI] [PubMed] [Google Scholar]
- 111.Van Dijk C. G., Oosterhuis N. R., Xu Y. J., et al. Distinct endothelial cell responses in the heart and kidney microvasculature characterize the progression of heart failure with preserved ejection fraction in the obese ZSF1 rat with Cardiorenal metabolic Syndrome. Circulation: Heart Failure. 2016;9(4, article e002760) doi: 10.1161/CIRCHEARTFAILURE.115.002760. [DOI] [PubMed] [Google Scholar]
- 112.Brouwers F. P., de Boer R. A., van der Harst P., et al. Incidence and epidemiology of new onset heart failure with preserved versus reduced ejection fraction in a community-based cohort: 11 year follow-up of PREVEND. European Heart Journal. 2013;34(19):1424–1431. doi: 10.1093/eurheartj/eht066. [DOI] [PubMed] [Google Scholar]
- 113.Rostand S., Drueke T. Parathyroid hormone, vitamin D, and cardiovascular disease in chronic renal failure. Kidney International. 1999;56(2):383–392. doi: 10.1046/j.1523-1755.1999.00575.x. [DOI] [PubMed] [Google Scholar]
- 114.Chitalia N., Recio-Mayoral A., Kaski J., Banerjee D. Vitamin D deficiency and endothelial dysfunction in non-dialysis chronic kidney disease patients. Atherosclerosis. 2012;220(1):265–268. doi: 10.1016/j.atherosclerosis.2011.10.023. [DOI] [PubMed] [Google Scholar]
- 115.Jie K., Zaikova M., Bergevoet M., et al. Progenitor cells and vascular function are impaired in patients with chronic kidney disease. Nephrology Dialysis Transplantation. 2010;25(6):1875–1882. doi: 10.1093/ndt/gfp749. [DOI] [PubMed] [Google Scholar]
- 116.Oberleithner H., Riethmuller C., Schillers H., Mac Gregor G. A., de Wardener H. E., Hausberg M. Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proceedings of the National Academy of Sciences. 2007;104(41):16281–16286. doi: 10.1073/pnas.0707791104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Edelmann F., Gelbrich G., Düngen H.-D., et al. Exercise training improves exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction (ex-DHF) Journal of the American College of Cardiology. 2011;58(17):1780–1791. doi: 10.1016/j.jacc.2011.06.054. [DOI] [PubMed] [Google Scholar]
- 118.Kitzman D. W., Brubaker P. H., Morgan T. M., Stewart K. P., Little W. C. Exercise training in older patients with heart failure and preserved ejection fraction: a randomized, controlled, single-blind trial. Circulation: Heart Failure. 2010;3(6):659–667. doi: 10.1161/CIRCHEARTFAILURE.110.958785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kitzman D. W., Brubaker P., Morgan T., et al. Effect of caloric restriction or aerobic exercise training on peak oxygen consumption and quality of life in obese older patients with heart failure with preserved ejection fraction. Journal of the American Medical Association. 2016;315(1):p. 36. doi: 10.1001/jama.2015.17346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Alves A. J., Ribeiro F., Goldhammer E., et al. Exercise training improves diastolic function in heart failure patients. Medicine and Science in Sports and Exercise. 2012;44(5):776–785. doi: 10.1249/MSS.0b013e31823cd16a. [DOI] [PubMed] [Google Scholar]
- 121.Smart N. A., Haluska B., Jeffries L., Leung D. Exercise training in heart failure with preserved systolic function: a randomized controlled trial of the effects on cardiac function and functional capacity. Congestive Heart Failure. 2012;18(6):295–301. doi: 10.1111/j.1751-7133.2012.00295.x. [DOI] [PubMed] [Google Scholar]
- 122.Gary R. A., Sueta C. A., Dougherty M., et al. Home-based exercise improves functional performance and quality of life in women with diastolic heart failure. Heart Lung. 2004;33(4):210–218. doi: 10.1016/j.hrtlng.2004.01.004. [DOI] [PubMed] [Google Scholar]
- 123.Chan E., Giallauria F., Vigorito C., Smart N. A. Exercise training in heart failure patients with preserved ejection fraction: a systematic review and meta-analysis. Monaldi Archive of Chest Diseases. 2016;86(1-2):p. 759. doi: 10.4081/monaldi.2016.759. [DOI] [PubMed] [Google Scholar]
- 124.Fukuta H., Goto T., Wakami K., Ohte N. Effects of drug and exercise intervention on functional capacity and quality of life in heart failure with preserved ejection fraction: A meta-analysis of randomized controlled trials. European Journal of Preventive Cardiology. 2016;23(1):78–85. doi: 10.1177/2047487314564729. [DOI] [PubMed] [Google Scholar]
- 125.Pandey A., Parashar A., Kumbhani D. J. Exercise training in patients with heart failure and preserved ejection fraction: meta-analysis of randomized control trials. Circulation: Heart Failure. 8(1):33–40. doi: 10.1161/CIRCHEARTFAILURE.114.001615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wisløff U., Støylen A., Loennechen J. P., et al. Superior cardiovascular effect of aerobic interval training versus moderate continuous training in heart failure patients: a randomized study. Circulation. 2007;115(24):3086–3094. doi: 10.1161/CIRCULATIONAHA.106.675041. [DOI] [PubMed] [Google Scholar]
- 127.Ellingsen Ø., Halle M., Conraads V. M., et al. High intensity interval training in heart failure patients with reduced ejection fraction. Circulation. 2017;135(9):839–849. doi: 10.1016/j.jtemb.2017.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Angadi S. S., Mookadam F., Lee C. D., Tucker W. J., Haykowsky M. J., Gaesser G. A. High-intensity interval training versus moderate-intensity continuous exercise training in heart failure with preserved ejection fraction: a pilot study. Journal of Applied Physiology. 2015;119(6):753–758. doi: 10.1152/japplphysiol.00518.2014. [DOI] [PubMed] [Google Scholar]
- 129.Suchy C., Massen L., Rognmo O., et al. Optimising exercise training in prevention and treatment of diastolic heart failure (OptimEx-CLIN): rationale and design of a prospective, randomised, controlled trial. European Journal of Preventive Cardiology. 2014;21(2S):18–25. doi: 10.1177/2047487314552764. [DOI] [PubMed] [Google Scholar]
- 130.Adams V., Linke A., Kränkel N., et al. Impact of regular physical activity on the NAD(P)H oxidase and angiotensin receptor system in patients with coronary artery disease. Circulation. 2005;111(5):555–562. doi: 10.1161/01.CIR.0000154560.88933.7E. [DOI] [PubMed] [Google Scholar]
- 131.Conraads V. M., Van Craenenbroeck E. M., De Maeyer C., Van Berendoncks A. M., Beckers P. J., Vrints C. J. Unraveling new mechanisms of exercise intolerance in chronic heart failure. Role of exercise training. Heart Failure Reviews. 2013;18(1):65–77. doi: 10.1007/s10741-012-9324-0. [DOI] [PubMed] [Google Scholar]
- 132.Urbich C., Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circulation Research. 2004;95(4):343–353. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 133.Van Craenenbroeck E. M., Conraads V. M. Mending injured endothelium in chronic heart failure: a new target for exercise training. International Journal of Cardiology. 2013;166(2):310–314. doi: 10.1016/j.ijcard.2012.04.106. [DOI] [PubMed] [Google Scholar]
- 134.Van Craenenbroeck E. M., Beckers P. J., Possemiers N. M., et al. Exercise acutely reverses dysfunction of circulating angiogenic cells in chronic heart failure. European Heart Journal. 2010;31(15):1924–1934. doi: 10.1093/eurheartj/ehq058. [DOI] [PubMed] [Google Scholar]
- 135.Van Craenenbroeck E. M., Hoymans V. Y., Beckers P. J., et al. Exercise training improves function of circulating angiogenic cells in patients with chronic heart failure. Basic Research in Cardiology. 2010;105(5):665–676. doi: 10.1007/s00395-010-0105-4. [DOI] [PubMed] [Google Scholar]
- 136.Sandri M., Viehmann M., Adams V., et al. Chronic heart failure and aging—effects of exercise training on endothelial function and mechanisms of endothelial regeneration: results from the Leipzig exercise intervention in chronic heart failure and aging (LEICA) study. European Journal of Preventive Cardiology. 2016;23(4):349–358. doi: 10.1177/2047487315588391. [DOI] [PubMed] [Google Scholar]
- 137.Trippel T. D., Holzendorf V., Halle M., et al. Ghrelin and hormonal markers under exercise training in patients with heart failure with preserved ejection fraction: results from the ex-DHF pilot study. ESC Heart Failure. 2017;4(1):56–65. doi: 10.1002/ehf2.12109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hambrecht R., Wolf A., Gielen S., et al. Effect of exercise on coronary endothelial function in patients with coronary artery disease. The New England Journal of Medicine. 2000;342(7):454–460. doi: 10.1056/NEJM200002173420702. [DOI] [PubMed] [Google Scholar]
- 139.Conraads V. M., Pattyn N., De Maeyer C., et al. Aerobic interval training and continuous training equally improve aerobic exercise capacity in patients with coronary artery disease: the SAINTEX-CAD study. International Journal of Cardiology. 2015;179:203–210. doi: 10.1016/j.ijcard.2014.10.155. [DOI] [PubMed] [Google Scholar]
- 140.Higashi Y., Sasaki S., Sasaki N., et al. Daily aerobic exercise improves reactive hyperemia in patients with essential hypertension. Hypertension. 1999;33(1, Part 2):591–597. doi: 10.1161/01.hyp.33.1.591. [DOI] [PubMed] [Google Scholar]
- 141.Haykowsky M. J., Brubaker P. H., Stewart K. P., Morgan T. M., Eggebeen J., Kitzman D. W. Effect of endurance training on the determinants of peak exercise oxygen consumption in elderly patients with stable compensated heart failure and preserved ejection fraction. Journal of the American College of Cardiology. 2012;60(2):120–128. doi: 10.1016/j.jacc.2012.02.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fu T.-C., Yang N.-I., Wang C.-H., et al. Aerobic interval training elicits different hemodynamic adaptations between heart failure patients with preserved and reduced ejection fraction. American Journal of Physical Medicine & Rehabilitation. 2016;95(1):15–27. doi: 10.1097/PHM.0000000000000312. [DOI] [PubMed] [Google Scholar]
- 143.Corretti M. C., Anderson T. J., Benjamin E. J., et al. Guidelines for the ultrasound assessment of endothelial-dependent flow- mediated vasodilation of the brachial artery: a report of the International brachial artery reactivity task force. Journal of the American College of Cardiology. 2002;39(2):257–265. doi: 10.1016/s0735-1097(01)01746-6. [DOI] [PubMed] [Google Scholar]
- 144.Legallois D., Belin A., Nesterov S. V., et al. Cardiac rehabilitation improves coronary endothelial function in patients with heart failure due to dilated cardiomyopathy: a positron emission tomography study. European Journal of Preventive Cardiology. 2016;23(2):129–136. doi: 10.1177/2047487314565739. [DOI] [PubMed] [Google Scholar]
- 145.Shah S. J., Katz D. H., Deo R. C. Phenotypic Spectrum of heart failure with preserved ejection fraction. Heart Failure Clinics. 2014;10(3):407–418. doi: 10.1016/j.hfc.2014.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Shah S. J., Kitzman D. W., Borlaug B. A., et al. Phenotype-specific treatment of heart failure with preserved ejection fraction. Circulation. 2016;134(1):73–90. doi: 10.1161/CIRCULATIONAHA.116.021884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shah S. J., Katz D. H., Selvaraj S., et al. Phenomapping for novel classification of heart failure with preserved ejection fraction. Circulation. 2014;131(3):269–279. doi: 10.1161/CIRCULATIONAHA.114.010637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Obokata M., Reddy Y. N. V., Pislaru S. V., Melenovsky V., Borlaug B. A. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation. 2017 doi: 10.1161/CIRCULATIONAHA.116.026807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Pieske B., Butler J., Filippatos G., et al. Rationale and design of the SOluble guanylate Cyclase stimulatoR in heArT failurE Studies (SOCRATES) European Journal of Heart Failure. 2014;16(9):1026–1038. doi: 10.1002/ejhf.135. [DOI] [PubMed] [Google Scholar]
- 150.Hundley W. G., Bayram E., Hamilton C. A., et al. Leg flow-mediated arterial dilation in elderly patients with heart failure and normal left ventricular ejection fraction. American Journal of Physiology. Heart and Circulatory Physiology. 2006;292(3):H1427–H1434. doi: 10.1152/ajpheart.00567.2006. [DOI] [PubMed] [Google Scholar]
- 151.Kishimoto S., Kajikawa M., Maruhashi T., et al. Endothelial dysfunction and abnormal vascular structure are simultaneously present in patients with heart failure with preserved ejection fraction. International Journal of Cardiology. 2017;231:181–187. doi: 10.1016/j.ijcard.2017.01.024. [DOI] [PubMed] [Google Scholar]
- 152.Vitiello D., Harel F., Touyz R. M., et al. Changes in cardiopulmonary reserve and peripheral arterial function concomitantly with subclinical inflammation and oxidative stress in patients with heart failure with preserved ejection fraction. International Journal of Vascular Medicine. 2014;2014:8. doi: 10.1155/2014/917271.917271 [DOI] [PMC free article] [PubMed] [Google Scholar]