

Abstract
Proximal tubule (PT) dysfunction, including tubular proteinuria, is a significant complication in young sickle cell disease (SCD) that can eventually lead to chronic kidney disease. Hemoglobin (Hb) dimers released from red blood cells upon hemolysis are filtered into the kidney and internalized by megalin/cubilin receptors into PT cells. The PT is especially sensitive to heme toxicity, and tubular dysfunction in SCD is thought to result from prolonged exposure to filtered Hb. Here we show that concentrations of Hb predicted to enter the tubule lumen during hemolytic crisis competitively inhibit the uptake of another megalin/cubilin ligand (albumin) by PT cells. These effects were independent of heme reduction state. The Glu7Val mutant of Hb that causes SCD was equally effective at inhibiting albumin uptake compared with wild-type Hb. Addition of the Hb scavenger haptoglobin (Hpt) restored albumin uptake in the presence of Hb, suggesting that Hpt binding to the Hb αβ dimer-dimer interface interferes with Hb binding to megalin/cubilin. BLAST searches and structural modeling analyses revealed regions of similarity between Hb and albumin that map to this region and may represent sites of Hb interaction with megalin/cubilin. Our studies suggest that impaired endocytosis of megalin/cubilin ligands, rather than heme toxicity, may be the cause of tubular proteinuria in SCD patients. Additionally, loss of these filtered proteins into the urine may contribute to the extra-renal pathogenesis of SCD.
Keywords: megalin, proteinuria, proximal tubule, sickle cell disease, vitamin D
sickle cell disease (SCD) is a devastating disease resulting from a single mutation (Glu7Val) in hemoglobin (Hb) that causes red blood cells to assume a rigid curved shape that blocks their passage through the vasculature. Obstruction of capillaries by sickled red blood cells results in ischemia, severe pain, and necrosis. Additionally, red blood cells (RBCs) in SCD patients are susceptible to hemolysis, resulting in chronically elevated plasma levels of free Hb that can skyrocket during hemolytic crises (26). Free Hb in the circulation can scavenge nitric oxide (NO) produced by endothelial cells, leading to vasoconstriction that compounds vaso-occlusion (34). Exposure of cells to heme proteins also triggers the production of cytotoxic reactive oxygen species (34).
With the development of treatment regimens to increase life expectancy, kidney manifestations of SCD have become increasingly appreciated. There are numerous renal complications in SCD, including glomerulopathy, acute kidney injury, chronic kidney disease, impaired urinary concentrating ability, and distal nephron dysfunction. Kidney disease currently accounts for >15% of mortality in SCD patients (20). These complications are due in part to the propensity of red blood cells to sickle in the hypoxic renal medulla. However, exposure of kidney cells to Hb liberated during hemolysis also plays an important role in the progression of renal disease. Released Hb dimers (consisting of α- and β-globin chains, each with molecular mass ~16 kDa) are readily filtered into the tubule lumen with a fractional filtration coefficient of 0.03 (18). At the normal plasma level of Hb of 3 mg/dl (2 µM), the concentration in the glomerular ultrafiltrate entering the kidney tubule lumen is very low, ~60 nM. However, plasma concentrations of Hb are chronically about tenfold higher in SCD patients, and during hemolytic crisis, the concentration of plasma Hb can approach 1 g/dl, resulting in tubular concentrations above 15 µM (21).
Filtered Hb is taken up by the multiligand receptors megalin and cubilin, which are abundantly expressed in the S1 segment of the kidney proximal tubule (7). Previous studies show that Hb binds to megalin and cubilin with relatively high affinity [1.7 µM and 4.1 µM, respectively (11)]. Megalin and cubilin also bind with comparable affinities to a large number of other filtered low-molecular-weight (LMW) proteins and other ligands, including vitamin D binding protein, intrinsic factor-cobalamine (vitamin B12), and parathyroid hormone (10). In addition, megalin and cubilin take up the low level of albumin that normally escapes the glomerular filtration barrier. Disruption of the apical endocytic pathway leads to tubular proteinuria (aka LMW proteinuria), that if left unchecked can trigger inflammation and fibrosis resulting in end-stage renal disease (22).
The PT is known to be especially sensitive to heme toxicity, and cytoprotective responses (upregulated expression of ferritin, ferroportin, heme-oxygenase I, heme oxygenase II, Hpt, and hemopexin) have been well characterized in response to heme-induced injury (19, 31). Consistent with this, tubular proteinuria has been reported in a significant fraction of SCD patients, and particularly in younger patients (3, 16, 17). These patients also exhibit increased excretion of urinary biomarkers characteristic of tubular injury (27). Tubular proteinuria in these patients frequently occurred independently of glomerular dysfunction, suggesting that PT injury is an initiating step in the cascade leading to chronic kidney disease in SCD patients.
PT function, including the uptake of filtered megalin/cubilin ligands, is highly responsive to changes in fluid shear stress that accompany tubular flow (25, 32). Because NO mediates mechanosensitive responses in endothelial cells, we wondered whether Hb released into the tubule lumen during hemolytic crises might scavenge NO to impair apical endocytosis. To test this, we assessed whether exposing PT cells to levels of Hb expected during SCD crisis affects uptake of albumin. We found that Hb inhibits albumin uptake by PT cells in a dose-dependent manner. Surprisingly, the effect of Hb is independent of any effect on NO and instead results from direct competition for uptake by megalin/cubilin receptors. Impaired uptake of normally filtered megalin/cubilin ligands uptake during hemolytic crisis may explain clinical manifestations of SCD of unknown etiology.
MATERIALS AND METHODS
Cell culture.
All cell culture reagents were from Sigma unless otherwise specified. Opossum kidney (OK) cells (Didelphis virginiana, adult female, kidney cortex) were cultured in DMEM/F12 medium with 10% FBS (Atlanta Biologicals) and 5 mM GlutaMAX (GIBCO). HK-2 cells (Homo sapiens, adult male, cortex/proximal tubules, papilloma immortalized) were cultured in DMEM/F12 with 5 µg/ml insulin, 0.02 µg/ml dexamethasone, 0.01 µg/ml selenium, 5 µg/ml transferrin, 2 mM l-glutamine, and 10% FBS (Atlanta Biologicals).
Quantitation and imaging of endocytosis.
HK-2 (5 × 105) or OK (4 × 105) cells were plated in triplicate samples on 12-mm Transwell (0.4 µm pore) polycarbonate membrane inserts (Corning) in a 12-well plate, with 0.5 ml apical medium and 1.5 ml basolateral medium. The following day, cells were transferred to an orbital shaker set at 74–146 rpm in the incubator and allowed to grow for an additional 3-4 days with daily medium changes. Unpublished studies in our lab demonstrate that chronic exposure to orbital shear stress enhances cell differentiation and endocytic capacity. Although endocytic capacity was lower, we found similar results to those reported here using OK cells cultured under static conditions.
For experiments measuring albumin uptake, cells were incubated with apically added 0.6 µM Alexa Fluor 647-BSA (Thermo Fisher Scientific) and unlabeled Hb or l-NAME (30 min pretreatment) as indicated in DMEM/F12 medium with 25 mM HEPES (GIBCO) for 1 h at 37°C under continuous orbital rotation. For endocytosis studies in Fig. 4B, OxyHb and Hpt (Athens Research and Technology) were added to cells as indicated 30 min prior to addition of albumin. In experiments measuring uptake of Hb uptake, cells were incubated with 2 µM Alexa Fluor 568-conjugated Hb for 1 h at 37°C under orbital shear stress in the presence of unlabeled albumin or OxyHb as specified. To quantify the uptake of fluorescent ligands, filters were washed five times with cold PBS containing Ca2+ and Mg2+, solubilized in 300 µl 20 mM MOPS, pH 7.4/0.1% Triton X-100 for 30 min shaking at 4°C, and fluorescence was quantified using the GloMax Multi-Detection System (Promega). Cells on filters used for imaging were washed, fixed in 4% paraformaldehyde, and imaged using a Leica TCS SP5 confocal microscope. Maximum projections of confocal stacks were created in FIJI.
Fig. 4.
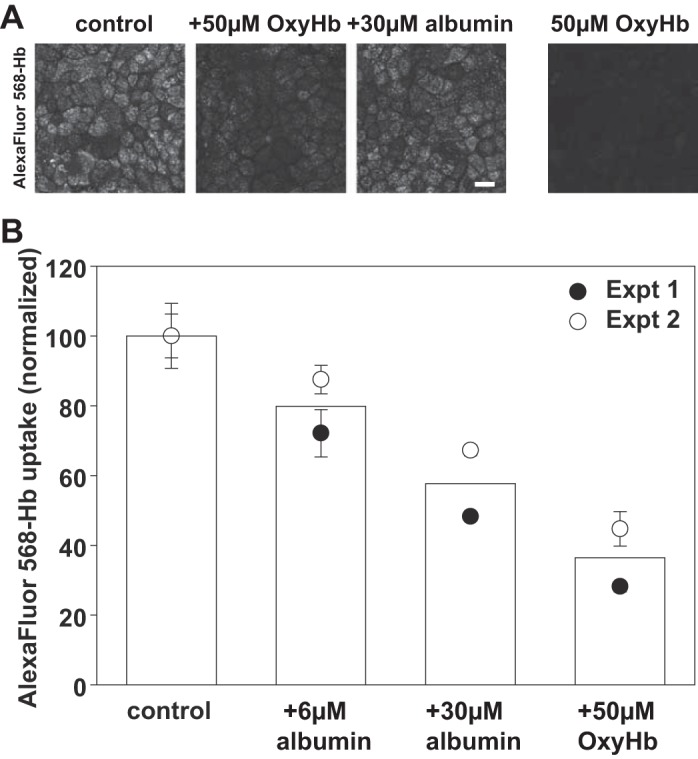
Albumin inhibits endocytosis of hemoglobin by OK cells. A: OK cells were incubated with apically added 2 µM Alexa Fluor 568-Hb in the presence or absence of 50 µM unlabeled OxyHb or 30 µM albumin for 1 h at 37°C, then fixed and processed for immunofluorescence. As a control to confirm that OxyHb fluorescence is not detected in our studies, cells were incubated for 1 h with 50 µM unlabeled OxyHb (right-hand panel) Scale bar, 25 µm. B: OK cells were incubated with apically added 2 µM Alexa Fluor 568-Hb in the presence or absence of the incubated concentrations of albumin or unlabeled Hb for 1 h at 37°C prior to quantitation of cell-associated Hb fluorescence. Data from two experiments performed in triplicate are shown. Background fluorescence in cells incubated only with 50 µM unlabeled OxyHb was <10% that of the control values.
Heme oxygenase 1 expression.
HK-2 (5 × 105) cells were cultured as above in the presence of 50 µM OxyHb for the indicated periods, then washed, solubilized, and lysates blotted using rabbit polyclonal anti HO-1 antibody (1:1,500; Abcam ab137749) and mouse monoclonal β-actin antibody (1:5,000; Sigma A1978).
Hemoglobin preparation and quantitation.
Hemoglobin A (HbA) was isolated from expired RBC units as described (14, 29). Sickle hemoglobin (HbS) was obtained from Sigma. Hb concentration and oxidation state was determined by spectral deconvolution using HbA standard spectra for met, oxy, and deoxy species as previously reported (14, 29). Hb concentrations were calculated per mole of heme. Hb was conjugated to Alexa Fluor 568 using the Protein Labeling Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions.
Sequence and structure analysis.
Sequences of human Hbα, Hbβ, and albumin were retrieved from UniProt (Hb α P69905, Hb β P68871, Albumin P02768). We compared the solvent accessible surfaces of the hemoglobin dimer in the absence and in the presence of Hpt as observed in the Hpt-Hb complex (PDB: 4F4O) (4). The sequences for the individual helices that form the main interactions with Hpt (Hb α helices G and H, and Hb β helices G and H) were aligned against the sequence of human albumin using the BLAST and CLUSTAL W software (2, 30). The sequences identified in human albumin were compared with the available structure of human albumin (PDB: 3SQJ) (13) to evaluate secondary structure conservation. Protein structures and electrostatic surface potentials were generated with PyMOL Molecular Graphics System (2002) (DeLano Scientific, San Carlos, CA).
RESULTS
Hemoglobin inhibits receptor-mediated albumin uptake by proximal tubule cells.
To test whether the NO scavenging by Hb affects PT endocytosis, we incubated polarized OK cells for 1 h with apically added 0.6 µM Alexa Fluor 647-albumin in the presence or absence of 50 µM OxyHb, MetHb, or CNMetHb. These three forms of Hb have different NO scavenging potential, with OxyHb being significantly more potent (9). As an additional control, we also pretreated cells for 30 min with the NOS inhibitor l-NAME (l-NG-nitroarginine methyl ester, 100 µM) before adding fluorescent albumin. Cells were then washed extensively and cell-associated albumin was visualized in fixed cells by confocal microscopy (Fig. 1A) or quantified by spectrofluorimetry (Fig. 1B). As we previously demonstrated, Alexa Fluor 647-albumin readily accumulated in intracellular vesicular compartments in OK cells (25). Addition of any of the three forms of Hb during the albumin incubation profoundly inhibited the uptake of albumin by PT cells. In contrast, l-NAME had no apparent effect on albumin uptake. Quantitation of albumin uptake by spectrofluorimetry in multiple experiments confirmed these qualitative observations (Fig. 1B). OxyHb also inhibited albumin uptake in human proximal tubule HK-2 cells, which also express megalin/cubilin but have a markedly lower endocytic capacity than OK cells (Fig. 1C).
Fig. 1.

Hemoglobin inhibits apical uptake of albumin by PT cells. A: filter-grown OK cells were preincubated for 30 min at 37°C with l-NAME (100 µM) where indicated and then exposed to 0.6 µM apically added Alexa Fluor 647-albumin in the presence or absence of 50 µM MetHb, CNMet-Hb, or OxyHb for 1 h at 37°C. After extensive washing, cells were fixed and processed for immunofluorescence to visualize cell-associated albumin. Representative fields are shown. Scale bar, 25 µm. B: cells incubated as above were solubilized, and cell-associated albumin was quantified by spectrofluorimetry. Mean albumin uptake in control cells was set at 100 to facilitate comparison between experiments. The results from several independent experiments (means ± SD of triplicate samples) are plotted, with each experiment represented by a different symbol. The mean uptake for a given condition is represented by the bar. C: filter-grown human HK-2 cells were incubated for 1 h at 37°C with 0.6 µM Alexa Fluor 647-albumin in the presence or absence of 50 µM OxyHb. Cell-associated albumin was quantified as above, and the mean ± SD of three independent experiments each performed in triplicate is plotted.
The insensitivity of albumin uptake to l-NAME and the equivalently robust inhibition we observed using Hb forms with different NO scavenging capabilities suggest that the effect of Hb on albumin uptake is independent of NO and independent of the heme reduction state. We hypothesized that Hb may be directly competing with albumin for binding to megalin/cubilin. Hb has been demonstrated using surface plasmon-reference analysis to bind to megalin and cubilin with affinities of 1.7 µM and 4.1 µM, respectively (11). In comparison, albumin binds to OK apical membranes with a Kd of 0.3 µM (12).
Normalized data from multiple experiments demonstrated a dose-dependent effect of OxyHb on albumin uptake with an estimated half-maximal inhibitory concentration of ~5 µM (Fig. 2). OxyHbS containing the SCD-causing mutation Glu7Val inhibited with a similar dose response. However, we still observed ~15% residual albumin uptake even when high concentrations of OxyHb (up to 250 µM) were added.
Fig. 2.
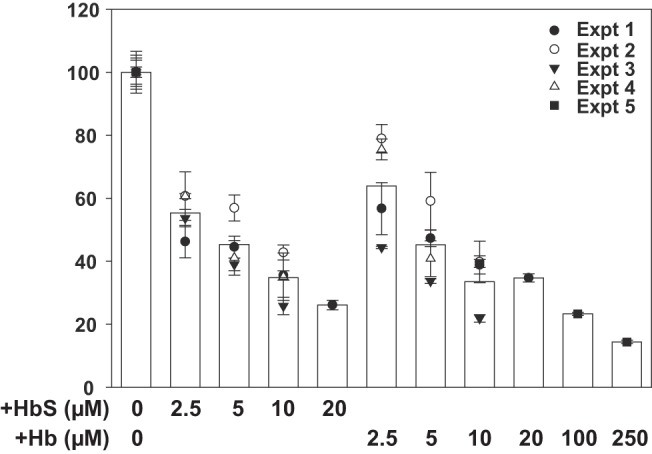
Dose response of hemoglobin and sickle cell hemoglobin S inhibition of albumin uptake. Triplicate filters of cells were preincubated with the indicated concentration of OxyHb or OxyHbS prior to addition of Alexa Fluor 647-albumin for 1 h and quantitation of albumin uptake. Cell-associated albumin in control untreated cells was normalized to 100 to enable combining data from multiple independent experiments, each represented by a distinct symbol. Based on this dose response, the half-maximal concentration at which OxyHb inhibits uptake of albumin is ~5 µM.
Western blotting confirmed that prolonged exposure of human PT cells to Hb caused a dramatic elevation in heme-oxygenase 1 as previously reported; however little if any upregulation was observed within 4 h of incubation (Fig. 3A). To confirm that the inhibitory effect of Hb on albumin endocytosis was not due to cellular toxicity, we preincubated cells with OxyHb for up to 5 days with 10–50 µM OxyHb, then washed the cells and examined the effect on endocytosis of Alexa Fluor 647-albumin in the absence of competing Hb. As shown in Fig. 3B, preincubation with Hb had no effect on albumin uptake. Consistent with our previous results above, inclusion of 10 µM OxyHb in the apical medium during the 1 h uptake period reduced albumin endocytosis by >60%. In contrast, when OxyHb was added to the basolateral medium of the Transwell filter supports, it had no effect on albumin uptake (not shown).
Fig. 3.

Hemoglobin acutely inhibits receptor-mediated endocytosis in proximal tubule cells. A: HK-2 cells were incubated with 50 µM OxyHb for 4 h or 3 days as indicated, then solubilized and blotted to detect heme oxygenase 1 (HO-1) and β-actin (as a loading control) Quantitation of HO-1 expression (avg ± range) relative to control and normalized to β-actin in two independent experiments is shown next to the blot. B: filter-grown OK cells were preincubated at 37°C with apically added OxyHb (10–50 µM) for 18 h to 5 days as indicated prior to extensive washout and subsequent addition of Alexa Fluor 647-albumin for 1 h at 37°C in the absence of OxyHb. As a positive control, OxyHb (10 µM) was included during the albumin uptake period; untreated cells incubated with Alexa Fluor 647-albumin were used as a negative control. Fluorescent cell-associated albumin was quantified by spectrofluorimetry. Data (means ± SD) from three independent experiments each performed in triplicate are shown.
Albumin inhibits hemoglobin uptake by proximal tubule cells.
Hb has previously been shown to be internalized by cells in the PT via a megalin-dependent pathway (11). In those studies, addition of BSA did not inhibit Hb binding, suggesting that albumin and Hb may interact with distinct sites on megalin/cubilin. To examine this in OK cells, we incubated OK cells with 1 µM apically added Alexa Fluor 568-OxyHb for 1 h, then fixed and imaged the cells. As shown in Fig. 4A, Hb was internalized into vesicular compartments similar to those observed with fluorescent albumin (Fig. 1A). As expected for a receptor-mediated event, uptake was abolished by inclusion of excess unlabeled Hb during the incubation period (Fig. 4A). We also observed significant inhibition of Hb uptake upon inclusion of 30 µM albumin (Fig. 4A). To test this further, we examined the dose dependence of albumin inhibition of Hb uptake using our spectrofluorimetry assay. In these experiments we found that 50 µM unlabeled Hb inhibited the uptake of fluorescently conjugated Hb by ~75% and 30 µM albumin inhibited Hb uptake by ~50% (Fig. 4B).
Haptoglobin inhibits Hb uptake by PT cells and restores albumin endocytosis.
Hpt is a large (unfiltered) protein in serum that binds with very high affinity (estimated Kd > 10−12 M) to the dimer-dimer interface of Hb (4) (1, 5). We found that 10 µM Hpt inhibited uptake of Alexa Fluor 568-OxyHb by OK cells (Fig. 5A). We used our spectrofluorimetry assay to confirm this quantitatively. However, because of the prohibitive cost of Hpt, we performed these assays with low concentrations of OxyHb (7.5 µM) and stoichiometric amounts of Hpt. As shown in Fig. 5B, addition of 7.5 µM OxyHb to OK cells inhibited the uptake of Alexa Fluor 647-albumin by ~30%. Preincubation of 7.5 µM OxyHb for 30 min with 7.5 µM Hpt restored albumin uptake to nearly control levels (Fig. 5B). Hpt also reversed the inhibitory effect of HbS on albumin uptake with comparable efficacy (not shown). Addition of 7.5 µM Hpt without Hb resulted in a minor but not statistically significant reduction in albumin uptake, suggesting that Hpt itself may weakly inhibit albumin uptake as well.
Fig. 5.
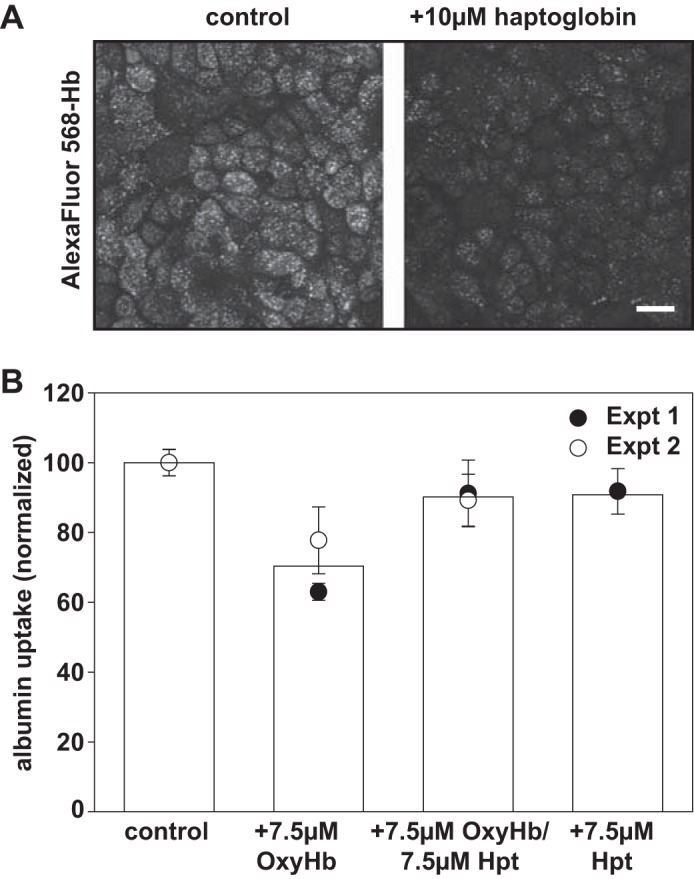
Haptoglobin inhibits hemoglobin uptake and restores albumin endocytosis by OK cells. A: OK cells were incubated with 2 µM Alexa Fluor 568-OxyHb for 1 h at 37°C in the presence or absence of 10 µM haptoglobin, then washed, fixed, and processed for immunofluorescence to visualize cell-associated Hb. Scale bar, 25 µm. B: filter-grown OK cells were incubated with 40 µg/ml Alexa Fluor 647-albumin in the presence of OxyHb (7.5 µM) and/or 7.5 µM haptoglobin (Hpt) as indicated. After extensive washing, cells were solubilized and fluorescent cell-associated albumin was quantified. Data (means ± SD) from two independent experiments each performed in triplicate are shown.
Modeling the hemoglobin interaction site with megalin.
As shown above, our results indicate that low concentrations of Hb (<10 µM) can impair albumin uptake. In addition, stoichiometric amounts of Hpt prevent this inhibition. The structure of the Hb-Hpt complex is known, involving two Hpt molecules and a Hb dimer (4). Thus it is reasonable to hypothesize that the binding site of Hb to megalin/cubilin may no longer be exposed upon the formation of the Hb-Hpt complex. In addition, the competitive effect of albumin suggests that the binding sites used by albumin and Hb may share similar properties. Based on these premises we searched for Hb motifs that met the following conditions: 1) are solvent-exposed in the dimer Hb structure, 2) are involved in the Hb-Hpt interface, 3) share sequence and structural similarity to motifs in albumin, 4) share electrostatic surface similarities to motifs in albumin.
The interaction of Hb and Hpt covers a large (2,954 Å2) surface with a number of Hb motifs involved (4). The regions of Hb involved in the interface include the helices G and H in Hb α and the helices C, G, and H of Hb β (4). As albumin and Hb are all-α-helical proteins, we used individual Hb α-helices as our search motif.
The Hb helical fragments involved in Hpt binding were aligned to human albumin sequence to search for comparable albumin motifs. Portions of albumin showing sequence homology to the Hb sequences were inspected for similar secondary structure. Three regions of albumin with sequence homology and similar secondary structure were identified (Fig. 6). Based on these results we conclude that helices G and H in Hb may be involved in the interaction with megalin/cubilin.
Fig. 6.

Sequence and structure comparison of potential megalin/cubilin binding regions in hemoglobin and albumin. Hb helical fragments involved in haptoglobin binding were aligned to human albumin sequence to search for comparable albumin motifs (see materials and methods for details). Three sequences in albumin were identified that have sequence and structural similarity to Hb. Top: alignment of Hb α- and β-helix H to albumin residues 244–271. The sequence alignment is shown on top. Aligned sequences are shown in the structures of albumin and Hb αβ-dimer and indicated by a red box. Helical elements are shown in red (albumin, hemoglobin α) and salmon (hemoglobin β); Middle: alignment of Hb α- and β-helix H to albumin residues 561–588. The sequence alignment is shown on top. Aligned sequences are shown in the structures of albumin and Hb αβ-dimer and indicated by a red box. Helical elements are shown in pink (albumin) red (hemoglobin α), and salmon (hemoglobin β) Bottom: alignment of Hb α- and β-helix G to albumin residues 87–105. The sequence alignment is shown on top. Aligned sequences are shown in the structures of albumin and Hb αβ-dimer and indicated by a blue box. Helical elements are shown in dark blue (albumin, hemoglobin α) and cyan (hemoglobin β).
DISCUSSION
Our results demonstrate that binding of Hb to megalin/cubilin competes directly for the uptake of albumin by PT cells. We observed potent inhibition of albumin uptake by concentrations of Hb predicted to enter the tubule lumen in patients during hemolytic crisis. We hypothesize that competition for ligand binding by excess Hb in the tubule lumen, rather than cytotoxic responses to heme, are the cause of tubular proteinuria frequently observed in SCD patients. Additionally, loss of these ligands may contribute to impaired vitamin homeostasis in SCD patients.
We found that 5 µM Hb and HbS inhibited albumin uptake by ~50%; however, we were unable to fully prevent uptake even at much higher concentrations of Hb. This suggests that albumin binds to multiple sites on megalin/cubilin, only some of which are inhibited by Hb. This is likely given that these receptors contain multiple ligand binding domains to engage a broad array of filtered ligands (10). Additionally, non-receptor-mediated mechanisms may contribute to the small amount of residual albumin uptake at high Hb concentrations.
Our data suggest that the interaction site for Hb with megalin/cubilin overlaps the Hpt binding site within the dimer-dimer interface. Based on this finding and on BLAST searches for regions of homology between albumin and Hb that map to this interface, we identified three sequences in albumin as putative binding motifs for megalin/cubilin, due to their sequence and structure similarities with Hb domains that are involved in Hpt binding.
A surprising finding is that whereas PT cells are highly sensitive to Hb, we did not observe any apparent toxicity in OK or HK-2 cells even after prolonged exposure to OxyHb. Control studies confirmed upregulation of heme oxygenase 1 in HK-2 cells exposed to OxyHb as previously reported (6). However, preincubation of OK cells with 50 µM OxyHb for up to 5 days did not impair subsequent albumin uptake. It is possible that other functions of our cells are compromised by exposure to Hb.
Our findings have potential implications for our understanding the pathogenesis of SCD. Hb αβ-dimers in the plasma are readily filtered into the tubule lumen in the absence of glomerular injury and reach concentrations during hemolytic crisis that would significantly impair the reabsorption of albumin and other megalin/cubilin ligands. Thus, our finding may explain why a significant fraction of young SCD patients exhibit LMW proteinuria independent of glomerular damage (3, 17). Prolonged exposure of PT cells to Hb is likely to overwhelm protective responses and lead to tubular injury that may initiate and/or contribute to the progression of chronic kidney disease/end-stage renal disease in SCD patients.
Our results suggest a possible explanation for why children with severe manifestations of SCD have abnormally low levels of 25-hydroxyvitamin D3 [25(OH)D3] (23) and low bone mineral density (15). Defective uptake of filtered ligands by megalin/cubilin in the PT is known to affect serum vitamin levels. This is especially evident in the case of vitamin D3, which is taken up by megalin/cubilin in its inactive, insoluble form [25(OH)D3] bound to vitamin D binding protein (VDBP), converted to the active 1,25(OH)2D3 form, and released from the basolateral surface of PT cells into the bloodstream (33). For example, patients with Dent disease, caused by mutations in a PT hydrogen-chloride antiporter that plays a role in apical endocytosis, have low levels of vitamin D3 and frequently develop osteomalacia and hypophosphatemic rickets (28). Cubilin-deficient dogs also have lower serum levels of mono- and dihydroxylated vitamin D3 metabolites (24). Although we did not test whether Hb competes for uptake of VDBP, it is notable that VDBP is highly homologous to albumin (8). Our studies provide a potential mechanism to explain the early steps in the development of kidney disease and suggest the possibility that selectively targeting the interaction of Hb with megalin/cubilin may have therapeutic value beyond simply preserving PT function in SCD patients
GRANTS
This work was supported by a P3HVB pilot grant from the Pittsburgh Heart Lung and Blood Vascular Medicine Institute, by National Institutes of Health (NIH) Grants R01 DK101484 (to O. A. Weisz) and R21 ES027390 (to J. Tejero), and by the Pittsburgh Center for Kidney Research (P30 DK079307). M. L. Eshbach was supported in part by NIDDK Grant T32 DK061296.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
M.L.E., A.K., J.T., and O.A.W. conceived and designed research; M.L.E., A.K., and Y.R. performed experiments; M.L.E., A.K., Y.R., J.T., and O.A.W. analyzed data; M.L.E., A.K., Y.R., J.T., and O.A.W. interpreted results of experiments; M.L.E., J.T., and O.A.W. prepared figures; M.L.E., J.T., and O.A.W. drafted manuscript; M.L.E., A.K., Y.R., J.T., and O.A.W. approved final version of manuscript; J.T. and O.A.W. edited and revised manuscript.
ACKNOWLEDGMENTS
We are grateful to Mark Gladwin, Peter Friedman, Tom Nolin, Sruti Shiva, and Ossama Kashlan for helpful discussions and to Solomon Ofori-Acquah for the generous gift of haptoglobin.
REFERENCES
- 1.Alayash AI. Haptoglobin: old protein with new functions. Clin Chim Acta 412: 493–498, 2011. doi: 10.1016/j.cca.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 2.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402, 1997. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Alvarez O, Montane B, Lopez G, Wilkinson J, Miller T. Early blood transfusions protect against microalbuminuria in children with sickle cell disease. Pediatr Blood Cancer 47: 71–76, 2006. doi: 10.1002/pbc.20645. [DOI] [PubMed] [Google Scholar]
- 4.Andersen CB, Torvund-Jensen M, Nielsen MJ, de Oliveira CL, Hersleth HP, Andersen NH, Pedersen JS, Andersen GR, Moestrup SK. Structure of the haptoglobin-haemoglobin complex. Nature 489: 456–459, 2012. doi: 10.1038/nature11369. [DOI] [PubMed] [Google Scholar]
- 5.Chiancone E, Alfsen A, Ioppolo C, Vecchini P, Agrò AF, Wyman J, Antonini E. Studies on the reaction of haptoglobin with haemoglobin and haemoglobin chains. I. Stoichiometry and affinity. J Mol Biol 34: 347–356, 1968. doi: 10.1016/0022-2836(68)90258-1. [DOI] [PubMed] [Google Scholar]
- 6.Chintagari NR, Nguyen J, Belcher JD, Vercellotti GM, Alayash AI. Haptoglobin attenuates hemoglobin-induced heme oxygenase-1 in renal proximal tubule cells and kidneys of a mouse model of sickle cell disease. Blood Cells Mol Dis 54: 302–306, 2015. doi: 10.1016/j.bcmd.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christensen EI, Birn H, Storm T, Weyer K, Nielsen R. Endocytic receptors in the renal proximal tubule. Physiology (Bethesda) 27: 223–236, 2012. doi: 10.1152/physiol.00022.2012. [DOI] [PubMed] [Google Scholar]
- 8.Cooke NE, David EV. Serum vitamin D-binding protein is a third member of the albumin and alpha fetoprotein gene family. J Clin Invest 76: 2420–2424, 1985. doi: 10.1172/JCI112256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Donadee C, Raat NJ, Kanias T, Tejero J, Lee JS, Kelley EE, Zhao X, Liu C, Reynolds H, Azarov I, Frizzell S, Meyer EM, Donnenberg AD, Qu L, Triulzi D, Kim-Shapiro DB, Gladwin MT. Nitric oxide scavenging by red blood cell microparticles and cell-free hemoglobin as a mechanism for the red cell storage lesion. Circulation 124: 465–476, 2011. doi: 10.1161/CIRCULATIONAHA.110.008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eshbach ML, Weisz OA. Receptor-mediated endocytosis in the proximal tubule. Annu Rev Physiol 79: 425–448, 2017. doi: 10.1146/annurev-physiol-022516-034234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gburek J, Verroust PJ, Willnow TE, Fyfe JC, Nowacki W, Jacobsen C, Moestrup SK, Christensen EI. Megalin and cubilin are endocytic receptors involved in renal clearance of hemoglobin. J Am Soc Nephrol 13: 423–430, 2002. [DOI] [PubMed] [Google Scholar]
- 12.Gekle M, Mildenberger S, Freudinger R, Silbernagl S. Functional characterization of albumin binding to the apical membrane of OK cells. Am J Physiol Renal Physiol 271: F286–F291, 1996. [DOI] [PubMed] [Google Scholar]
- 13.He Y, Ning T, Xie T, Qiu Q, Zhang L, Sun Y, Jiang D, Fu K, Yin F, Zhang W, Shen L, Wang H, Li J, Lin Q, Sun Y, Li H, Zhu Y, Yang D. Large-scale production of functional human serum albumin from transgenic rice seeds. Proc Natl Acad Sci USA 108: 19078–19083, 2011. doi: 10.1073/pnas.1109736108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, Huang KT, Ho C, Hogg N, Schechter AN, Gladwin MT. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest 115: 2099–2107, 2005. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lal A, Fung EB, Pakbaz Z, Hackney-Stephens E, Vichinsky EP. Bone mineral density in children with sickle cell anemia. Pediatr Blood Cancer 47: 901–906, 2006. doi: 10.1002/pbc.20681. [DOI] [PubMed] [Google Scholar]
- 16.Lonsdorfer A, Comoe L, Yapo AE, Lonsdorfer J. Proteinuria in sickle cell trait and disease: an electrophoretic analysis. Clin Chim Acta 181: 239–247, 1989. doi: 10.1016/0009-8981(89)90229-5. [DOI] [PubMed] [Google Scholar]
- 17.Marsenic O, Couloures KG, Wiley JM. Proteinuria in children with sickle cell disease. Nephrol Dial Transplant 23: 715–720, 2008. doi: 10.1093/ndt/gfm858. [DOI] [PubMed] [Google Scholar]
- 18.Monke JV, Yuile CL. The renal clearance of hemoglobin in the dog. J Exp Med 72: 149–165, 1940. doi: 10.1084/jem.72.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nath KA, Grande JP, Farrugia G, Croatt AJ, Belcher JD, Hebbel RP, Vercellotti GM, Katusic ZS. Age sensitizes the kidney to heme protein-induced acute kidney injury. Am J Physiol Renal Physiol 304: F317–F325, 2013. doi: 10.1152/ajprenal.00606.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nath KA, Hebbel RP. Sickle cell disease: renal manifestations and mechanisms. Nat Rev Nephrol 11: 161–171, 2015. doi: 10.1038/nrneph.2015.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Naumann HN, Diggs LW, Barreras L, Williams BJ. Plasma hemoglobin and hemoglobin fractions in sickle cell crisis. Am J Clin Pathol 56: 137–147, 1971. doi: 10.1093/ajcp/56.2.137. [DOI] [PubMed] [Google Scholar]
- 22.Nielsen R, Christensen EI. Proteinuria and events beyond the slit. Pediatr Nephrol 25: 813–822, 2010. doi: 10.1007/s00467-009-1381-9. [DOI] [PubMed] [Google Scholar]
- 23.Nolan VG, Nottage KA, Cole EW, Hankins JS, Gurney JG. Prevalence of vitamin D deficiency in sickle cell disease: a systematic review. PLoS One 10: e0119908, 2015. doi: 10.1371/journal.pone.0119908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nykjaer A, Fyfe JC, Kozyraki R, Leheste JR, Jacobsen C, Nielsen MS, Verroust PJ, Aminoff M, de la Chapelle A, Moestrup SK, Ray R, Gliemann J, Willnow TE, Christensen EI. Cubilin dysfunction causes abnormal metabolism of the steroid hormone 25(OH) vitamin D(3). Proc Natl Acad Sci USA 98: 13895–13900, 2001. doi: 10.1073/pnas.241516998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raghavan V, Rbaibi Y, Pastor-Soler NM, Carattino MD, Weisz OA. Shear stress-dependent regulation of apical endocytosis in renal proximal tubule cells mediated by primary cilia. Proc Natl Acad Sci USA 111: 8506–8511, 2014. doi: 10.1073/pnas.1402195111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serjeant GR. The natural history of sickle cell disease. Cold Spring Harb Perspect Med 3: a011783, 2013. doi: 10.1101/cshperspect.a011783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sundaram N, Bennett M, Wilhelm J, Kim MO, Atweh G, Devarajan P, Malik P. Biomarkers for early detection of sickle nephropathy. Am J Hematol 86: 559–566, 2011. doi: 10.1002/ajh.22045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szczepanska M, Zaniew M, Recker F, Mizerska-Wasiak M, Zaluska-Lesniewska I, Kilis-Pstrusinska K, Adamczyk P, Zawadzki J, Pawlaczyk K, Ludwig M, Sikora P. Dent disease in children: diagnostic and therapeutic considerations. Clin Nephrol 84: 222–230, 2015. doi: 10.5414/CN108522. [DOI] [PubMed] [Google Scholar]
- 29.Tejero J, Basu S, Helms C, Hogg N, King SB, Kim-Shapiro DB, Gladwin MT. Low NO concentration dependence of reductive nitrosylation reaction of hemoglobin. J Biol Chem 287: 18262–18274, 2012. doi: 10.1074/jbc.M111.298927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680, 1994. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tracz MJ, Alam J, Nath KA. Physiology and pathophysiology of heme: implications for kidney disease. J Am Soc Nephrol 18: 414–420, 2007. doi: 10.1681/ASN.2006080894. [DOI] [PubMed] [Google Scholar]
- 32.Weinbaum S, Duan Y, Satlin LM, Wang T, Weinstein AM. Mechanotransduction in the renal tubule. Am J Physiol Renal Physiol 299: F1220–F1236, 2010. doi: 10.1152/ajprenal.00453.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Willnow TE, Nykjaer A. Pathways for kidney-specific uptake of the steroid hormone 25-hydroxyvitamin D3. Curr Opin Lipidol 13: 255–260, 2002. doi: 10.1097/00041433-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 34.Wood KC, Hsu LL, Gladwin MT. Sickle cell disease vasculopathy: a state of nitric oxide resistance. Free Radic Biol Med 44: 1506–1528, 2008. doi: 10.1016/j.freeradbiomed.2008.01.008. [DOI] [PubMed] [Google Scholar]