

Abstract
Freestanding fibrous matrices with proper protein composition and desirable mechanical properties, stability, and biocompatibility are in high demand for tissue engineering. Electrospun (E-spun) collagen–silk composite fibers are promising tissue engineering scaffolds. However, as-spun fibers are mechanically weak and unstable. In this work, we applied glutaraldehyde (GA) vapor treatment to improve the fiber performance, and the effect on the properties of E-spun collagen–silk fibers was studied systematically. GA treatment was found to affect collagen and silk distinctively. Whereas GA chemically links collagen peptides, it induces conformational transitions to enrich β-sheets in silk. The combined effects impose a control of the mechanical properties, stability, and degradability of the composite fibers, which are dependent on the extent of GA treatment. In addition, a mild treatment of the fibers did not diminish cell proliferation and viability. However, overly treated fibers demonstrated reduced cell–matrix adhesion. The understanding of GA treatment effects on collagen, silk, and the composite fibers enables effective control and fine tuning of the fiber properties to warrant their diverse in vitro and in vivo applications.
Introduction
Tissue engineering scaffolds with appropriate chemical composition, topographical features, mechanical properties, stability, and degradability are essential to support cell development and tissue remodeling. Among a variety of techniques for fabricating biomaterial scaffolds, electrospinning has gained considerable interest because the process is remarkably efficient, rapid, and inexpensive, and the electrospun (E-spun) nanofibrous architecture is similar to the naturally occurring protein fibrils in the extracellular environment.1,2 As a major component of the extracellular matrix, collagen type I has been widely adopted for electrospinning to generate fibers with diameters ranging from less than 100 nm to a few microns mimicking native collagen fibrils.3−5 The interaction between collagen type I and cell membrane proteins, such as β-1 integrin, is known to regulate cell attachment, growth, and differentiation.6−8 The properties of E-spun collagen scaffolds were often manipulated by incorporating various synthetic and/or natural materials.9−11 Dragline silk protein has been widely considered a promising biomaterial due to its superior mechanical strength and good in vitro and in vivo biocompatibility.12,13 The unique combination of highly organized β-sheet crystalline domains of an alanine-rich motif and amorphous matrix of a glycine-rich motif in silk is accountable for its high tensile strength and extensibility.14,15
In a previous work, we established the method to fabricate unidirectionally aligned collagen–silk composite microfibers using a home-built electrospinning system.16 The aligned fibers mimicked the locally oriented collagen fibers in native tissues. The incorporation of synthetic spider dragline silk proteins in collagen significantly enhanced the mechanical strength and stability of the E-spun fibers. Composite fibers with a low silk content (15 or 30% silk) were considered the most favorable scaffolds for potential neural tissue engineering application due to the optimal balance in biochemical and biophysical properties to support rapid neural differentiation of stem cells. However, unlike native collagen fibrils that are stable in aqueous media, the E-spun collagen fibers were found unstable in water, tissue fluid, or blood.17−19 Different from native silk fibers, the as-spun silk fibers were much weaker. Posttreatments, such as chemical cross-linking for collagen17,20−23 and solvent-induced crystallization of β sheets for silk fibers,24−28 were often carried out to improve the fiber performance. To our best knowledge, posttreatment methods to suit collagen–silk composite fibers have not been explored extensively.
Glutaraldehyde (GA) is commonly used as a cross-linking agent for collagen-based biomaterials.29,30 Among various cross-linking protocols, GA vapor treatment is most widely used because it is easy to control and can avoid the collapse of the E-spun fiber matrix during cross-linking in an aqueous environment.3,17,23,31 It was also found by us and other groups that GA treatment can enhance the mechanical properties of silk-based materials.32−34 However, the protocols of GA vapor treatment established by different groups vary remarkably, with the concentration ranging from 0.5 to 50% and the treatment time from 12 h to 4 days.3,17,23,31,35 Such a large variation can lead to huge discrepancies in the properties of E-spun fibers and make results obtained by different groups inconsistent and incomparable. It is imperative to perform a systematic study to delve into the mechanism of GA treatment on collagen and silk and to examine the effectiveness of such a treatment on E-spun collagen–silk composite fibers.
In this work, we aim to evaluate the effect of GA vapor treatment on the mechanical properties, stability and biocompatibility of E-spun collagen–silk composite fibers. The study is focused on composite fibers with 30% silk (CS30) due to their superior physical and biological properties as a cell culture matrix,16 with pure collagen and pure silk fibers as controls to delineate the mechanism of the treatment effects. The relationship between the effect and the extent of GA treatment has been examined by varying the treatment time while maintaining the concentration of GA at 20% and the temperature at 25 °C. The systematic study provides the guideline to effectively tune the properties of fibers with suitable protein compositions by a simple GA treatment to achieve desirable matrices for various applications.
Results
Effect of GA Treatment on Fiber Morphology and Fluorescence
GA vapor treatment turned the white and fluffy E-spun fiber matrices into brown and firm materials (Figure S1). In this process, the GA vapor treatment induced changes in the fiber dimension and fiber morphology. The treatment time-dependent changes of CS30 fibers are shown in Figure 1. As illustrated in the large-scale optical images (Figure 1A–C), the fibers are largely parallel and densely aligned. The AFM images (insets) show that with no or 6 h of GA treatment, the fibers are cylindrical and smooth and are uniform along the fiber axis. Extended GA treatment (48 h) caused a roughly 8% decrease in the fiber width and frequent coalescence of adjacent or joint fibers (circled region in Figure 1C, inset). Similar changes were more frequently observed on pure collagen fibers but rarely observed on pure silk fibers. It suggests that overtreatment has a greater impact on collagen than on silk. This is evidenced by the 13.6 and 3.8% decrease in the fiber width of pure collagen and pure silk, respectively, after 48 h of GA treatment (Figure 1H). The percentage of fiber weight change after the 48 h GA treatment was also monitored to evaluate the amount of GA molecules incorporated into different types of fibers. As shown in Figure 1G, light GA treatment (2 and 6 h) caused less than 1.0% change for all fiber types. A 48 h GA vapor treatment led to the weight gain by 12.1, 6.5, and 3.2% for collagen, CS30, and silk fibers, respectively. Thus, collagen-rich fibers can uptake a significantly larger amount of GA after extensive GA treatment.
Figure 1.

Effect of GA vapor treatment on CS30 E-spun fibers. (A–C) Bright-field images illustrating unidirectionally aligned fibers with 0, 6, and 48 h treatments. The insets are AFM images showing the decrease in fiber width with GA treatment time. The red circle highlights the coalesced fibers. (D–F) Fluorescence images illustrating the increase of fluorescence intensity with GA treatment time. Bar size for bright-field and fluorescence images: 100 μm. Bar size for AFM images: 5 μm. (G) Comparison of the percentage of fiber weight change for CS30, pure collagen (C), and pure silk (S) fibers, indicating the variation in the amount of GA incorporated in each type of fibers. (H) Comparison of the fiber width for CS30, pure collagen (C), and pure silk (S) fibers and the changes with GA treatment time. Fiber widths were measured from AFM images. Error bars indicate standard errors.
As-spun CS30 fibers displayed weak fluorescence, which is attributed to the autofluorescence of collagen.16 Stronger green fluorescence was observed after the fibers were treated with GA vapor, and the fluorescence intensity increased with the GA treatment time (Figure 1D–F). The same trend of changes was observed in pure collagen but not in pure silk. Thus, we premise that the enhanced fluorescence is due to a higher level of cross-linking in the collagen component of composite fibers. This was corroborated by the increased fluorescence intensity of the 520–560 nm band in the fluorescence spectra of collagen fibers with increased GA treatment time (Figure S2). We ascribe the increased fluorescence to the formation of a Schiff base (C=N) between the collagen and polymerized GA molecules,36 as well as the increased fiber stiffness, which restricts intramolecular rotation leading to the enhancement of fluorescence emission.37 This is consistent with the results from Fourier transform infrared (FTIR) spectroscopic analysis, free amine quantification, and measurements of fiber mechanics (see below).
Degree of GA Cross-linking in Different Types of Fibers
GA cross links proteins through its aldehyde groups’ reaction with primary amines to form the Schiff base.38,39,36,40 The degree of GA cross-linking was determined by the decrease of the number of free amine groups evaluated by a 2,4,6-trinitrobenzenesulfonic acid (TNBS) assay. As shown in Figure 2, before GA treatment, collagen, CS30, and silk fibers had a free amine group content of 33, 24, and 7 per 1000 amino acids, respectively. According to the amino acid sequences, collagen has about 38 free amines per 1000 amino acids, counting lysine and hydroxylysine as well as the terminal amines. This is in good agreement with the result of the TNBS assay. Synthetic silk protein has free amines at the terminals only.41 On the basis of the protein size, in theory, there are approximately 2 free amines per 1000 amino acids. The descrepancy between the theoretical and experimental data is likely due to the fact that the goat-derived silk proteins are often truncated into smaller peptide pieces during purifications,26 leading to a greater number of free amines per 1000 amino acids. The derived free amine abundance for CS30 (27 per 1000 amino acids) agrees well with the experimental data. Figure 2 shows that the number of free amines in collagen and CS30 decreased dramatically in the first 12 h of GA treatment and then plateaued at less than 7 per 1000 amino acids afterward. The silk protein has much fewer free amine groups. As a result, the free amine content of silk dropped slightly from 7 to 4 per 1000 amino acids even after 48 h of treatment. Thus, the Schiff base formation reaction, induced by GA cross-linking, is more prominent in collagen and CS30 than that in silk. The degree of cross-linking directly affects fiber mechanics, stability, and degradability.
Figure 2.
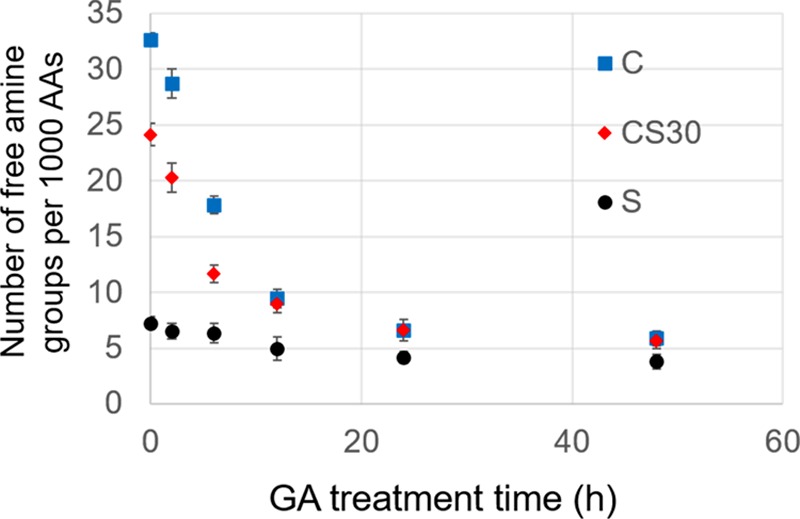
Characterization of the free amine content as a function of GA treatment time by a TNBS assay. Comparison is made between CS30, collagen (C), and silk (S) fibers. Data were summarized from three trials. Error bars indicate standard errors.
Effect of GA Treatment on Fiber Mechanics
The mechanical properties of E-spun fibers were characterized by stress–strain tests. As shown in Figure 3A, untreated collagen, silk, and composite fibers responded differently when subjected to stretching along the fiber direction. Compared with collagen fibers, silk fibers can sustain remarkably higher tensile stress with greater Young’s modulus; however, they break abruptly upon extension. Consistent with our previous results,16 both the ultimate tensile strength and Young’s modulus increase monotonically with the percentage of silk in the collagen–silk composite fibers, whereas the ultimate strain increases with the percentage of collagen in the fibers. Thus, silk-rich fibers have higher tensile strength, whereas collagen-rich fibers are more stretchable. Fiber toughness was evaluated by the integral of the stress–strain curves. As shown in the inset of Figure 3A, fiber toughness is optimized at 6.57 MJ/m3 in CS30 composite fibers. Thus, CS30 is superior to both the pure materials in fiber mechanics.
Figure 3.

Variation of fiber mechanics with GA treatment time for collagen (C), CS30, CS60, and silk (S) fibers. (A) Typical stress–strain curves of untreated fibers. The inset illustrates the variation of fiber toughness with silk percentage in the fibers. (B–E) Variation of the ultimate tensile strength (B), ultimate tensile strain (C), Young’s modulus (D), and toughness (E) of the fibers with GA treatment time.
The effect of GA treatment on fiber mechanical properties is shown in Figure 3B–E. Short-term treatments (<6 h) led to marked increase in ultimate tensile strength, strain, Young’s modulus, and fiber toughness for all types of fibers. In addition, the GA treatment prompted a greater increase in ultimate tensile strength and Young’s modulus in silk-rich fibers and a greater increase in fiber strain in collagen-rich fibers. Longer-term GA treatments induced decreases in the properties of all types of fibers, particularly in ultimate stress and strain. As a result, the toughness of collagen, CS30, and silk fibers reached the highest values at 6 h of treatment, with CS30 being the strongest (27.1 MJ/m3). CS60 fibers reached the highest toughness of 25.6 MJ/m3 at 12 h of treatment.
GA Treatment-Induced Protein Structural Changes
The enhanced fiber mechanics by GA treatment suggests that GA interacts with collagen and silk strongly and distinctively. The secondary structure of the protein is directly relevant to fiber mechanics.42 The change of the protein secondary structure was monitored by FTIR spectra at the amide I and amide II regions. As shown in Figure 4A, the amide I band of pure collagen fibers is centered at 1655 cm–1, characteristic for the α helices dominant in collagen.49 The peak remains unchanged in position and relative intensity at different times of the GA treatment, suggesting that GA treatment has a negligible effect on the secondary structure of collagen. With the increase of the GA treatment time, a gradual increase of the peak at 1588 cm–1 was observed in collagen-rich fibers (C and CS30), coinciding with the increase of fiber fluorescence (Figure 1D–F). The peak is negligible in the spectra of silk with or without GA treatment. It is assigned to the C=N bond of the Schiff base formed from the reaction between the aldehyde groups of GA and the primary amine groups of proteins.36 A peak at 1720 cm–1 was also seen to increase with the GA treatment time in all types of fibers. The peak is present in the spectrum of GA and is assigned to C=O stretching in aldehydes.43 Although the intensity of both 1588 and 1720 cm–1 peaks increases with the GA treatment time, the increase is more dramatic after longer treatment times. It suggests the presence of excessive GA molecules, which likely leads to further GA polymerization due to aldol condensation reactions. The marked increase of the two peaks in collagen over silk is consistent with the observation that collagen uptakes more GA molecules than silk after long-term GA treatment.
Figure 4.

(A) FTIR spectra of E-spun collagen (C), CS30, CS60, and silk (S) fibers in the amide I and amide II region (1485–1750 cm–1), collected at various GA treatment times. Blue arrows highlight the changes of the designated peaks with GA treatment time. (B) Variation of the β-sheet to helix/coil ratio of the silk content in the E-spun fibers with GA treatment time. The ratios were calculated by quantitative analysis of signature peaks derived from peak deconvolution of the amide I band (see Figure S3).
In pure silk, the peaks centered at 1655 and 1540 cm–1 in Figure 4A are assigned to amide I and amide II, respectively, which are characteristic for helices and random coils.13 After GA treatment, shoulder peaks appeared at 1630 and 1528 cm–1, and the intensity increased with the GA treatment time (Figure S3). They are signature peaks for β-sheets in silk proteins.32,44 It suggests that the treatment induced a secondary structural transition from helical or coiled conformation to β-sheets. The transition was quantitatively analyzed by peak deconvolution of the amide I band (see Figure S4) following the method established by Yu et al.45−47 The amide I band was chosen because it is particularly sensitive to the protein secondary structure, whereas the amide II band involves complex vibrations of multiple groups.45 After peak fitting, integrals of the 1655 and 1630 cm–1 peaks were used to quantify the degree of helix/coil to β-sheet transition as illustrated in Figure 4B. In the absence of GA treatment, silk proteins in all types of fibers show a β-sheet to coil ratio of around 0.4, implying the dominant helical or coiled structures in the protein polypeptides. This ratio markedly increases to above 0.7 after 2 h of treatment, continuously rises to 0.8 after 6 h of treatment, and plateaus at 0.9 after longer treatment times. Thus, a short-term GA treatment is adequate to induce an effective secondary structural transition in silk.
Effect of GA Treatment on Fiber Stability
A tissue engineering scaffold is expected to be stable to support the cell culture over a period of time. We examined the morphological changes of fibers after they were immersed in phosphate-buffered saline (PBS) for 2 days under cell culture conditions. As shown in the AFM images in Figure 5, without GA treatment, collagen and CS30 fibers became flat and sometimes curled and swelled dramatically along with the appearance of rough and irregular surface features, indicating severe fiber deterioration. Coinciding with the morphological changes, Young’s modulus reduced by 78% for collagen fibers and 60% for CS30 fibers (measured by an AFM nanoindentation method, data not shown). With the 6 h GA treatment, however, both collagen and CS30 fibers preserved the fiber morphology and elasticity even after 2 days of incubation in PBS. Silk fibers, regardless of the GA treatment, showed negligible changes in fiber morphology and mechanics under the same conditions, demonstrating remarkable stability.
Figure 5.

Characterization of fiber stability. (A–F) AFM images of collagen, CS30, and silk fibers after they were immersed in PBS for two days. (A–C) Untreated fibers; (D–F) fibers with 6 h of GA treatment. Bar size: 5 μm. (G) Variation of the fiber swelling ratio with GA treatment time for collagen (C), silk (S), and CS30 fibers. (H) Variation of the weight loss ratio with GA treatment time to evaluate the level of protein dissolution. For G and H, fibers were immersed in DI water for 2 h before the measurements.
The morphological instability of collagen and CS30 fibers is likely relevant to water uptake. To quantify the effect of GA treatment on water uptake, the swelling ratio of fibers with GA treatment time was measured after the fibers were immersed in DI water for 2 h. As shown in Figure 5G, untreated collagen fibers can uptake a significantly higher amount of water (a swelling ratio of 561%) than CS30 (172%) and silk (88%) fibers. With the increase of the GA treatment time, the swelling ratio of collagen dropped exponentially and reached 36% after the 24 h treatment. The GA treatment also impacted silk fibers, causing the swelling ratio to decrease to 24% after only 2 h of treatment. A longer treatment time induced negligible changes. Although collagen is dominant in CS30 fibers, CS30 fibers uptake much less water than collagen, and a 6 h GA treatment effectively reduced the swelling ratio to 27%.
A large amount of water uptake by the fibers can cause protein dissolution, leading to weight loss and structural defects of the fibers and hence a decrease in fiber mechanics. By comparing the dry weight of each sample before and after the 2 h incubation in water, the percentage of weight loss was calculated, and its change with GA treatment time is shown in Figure 5H. The weight loss of untreated collagen, CS30, and silk fibers was 22.1, 12.2, and 7.2%, respectively. The values dropped rapidly within 6 h of GA treatment for all types of fibers and plateaued after 12 h of treatment. The result suggests that the fibers became more resistant to water dissolution after GA treatment.
Taken together, GA treatment reduced water uptake by the fibers and hence reduced the protein dissolution, leading to more stable fibers. We found that CS30 fibers with a 6–12 h GA treatment remained stable for up to 7 days in an aqueous environment; thus, they are suitable candidates as scaffolds to support in vitro cell culture.
Biocompatibility of GA-Treated Fibers
To examine the biocompatibility of the fibrous matrices, we cultured cells on collagen, CS30, and silk fibers with various GA treatment times. Human decidua parietalis placental stem cells (hdpPSCs) were chosen in the study due to their relatively short doubling time and sensitive responses to matrix properties according to our previous studies.8,16,48Figure 6 shows optical images of the cells after a 1-day culture on the matrices. Cells on the 0 and 6 h GA-treated collagen and CS30 fibers were shown to tightly adhere to and were well polarized along the fibers with high length-to-width ratios. However, cells on the 48 h GA-treated fibers were less polarized, and many were round. Cells were much less polarized and more cells were round or polygonal on silk fibers regardless of the GA treatment time. The less polarized morphology of cells on silk fibers and on overly treated collagen and CS30 fibers infers a weaker cell–fiber interaction.
Figure 6.

Optical images of hdpPSCs grown on collagen, CS30, and silk fiber matrices with 0, 6, and 48 h GA treatments. Images were taken at day 1 postplating. Bar size: 100 μm.
The strength of cell adhesion on various fiber matrices was evaluated by a trypsin deadhesion assay at 12 h postplating, with a glass substrate as a control. A longer deadhesion time implies a stronger cell–matrix adhesion. As shown in Figure 7A, with the increase of the collagen percentage in composite fibers, the cell adhesion is stronger. This is consistent with the fact that collagen promotes cell adhesion with its integrin-binding sites, whereas silk proteins do not have a specific motif binding to the cell surface proteins. The 6 h GA treatment slightly improved or retained cell adhesion on the fibers; however, the 48 h treatment reduced cell adhesion on all types of fibers likely due to the denatured, rigid, and stiff fibers induced by the overtreatment (see Figure 3). The result suggests that lightly treated collagen and CS30 fibers are favorable for cell attachment.
Figure 7.

Characterization of hdpPSCs adhesion, proliferation, and viability of various fiber matrices. (A) Cell–matrix adhesion characterized by the trypsin deadhesion time constant (t0.5) at 12 h postplating of cells on fibers with 0, 6, and 48 h GA treatments. (B) Profiles of cell proliferation on collagen, CS30, and silk fibers at various GA treatment times, characterized by the MTS assay for cells at days 1, 4, and 7 postplating. (C) Cell viability examined by the MTS assay for cells on fiber-loaded Petri dishes after a 1-day culture.
Cell proliferation profiles on various matrices were examined by the MTS assay. As shown in Figure 7B, cells on all types of fibers proliferated rapidly. The proliferation rate slightly decreased with the increase of the GA treatment time (Figure S5), and the doubling time ranged from 3.1 days for untreated fibers to 3.5 days for fibers with 48 h of treatment. At day 1 postplating, the number of cells on untreated collagen and CS30 fibers was similar but was 27% higher than that on silk fibers. The cell number barely changed with the less than 6 h GA treatment but decreased noticeably on collagen and CS30 fibers with longer-term treatment (e.g., a decrease of 35% on collagen and 31% on CS30 after the 48 h treatment). The difference is due to the distinction in the strength of cell–matrix adhesion among the fibers (Figure 7A), leading to the difference in initial cell attachment. The cell viability test revealed no significant difference on fibers with different compositions and GA treatment times (Figure 7C). Even on overly treated fibers, the overall cell viability was above 94%. Presumably, glycine and 70% ethanol rinsing after the GA treatment effectively quenched the toxic residuals of GA and sterilized the fibers. Note that this process had a negligible effect on the fiber structure and fiber mechanics according to our AFM analysis. The results suggest that although the fibers differ in cell adhesion, they all adequately support cell culture.
As a tissue engineering scaffold, the fibers must be degradable. The degradability of the fibers was examined by an in vitro collagenase degradation assay. As shown in Figure 8A, the susceptibility of CS30 fibers to collagenase digestion was characterized by the decrease in the remaining weight of the fibers with the digestion time. Apparently, degradation of untreated fibers is much faster than that of GA-treated fibers. After 24 h of enzyme digestion, the remaining weight ratio of CS30 fibers with 2, 6, and 48 h of GA treatments was 0.202, 0.635, and 0.888, respectively, whereas the untreated fibers were completely degraded. Fibers with 48 h of treatment showed high resistance against enzyme degradation. The degradability of collagen, CS30 and, silk fibers is compared in Figure 8B after 2 h of enzyme digestion. The weight loss of silk fibers was less than 10% regardless of the GA treatment time. On the contrary, collagen and CS30 fibers were readily degraded, and the degradation was hampered by GA treatment in a time-dependent manner. Therefore, by adjusting the fiber composition and the GA treatment time, scaffolds with desirable degradability can be achieved.
Figure 8.

Characterization of fiber degradation by the change of the remaining weight ratio of fibers. (A) Comparison of the degradation kinetics of CS30 fibers at different GA treatment times. (B) Comparison of fiber degradation with GA treatment time in collagen (C), CS30, and silk (S) fibers. All fibers were digested by collagenase for 2 h before the measurement.
Discussion
In this work, we demonstrated that a short-term GA treatment can not only enhance the mechanical properties and stability of E-spun collagen–silk composite fibers but also impose controls on fiber degradability while the fibers retain biocompatibility. Thus, the treated fibers are excellent candidates to be employed as tissue engineering scaffolds.
Owing to the distinctive structures of collagen and silk proteins, GA treatment affects the fibers differently. GA cross links collagen peptides through reactions of the aldehyde groups of GA with the free amine groups of lysine or hydroxylysine residues of the polypeptide chains to form Schiff base structures.38,39 This was confirmed by the dramatic decrease of the free amine groups with GA treatment time (Figure 2), as well as the increase of the IR peak at 1588 cm–1, which indicates the formation of a Schiff base (Figure 4A). It was also evidenced by the increased fiber fluorescence, which is attributed to the formation of the Schiff base as well as the increased fiber stiffness that restricts intramolecular rotation of the fluorophores, leading to the enhancement of emission.37 Native collagen is made up of strands of triple α helices. The FTIR spectroscopic analysis suggests that the α helix is the dominant structure in as-spun collagen fibers, consistent with the GXY repeating sequences in collagen peptides. By electrospinning, the molecules were aligned preferentially in the axial direction;49,50 however, they were loosely packed. GA treatment generated cross-links via intra- and inter-molecular covalent bonds that bundled peptides within the E-spun fibers together, evidenced by the reduced fiber width, to produce more closely stacked structures resembling that of native collagen fibers.21 The cross-links restrain the slippage between molecules, rendering a strong resistance against uniaxial stretching along the fibers.35 Thus, GA treatment augmented the ultimate strain of collagen fibers most significantly, while also increasing the ultimate tensile strength, ultimate stress, and Young’s modulus. Although GA treatment effectively tightened the collagen peptides in fibers, the secondary structure was largely unaffected, evidenced by the FTIR spectra.
Unlike collagen, dragline silk proteins do not have lysine or hydroxylysine residues, and they are relatively hydrophobic. The glycine-rich regions (GGX or GPGXX motifs) likely form coiled structures, and the polyalanine regions, if arranged properly, form β-sheet structures. The FTIR spectrum of the as-spun silk fibers implies a random coil dominant structure, indicating the improper folding of the silk proteins. The GA vapor induced the internal transition of the secondary structure of silk proteins from random coils to ordered β-sheets,32,33 proved by the quantitative analysis of the amide I band in the FTIR spectra (Figure 4). It has been reported that a low pH can induce a conformational change of silk proteins from a disordered spidroin into the β-sheet rich structures.51 The GA vapor created a mildly acidic environment. In addition, GA acted as a polymer plasticizer and generated a local hydrophilic environment, which may drive the hydrophobic polyalanine to fold–unfold and to stack into β-sheet domains.46 In native dragline silk, crystalline β-sheet domains of alanine-rich motifs are responsible for the exceptionally high tensile strength of silk. Posttreatment methods of artificial silk fibers, such as mechanical stretching in ethanol/water bath, water vapor annealing, and acid/cation treatment, have been applied to induce β-sheet formation to enhance the strength of the fibers.24,26−28,51 The GA vapor treatment in this study provides an alternative simple and mild treatment method to elevate β-sheet formation and hence greatly improve the tensile strength and Young’s modulus of the silk fibers.
In as-spun composite fibers (e.g., CS30), the glycine-rich regions of silk protein (relatively hydrophilic) likely complex with the GXY motifs of collagen via hydrogen bonding and electrostatic interaction, leaving the hydrophobic polyalanine regions of the silk protein relatively free. When GA treatment was applied, due to the hydrophobic nature and the lack of free amines of the polyalanine motifs, we infer that the cross-linking reactions predominantly occurred at the integrated regions of collagen and silk, and their helix and coil structures were largely retained. Such reinforced interactions between biopolymers can effectively reduce the space between neighboring molecules and hence effectively prevent water uptake and protein dissolution (Figure 1G,H), yielding increased fiber stability,23 which is essential for the fibers to provide consistent biophysical and biochemical cues to support cellular behaviors. However, GA treatment can lead to the diffusion of small GA molecules to the polyalanine region of the silk protein, driving the conformational change of polyalanine from random coils to β-sheet domains. Thus, the dual effects of GA treatment on collagen and silk are augmented in the composite fibers to make a profound impact on reinforcing fiber strength, strain, and elasticity52 to achieve resistance to both fracture and rupture. The fibers were also shown to adequately support cell adhesion and proliferation and can be degraded by enzymes in a controllable manner. Thus, the GA-treated CS30 fiber is an excellent candidate of tissue engineering scaffolds.
Nevertheless, a longer-term (>12 h) GA treatment is undesirable. Our data in Figure 3 have shown that a long-term GA treatment induced a marked decrease in the strain of collagen fibers and a noticeable decrease in the tensile strength of silk fibers, leading to corresponding changes in CS30 fibers and, consequently, a decrease in the fiber toughness. Figures 1G, 2, and 4 collectively indicate that a long-term GA treatment resulted in an excess of GA molecules deposited on the fibers. Collagen can uptake more GA molecules than silk because of its hydrophilic nature. These molecules may self-react to form unsaturated polymers53 in the fiber matrix and generate overly cross-linked fibers to impede the free extension of the molecules leading to reduced fiber strain. Excessive GA molecules may also fill up the free space in silk fibers, creating steric hindrance to prevent an effective random coil to β-sheet transition and lead to a reduced fiber tensile strength. In contrast, initial GA cross-linking involves one GA binding with two free amine groups. When the amine groups are abundant in the fibers (short-term treatment), the binding is local and quick, and the effect is immediate. Over a long-term treatment, the free amine groups become fewer and the GA molecules become excessive. In this case, the amine groups are farther apart on average, and cross-linking is achieved by extending the chain via GA polymerization. The GA polymer network formed over time prevents the penetration of GA molecules to further enhance the fiber mechanics. This is consistent with the observation that GA treatment in the initial six hours greatly affected the fiber properties; however, most effects were alleviated with long-term treatment. The study of cell adhesion (Figure 7A) suggests that a long-term GA treatment reduced cell adhesion. This is likely due to GA polymerization, which shades the integrin-binding site on collagen and reduces the cell–matrix interaction. The increased fiber stiffness by a long-term GA treatment may also contribute to the decreased cell–matrix interaction. Despite the decrease in the number of initially attached cells, cell viability and proliferation were barely affected even with long-term GA treatment. Importantly, GA treatment was shown to reduce the degradation rate of the collagen–silk composite fibers. Thus, GA vapor treatment provides a simple, mild, yet effective way to fine-tune the mechanics, stability, and degradability of fibers with a desirable composition serving for a designated biological function.
Although the GA vapor treatment is proven to be an effective approach to rectify the E-spun composite fibers, many other posttreatment methods, such as additional physical or chemical cross-linking techniques to stabilize collagen, water bath stretching, and alcohol vapor treatment to enhance the mechanical properties of silk, are yet to be explored. Ultimately, systematic studies are expected to reveal the mechanisms of the treatment effects and guide us to tailor a method, likely a combination of the methods and many others yet to be discovered, to achieve optimal performance of the biocompatible scaffolds with appropriate remodeling characteristics and mechanical properties.
Conclusions
Our study has shown that the GA vapor treatment greatly impacts the E-spun collagen–silk composite fibers. GA chemically links collagen peptides through reactions between its aldehyde groups and free amino groups of lysine or hydroxylysine residues. It also induces the enrichment of the β-sheet character in silk. Consequently, a short-term treatment significantly enhanced the tensile strength, elasticity, stretchability, and stability of the fibers, whereas overtreatment caused the plasticity and fragility as well as reduced cell–matrix adhesion. A 6 h GA vapor treatment of CS30 fibers was shown to lead to an optimal balance of physical properties and biocompatibility. The time-dependent effect of GA treatment offers an exceptional way to conveniently tune the properties of E-spun fibers without changing its protein compositions.
Freestanding tissue engineering scaffolds are highly desirable for in vitro and in vivo applications. They are required to be sufficiently flexible, tough, stable, and biocompatible for effortless handling and for adequately supporting cell development. The systematic study of the effects of the GA vapor treatment provides a deeper understanding of the treatment mechanisms and makes it possible to control the topographic features, mechanical properties, stability, cell adhesion, and biodegradation rates of E-spun collagen–silk fibers by adjusting only the posttreatment protocols for versatile applications.
Experimental Section
Materials
Collagen type I from calf skin was purchased from MP Biomedicals (Solon, OH). Major ampullate spidroin proteins 1 and 2 (MaSp 1 and MaSp 2) of dragline spider silk were purified from the milk of transgenic goats and mixed at a MaSp 1/MaSp 2 ratio of 4:1 to obtain optimized mechanical properties.25,41 Collagen and silk proteins were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) (Fisher Scientific, Pittsburgh, PA) separately. The collagen and silk proteins were mixed to make solutions containing silk at 0% (pure collagen), 30% (CS30), 60% (CS60), and 100% (pure silk) while the total protein concentration was maintained at 80 mg/mL. GA (Fisher Scientific, Pittsburgh, PA) was diluted to 20% v/v in water, and its vapor was used for E-spun fiber posttreatment.
Preparation of Collagen–Silk Composite Fibers
Aligned freestanding collagen–silk composite fibers were prepared using a home-built electrospinning system, as described in the previous work.16 The spinning conditions were optimized to obtain fibers with a desired dimension, density, and alignment. The parameters are summarized in Table S1.
The as-spun fibers were dried in a vacuum oven at 30 °C for 24 h to remove any solvent (HFIP) residue and then exposed to water vapor overnight as a calibration of all fibers before any test or treatment. GA treatment was carried out by exposing the fibers to the vapor of 20% (v/v) GA at room temperature for 0, 2, 6, 12, 24, 48, or 96 h. Fibers for cell culture were immersed in a 0.1 M glycine aqueous solution for 1 h to abate the toxicity of residue aldehyde groups to cells, followed by sterilization using 70% (v/v) ethanol and overnight UV exposure.
Atomic Force Microscopic Imaging and Optical Imaging
Fiber morphology was characterized by using a multimode Nanoscope IIIa atomic force microscope (AFM; Veeco Metrology, Santa Barbara, CA) equipped with a J-scanner. The fiber width was analyzed using the NanoScope Analysis software. To examine fiber stability in a cell culture environment, the images were collected after the fibers with various GA treatment times were immersed in 1× PBS buffer and stored in the cell culture incubator (37 °C, 10% CO2) for 48 h.
A Nikon TE2000-U microscope was used to collect optical images to examine the fiber alignment and cell growth on the fibers. The effect of the GA treatment on fiber fluorescence was studied using the same system at the excitation and emission wavelengths of 470 and 520 nm, respectively.
Fiber Weight Measurement
E-spun collagen, CS30, or silk fibers (3–5 mg) before and after GA treatment were vacuum-dried overnight at 37 °C and weighed using a thermogravimetric analyzer (Mettler-Toledo, Columbus, OH) at a resolution of 0.001 mg to evaluate the amount of GA uptake by various fibers.
To evaluate fiber stability, the swelling ratio of fibers after incubation in water was examined. In this case, the weight of each dry sample (W0) was measured before it was soaked in deionized water at 37 °C for 2 h. After removing excess water, the weight of each sample (W1) was measured. The swelling ratio was calculated by (W1 – W0)/W0 × 100%. To evaluate protein dissolution, the wet samples were dried in a vacuum oven and weighed (W2). The weight loss was calculated by (W0 – W2)/W0 × 100%.
Determination of Free Amine Group Content
The as-spun fibers were subjected to a TNBS assay.54 In brief, the fibers were first immersed in 1 mL of 4% (wt) NaHCO3 solution for 2 h at room temperature. Then, 1 mL of freshly prepared 0.1% (wt) TNBS in 4% (wt) NaHCO3 was added, and the mixture was heated at 40 °C for 2 h. After 3 mL of 6 N HCl was added, the solution was heated at 60 °C for 1.5 h, followed by 3 min sonication and a further 1 h incubation to allow complete dissolution of the fibers. After cooling down to room temperature, the resulting solution was diluted to 10 mL to measure the absorbance at 340 nm using a Beckman Coulter DU800 spectrophotometer (Brea, CA). Control samples for each type of fibers were prepared by adding HCl before the addition of TNBS. By generating a standard curve of absorbance versus the number of free amine groups using glycine, the free amine group content of the fibers was determined and presented as the number of free amine groups per 1000 amino acids (n/1000).
Infrared Spectroscopy and Peak Deconvolution
Infrared spectra of E-spun fibers were collected using a Thermo Nicolet Nexus 470 FTIR Spectrometer (Thermo Electron Co., Madison, WI). After treatment and overnight drying in a vacuum desiccator, freestanding fibers, prefixed on aluminum frames, were directly exposed to the IR beam and scanned in the range of 400–4000 cm–1, with a nominal resolution of 4 cm–1. After linear baseline correction of each spectrum, the absorbance of the amide I band in the 1590–1710 cm–1 region was normalized into the 0–1 scale. The normalized spectra were then subjected to a peak deconvolution procedure using “peak analyzer” of Origin Software (see details in the Supporting Information). The percentage of the integral of each fitting peak was used to assess the proportion of the protein in the corresponding configuration.
Mechanical Testing
Tensile stress–strain curves of E-spun fibers were obtained using an MTS Synergie 100 system (Test Resources Inc., Shakopee, MN), as described in the previous work.16 In short, aligned freestanding fibers with various GA treatment times were collected across an 8 mm gap of an aluminum frame. Optical images (10×) were taken to estimate fiber density, and the average cross-sectional area of individual fibers was determined by AFM. The fibers were then tested at a stretching rate of 1 mm/min and a data acquisition rate of 120 Hz to measure the ultimate stress, ultimate strain, and Young’s modulus from the stress–strain curves. Fiber toughness, the integral of the area beneath the stress–strain curve, was calculated using the Origin software. The data were derived from more than five measurements for each sample type.
Cell Proliferation and Viability Tests
Undifferentiated hdpPSCs were maintained in the phenol red-free RPMI-1640 medium (Invitrogen, Carlsbad, CA) supplemented with 0.1 mM sodium pyruvate, 100 U/mL penicillin–streptomycin (Sigma-Aldrich, St. Louis, MO), and 10% charcoalstripped fetal bovine serum (S-FBS).8,55 E-spun fibers were collected on 4 mm × 4 mm precleaned glass substrates. After the posttreatments and sterilization, the fiber-loaded substrates were placed in the wells of a 96-well plate with a flat bottom (Corning, Corning, NY).
In each well, 9.6 × 103 cells were seeded. To assure that only cells on the fiber-loaded substrates were subject to the test, the substrates with cells at days 1, 4, and 7 of culture were transferred to a new 96-well plate before 120 μL of the medium containing 20 μL of CellTiter 96 Aqueous One Solution Reagent (Promega, Fitchburg, WI) was added to each well. After incubation at 37 °C for 2 h, 100 μL of the solution from each well was taken to measure the absorbance at 490 nm (ELx808 Absorbance Reader, BioTek, VT) to examine the cell proliferation. Data were displayed after subtracting the background absorbance generated by the cell culture medium.
To evaluate the effect of the GA treatment on cell viability, the cells after a 1-day culture on the fiber-loaded Petri dishes were subjected to a CellTiter 96 Aqueous Assay (Promega, Fitchburg, WI). Absorbance of cells cultured on the Petri dish in the absence of fibers was used as a control to derive the percentage of metabolically active cells on various matrices.
Trypsin Deadhesion Assay
Cell adhesion was examined using a trypsin deadhesion assay, as previously described.16 Briefly, hdpPSCs after a 12 h culture were washed with PBS and trypsinized. By taking in situ optical images (20×) every 15 s, the reduction of the cell–substrate contact area was monitored. The change of the area with time was plotted and fit to the Boltzmann sigmoid equation to derive constant t0.5, which represents the time point when the area change is 50% of the total area reduction, and is used to evaluate cell adhesion.56,57
In Vitro Biodegradation
Preweighed E-spun collagen, CS30, and silk fibers (3–5 mg) were placed in 1.5 mL of centrifuge tubes containing 1 mL of 100 unit/mL type I collagenase (Gibco, Big Cabin, OK) in HBSS solution (Gibco, Big Cabin, OK) and incubated in a shaker (37 °C, 120 rpm) for a specific period of digestion time. The samples were then centrifuged (12 000 rpm, 3 min) and washed with ultrapure water thrice before they were dried in a vacuum oven (37 °C) and weighed again. The degree of fiber degradation was presented by the ratio of the remaining dry weight (Wd) to the initial dry weight of E-spun fibers (W0), that is, Wd/W0.
Acknowledgments
We thank Russ Janota for his assistance in testing fiber mechanics, Dr. Zuzana Strakova for kindly providing the human decidua parietalis placental stem cells (hdpPSCs), and Dr. Carlo Segre for helping us set up the home-built electrospinning system. This research was partly supported by NIH (R01 NS047719) and the Utah Science Technology and Research (USTAR) initiative fund.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00290.
Optimization of electrospinning conditions; fluorescence spectra of E-spun collagen fibers after GA treatment; the color change of CS30 fibers with GA treatment time; differential FTIR spectra of collagen, CS30, CS60, and silk fibers after various GA treatment times; deconvolution of the amide I band in normalized FTIR spectra; proliferation of cells grown on collagen, CS30, and silk fibers after various GA treatment times (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Li W. J.; Laurencin C. T.; Caterson E. J.; Tuan R. S.; Ko F. K. Electrospun nanofibrous structure: a novel scaffold for tissue engineering. J. Biomed. Mater. Res. 2002, 60, 613–621. 10.1002/jbm.10167. [DOI] [PubMed] [Google Scholar]
- Ma Z.; Kotaki M.; Inai R.; Ramakrishna S. Potential of nanofiber matrix as tissue-engineering scaffolds. Tissue Eng. 2005, 11, 101–109. 10.1089/ten.2005.11.101. [DOI] [PubMed] [Google Scholar]
- Matthews J. A.; Wnek G. E.; Simpson D. G.; Bowlin G. L. Electrospinning of collagen nanofibers. Biomacromolecules 2002, 3, 232–238. 10.1021/bm015533u. [DOI] [PubMed] [Google Scholar]
- Telemeco T. A.; Ayres C.; Bowlin G. L.; Wnek G. E.; Boland E. D.; Cohen N.; Baumgarten C. M.; Mathews J.; Simpson D. G. Regulation of cellular infiltration into tissue engineering scaffolds composed of submicron diameter fibrils produced by electrospinning. Acta Biomater. 2005, 1, 377–385. 10.1016/j.actbio.2005.04.006. [DOI] [PubMed] [Google Scholar]
- Zhong S.; Teo W. E.; Zhu X.; Beuerman R. W.; Ramakrishna S.; Yung L. Y. An aligned nanofibrous collagen scaffold by electrospinning and its effects on in vitro fibroblast culture. J. Biomed. Mater. Res., Part A 2006, 79, 456–463. 10.1002/jbm.a.30870. [DOI] [PubMed] [Google Scholar]
- Lin H. J.; O’Shaughnessy T. J.; Kelly J.; Ma W. Neural stem cell differentiation in a cell-collagen-bioreactor culture system. Brain Res. Dev. Brain Res. 2004, 153, 163–173. 10.1016/j.devbrainres.2004.08.010. [DOI] [PubMed] [Google Scholar]
- Sridharan I.; Kim T.; Wang R. Adapting collagen/CNT matrix in directing hESC differentiation. Biochem. Biophys. Res. Commun. 2009, 381, 508–512. 10.1016/j.bbrc.2009.02.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan I.; Kim T.; Strakova Z.; Wang R. Matrix-specified differentiation of human decidua parietalis placental stem cells. Biochem. Biophys. Res. Commun. 2013, 437, 489–495. 10.1016/j.bbrc.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. P.; Chang G. Y.; Chen J. K. Electrospun collagen/chitosan nanofibrous membrane as wound dressing. Colloids Surf., A 2008, 313–314, 183–188. 10.1016/j.colsurfa.2007.04.129. [DOI] [Google Scholar]
- Boland E. D.; Matthews J. A.; Pawlowski K. J.; Simpson D. G.; Wnek G. E.; Bowlin G. L. Electrospinning collagen and elastin: preliminary vascular tissue engineering. Front. Biosci. 2004, 9, 1422–1432. 10.2741/1313. [DOI] [PubMed] [Google Scholar]
- Schnell E.; Klinkhammer K.; Balzer S.; Brook G.; Klee D.; Dalton P.; Mey J. Guidance of glial cell migration and axonal growth on electrospun nanofibers of poly-epsilon-caprolactone and a collagen/poly-epsilon-caprolactone blend. Biomaterials 2007, 28, 3012–3025. 10.1016/j.biomaterials.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Altman G. H.; Diaz F.; Jakuba C.; Calabro T.; Horan R. L.; Chen J.; Lu H.; Richmond J.; Kaplan D. L. Silk-based biomaterials. Biomaterials 2003, 24, 401–416. 10.1016/S0142-9612(02)00353-8. [DOI] [PubMed] [Google Scholar]
- Bini E.; Foo C. W.; Huang J.; Karageorgiou V.; Kitchel B.; Kaplan D. L. RGD-functionalized bioengineered spider dragline silk biomaterial. Biomacromolecules 2006, 7, 3139–3145. 10.1021/bm0607877. [DOI] [PubMed] [Google Scholar]
- Lewis R. V. Spider silk: ancient ideas for new biomaterials. Chem. Rev. 2006, 106, 3762–74. 10.1021/cr010194g. [DOI] [PubMed] [Google Scholar]
- Teulé F.; Addison B.; Cooper A. R.; Ayon J.; Henning R. W.; Benmore C. J.; Holland G. P.; Yarger J. L.; Lewis R. V. Combining flagelliform and dragline spider silk motifs to produce tunable synthetic biopolymer fibers. Biopolymers 2012, 97, 418–431. 10.1002/bip.21724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu B.; Li W.; Lewis R. V.; Segre C. U.; Wang R. E-spun composite fibers of collagen and dragline silk protein: fiber mechanics, biocompatibility, and application in stem cell differentiation. Biomacromolecules 2015, 16, 202–213. 10.1021/bm501403f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rho K. S.; Jeong L.; Lee G.; Seo B. M.; Park Y. J.; Hong S. D.; Roh S.; Cho J. J.; Park W. H.; Min B. M. Electrospinning of collagen nanofibers: effects on the behavior of normal human keratinocytes and early-stage wound healing. Biomaterials 2006, 27, 1452–1461. 10.1016/j.biomaterials.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Buttafoco L.; Kolkman N. G.; Engbers-Buijtenhuijs P.; Poot A. A.; Dijkstra P. J.; Vermes I.; Feijen J. Electrospinning of collagen and elastin for tissue engineering applications. Biomaterials 2006, 27, 724–734. 10.1016/j.biomaterials.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Zeugolis D. I.; Khew S. T.; Yew E. S.; Ekaputra A. K.; Tong Y. W.; Yung L. Y.; Hutmacher D. W.; Sheppard C.; Raghunath M. Electro-spinning of pure collagen nano-fibres - just an expensive way to make gelatin?. Biomaterials 2008, 29, 2293–2305. 10.1016/j.biomaterials.2008.02.009. [DOI] [PubMed] [Google Scholar]
- Berglund J. D.; Mohseni M. M.; Nerem R. M.; Sambanis A. A biological hybrid model for collagen-based tissue engineered vascular constructs. Biomaterials 2003, 24, 1241–1254. 10.1016/S0142-9612(02)00506-9. [DOI] [PubMed] [Google Scholar]
- Grover C. N.; Gwynne J. H.; Pugh N.; Hamaia S.; Farndale R. W.; Best S. M.; Cameron R. E. Crosslinking and composition influence the surface properties, mechanical stiffness and cell reactivity of collagen-based films. Acta Biomater. 2012, 8, 3080–3090. 10.1016/j.actbio.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih Y. R.; Chen C. N.; Tsai S. W.; Wang Y. J.; Lee O. K. Growth of mesenchymal stem cells on electrospun type I collagen nanofibers. Stem Cells 2006, 24, 2391–2397. 10.1634/stemcells.2006-0253. [DOI] [PubMed] [Google Scholar]
- Lin H. Y.; Kuo Y. J.; Chang S. H.; Ni T. S. Characterization of electrospun nanofiber matrices made of collagen blends as potential skin substitutes. Biomed. Mater. 2013, 8, 025009 10.1088/1748-6041/8/2/025009. [DOI] [PubMed] [Google Scholar]
- Albertson A. E.; Teule F.; Weber W.; Yarger J. L.; Lewis R. V. Effects of different post-spin stretching conditions on the mechanical properties of synthetic spider silk fibers. J. Mech. Behav. Biomed. Mater. 2014, 29, 225–234. 10.1016/j.jmbbm.2013.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker C. L.; Jones J. A.; Bringhurst H. N.; Copeland C. G.; Addison J. B.; Weber W. S.; Mou Q.; Yarger J. L.; Lewis R. V. Mechanical and physical properties of recombinant spider silk films using organic and aqueous solvents. Biomacromolecules 2014, 15, 3158–3170. 10.1021/bm5007823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones J. A.; Harris T. I.; Tucker C. L.; Berg K. R.; Christy S. Y.; Day B. A.; Gaztambide D. A.; Needham N. J.; Ruben A. L.; Oliveira P. F.; Decker R. E.; Lewis R. V. More than just fibers: an aqueous method for the production of innovative recombinant spider silk protein materials. Biomacromolecules 2015, 16, 1418–1425. 10.1021/acs.biomac.5b00226. [DOI] [PubMed] [Google Scholar]
- Meinel A. J.; Kubow K. E.; Klotzsch E.; Garcia-Fuentes M.; Smith M. L.; Vogel V.; Merkle H. P.; Meinel L. Optimization strategies for electrospun silk fibroin tissue engineering scaffolds. Biomaterials 2009, 30, 3058–3067. 10.1016/j.biomaterials.2009.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X.; Shmelev K.; Sun L.; Gil E. S.; Park S. H.; Cebe P.; Kaplan D. L. Regulation of silk material structure by temperature-controlled water vapor annealing. Biomacromolecules 2011, 12, 1686–1696. 10.1021/bm200062a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chvapil M.; Gibeault D.; Wang T. F. Use of chemically purified and cross-linked bovine pericardium as a ligament substitute. J. Biomed. Mater. Res. 1987, 21, 1383–1393. 10.1002/jbm.820211204. [DOI] [PubMed] [Google Scholar]
- Nimni M. E.; Cheung D.; Strates B.; Kodama M.; Sheikh K. Chemically modified collagen: a natural biomaterial for tissue replacement. J. Biomed. Mater. Res. 1987, 21, 741–771. 10.1002/jbm.820210606. [DOI] [PubMed] [Google Scholar]
- Hartman O.; Zhang C.; Adams E. L.; Farach-Carson M. C.; Petrelli N. J.; Chase B. D.; Rabolt J. F. Microfabricated electrospun collagen membranes for 3-D cancer models and drug screening applications. Biomacromolecules 2009, 10, 2019–2032. 10.1021/bm8012764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo I. S.; Oh J. E.; Jeong L.; Lee T. S.; Lee S. J.; Park W. H.; Min B. M. Collagen-based biomimetic nanofibrous scaffolds: preparation and characterization of collagen/silk fibroin bicomponent nanofibrous structures. Biomacromolecules 2008, 9, 1106–1116. 10.1021/bm700875a. [DOI] [PubMed] [Google Scholar]
- Wang Y. X.; Qin Y. P.; Kong Z. J.; Wang Y. J.; Ma L. Glutaraldehyde Cross-Linked Silk Fibroin Films for Controlled Release. Adv. Mater. Res. 2014, 887–888, 541–546. 10.4028/www.scientific.net/AMR.887-888.541. [DOI] [Google Scholar]
- Teng W.; Cappello J.; Wu X. Recombinant silk-elastinlike protein polymer displays elasticity comparable to elastin. Biomacromolecules 2009, 10, 3028–3036. 10.1021/bm900651g. [DOI] [PubMed] [Google Scholar]
- Yang L.; Fitie C. F.; van der Werf K. O.; Bennink M. L.; Dijkstra P. J.; Feijen J. Mechanical properties of single electrospun collagen type I fibers. Biomaterials 2008, 29, 955–962. 10.1016/j.biomaterials.2007.10.058. [DOI] [PubMed] [Google Scholar]
- Shin D. H.; Heo M. B.; Lim Y. T. Self-assembled polyelectrolyte nanoparticles as fluorophore-free contrast agents for multicolor optical imaging. Molecules 2015, 20, 4369–4382. 10.3390/molecules20034369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupane L. N.; Oh E. T.; Park H. J.; Lee K. H. Selective and Sensitive Detection of Heavy Metal Ions in 100% Aqueous Solution and Cells with a Fluorescence Chemosensor Based on Peptide Using Aggregation-Induced Emission. Anal. Chem. 2016, 88, 3333–3340. 10.1021/acs.analchem.5b04892. [DOI] [PubMed] [Google Scholar]
- Cheung D. T.; Nimni M. E. Mechanism of crosslinking of proteins by glutaraldehyde II. Reaction with monomeric and polymeric collagen. Connect. Tissue Res. 1982, 10, 201–216. 10.3109/03008208209034419. [DOI] [PubMed] [Google Scholar]
- Lubig R.; Kusch P.; Roper K.; Zahn H. On the Mechanism Of Protein Crosslinking with Glutaraldehyde. Monatsh. Chem. 1981, 112, 1313–1323. 10.1007/BF00904683. [DOI] [Google Scholar]
- Damink L. H. H. O.; Dijkstra P. J.; Vanluyn M. J. A.; Vanwachem P. B.; Nieuwenhuis P.; Feijen J. Glutaraldehyde as a Cross-Linking Agent for Collagen-Based Biomaterials. J. Mater. Sci.: Mater. Med. 1995, 6, 460–472. 10.1007/BF00123371. [DOI] [Google Scholar]
- An B.; Jenkins J. E.; Sampath S.; Holland G. P.; Hinman M.; Yarger J. L.; Lewis R. Reproducing natural spider silks’ copolymer behavior in synthetic silk mimics. Biomacromolecules 2012, 13, 3938–3948. 10.1021/bm301110s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw M. J.; Cheung M. C.; Ehrlich D. J.; Smith M. L. Using molecular mechanics to predict bulk material properties of fibronectin fibers. PLoS Comput. Biol. 2012, 8, e1002845 10.1371/journal.pcbi.1002845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Reis E. F.; Campos F. S.; Lage A. P.; Leite R. C.; Heneine L. G.; Vasconcelos W. L.; Lobato Z. I. P.; Mansur H. S. Synthesis and characterization of poly (vinyl alcohol) hydrogels and hybrids for rMPB70 protein adsorption. Mater. Res. 2006, 9, 185–191. 10.1590/S1516-14392006000200014. [DOI] [Google Scholar]
- Arrondo J. L.; Goni F. M. Structure and dynamics of membrane proteins as studied by infrared spectroscopy. Prog. Biophys. Mol. Biol. 1999, 72, 367–405. 10.1016/S0079-6107(99)00007-3. [DOI] [PubMed] [Google Scholar]
- Yu P. Protein secondary structures (alpha-helix and beta-sheet) at a cellular level and protein fractions in relation to rumen degradation behaviours of protein: a new approach. Br. J. Nutr. 2005, 94, 655–665. 10.1079/BJN20051532. [DOI] [PubMed] [Google Scholar]
- Kreplak L.; Doucet J.; Dumas P.; Briki F. New aspects of the alpha-helix to beta-sheet transition in stretched hard alpha-keratin fibers. Biophys. J. 2004, 87, 640–647. 10.1529/biophysj.103.036749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramanti E.; Catalano D.; Forte C.; Giovanneschi M.; Masetti M.; Veracini C. A. Solid state C-13 NMR and FT-IR spectroscopy of the cocoon silk of two common spiders. Spectrochim. Acta, Part A 2005, 62, 105–111. 10.1016/j.saa.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Kim T.; Sridharan I.; Zhu B.; Orgel J.; Wang R. Effect of CNT on collagen fiber structure, stiffness assembly kinetics and stem cell differentiation. Mater. Sci. Eng., C 2015, 49, 281–289. 10.1016/j.msec.2015.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu S. Y.; Wu Q. L.; Ren J.; Vancso G. J. Mechanical properties of a single electrospun fiber and its structures. Macromol. Rapid Commun. 2005, 26, 716–720. 10.1002/marc.200400667. [DOI] [Google Scholar]
- Reneker D. H.; Chun I. Nanometre diameter fibres of polymer, produced by electrospinning. Nanotechnology 1996, 7, 216–223. 10.1088/0957-4484/7/3/009. [DOI] [Google Scholar]
- Dicko C.; Kenney J. M.; Knight D.; Vollrath F. Transition to a beta-sheet-rich structure in spidroin in vitro: the effects of pH and cations. Biochemistry 2004, 43, 14080–14087. 10.1021/bi0483413. [DOI] [PubMed] [Google Scholar]
- Wang M. C.; Pins G. D.; Silver F. H. Collagen fibres with improved strength for the repair of soft tissue injuries. Biomaterials 1994, 15, 507–512. 10.1016/0142-9612(94)90016-7. [DOI] [PubMed] [Google Scholar]
- Migneault I.; Dartiguenave C.; Bertrand M. J.; Waldron K. C. Glutaraldehyde: behavior in aqueous solution, reaction with proteins, and application to enzyme crosslinking. BioTechniques 2004, 37, 790–802. [DOI] [PubMed] [Google Scholar]
- Nagai N.; Yunoki S.; Suzuki T.; Sakata M.; Tajima K.; Munekata M. Application of cross-linked salmon atelocollagen to the scaffold of human periodontal ligament cells. J. Biosci. Bioeng. 2004, 97, 389–394. 10.1016/S1389-1723(04)70224-8. [DOI] [PubMed] [Google Scholar]
- Strakova Z.; Livak M.; Krezalek M.; Ihnatovych I. Multipotent properties of myofibroblast cells derived from human placenta. Cell Tissue Res. 2008, 332, 479–488. 10.1007/s00441-008-0604-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen S.; Kumar S. Cell-Matrix De-Adhesion Dynamics Reflect Contractile Mechanics. Cell. Mol. Bioeng. 2009, 2, 218–230. 10.1007/s12195-009-0057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbitt M. W.; Kloxin A. M.; Dyamenahalli K. U.; Anseth K. S. Controlled two-photon photodegradation of PEG hydrogels to study and manipulate subcellular interactions on soft materials. Soft Matter 2010, 6, 5100–5108. 10.1039/c0sm00174k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.