

Abstract
The relative ease of Mössbauer spectroscopy and of density functional theory (DFT) calculations encourages the use of Mössbauer parameters as a validation method for calculations, and the use of calculations as a double check on crystallographic structures. A number of studies have proposed correlations between the computationally determined electron density at the iron nucleus and the observed isomer shift, but deviations from these correlations in low-valent iron β-diketiminate complexes encouraged us to determine a new correlation for these compounds. The use of B3LYP/def2-TZVP in the ORCA platform provides an excellent balance of accuracy and speed. We provide here not only this new correlation and a clear guide to its use but also a systematic analysis of the limitations of this approach. We also highlight the impact of crystallographic inaccuracies, DFT model truncation, and spin states, with intent to assist experimentalists to use Mössbauer spectroscopy and calculations together.
Introduction
Since the discovery of recoilless nuclear resonance absorption of γ-rays in 1957 by Rudolf Mössbauer,1 the spectroscopy bearing his name has become a powerful technique in transition metal and bioinorganic chemistry to obtain information regarding the electronics and magnetic properties of metal centers.2 The Mössbauer effect has been observed for more than 80 nuclides, but 57Fe Mössbauer spectroscopy is the most common.3−6 Mössbauer spectroscopy using the 57Fe nucleus has become ubiquitous in both bioinorganic and coordination chemistry because of the ability to selectively probe Fe environments in a sample, even in the presence of large protein scaffolds or complex ligands that have numerous other atoms.5 Each unique iron environment typically exhibits a doublet in a Mössbauer spectrum when the applied field is small (“zero field”) (Figure 1). Fitting the zero-field doublet yields two parameters, the isomer shift (δ) and the absolute value of the quadrupole splitting (|ΔEQ|).3
Figure 1.
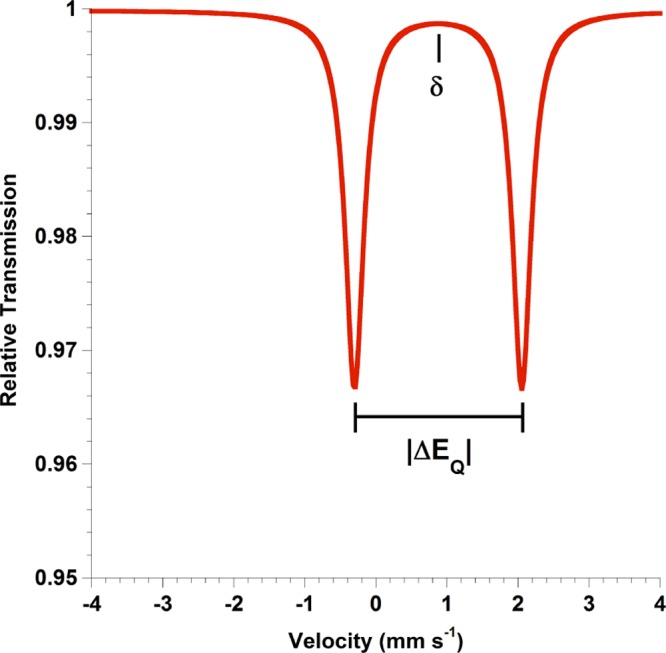
Diagram of a typical zero-field Mössbauer spectrum with isomer shift (δ) and quadrupole splitting parameters (|ΔEQ|) indicated.
The isomer shift, δ, which lies at the midpoint of the doublet, reflects the electron density at the iron nucleus and is often used to interpret the oxidation state and spin state of the iron center. For example, high oxidation states tend to give lower isomer shifts in high-spin compounds, and for the same oxidation state, low-spin compounds tend to give lower isomer shifts than high-spin compounds.3 However, the ligand field has a substantial influence on the isomer shift, and additional trends are evident based on the ligand identities (more covalency tends to give lower δ) and the geometry at the metal (lower coordination numbers tend to give lower δ). These trends have been usefully summed up by the generalization that shorter bonds give lower δ, a principle that unites the trends with oxidation state, coordination number, and spin state.3 However, due to the number of (often competing) influences, it may be difficult to predict the isomer shift adequately with these qualitative relationships.
The quadrupole splitting, which represents the peak-to-peak separation of the doublet, reflects the asymmetry in electron distribution around the nucleus and therefore can offer insight into the occupancy of the 3d orbitals. For high-symmetry iron centers with octahedral and tetrahedral coordination environments, interpretation of the quadrupole splitting can be straightforward, but in more complex systems with less symmetry and orbital mixing, interpretation is typically more complicated.3
The complexity of interpreting the spectral values for understanding of electronics and bonding has led chemists to use density functional theory (DFT) computations to predict Mössbauer parameters for a given structure.6,7 The ability to accurately calculate the experimental values has also become a valuable way to validate a computational model.7,8 Computation of the isomer shift and quadrupole splitting for an iron site requires calculation of the electron density and the electric field gradient (EFG) at the iron nucleus, respectively. Calculating the isomer shift is problematic because of systematic errors of common basis sets in describing the electron density very close to the iron nucleus. As a result, calculation of isomer shift values generally depends on an empirically determined correlation between the calculated electron density and the experimental isomer shift.6,9 These fits are typically linear, but the fit parameters only apply when using a specific set of computational parameters (functional, basis set, relativistic corrections). The calculated EFG yields a predicted quadrupole splitting through the relationship given in eq 1, where e is the charge of an electron, Q is the quadrupole moment of the 57Fe nucleus (0.16(1) barn,3 and Vzz is the major component of the EFG. Comparison of these to experiment is complicated by the fact that the sign of the experimental quadrupole splitting is not determined in a zero-field measurement, and thus most researchers simply use the absolute value of the quadrupole splitting for comparison to experiment.
| 1 |
Though the number of DFT studies on Mössbauer parameters is too large to describe them all, a few are particularly worth noting. Oldfield et al. established an early precedent for predicting Mössbauer parameters of paramagnetic complexes in various spin states as well as the use of relativistic corrections for improving isomer shift predictions.6d Noodleman et al. examined different classes of iron complexes and proteins with particular focus on iron-nitrosyl and iron-sulfur species, and extended their results to prediction of intermediates in metalloproteins, such as ribonucleotide reductase.6c,6f,6g,7e,9f Neese et al. compared pure and hybrid functionals along with various basis sets for the prediction of Mössbauer parameters for simple iron compounds.6b,6e,9b Filatov et al. reported a new method based on energy derivatives due to the excitation of the 57Fe nucleus enabling isomer shift calculations without a correlation with experimental results.10 Friesner et al. benchmarked many functionals and basis sets on a broad range of iron compounds, established the use of X-ray structures for Mössbauer parameter predictions, and extended their results to assessing the validity of proposed methane monooxygenase intermediates.9c Pápai et al. published an extensive benchmark study using a wide range of iron compounds, and compared GGA and hybrid functionals, STO and GTO basis sets, and effects of solvation models on Mössbauer parameter predictions.9d They also discussed potential difficulties with the correlation of computation to experiment, including low-lying excited states, geometrical isomerism, and spin state changes. More recent reports have focused on expansion of these ideas to new ligand systems, inclusion of experimental error in the development of the correlation fits, as well as new methods for structure optimization, including systems requiring periodic boundary conditions.11
We became interested in computing Mössbauer parameters for reduced β-diketiminate supported iron complexes to validate calculations on their electronic structures.8 These showed us that the existing correlations have few examples of multimetallic complexes, redox-active ligands, low-valent, and low-coordinate iron complexes. Additionally, the established correlations use continuum solvation models with the polarity of water9b or methanol,9d whereas the iron complexes of interest to our group are often studied experimentally in less polar solvents, such as toluene. When using the isomer shift correlation reported by Neese and co-workers, we often calculated isomer shifts that had systematic deviations from experiment (0.1–0.2 mm s−1 lower in the calculations) despite the optimized geometries agreeing quite well with the crystal structure geometries.8a,8e The need to address these issues and the large number of zero-field Mössbauer spectra that we have now collected on related systems encouraged us to seek a more applicable correlation as well as the most facile method for the calculations. Others have also noted that the isomer shift correlations in the literature often need to be “tuned” to different kinds of environments, for example iron-sulfur clusters.12
Herein, we report a revised set of correlation parameters between the calculated electron density and the experimental isomer shift. We use ORCA, a free platform for computations that has implemented a simple readout for Mössbauer parameters,13 and utilize the common functional B3LYP, which has been validated in many earlier studies. Importantly, the new correlation comes from a representative subset of β-diketiminate complexes as a training set, and we test the effectiveness of this correlation for prediction of Mössbauer parameters using a test set of different complexes. Furthermore, we test the correlation more generally using other low-valent iron complexes with different ligand types having various donors (carbon donors, phosphines, thiolates, and hydrides). The power of the same computations for prediction of quadrupole splitting values is also evaluated for all complexes. As experimentalists ourselves, we were also motivated to evaluate the tools that are often used to make computations feasible for the nonspecialist. Thus, we describe the effects of ligand truncation in the computations, and the effect of the spin state on predicted isomer shift and quadrupole splitting values in selected examples.
Results
Nine iron β-diketiminate complexes were chosen as a training set to calibrate the relationship between the calculated electron densities and the experimental isomer shifts. The complexes were specifically chosen because they had very high-quality X-ray structures with no disorder and span a variety of oxidation states, spin states, and coordination numbers. We purposely chose a small representative training set because we wanted to be able to test the quality of this correlation with independent compounds and structures that are not part of the training set. A list of the training set complexes along with coordination number, formal oxidation state, spin state, and experimental isomer shifts are given in Table 1, and the labeling scheme is given in Figure 2. The isomer shift is temperature dependent due to the second-order Doppler shift, which is often minimal (<0.02 mm s–1) at liquid N2 temperatures and below, however the exact contribution depends on the effective mass of the absorber and bonding environment.3,14 Due to the lack of a generality of this dependence, we chose to include data irrespective of the temperature of measurement. Only literature spectra referenced to α-Fe at 298–300 K were chosen.
Table 1. Training Set for Establishing a Correlation between Computed Electron Densities (ρ) and Experimental Isomer Shift.
| compound | C.N.a | formal ox. stateb | Sc | exp. δ (mm s−1)d | refe |
|---|---|---|---|---|---|
| K2[LMeFeNNFeLMe] | 3 | 0 | 2 | 0.47 | (15) |
| LMeFeBr(THF) | 4 | +2 | 2 | 0.89 | (16) |
| MeLMe,Me2Fe | 4 | +2 | 2 | 0.84 | (22) |
| MeLMe,MeFeCl2 | 4 | +3 | 5/2 | 0.33 | (17) |
| MeLMe,MeFe(Cl)2K(18-crown-6) | 4 | +2 | 2 | 0.88 | (17) |
| MeLMe,MeFe(μ-S)2FeMeLMe,Me | 4 | +3 | 0 | 0.28 | (18) |
| LMeFe(η6-C6H6) | 5 | +1 | 1/2 | 0.70 | (16) |
| MeLMe,MeFe(CNXyl)3f | 5 | +1 | 1/2 | 0.17 | (19) |
| MeLMe,MeFe(CO)3 | 5 | +1 | 1/2 | 0.12 | (19) |
Coordination number of iron.
Formal oxidation state of the iron center(s) based on charge counting.
Experimental spin of the system.
Experimental isomer shifts were collected at 80 K with a small applied magnetic field of 0.07 T referenced to α-Fe or iron foil at 298–300 K.
Reference containing crystal structure and Mössbauer parameters.
CNXyl = 2,6-dimethylphenyl isocyanide.
Figure 2.

Structure and labeling scheme of β-diketiminate ligands LMe, LtBu, and MeLMe,Me.
We chose to fit the training set results with a single line for each basis set choice (eq 2), a strategy that was employed by Neese and by Pápai to generalize single correlations without need to select fit parameters based on oxidation state of the metal center.9b,9d Some other studies break down the test sets into smaller groups using parameters such as oxidation state and ligand sphere, and then establish a linear correlation for each subset,9c but we disfavor this strategy because it cannot be applied to complexes with redox-active ligands or with unusual environments in which there is no independent, unambiguous determination of the oxidation state. Also note that many of the complexes examined in this report contain π-accepting ligands or redox-active ligands that can drastically affect the electron density at the iron center. In these cases, Mössbauer spectroscopy has been useful in assigning the “true” oxidation state of the metal center over the formal oxidation state established by charge counting, which can differ substantially.9c
| 2 |
In eq 2, δ is the isomer shift, ρ(0) is the calculated electron density at Fe, α and β are calibration constants with C as an unrefined constant that is subtracted from ρ(0) to avoid a fit where the β parameter is unreasonably large. Parameters for the fits developed from the training set of compounds, the square of the correlation coefficient (R2), the mean absolute error (MAE), and the maximum deviation from experiment (max. dev.), are given in Table 2 and shown in Figure 3. The fit values of α are within error of the experimental value of 0.31 ± 0.04 a03 mm s–1, which has been estimated from the change in electron capture rate of 52Fe in different iron compounds.20
Table 2. Fit Parameters and Statistics for the Linear Fit of the Training Set Compounds for the Prediction of Isomer Shifts, Using the B3LYP Functional.
Figure 3.

Plots of calculated electron density at Fe against the experimentally observed isomer shifts for the training set compounds for def2-TZVP (top) and CP(PPP) (bottom). Data are shown in red and the linear fits are shown in black with given fit parameters (α and β) as well as the square of the correlation coefficient (R2).
With the correlation established and fits for eq 2 showing excellent agreement with the experimental isomer shifts, we tested the predictive power of the new correlations toward other compounds reported by our group as well as the generality toward other ligand classes, the ability of the correlation to evaluate different spin states, and the efficacy in predicting Mössbauer parameters for spectra collected at temperatures other than 80 K. These “test” compounds and their corresponding experimental isomer shifts are shown in Table 3 along with the isomer shifts calculated using the fit parameters established above for both def2-TZVP and CP(PPP).
Table 3. Identity and Parameters of Test Compounds for Prediction of Isomer Shift Values.
| compound | C.N.a | formal ox. state | Sb | exp. δc | calc. δ def2-TZVPd | calc. δ CP(PPP)e | T (K)f | ref |
|---|---|---|---|---|---|---|---|---|
| LMeFe(NCtBu)2g | 4 | +1 | 3/2 | 0.72 | 0.72 | 0.73 | 80 | (8c) |
| LMeFe(tBu-Py)2h | 4 | +1 | 3/2 | 0.79 | 0.82 | 0.83 | 80 | (8d) |
| LtBuFeCH3 | 3 | +2 | 2 | 0.48 | 0.47 | 0.48 | 4.2 | (21) |
| LtBuFeCl | 3 | +2 | 2 | 0.74 | 0.76 | 0.77 | 4.2 | (21) |
| LtBuFeNHtolyli | 3 | +2 | 2 | 0.71 | 0.65 | 0.66 | 4.2 | (21) |
| LtBuFe(HCCPh) | 3 | +1 | 3/2 | 0.44 | 0.48 | 0.49 | 150 | (21) |
| [LtBuFeH][K(18-crown-6)] | 3 | +1 | 3/2 | 0.47 | 0.46 | 0.47 | 80 | (8b) |
| [LtBuFeH][K(crypt)]j | 3 | +1 | 3/2 | 0.40 | 0.40 | 0.41 | 80 | (8b) |
| MeLMe,MeFe(η6-C6H6) | 5 | +1 | 1/2 | 0.68 | 0.77 | 0.77 | 80 | (19) |
| MeLMe,MeFe(η5-Ind)k | 5 | +2 | 1 | 0.67 | 0.70 | 0.71 | 80 | (22) |
| MeLMe,MeFe(Py)(μ-N2C10H10)(Py)FeLl | 4 | +2 | 4 | 0.76 | 0.70, 0.71 | 0.71, 0.72 | 80 | (8e) |
| MeLMe,MeFe(Py)(μ-Py)FeMeLMe,Mel | 4, 4 | +2, +2 | 3 | 0.67, 0.76 | 0.71, 0.73 | 0.71, 0.74 | 80 | (8e) |
| LMeFe(AdNNNNAd)m | 4 | +2 | 3/2 | 0.69 | 0.62 | 0.63 | 80 | (8a) |
| LS,SFe(THF)2n | 4 | +2 | 2 | 0.89 | 0.79 | 0.80 | 80 | (23) |
| (SiPiPr3)FeCOSi(CH3)3o | 5 | 0 | 0.061 | 0.10 | 0.11 | 80 | (24) | |
| Cr(iPrNPPh2)3Fe-PMe3p | 5 | 3/2 | 0.25 | 0.27 | 0.28 | 110 | (25) | |
| [Cp*Fe(S2Ph)(N2H2)FeCp*]PF6q | 6 | +2.5 | 1/2 | 0.29 | 0.47 | 0.47 | 80 | (26) |
| Cp2Fer | 6 | +2 | 0 | 0.53 | 0.68 | 0.69 | 80 | (27) |
| PNPiPrFeCl2s | 5 | +2 | 2 | 0.86 | 0.85 | 0.86 | 80 | (28) |
| PNPtBuFeCl2t | 5 | +2 | 2 | 0.99 | 0.89 | 0.91 | 80 | (28) |
| [(IMes)2FeCl]u | 3 | +2 | 2 | 0.65 | 0.52 | 0.54 | 80 | (29) |
| [(IMes)(Me2-cAAC)-FeCl]u,v | 3 | +2 | 2 | 0.52 | 0.45 | 0.47 | 80 | (29) |
| [(Me2-cAAC)2FeCl]v | 3 | +2 | 2 | 0.49 | 0.34 | 0.35 | 80 | (29) |
| [(cylDep)2Fe][BArF4]w | 2 | +1 | 3/2 | 0.48 | 0.52 | 0.52 | 80 | (30) |
| [(sIDep)2Fe][BArF4]x | 2 | +1 | 3/2 | 0.55 | 0.47 | 0.47 | 80 | (30) |
C.N. is the coordination number of the Fe center.
Total spin of the system.
Experimental isomer shift referenced to α-Fe or iron foil at 298–300 K.
Collection temperature for experimental parameters.
Three unique starting geometries were used and the results averaged. “NCtBu” is tert-butylnitrile.
“tBu-Py” is 4-tert-butylpyridine.
“NHtolyl” is 4-methylanilide.
“crypt” is 2.2.2-cryptand.
“Ind” is indenyl.
“Py” is pyridine. “N2C10H10” is 4H,4′H-[4,4′-bipyridine]-1,1′-diide.
“AdNNNNAd” is N,N‴-bis(adamantyl)tetrazene.
“LS,S” is 4′,6‴-difluoro-2,2′′′′,4,4′′′′,6,6′′′′-hexaisopropyl-[1,1′,3′,1″,3″,1‴,3‴,1′′′′-quinquephenyl]-2,2′-dithiolate.
1,1′,1″-Tris(2-(diisopropylphosphino)phenyl)silane.
N-Isopropyl-1,1-diphenylphosphanamide.
Cp* is 1,2,3,4,5-pentamethylcyclopentadienyl anion and S2Ph is 1,2-benzenedithiolate.
Cp is the cyclopentadienyl anion.
Bis(2-(diisopropylphosphaneyl)ethyl)amine.
Bis(2-(di-tert-butylphosphaneyl)ethyl)amine.
IMes is 1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene.
Me2-cAAC is 3,3,5,5-tetramethyl-1-(2′,6′-diisopropylphenyl)pyrrolidine-2-ylidene.
cylDep is 1,3-bis-2′6′-diethylphenyl)-4,5-(CH2)4-imidazolin-2-ylidene.
sIDep is 1,3-bis-(2′,6′-diethylphenyl)-imidazolin-2-ylidene.
Uncertainty statistics for the isomer shift predictions are given in Table 4, which gives both the MAE and the standard deviation. We also give the largest deviation seen in the test set, which gives the “worst-case” scenario. These data show no clear empirical preference for CP(PPP) over def2-TZVP as the errors for both basis sets are comparable for all classes of compounds examined. Neese and Pápai similarly have seen little improvement in statistics when using CP(PPP), but favor the larger basis set because it is specially designed for core properties.9b,9d Despite the theoretical justification for the CP(PPP) basis set, def2-TZVP performed comparably to CP(PPP) over the set of compounds examined with a 0.001 mm s–1 difference in MAE, which is well within the accepted experimental error (±0.02 mm s–1) for Mössbauer spectroscopy. The computations using CP(PPP) as the basis set on iron took on average 1.46 times longer than when def2-TZVP was used on all atoms. Considering the savings of computational resources along with the comparable accuracy of the two basis sets, we find that def2-TZVP is the most efficient choice for computational predictions of the Mössbauer isomer shift.
Table 4. Error Comparison of def2-TZVP versus CP(PPP) for Prediction of 57Fe Mössbauer Isomer Shift Parameter.
| Def2-TZVP |
CP(PPP) |
|||||
|---|---|---|---|---|---|---|
| MAEa (mm s–1) | std. dev.b (mm s–1) | largest deviationc (mm s–1) | MAEa (mm s–1) | std. dev.b (mm s–1) | largest deviationc (mm s–1) | |
| training set | 0.038 | 0.050 | 0.11 | 0.040 | 0.053 | 0.13 |
| all β-diketiminated | 0.035 | 0.045 | 0.11 | 0.035 | 0.045 | 0.13 |
| othere | 0.097 | 0.12 | 0.18 | 0.091 | 0.11 | 0.18 |
| all compoundsf | 0.054 | 0.075 | 0.18 | 0.053 | 0.073 | 0.18 |
MAE between predicted and experimental values.
Standard deviation (Std. Dev.) of errors between predicted and experimental values.
Largest deviation between predicted and experimental values.
Statistics for all β-diketiminate supported complexes.
Statistics for non-β-diketiminate complexes.
Statistics for all compounds.
We were gratified to discover that this new correlation performed much better for compounds that had been problematic when using the correlation from Neese.9b For example, in a recent study of diketiminate compounds with bridging pyridine ligands,8e two compounds were addressed computationally. In one, the experimental isomer shift of 0.76 mm s–1 was far from the previously calculated value of 0.59 mm s–1, but the method described here predicted a closer value of 0.70 mm s–1 (Table 3, row 11). In a diiron compound with inequivalent iron centers, the experimental isomer shifts of 0.76 and 0.67 mm s–1 were predicted to be 0.62 and 0.52 mm s–1 using the literature correlation, whereas the method described here predicted much closer values of 0.73 and 0.71 mm s–1 (Table 3, row 12).
Prediction of Quadrupole Splitting
Previous studies observed larger errors in predicted quadrupole splitting values as the magnitude of the quadrupole splitting increases.9c,9d Our calculations show similar scattering at larger quadrupole splitting values (Table 5), and therefore we have included the percent error statistics with the caveat that this measure downplays large errors in large values in Table 6. Overall, both basis sets performed equally well at predicting the quadrupole splitting for the compounds examined. Given the much greater computational resources needed for CP(PPP) versus def2-TZVP, we again favor def2-TZVP for computational predictions.
Table 5. Experimental and Calculated Quadrupole Splitting Parameters.
| compound | exp. |ΔEQ| (mm s–1)a | def2-TZVP |ΔEQ| (mm s–1)b | CP(PPP) |ΔEQ| (mm s–1)c | ref |
|---|---|---|---|---|
| K2[LMeFeNNFeLMe] | 2.48 | 2.37, 2.33 | 2.22, 2.18 | (15) |
| LMeFeBr(THF) | 2.36 | 2.19 | 2.34 | (16) |
| LMeFe(η6-C6H6) | 0.74 | 0.93 | 1.14 | (19) |
| MeLMe,Me2Fe | 1.80 | 1.48 | 1.69 | (19) |
| MeLMe,MeFe(Cl)2 | 1.23 | 1.00 | 1.05 | (18) |
| MeLMe,MeFe(Cl)2K(18-crown-6) | 2.10 | 2.07 | 2.27 | (17) |
| MeLMe,MeFe(μ-S)2FeMeLMe,Me | 1.14 | 1.18, 1.18 | 1.15, 1.15 | (18) |
| MeLMe,MeFe(CNXyl)3d | 0.81 | 0.76 | 0.80 | (19) |
| MeLMe,MeFe(CO)3 | 0.77 | 0.79 | 0.89 | (19) |
| LMe Fe(NCtBu)2e | 0.72 | 1.06 | 1.20 | (8c) |
| LMeFe(tBu-Py)2f | 0.59 | 0.59 | 0.56 | (8b) |
| LtBuFeCH3 | 1.74 | 2.14 | 2.10 | (21) |
| LtBuFeCl | 1.61 | 2.05 | 2.05 | (21) |
| LtBuFeNHtolylg | 1.42 | 1.72 | 1.66 | (21) |
| LtBuFe(HCCPh) | 2.05 | 2.53 | 2.44 | (21) |
| [LtBuFeH][K(18-crown-6)] | 1.84 | 2.21 | 2.25 | (8b) |
| [LtBuFeH][K(crypt)]h | 1.93 | 2.25 | 2.36 | (8b) |
| MeLMe,MeFe(η6-C6H6) | 0.69 | 0.87 | 1.08 | (19) |
| MeLMe,MeFe(η5-Ind)i | 1.07 | 1.02 | 0.89 | (22) |
| MeLMe,MeFe(Py)(μ-N2C10H10)(Py)FeMeLMe,Mej | 1.38 | 1.12, 1.10 | 1.26, 1.26 | (8e) |
| MeLMe,MeFe(Py)(μ-Py)FeMeLMe,Mej | 1.29, 1.66 | 1.14, 1.22 | 1.04, 1.37 | (8e) |
| LMeFe(AdNNNNAd)k | 1.32 | 1.53 | 1.77 | (8a) |
| LS,SFe(THF)2l | 3.77 | 3.81 | 3.99 | (23) |
| (SiPiPr3)FeCOSi(CH3)3m | 1.115 | 1.12 | 1.41 | (24) |
| Cr(iPrNPPh2)3Fe-PMe3n | 0.31 | 1.07 | 1.11 | (25) |
| [Cp*Fe(S2Ph)(N2H2)FeCp*]PF6o | 0.74 | 0.80, 0.80 | 0.82, 0.82 | (26) |
| Cp2Fep | 2.41 | 2.61 | 2.97 | (27) |
| PNPiPrFeCl2q | 2.89 | 3.25 | 3.36 | (28) |
| PNPtBuFeCl2r | 2.69 | 3.09 | 3.22 | (28) |
| [(IMes)2FeCl]s | 2.63 | 2.23 | 2.30 | (29) |
| [(IMes)(Me2-cAAC)-FeCl]s,t | 2.03 | 2.94 | 2.99 | (29) |
| [(Me2-cAAC)2-FeCl]t | 2.04 | 1.84 | 1.69 | (29) |
| [(cylDep)2Fe][BArF4]u | 5.75 | 5.43 | 5.40 | (30) |
| [(sIDep)2Fe][BArF4]v | 6.82 | 6.00 | 5.99 | (30) |
Experimentally observed quadrupole splitting.
Quadrupole splitting calculated using the def2-TZVP basis set on all atoms.
Quadrupole splitting calculated using the CP(PPP) basis set on iron.
“CNXyl” is 2,5-dimethylphenylisocyanide.
“NCtBu” is tert-butylnitrile.
“tBu-Py” is 4-tert-butylpyridine.
“NHtolyl” is 4-methylanilide.
“crypt” is 2.2.2-cryptand.
“Ind” is indenyl.
“Py” is pyridine. “N2C10H10” is 4H,4′H-[4,4′-bipyridine]-1,1′-diide.
“AdNNNNAd” is N,N‴-bis(adamantly)tetrazene.
“LS,S” is 4′,6‴-difluoro-2,2′′′′,4,4′′′′,6,6′′′′-hexaisopropyl-[1,1′,3′,1″,3″,1‴,3‴,1′′′′-quinquephenyl]-2,2′-dithiolate.
1,1′,1″-Tris(2-(diisopropylphosphino)phenyl)silane.
N-Isopropyl-1,1-diphenylphosphanamide.
Cp* is 1,2,3,4,5-pentamethylcyclopentadienyl anion. S2Ph is 1,2-benzenedithiolate.
Cp is the cyclopentadienyl anion.
Bis(2-(diisopropylphosphaneyl)ethyl)amine.
Bis(2-(di-tert-butylphosphaneyl)ethyl)amine.
IMes is 1,3-bis(2,4,6-trimethylphenyl)imidazole-2-ylidene.
Me2-cAAC is 3,3,5,5-tetramethyl-1-(2′,6′-diisopropylphenyl)pyrrolidine-2-ylidene.
cylDep is 1,3-bis-2′6′-diethylphenyl)-4,5-(CH2)4-imidazolin-2-ylidene.
sIDep is 1,3-bis-(2′,6′-diethylphenyl)-imidazolin-2-ylidene.
Table 6. Error Comparison of def2-TZVP versus CP(PPP) for Prediction of 57Fe Mössbauer |ΔEQ| Parameters.
| Def2-TZVP |
CP(PPP) |
|||||||
|---|---|---|---|---|---|---|---|---|
| MAEa (mm s–1) | std. dev.b (mm s–1) | largest deviationc (mm s–1) | % errord | MAEa (mm s–1) | std. dev.b (mm s–1) | largest deviationc (mm s–1) | % errord | |
| training set | 0.12 | 0.14 | 0.32 | 4.9 | 0.15 | 0.20 | 0.40 | 5.9 |
| β-diketiminate complexes | 0.27 | 0.36 | 0.84 | 11.0 | 0.28 | 0.35 | 0.70 | 11.1 |
| non-β-diketiminate complexes | 0.35 | 0.47 | 0.91 | 14.0 | 0.45 | 0.52 | 0.96 | 18.1 |
| all compounds | 0.30 | 0.40 | 0.91 | 12.0 | 0.33 | 0.41 | 0.96 | 13.4 |
MAE between predicted and experimental values.
Standard deviation of errors between predicted and experimental values.
Largest deviation between predicted and experimental values.
Average percent error between predicted and experimental values.
Testing the Use of Crystallographic Geometries versus Optimized Geometries
Previously, Friesner and co-workers examined the calculation of Mössbauer parameters, comparing between using the geometry from X-ray crystallography versus using a DFT optimized structure.9c They concluded that as long as the X-ray structure and DFT geometry were in good agreement, the predicted Mössbauer parameters derived from each should not be significantly different. On the other hand, Pápái and co-workers state that the error in X-ray structures due to thermal motion can affect the calculation of Mössbauer parameters and support the use of DFT optimized geometries.9d Nemykin and Noodleman have employed both methods, with emphasis on the need for geometry optimization to closely match the X-ray structures.6c,6f,6g,7e,9a,9f Common issues not addressed in these studies are the impact of crystal packing effects on the molecular geometry at iron and disorder in the X-ray structures. Our experience is that it is fairly common to have two polymorphs of the same complex or two different crystal structures of the same complex with different solvents of crystallization. Additionally, it is frequently observed that there are two crystallographically independent molecules in an X-ray crystal structure, and sometimes the bond distances/angles differ between the inequivalent molecules. Disorder of ligands can also lead to multiple observed molecular configurations for the same compound. Using X-ray coordinates as a report of molecular geometry also introduces another problem because X-ray diffraction measures electron density rather than nuclear positions, and thus there is systematic shortening of all distances to H atoms (by ∼0.1 Å) in the refined structures.31 Below, we show that these crystallographic issues can lead to large inaccuracies in computationally predicted parameters if a full geometry optimization is not performed.
To evaluate this issue, we computationally examined LMeFe(NCtBu)2, a compound reported previously by Cowley et al.8c It is an ideal test case for the influence of crystal packing because the asymmetric unit in the X-ray crystal structure contained three chemically identical but crystallographically independent molecules of LMeFe(NCtBu)2. In addition to performing point calculations on the crystallographic geometries with only the H atom positions optimized, we also performed a full geometry optimization on each independent molecule. For each of the six resulting geometries, the electron density at the nucleus and quadrupole splitting were calculated (Table 7). (It is most appropriate to compare the electron densities rather than converting them to isomer shifts as the correlation fit parameters reported above are from a fully optimized training set. The predicted isomer shifts are also given to demonstrate the variation.)
Table 7. Comparison of Calculated Parameters for Multiple Geometries from a Single Crystal Using Fully Optimized DFT Geometries and H-Atom Only Optimized Geometries.
| structure | exp. IS (mm s–1) | calc. ρ(0)-13780 def2-TZVPa | calc. ρ(0)-14760 CP(PPP)a | exp. |ΔEQ| (mm s–1) | calc. |ΔEQ| def2-TZVP | calc. |ΔEQ| CP(PPP) |
|---|---|---|---|---|---|---|
| LMeFe(NCtBu)2 – 1 H-only | 0.72 | 1.054 (0.843) | 2.129 (0.851) | 1.87 | 1.09 | 1.26 |
| LMeFe(NCtBu)2 – 2 H-only | 1.225 (0.788) | 2.310 (0.797) | 0.85 | 1.04 | ||
| LMeFe(NCtBu)2 – 3 H-only | 1.076 (0.836) | 2.153 (0.844) | 0.90 | 1.08 | ||
| LMeFe(NCtBu)2 – 1 | 1.438 (0.720) | 2.536 (0.729) | 1.08 | 1.22 | ||
| LMeFe(NCtBu)2 – 2 | 1.429 (0.723) | 2.528 (0.732) | 1.06 | 1.20 | ||
| LMeFe(NCtBu)2 – 3 | 1.436 (0.721) | 2.533 (0.730) | 1.04 | 1.18 |
The numbers in parentheses are the calculated isomer shifts according to our computational method.
For the fully optimized structures the calculated ρ(0) using both def2-TZVP and CP(PPP) vary by less than 0.01 a0−3 among the three structures, as would be expected since these structures should optimize to the same energy minimum regardless of the starting geometry. This would lead to a difference of less than 0.003 mm s–1 in the calculated isomer shift, well within the uncertainty of the correlation (±0.05 mm s−1) as well as the uncertainty of the measured isomer shift (±0.02 mm s−1). For the structures with only H-atoms optimized, on the other hand, there is significant variation in the computed electron densities of nearly 0.2 a0–3. This would correspond to changes in the calculated isomer shift of 0.05 mm s–1 using the correlations reported here, which is on the order of the error in the isomer shift prediction. Thus, optimizing only H atoms can introduce a significant source of error in cases wherein multiple molecules are present in the asymmetric unit or disorder gives multiple conformations.
Similarly, the calculated quadrupole splitting varied less in the fully optimized geometries (0.04 mm s–1) than in the H-atom only optimized geometries (0.24 mm s–1). Again, the magnitude of the error in the predicted quadrupole splitting was similar to the uncertainty in our best correlation (0.27 mm s–1). In both parameters, less variation is observed for fully optimized geometries leading to the conclusion that a full geometry optimization is best, because systematic error is introduced by selection of a random molecule within the asymmetric unit or a particular polymorph.
It is also useful to comment on the accuracy of X-ray crystallographic structures. Despite the ubiquity of X-ray crystallography and the high level of faith that the inorganic chemistry community places in crystallographic results, it is important to remember that crystallographic analysis gives metrical parameters with an accuracy that is poorer than the “estimated standard deviations” that arise from the refinement protocol.32 Even more serious systematic deviations have been noted in some cases.33 Treating Mössbauer spectroscopy as an independent check of geometries is therefore valuable. In the case discussed above, the Mössbauer spectrum of the crystalline solid shows narrow lines and Lorentzian line shapes that indicate a relatively unvarying geometry in the crystal, though the bond distances around Fe vary by 0.02 Å and orientations of the nitriles vary by 90° in the crystallographic structure. Our computations show that these would lead to a variation in Mössbauer parameters of ∼0.05 mm s–1, which would have led to a ∼20% increase in the broadness of the right line of the spectra, as indicated by simulations (Figures S-3 and S-4). The fact that this broadening was not observed implies that the deviations between the geometries of independent molecules in the X-ray crystal structure are not real despite the good R value of 5.4% of the crystal structure. These results emphasize the importance of confirming crystallographic conclusions using an independent measure, whether spectroscopic or computational (or both). Of course, using the DFT-optimized structure is not without danger either, as DFT optimization can give inaccurate geometries, particularly in cases of low-lying excited states, ambiguous spin states, and shallow potential energy surfaces.
Ferrocene presents another interesting test case because the Cambridge Structural Database contains 27 different crystal structures of this molecule, including a polymorph with staggered Cp rings (“staggered 1 & 2”) with two refinements of the same data, a second similar polymorph (“staggered 3”), and a separate structure with eclipsed Cp rings (“eclipsed 1”).34 Because we were unsure of the effect of crystal packing, we independently collected both solid and solution Mössbauer spectra on purified ferrocene samples at 80 K (Figures S-1 and S-2), and both spectra were virtually identical with isomer shifts of 0.53 mm s–1 and quadrupole splittings of 2.41 mm s–1 (2.40 mm s–1 for frozen toluene solution). These match well with the solid-state parameters reported by Foyt et al. at 78 K.27 The minimal difference between the solid-state spectrum and frozen solution spectrum suggests that there is little influence of crystal packing effects on the Mössbauer parameters. Despite the similarity of the experimental parameters for solid-state and solution samples, we observed variation in the H-atom only optimized structures on the order of 0.05 mm s–1 but only 0.001 mm s–1 variation in the fully optimized DFT structures (Table 8). The quadrupole splitting for the H-atom only optimizations varied by nearly 0.30 mm s–1, whereas in the fully optimized structures they varied by only 0.01 mm s–1. Interestingly, calculations on atomic positional parameters from two different reported refinements of the same neutron diffraction data34 (staggered 1 & 2) led to isomer shifts that differed by more than 0.04 mm s–1 with both basis sets. These results are consistent with those of the nitrile complex described above, and further support the conclusion that use of an unoptimized geometry from an X-ray crystal structure introduces a source of error when predicting Mössbauer parameters.
Table 8. Comparison of Calculated Parameters for Multiple Geometries from a Single Crystal Using Fully Optimized DFT Geometries and H-Atom Only Optimized Geometries.
| structure | exp. IS (mm s–1) | calc. ρ(0)-13780 def2-TZVPa | calc. ρ(0)-14760 CP(PPP)a | exp. |ΔEQ| (mm s–1) | calc. |ΔEQ| def2-TZVP | calc. |ΔEQ| CP(PPP) |
|---|---|---|---|---|---|---|
| FeCp2 staggered 1 H-onlyb,c | 0.53 | 1.621 (0.661) | 2.740 (0.668) | 2.41 | 2.64 | 2.98 |
| FeCp2 staggered 2 H-onlyb,c | 1.491 (0.703) | 2.603 (0.709) | 2.86 | 3.22 | ||
| FeCp2 staggered 3 H-onlyd | 1.477 (0.707) | 2.589 (0.713) | 2.91 | 3.27 | ||
| FeCp2 staggered 1b,c | 1.555 (0.682) | 2.674 (0.688) | 2.60 | 2.97 | ||
| FeCp2 staggered 2b,c | 1.555 (0.683) | 2.671 (0.689) | 2.61 | 2.97 | ||
| FeCp2 staggered 3d | 1.554 (0.683) | 2.671 (0.689) | 2.61 | 2.97 | ||
| FeCp2 eclipsed 1 H-onlye | 1.441 (0.719) | 2.548 (0.726) | 2.71 | 3.09 | ||
| FeCp2 eclipsed 1e | 1.598 (0.669) | 2.713 (0.676) | 2.49 | 2.87 |
Values in parentheses are the predicted isomer shifts based on the parameters for the correlations given above. Three decimals places are given to demonstrate the similarity for the DFT fully optimized structures and are not significant.
Structures from ref (34a).
Structures are from two refinement methods of neutron diffraction data.
Structure from ref (34b).
Structure from ref (34c).
We note that for Cp2Fe, both the optimized and nonoptimized structures gave a substantial difference in the experimental versus computed isomer shifts (0.53 vs 0.68 mm s–1) (see below). This is not from a problem with the reported experimental Mössbauer parameters because we independently verified the Mössbauer spectrum (see Supporting Information). We also evaluated whether an eclipsed structure of ferrocene could give rise to the difference. Both an H-atom only optimization and a full DFT optimization of an eclipsed D5h structure led to isomer shift values very similar to those computed for the staggered structure (<0.02 mm s–1 difference) consistent with the results reported by Nemykin for the two conformers.9a Therefore, the structure does not account for the 0.15 mm s–1 difference between theory and experiment. We do not yet have an explanation for this large deviation, as Nemykin was able to reproduce the experimental isomer shift using B3LYP with Wacheter’s full electron basis set.9a
Overall, despite the low accuracy of the computations for the prediction of the isomer shift for ferrocene and the quadrupole splitting for LMeFe(NCtBu)2, in each case, the variation in predicted Mössbauer parameters observed in H-atom optimized structures had lower precision than the fully optimized structures. These two case studies demonstrate the importance of full optimization prior to prediction of parameters.
Limitations of the Computational Protocol
Cp and associated derivatives are prevalent ligands throughout inorganic and organometallic chemistry. Ferrocene, a classic example of such Cp compounds, has been examined by DFT calculations previously and attempts to predict the isomer shift have led to deviations from experiment for other authors as well.9a,9d Using the protocols in this report, we examined ferrocene as well as a recent coordination compound [Cp*Fe(S2Ph)(N2H2)FeCp*]− that contains pentamethylcyclopentadienyl (Cp*). In both cases, the fully optimized geometry matched well with the reported crystal structure and no significant deviations in bond distances were observed. Despite geometric agreement between experimental and computed structures, the predicted isomer shifts for these compounds were overestimated by computations by >0.15 mm s–1, whereas the predicted quadrupole splittings generally compared quite well with experiment (<10% deviation) (Table 9). On the basis of these results, we do not recommend use of these conditions for prediction of Mössbauer parameters for compounds with Cp-based ligands as there is a systematic error in the isomer shift when compared with that of the experiment. Pápai et al. found better agreement for ferrocene with use of Slater-type (STO) TZP all electron basis set regardless of the functional employed.9d,9h
Table 9. Comparison of Experimental and Predicted Parameters for Cp Iron Complexes.
| structure | ferrocene (staggered) | ferrocene (eclipsed) | [Cp*Fe(S2Ph)(N2H2)FeCp*][PF6] |
|---|---|---|---|
| exp. IS (mm s–1) | 0.53 | 0.29 | |
| δ def2-TZVP (mm s–1) | 0.68 | 0.67 | 0.47 |
| δ CP(PPP) (mm s–1) | 0.69 | 0.68 | 0.47 |
| exp. |ΔEQ| (mm s–1) | 2.41 | 0.74 | |
| |ΔEQ| def2-TZVP (mm s–1) | 2.61 | 2.49 | 0.80 |
| |ΔEQ| CP(PPP) (mm s–1) | 2.97 | 2.87 | 0.82 |
We did not observe consistent deviations for other ligand classes, but we note that low-coordinate N-heterocyclic carbene complexes (NHC) and cyclic alkyl amino carbene (cAAC) complexes showed sporadic larger deviations in both isomer shift (>0.1 mm s–1) and quadrupole splitting (>0.8 mm s–1). We also looked for systematic deviations associated with the formal oxidation state of iron and the coordination number of the iron (Tables 10 and 11). On the basis of formal oxidation states, the two basis sets examined have comparable accuracies across all oxidation states with predictions for iron(II) compounds having the greatest errors in predicted isomer shifts when using def2-TZVP. Previous studies have noted iron(II) compounds with S = 1 or 2 can have low-lying excited states or electronically degenerate ground states that can affect the calculated parameters,10 but we made no special efforts to account for these effects in this study. The results based on the coordination number at iron were comparable for both basis sets for all coordination numbers (2–6). Overall, def2-TZVP performed better than CP(PPP) for most coordination numbers for quadrupole splitting predictions, except 4-coordinate where CP(PPP) was slightly (1.6%) better.
Table 10. Error Statistics for Mössbauer Parameter Predictions Based on the Formal Oxidation State of Iron.
| Def2-TZVP |
CP(PPP) |
|||||
|---|---|---|---|---|---|---|
| formal ox. state | MAE δ (mm s–1)a | MAE |ΔEQ| (mm s–1)b | % error in |ΔEQ|c | MAE δ (mm s–1)a | MAE |ΔEQ| (mm s–1)b | % error in |ΔEQ|c |
| 0 (2)d | 0.031 | 0.088 | 3.5 | 0.028 | 0.284 | 11.4 |
| 1 (11)d | 0.032 | 0.321 | 13.0 | 0.036 | 0.365 | 14.7 |
| 2 (16)d | 0.081 | 0.291 | 11.7 | 0.054 | 0.325 | 13.1 |
| 3 (2)d | 0.046 | 0.102 | 4.1 | 0.069 | 0.070 | 2.8 |
MAE between predicted and experimental values in isomer shift.
MAE between predicted and experimental values in quadrupole splitting.
Average percent error between predicted and experimental values for quadrupole splitting.
Number of compounds examined with the specified formal oxidation state.
Table 11. Error Statistics for Mössbauer Parameter Predictions Based on the Coordination Number at Iron.
| Def2-TZVP |
CP(PPP) |
|||||
|---|---|---|---|---|---|---|
| C.N. at Fe | MAE δ (mm s–1)a | MAE |ΔEQ| (mm s–1)b | % error in |ΔEQ|c | MAE δ (mm s–1)a | MAE |ΔEQ| (mm s–1)b | % error in |ΔEQ|c |
| 2-coord. (2)d | 0.061 | 0.568 | 22.9 | 0.059 | 0.595 | 24.0 |
| 3-coord. (9)d | 0.036 | 0.371 | 14.9 | 0.031 | 0.406 | 16.4 |
| 4-coord. (11)d | 0.039 | 0.29 | 11.7 | 0.040 | 0.251 | 10.1 |
| 5-coord. (9)d | 0.047 | 0.223 | 9.0 | 0.045 | 0.329 | 14.3 |
MAE between predicted and experimental values in isomer shift.
MAE between predicted and experimental values in quadrupole splitting.
Average percent error between predicted and experimental values for quadrupole splitting.
Number of compounds examined with the specified C.N.
Distinguishing Spin States Using Mössbauer Correlations
The spin state of a metal center affects the electron distribution within the d-orbital manifold, which in turn influences the isomer shift and quadrupole splitting parameters. Therefore, in cases where the magnetization of a sample is not easily measured or the compound of interest is present in a mixture with other paramagnetic compounds, we hypothesized that DFT calculations can be used to compare computed Mössbauer parameters for various spin states with experimental parameters. Pápái has previously examined the accuracy of quadrupole splitting parameter predictions in spin-crossover compounds wherein two spin states have been experimentally observed.9d Long explored prediction of Mössbauer parameters for known high- and low-spin pyrazolylborate complexes and clustering of computed parameters was observed based on spin state.9g We examined the sensitivity of the predicted Mössbauer parameters toward changes in spin state for three of the complexes in the test set: one high spin, one intermediate spin, and one low spin. Each structure was optimized in the high spin, intermediate spin (when applicable), and low spin configurations, and then the Mössbauer parameters were computed (Table 12).
Table 12. Comparison of Computed 57Fe Mössbauer Parameters for Various Spin States for Three β-Diketiminate Complexes.
| compound | Sa | exp. δb | calc. δ def2-TZVP (mm s–1)c | calc. δ CP(PPP)/def2-TZVP (mm s–1)d | exp. |ΔEQ| (mm s–1)e | calc. |ΔEQ| def2-TZVP (mm s–1)f | calc. |ΔEQ| CP(PPP)/def2-TZVP (mm s–1)g | T (K)h | refj |
|---|---|---|---|---|---|---|---|---|---|
| LtBuFeCH3 | 1/2 | 0.22 | 0.24 | 1.11 | 1.40 | 80 | (21) | ||
| 3/2 | 0.39 | 0.40 | 2.33 | 2.28 | |||||
| 5/2 | 0.48 | 0.47 | 0.48 | 1.74 | 2.14 | 2.10 | |||
| MeLMe,MeFe(η5-Ind)k | 1/2 | 0.58 | 0.58 | 3.70 | 3.91 | 80 | (22) | ||
| 3/2 | 0.68 | 0.70 | 0.70 | 1.07 | 1.02 | 0.89 | |||
| 5/2 | 0.79 | 0.80 | 2.15 | 2.23 | |||||
| MeLMe,MeFe(CO)3 | 1/2 | 0.12 | 0.12 | 0.13 | 0.77 | 0.79 | 0.89 | 80 | (19) |
| 3/2 | 0.49 | 0.50 | 1.11 | 1.09 |
Spin state of the computed geometry.
Experimental isomer shift.
Experimental quadrupole splitting.
Calculated quadrupole splitting using the def2-TZVP basis set.
Calculated quadrupole splitting using the def2-TZVP/CP(PPP) basis set combination.
Collection temperature for the experimental parameters.
Reference with the experimental parameters and crystal structure.
“Ind” represents indenyl (C8H7).
Using our favored protocol with def2-TZVP as the basis set on iron, the isomer shift became more positive as the total spin increased as is expected.3 The average difference between computed isomer shifts between spin states was 0.17 mm s–1 with a minimum difference of 0.08 mm s–1, values that are 5 and 2 times the MAE for the correlation with β-diketiminate complexes described above. The average difference in quadrupole splitting was 1.11 mm s–1 for different spin states with a minimum difference of 0.18 mm s–1. The average difference is 4 times larger than the MAE for predicted quadrupole splittings in β-diketiminate complexes (0.27 mm s–1), whereas the minimum difference is about half of this error. In all three cases, comparison of the optimized geometry and computed Mössbauer parameters agree best with experiment for the spin state that has been experimentally determined. This suggests that computations can indeed aid in determining the spin state of a compound, and we encourage the use of this protocol for compounds whose magnetic susceptibility or spin state cannot be determined by more rigorous means.
Truncating Computational Models
Sterically bulky ligands are often used to control the geometry, solubility, or other properties of iron complexes. However, the size of these ligands greatly increases the computational cost. Therefore, it is common to use truncated computational models to increase the speed of calculations; we have pursued this route with many diketiminate complexes.35 As part of our evaluation, we tested whether truncation can harm the validity of the computed Mössbauer parameters. Specifically, we tested the influence of truncation on the accuracy of the predicted spectral parameters for three complexes with different bulky ligands (Figure 4 and Table 13).
Figure 4.

Complexes examined for effects of ligand truncation on Mössbauer parameter prediction. (a) LtBuFe(HCCPh), (b) (SiPiPr3)FeCOSi(CH3)3, (c) LS,SFe(THF)2.
Table 13. Calculated Mössbauer Parameters for Truncated Models of Select Compounds.
| compound | trunca | exp. δb | calc. δ def2-TZVP (mm s–1)c | calc. δ CP(PPP)/def2-TZVP (mm s–1)d | exp. |ΔEQ| (mm s–1)e | calc. |ΔEQ| def2-TZVP (mm s–1)f | calc. |ΔEQ| CP(PPP)/def2-TZVP (mm s–1)g | T (K)h | refj |
|---|---|---|---|---|---|---|---|---|---|
| LtBuFe(HCCPh) | none | 0.44 | 0.47 | 0.48 | 2.05 | 2.53 | 2.44 | 4.2 | 21 |
| 1 | 0.44 | 0.45 | 2.58 | 2.52 | |||||
| 2 | 0.44 | 0.45 | 2.73 | 2.59 | |||||
| 3 | 0.45 | 0.47 | 2.81 | 2.69 | |||||
| (SiPiPr3)FeCOSi(CH3)3 | none | 0.06 | 0.08 | 0.08 | 1.12 | 1.12 | 1.41 | 80 | 24 |
| 1 | −0.03 | −0.03 | 1.19 | 1.47 | |||||
| 2 | –0.038 | –0.033 | 1.25 | 1.55 | |||||
| 3 | 0.01 | 0.00 | 1.10 | 1.44 | |||||
| LS,SFe(THF)2 | none | 0.89 | 0.78 | 0.79 | 3.77 | 3.81 | 3.99 | 80 | 21 |
| 1 | 0.81 | 0.81 | 3.94 | 4.07 | |||||
| 2 | 0.82 | 0.82 | 3.80 | 3.92 |
Experimental isomer shift.
Experimental quadrupole splitting.
Calculated quadrupole splitting using the def2-TZVP basis set.
Calculated quadrupole splitting using the def2-TZVP/CP(PPP) basis set combination.
Collection temperature for the experimental parameters.
Reference with the experimental parameters and crystal structure.
Comparison between a DFT optimized geometry and the geometry of the solid-state molecular structure is the most commonly used metric for assessing the validity of a truncated DFT model. In these three examples, a variety of degrees of truncation did not lead to significant deviations from the X-ray structure and, with LS,SFe(THF)2, even led to metrics more similar to the crystal structure (Table 14). The predicted isomer shifts in two of the three cases exhibited very little change (≤0.04 mm s–1). The third case, (SiPiPr3)FeCOSi(CH3)3, exhibited the largest deviations upon truncation (∼0.1 mm s–1), which is on the order of errors observed for non-β-diketiminate compounds using the correlations established in this work. The predicted quadrupole splittings did not deviate greatly upon increased truncation for the three examples: it is less than the error for the quadrupole splitting predictions observed for the set of compounds. In each case, the computational time was an order of magnitude less for the greatest degree of truncation compared with that of the parent complex. Thus, the cost in accuracy from truncation appears to be limited relative to the great advantage in computational resources, but all truncation should be done with caution because there are sometimes deviations in the calculated parameters that do not appear to correlate with observed geometry deviations.
Table 14. Average First Coordination Sphere Bond and Angle Deviations from X-ray Structures for Truncated Models.
| compound | trunc.a | avg. bond dev.b (%) | avg. angle dev.c (%) | max bond dev.d (%) | max angle dev.e (%) |
|---|---|---|---|---|---|
| LtBuFe(HCCPh) | none | 1.7 | 2.8 | 2.5 | 3.8 |
| 1 | 2.1 | 3.1 | 2.8 | 4.4 | |
| 2 | 2.3 | 2.5 | 2.8 | 4.7 | |
| 3 | 2.2 | 2.1 | 3.5 | 4.0 | |
| (SiPiPr3)FeCOSi (CH3)3 | none | 0.9 | 1.4 | 1.6 | 4.8 |
| 1 | 2.3 | 4.4 | 4.0 | 21.2 | |
| 2 | 2.4 | 3.9 | 3.9 | 22.8 | |
| 3 | 2.6 | 4.0 | 4.0 | 24.6 | |
| LS,SFe(THF)2 | none | 2.3 | 3.7 | 3.7 | 12.7 |
| 1 | 1.9 | 3.3 | 3.9 | 11.9 | |
| 2 | 1.9 | 2.6 | 4.3 | 7.4 |
Average percent deviation of bonds in the first coordination sphere about iron from the crystal structure.
Average percent deviation of angles in the first coordination sphere about iron from the crystal structure.
Maximum percent deviation of bonds in the first coordination sphere about iron from the crystal structure.
Maximum percent deviation of angles in the first coordination sphere about iron from the crystal structure.
Conclusions
DFT calculations on β-diketiminate compounds with various spin states, geometries, and ligand spheres were used to establish correlations for the prediction of Mössbauer isomer shifts using a set of standard optimization conditions. Our results indicate that def2-TZVP and CP(PPP) are similar in their predictive ability, and the former is preferable due to greater computational expediency. Full geometry optimizations are essential. The predictive power of the correlations was tested with a test set of β-diketiminate complexes as well as a variety of other iron complexes using the same procedure. These correlations have a mean average error (MAE) of 0.035 mm s–1 for β-diketiminate complexes, an improvement from other previously reported conditions and 0.05 mm s–1 for all compounds tested, which is comparable to other correlations. Quadrupole splitting parameters were also calculated for the compounds tested showing good agreement with experiment with MAE of 0.30 mm s–1 for all compounds.
We also tested other factors, such as the use of X-ray geometries and truncation of computational models. We found DFT geometry optimization to be extremely important for obtaining reliable values in the cases we tried. However, it should be remembered that the optimized geometry can be predicted poorly by DFT, and so care should be taken. Comparison of experimental Mössbauer parameters with the computed results from different spin states successfully reproduced experiment in three of three test cases; thus, these computations are powerful in their ability to support spin-state assignment. Similarly, we examined the effects of truncation of large ligands, which showed that even major truncation led to <5% change in geometry metrics around the metal center and minimal changes in Mössbauer parameters predicted. Overall, we anticipate that the protocols established here will assist others for using Mössbauer spectroscopy coupled with computations for reliable characterization of new compounds.
Computational Details
DFT calculations were performed with the ORCA program package, version 3.0.3.13 Optimized geometries were computed using the BP86 functional.36 Atom-pairwise dispersion correction with the Becke–Johnson damping scheme (D3BJ),37 the scalar relativistic zero-order regular approximation (ZORA),38 and the scalar relativistically recontracted version of the Aldrichs triple-ζ basis set, def2-TZVP,39 were used on all atoms. Mössbauer parameters were calculated using the B3LYP40 functional with either all atoms modeled with the def2-TZVP basis set or a combination of the CP(PPP) for the iron atom with all other atoms modeled with def2-TZVP. It was important to test both because CP(PPP)6b,6e,9b has been reported as best for calculation of the electronic properties in independent benchmark studies by Neese and Pápai, but has significantly higher resource costs.9b,9d The conductor-like screening model41 (COSMO) was used to simulate a toluene solution (ε = 2.4). The choice of solvent model was used to best simulate the environment of organometallic complexes and β-diketiminate complexes in the solid state, wherein the aryl groups of the supporting ligands are most generally the closest contacts between molecules. Resolution of identity (RI) was used to approximate two electron integrals, but this was used only for geometry optimizations; the calculations of Mössbauer parameters did not use RI because we have observed substantial variations in calculated Mössbauer parameters when RI is used. Geometry optimization started from the X-ray crystallographic models and was tightly converged (TightOpt). The SCF calculations were tightly converged (TightSCF) with unrestricted spin (UKS). Numerical integrations during all DFT calculations were done on a dense grid (ORCA grid4), and for Mössbauer calculations a very dense grid (ORCA grid 7) was used on the iron atoms. The calculated structures were confirmed to be minima on the potential energy surfaces by the absence of imaginary frequencies after numerical frequency calculations on the optimized structures. Noncoordinating counterions and solvent were not included in the calculations because previous studies have demonstrated that these have a minimal effect on the calculated Mössbauer parameters.9b,9c,9e,42
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00595.
Mössbauer spectra of ferrocene, computational method, optimized structures, and input files (PDF)
This work was supported by the National Institutes of Health (GM065313 to P.L.H.; GM116463 to S.F.M.).
The authors declare no competing financial interest.
Supplementary Material
References
- Mössbauer R. L. Kernresonanzfluoreszenz von Gammastrahlung in 191Ir. Z. Phys. 1958, 151, 124–143. 10.1007/BF01344210. [DOI] [Google Scholar]
- a Martinho M.; Münck E.. 57Fe Mössbauer Spectroscopy in Chemistry and Biology. In Physical Inorganic Chemistry; John Wiley & Sons, Inc., 2010; pp 39–67. [Google Scholar]; b Münck E.; Stubna A.. Mössbauer Spectroscopy: Bioinorganic. In Comprehensive Coordination Chemistry II; McCleverty J. A., Meyer T. J., Eds.; Pergamon: Oxford, 2003; pp 279–286. [Google Scholar]; c Trautwein A. X.; Bill E.. Mössbauer Studies in Bioinorganic Chemistry. In Transition Metal Chemistry, Proceeding Workshop; Müller A., Diemann E., Eds.; Verlag Chem.: Weinheim, 1981; pp 239–263. [Google Scholar]
- a Gütlich P.; Bill E.; Trautwein A. X.. Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications; Springer-Verlag: Berlin, 2011; p 568. [Google Scholar]; b Raghavan P. Table of nuclear moments. At. Data Nucl. Data Tables 1989, 42, 189–291. 10.1016/0092-640X(89)90008-9. [DOI] [Google Scholar]
- a Burda K.; Stanek J. Application of Mössbauer Spectroscopy in Study of Selected Biochemical Processes. Acta Phys. Pol., A 2003, 103, 499–509. 10.12693/APhysPolA.103.499. [DOI] [Google Scholar]; b Ruby S. L.; Shenoy G. K. In Mössbauer Isomer Shifts for the 5s-5p Elements Beyond Tin: (Antimony, Tellurium, Iodine, Xenon); Elsevier, 1978; pp 617–59. [Google Scholar]; c Herber R. H. The Mössbauer Effect and Its Application in Chemistry. Adv. Chem. 1967, 68, 1–20. 10.1021/ba-1967-0068.ch001. [DOI] [Google Scholar]
- a Unno M.; Ikeda-Saito M.. Characterization of Metal Proteins. In Nanohybridization of Organic-Inorganic Materials; Muramatsu A.; Miyashita T., Eds.; Springer: Berlin, 2009; pp 193–217. [Google Scholar]; b Krebs C.; Bollinger J. M. Freeze-quench 57Fe-Mössbauer spectroscopy: trapping reactive intermediates. Photosynth. Res. 2009, 102, 295. 10.1007/s11120-009-9406-6. [DOI] [PubMed] [Google Scholar]; c Huynh B. H. Mössbauer spectroscopy. Methods Mol. Biol. 2011, 766, 221–235. 10.1007/978-1-61779-194-9_15. [DOI] [PubMed] [Google Scholar]
- a Nemykin V. N.; Kobayashi N.; Chernii V. Y.; Belsky V. K. Mössbauer, Crystallographic, and Density Functional Theoretical Investigation of the Electronic Structure of Bis-Ligated Low-Spin Iron(II) Phthalocyanines. Eur. J. Inorg. Chem. 2001, 733–743. . [DOI] [Google Scholar]; b Neese F. Prediction and interpretation of the 57Fe isomer shift in Mössbauer spectra by density functional theory. Inorg. Chim. Acta 2002, 337, 181–192. 10.1016/S0020-1693(02)01031-9. [DOI] [Google Scholar]; c Lovell T.; Han W.-G.; Liu T.; Noodleman L. A Structural Model for the High-Valent Intermediate Q of Methane Monooxygenase from Broken-Symmetry Density Functional and Electrostatics Calculations. J. Am. Chem. Soc. 2002, 124, 5890–5894. 10.1021/ja0121282. [DOI] [PubMed] [Google Scholar]; d Zhang Y.; Mao J.; Oldfield E. 57Fe Mössbauer Isomer Shifts of Heme Protein Model Systems: Electronic Structure Calculations. J. Am. Chem. Soc. 2002, 124, 7829–7839. 10.1021/ja011583v. [DOI] [PubMed] [Google Scholar]; e Sinnecker S.; Slep L. D.; Bill E.; Neese F. Performance of Nonrelativistic and Quasi-Relativistic Hybrid DFT for the Prediction of Electric and Magnetic Hyperfine Parameters in 57Fe Mössbauer Spectra. Inorg. Chem. 2005, 44, 2245–2254. 10.1021/ic048609e. [DOI] [PubMed] [Google Scholar]; f Han W.-G.; Liu T.; Lovell T.; Noodleman L. DFT calculations of 57Fe Mössbauer isomer shifts and quadrupole splittings for iron complexes in polar dielectric media: Applications to methane monooxygenase and ribonucleotide reductase. J. Comput. Chem. 2006, 27, 1292–1306. 10.1002/jcc.20402. [DOI] [PubMed] [Google Scholar]; g Hopmann K. H.; Ghosh A.; Noodleman L. Density Functional Theory Calculations on Mössbauer Parameters of Nonheme Iron Nitrosyls. Inorg. Chem. 2009, 48, 9155–9165. 10.1021/ic9008784. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Kurian R.; Filatov M. Calibration of 57Fe isomer shift from ab initio calculations: can theory and experiment reach an agreement?. Phys. Chem. Chem. Phys. 2010, 12, 2758–2762. 10.1039/b918655g. [DOI] [PubMed] [Google Scholar]; i Gubler J.; Finkelmann A. R.; Reiher M. Theoretical 57Fe Mössbauer Spectroscopy for Structure Elucidation of [Fe] Hydrogenase Active Site Intermediates. Inorg. Chem. 2013, 52, 14205–14215. 10.1021/ic4021349. [DOI] [PubMed] [Google Scholar]
- a Li M.; Bonnet D.; Bill E.; Neese F.; Weyhermüller T.; Blum N.; Sellmann D.; Wieghardt K. Tuning the Electronic Structure of Octahedral Iron Complexes [FeL(X)] (L = 1-Alkyl-4,7-bis(4-tert-butyl-2-mercaptobenzyl)-1,4,7-triazacyclononane, X = Cl, CH3O, CN, NO). The S = 1/2 ⇌ S = 3/2 Spin Equilibrium of [FeLPr(NO)]. Inorg. Chem. 2002, 41, 3444–3456. 10.1021/ic011243a. [DOI] [PubMed] [Google Scholar]; b Hu C.; Sulok C. D.; Paulat F.; Lehnert N.; Twigg A. I.; Hendrich M. P.; Schulz C. E.; Scheidt W. R. Just a Proton: Distinguishing the Two Electronic States of Five-Coordinate High-Spin Iron(II) Porphyrinates with Imidazole/ate Coordination. J. Am. Chem. Soc. 2010, 132, 3737–3750. 10.1021/ja907584x. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Borowski T.; Noack H.; Radoń M.; Zych K.; Siegbahn P. E. M. Mechanism of Selective Halogenation by SyrB2: A Computational Study. J. Am. Chem. Soc. 2010, 132, 12887–12898. 10.1021/ja101877a. [DOI] [PubMed] [Google Scholar]; d Tondreau A. M.; Milsmann C.; Lobkovsky E.; Chirik P. J. Oxidation and Reduction of Bis(imino)pyridine Iron Dicarbonyl Complexes. Inorg. Chem. 2011, 50, 9888–9895. 10.1021/ic200730k. [DOI] [PubMed] [Google Scholar]; e Zueva E. M.; Sameera W. M. C.; Piñero D. M.; Chakraborty I.; Devlin E.; Baran P.; Lebruskova K.; Sanakis Y.; McGrady J. E.; Raptis R. G. Experimental and Theoretical Mössbauer Study of an Extended Family of [Fe8(μ4-O)4(μ-4-R-px)12X4] Clusters. Inorg. Chem. 2011, 50, 1021–1029. 10.1021/ic101691q. [DOI] [PubMed] [Google Scholar]; f Han W.-G.; Sandala G. M.; Giammona D. A.; Bashford D.; Noodleman L. Mossbauer properties of the diferric cluster and the differential iron(II)-binding affinity of the iron sites in protein R2 of class Ia Escherichia coli ribonucleotide reductase: a DFT/electrostatics study. Dalton Trans. 2011, 40, 11164–11175. 10.1039/c1dt10950b. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Surawatanawong P.; Sproules S.; Neese F.; Wieghardt K. Electronic Structures and Spectroscopy of the Electron Transfer Series [Fe(NO)L2]z (z = 1+, 0, 1–, 2–, 3–; L = Dithiolene). Inorg. Chem. 2011, 50, 12064–12074. 10.1021/ic201565d. [DOI] [PubMed] [Google Scholar]; h Jayapal P.; Rajaraman G. On the controversy of metal ion composition on amine oxygenase (AurF): a computational investigation. Phys. Chem. Chem. Phys. 2012, 14, 9050–9053. 10.1039/c2cp40874k. [DOI] [PubMed] [Google Scholar]; i Christian G. J.; Ye S.; Neese F. Oxygen activation in extradiol catecholate dioxygenases - a density functional study. Chem. Sci. 2012, 3, 1600–1611. 10.1039/c2sc00754a. [DOI] [Google Scholar]; j Zall C. M.; Zherebetskyy D.; Dzubak A. L.; Bill E.; Gagliardi L.; Lu C. C. A Combined Spectroscopic and Computational Study of a High-Spin S = 7/2 Diiron Complex with a Short Iron–Iron Bond. Inorg. Chem. 2012, 51, 728–736. 10.1021/ic202384b. [DOI] [PubMed] [Google Scholar]; k Jaccob M.; Rajaraman G. A computational examination on the structure, spin-state energetics and spectroscopic parameters of high-valent FeIV = NTs species. Dalton Trans. 2012, 41, 10430–10439. 10.1039/c2dt31071f. [DOI] [PubMed] [Google Scholar]; l Dong G.; Shaik S.; Lai W. Oxygen activation by homoprotocatechuate 2,3-dioxygenase: a QM/MM study reveals the key intermediates in the activation cycle. Chem. Sci. 2013, 4, 3624–3635. 10.1039/c3sc51147b. [DOI] [Google Scholar]
- a Cowley R. E.; Bill E.; Neese F.; Brennessel W. W.; Holland P. L. Iron(II) Complexes with Redox-Active Tetrazene (RNNNNR) Ligands. Inorg. Chem. 2009, 48, 4828–4836. 10.1021/ic900001y. [DOI] [PubMed] [Google Scholar]; b Chiang K. P.; Scarborough C. C.; Horitani M.; Lees N. S.; Ding K.; Dugan T. R.; Brennessel W. W.; Bill E.; Hoffman B. M.; Holland P. L. Characterization of the Fe-H Bond in a Three-Coordinate Terminal Hydride Complex of Iron(I). Angew. Chem., Int. Ed. 2012, 51, 3658–3662. 10.1002/anie.201109204. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cowley R. E.; Christian G. J.; Brennessel W. W.; Neese F.; Holland P. L. A Reduced (β-Diketiminato)iron Complex with End-On and Side-On Nitriles: Strong Backbonding or Ligand Non-Innocence?. Eur. J. Inorg. Chem 2012, 479–483. 10.1002/ejic.201100787. [DOI] [Google Scholar]; d Dugan T. R.; Bill E.; MacLeod K. C.; Christian G. J.; Cowley R. E.; Brennessel W. W.; Ye S.; Neese F.; Holland P. L. Reversible C–C Bond Formation between Redox-Active Pyridine Ligands in Iron Complexes. J. Am. Chem. Soc. 2012, 134, 20352–20364. 10.1021/ja305679m. [DOI] [PubMed] [Google Scholar]; e Lewis R. A.; MacLeod K. C.; Mercado B. Q.; Holland P. L. Geometric and redox flexibility of pyridine as a redox-active ligand that can reversibly accept one or two electrons. Chem. Commun. 2014, 50, 11114–11117. 10.1039/C4CC05495D. [DOI] [PMC free article] [PubMed] [Google Scholar]; f McLaughlin M. P.; Retegan M.; Bill E.; Payne T. M.; Shafaat H. S.; Peña S.; Sudhamsu J.; Ensign A. A.; Crane B. R.; Neese F.; Holland P. L. Azurin as a Protein Scaffold for a Low-coordinate Nonheme Iron Site with a Small-molecule Binding Pocket. J. Am. Chem. Soc. 2012, 134, 19746–19757. 10.1021/ja308346b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Nemykin V. N.; Hadt R. G. Influence of Hartree–Fock Exchange on the Calculated Mössbauer Isomer Shifts and Quadrupole Splittings in Ferrocene Derivatives Using Density Functional Theory. Inorg. Chem. 2006, 45, 8297–8307. 10.1021/ic061176q. [DOI] [PubMed] [Google Scholar]; b Römelt M.; Ye S.; Neese F. Calibration of Modern Density Functional Theory Methods for the Prediction of 57Fe Mössbauer Isomer Shifts: Meta-GGA and Double-Hybrid Functionals. Inorg. Chem. 2009, 48, 784–785. 10.1021/ic801535v. [DOI] [PubMed] [Google Scholar]; c Bochevarov A. D.; Friesner R. A.; Lippard S. J. Prediction of 57Fe Mössbauer Parameters by Density Functional Theory: A Benchmark Study. J. Chem. Theory Comput. 2010, 6, 3735–3749. 10.1021/ct100398m. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Pápai M.; Vankó G. On Predicting Mössbauer Parameters of Iron-Containing Molecules with Density-Functional Theory. J. Chem. Theory Comput. 2013, 9, 5004–5020. 10.1021/ct4007585. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kurian R.; Filatov M. DFT Approach to the Calculation of Mössbauer Isomer Shifts. J. Chem. Theory Comput. 2008, 4, 278–285. 10.1021/ct700227s. [DOI] [PubMed] [Google Scholar]; f Sandala G. M.; Hopmann K. H.; Ghosh A.; Noodleman L. Calibration of DFT Functionals for the Prediction of 57Fe Mössbauer Spectral Parameters in Iron–Nitrosyl and Iron–Sulfur Complexes: Accurate Geometries Prove Essential. J. Chem. Theory Comput. 2011, 7, 3232–3247. 10.1021/ct200187d. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Remacle F.; Grandjean F.; Long G. J. A Density Functional Theory Calculation of the Electronic Properties of Several High-Spin and Low-Spin Iron(II) Pyrazolylborate Complexes. Inorg. Chem. 2008, 47, 4005–4014. 10.1021/ic701367b. [DOI] [PubMed] [Google Scholar]; h Trautwein A.; Regnard J. R.; Harris F. E.; Maeda Y. Isomer-Shift Calibrations Using Multivalent States of 57Fe in KMgF3. Phys. Rev. B 1973, 7, 947–951. 10.1103/PhysRevB.7.947. [DOI] [Google Scholar]; i Hedegård E. D.; Knecht S.; Ryde U.; Kongsted J.; Saue T. Theoretical 57Fe Mossbauer spectroscopy: isomer shifts of [Fe]-hydrogenase intermediates. Phys. Chem. Chem. Phys. 2014, 16, 4853–4863. 10.1039/c3cp54393e. [DOI] [PubMed] [Google Scholar]
- a Filatov M. On the calculation of Mössbauer isomer shift. J. Chem. Phys. 2007, 127, 084101 10.1063/1.2761879. [DOI] [PubMed] [Google Scholar]; b Filatov M. First principles calculation of Mössbauer isomer shift. Coord. Chem. Rev. 2009, 253, 594–605. 10.1016/j.ccr.2008.05.002. [DOI] [Google Scholar]
- a Casassa S.; Ferrari A. M. Calibration of 57Fe Mossbauer constants by first principles. Phys. Chem. Chem. Phys. 2016, 18, 10201–10206. 10.1039/C5CP07882B. [DOI] [PubMed] [Google Scholar]; b Grandjean F.; Long G. J. Comment on “Calibration of 57Fe Mossbauer constants by first principles”. Phys. Chem. Chem. Phys. 2016, 18, 26306–26309. 10.1039/c6cp02950g. [DOI] [PubMed] [Google Scholar]; c Čajan M.; Trávníček Z. Calculations of 57Fe Mössbauer parameters of mononuclear iron(II) N4 Schiff-base complexes by HF and DFT quantum-chemical approaches. Inorg. Chim. Acta 2012, 387, 412–419. 10.1016/j.ica.2012.02.039. [DOI] [Google Scholar]; d Čajan M.; Trávníček Z. Impact of solvent models and van der Waals corrections on DFT geometric and 57Fe Mössbauer parameters of trans-[FeCl2(iPrOH)4]. Inorg. Chim. Acta 2014, 423, 369–372. 10.1016/j.ica.2014.08.042. [DOI] [Google Scholar]
- Bjornsson R.; Neese F.; DeBeer S. Revisiting the Mössbauer Isomer Shifts of the FeMoco Cluster of Nitrogenase and the Cofactor Charge. Inorg. Chem. 2017, 56, 1470–1477. 10.1021/acs.inorgchem.6b02540. [DOI] [PubMed] [Google Scholar]
- Neese F. The ORCA program system. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2012, 2, 73–78. 10.1002/wcms.81. [DOI] [Google Scholar]
- Gupta G. P.; Lal K. C. Temperature Shift, Recoil-Free Fraction, and Force Constant in Mössbauer Studies. Phys. Status Solidi B 1972, 51, 233–239. 10.1002/pssb.2220510124. [DOI] [Google Scholar]
- McWilliams S. F.; Rodgers K. R.; Lukat-Rodgers G.; Mercado B. Q.; Grubel K.; Holland P. L. Alkali Metal Variation and Twisting of the FeNNFe Core in Bridging Diiron Dinitrogen Complexes. Inorg. Chem. 2016, 55, 2960–2968. 10.1021/acs.inorgchem.5b02841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan T. R.; Bill E.; MacLeod K. C.; Brennessel W. W.; Holland P. L. Synthesis, Spectroscopy, and Hydrogen/Deuterium Exchange in High-Spin Iron(II) Hydride Complexes. Inorg. Chem. 2014, 53, 2370–2380. 10.1021/ic4013137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez M. M.; Bill E.; Brennessel W. W.; Holland P. L. N2 Reduction and Hydrogenation to Ammonia by a Molecular Iron-Potassium Complex. Science 2011, 334, 780–783. 10.1126/science.1211906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reesbeck M. E.; Rodriguez M.; Brennessel W.; Mercado B.; Vinyard D.; Holland P. Oxidized and reduced [2Fe–2S] clusters from an iron(I) synthon. J. Biol. Inorg. Chem. 2015, 20, 875–883. 10.1007/s00775-015-1272-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod K. C.; Vinyard D. J.; Holland P. L. A Multi-iron System Capable of Rapid N2 Formation and N2 Cleavage. J. Am. Chem. Soc. 2014, 136, 10226–10229. 10.1021/ja505193z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladrière J.; Meykens A.; Coussement R.; Cogneau M.; Boge M.; Auric P.; Bouchez R.; Benabed A.; Godard J. Isomer Shift Calibration of 57Fe by Life-Time Variations in The Electron-Capture Decay of 52Fe. J. Phys. Colloq. 1979, 40, C2-20–C2-22. 10.1051/jphyscol:1979205. [DOI] [Google Scholar]
- Andres H.; Bominaar E. L.; Smith J. M.; Eckert N. A.; Holland P. L.; Münck E. Planar Three-Coordinate High-Spin FeII Complexes with Large Orbital Angular Momentum: Mössbauer, Electron Paramagnetic Resonance, and Electronic Structure Studies. J. Am. Chem. Soc. 2002, 124, 3012–3025. 10.1021/ja012327l. [DOI] [PubMed] [Google Scholar]
- MacLeod K. C.; McWilliams S. F.; Mercado B. Q.; Holland P. L. Stepwise N-H bond formation from N2-derived iron nitride, imide and amide intermediates to ammonia. Chem. Sci. 2016, 7, 5736–5746. 10.1039/C6SC00423G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Čorić I.; Mercado B. Q.; Bill E.; Vinyard D. J.; Holland P. L. Binding of dinitrogen to an iron-sulfur-carbon site. Nature 2015, 526, 96–99. 10.1038/nature15246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y.; Peters J. C. Silylation of Iron-Bound Carbon Monoxide Affords a Terminal Fe Carbyne. J. Am. Chem. Soc. 2011, 133, 4438–4446. 10.1021/ja109678y. [DOI] [PubMed] [Google Scholar]
- Kuppuswamy S.; Bezpalko M. W.; Powers T. M.; Wilding M. J. T.; Brozek C. K.; Foxman B. M.; Thomas C. M. A series of C3-symmetric heterobimetallic Cr-M (M = Fe, Co and Cu) complexes. Chem. Sci. 2014, 5, 1617–1626. 10.1039/c3sc52943f. [DOI] [Google Scholar]
- Li Y.; Li Y.; Wang B.; Luo Y.; Yang D.; Tong P.; Zhao J.; Luo L.; Zhou Y.; Chen S.; Cheng F.; Qu J. Ammonia formation by a thiolate-bridged diiron amide complex as a nitrogenase mimic. Nat. Chem. 2013, 5, 320–326. 10.1038/nchem.1594. [DOI] [PubMed] [Google Scholar]
- Good M. L.; Buttone J.; Foyt D. Mössbauer Spectroscopy of Metal Sandwich Compounds. Ann. N. Y. Acad. Sci. 1974, 239, 193–207. 10.1111/j.1749-6632.1974.tb25327.x. [DOI] [PubMed] [Google Scholar]
- Fillman K. L.; Bielinski E. A.; Schmeier T. J.; Nesvet J. C.; Woodruff T. M.; Pan C. J.; Takase M. K.; Hazari N.; Neidig M. L. Flexible Binding of PNP Pincer Ligands to Monomeric Iron Complexes. Inorg. Chem. 2014, 53, 6066–6072. 10.1021/ic5004275. [DOI] [PubMed] [Google Scholar]
- Mo Z.; Ouyang Z.; Wang L.; Fillman K. L.; Neidig M. L.; Deng L. Two- and three-coordinate formal iron(I) compounds featuring monodentate aminocarbene ligands. Org. Chem. Front. 2014, 1, 1040–1044. 10.1039/C4QO00175C. [DOI] [Google Scholar]
- Ouyang Z.; Du J.; Wang L.; Kneebone J. L.; Neidig M. L.; Deng L. Linear and T-Shaped Iron(I) Complexes Supported by N-Heterocyclic Carbene Ligands: Synthesis and Structure Characterization. Inorg. Chem. 2015, 54, 8808–8816. 10.1021/acs.inorgchem.5b01522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sheldrick G. A Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]; b Lusi M.; Barbour L. J. Determining Hydrogen Atom Positions for Hydrogen Bonded Interactions: A Distance-Dependent Neutron-Normalized Method. Cryst. Growth Des. 2011, 11, 5515–5521. 10.1021/cg201087s. [DOI] [Google Scholar]
- a Massa W.Crystal Structure Determination, Springer: New York, 2000; Chapter 11. [Google Scholar]; b Clegg W.; Blake A. J.; Gould R. O.; Main P.. Crystal Structure Analysis: Principles and Practice; Oxford Science Publications: Oxford, 2001; Chapter 14. [Google Scholar]
- a Yoon K.; Parkin G.; Rheingold A. L. Bond-stretch isomerism in the chlorooxomolybdenum complexes cis-mer-MoOCl2(PR3)3: a reinvestigation. J. Am. Chem. Soc. 1992, 114, 2210–2218. 10.1021/ja00032a041. [DOI] [Google Scholar]; b Yoon K.; Parkin G. Resolved and unresolved crystallographic disorder between cyano-[hydrotris(3-tert-butylpyrazolyl)borato]zinc and the halo analogs. Inorg. Chem. 1992, 31, 1656–1662. 10.1021/ic00035a026. [DOI] [Google Scholar]; c Murphy V. J.; Rabinovich D.; Parkin G. False Minima and the Perils of a Polar Axis in X-ray Structure Solutions: Molecular Structures of W(PMe3)4H2X2 (X = F, Cl, Br) and W(PMe3)4H2F2(H2O). J. Am. Chem. Soc. 1995, 117, 9762–9763. 10.1021/ja00143a022. [DOI] [Google Scholar]; d Murphy V. J.; Rabinovich D.; Hascall T.; Klooster W. T.; Koetzle T. F.; Parkin G. False Minima in X-ray Structure Solutions Associated with a “Partial Polar Ambiguity”: Single Crystal X-ray and Neutron Diffraction Studies on the Eight-Coordinate Tungsten Hydride Complexes, W(PMe3)4H2X2 (X = F, Cl, Br, I) and W(PMe3)4H2F(FHF). J. Am. Chem. Soc. 1998, 120, 4372–4387. 10.1021/ja974086g. [DOI] [Google Scholar]
- CCDC IDs: FEROCE07 & FEROCE08.; a Takusagawa F.; Koetzle T. F. A Neutron Diffraction Study of the Crystal Structure of Ferrocene. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1979, 35, 1074–1081. 10.1107/S0567740879005604. [DOI] [Google Scholar]; b Seiler P.; Dunitz J. D. The Disordered Crystal Structure of Ferrocene. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1979, 35, 1068–1074. 10.1107/S0567740879005598. [DOI] [Google Scholar]; c Seiler P.; Dunitz J. D. Low Temperature Crystallization of Orthorhombic Ferrocene: Structure Analysis at 98 K. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1982, 38, 1741–1745. 10.1107/S0567740882007080. [DOI] [Google Scholar]
- a Smith J. M.; Sadique A. R.; Cundari T. R.; Rodgers K. R.; Lukat-Rodgers G.; Lachicotte R. J.; Flaschenriem C. J.; Vela J.; Holland P. L. Studies of Low-Coordinate Iron Dinitrogen Complexes. J. Am. Chem. Soc. 2006, 128, 756–769. 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]; b Chiang K. P.; Bellows S. M.; Brennessel W. W.; Holland P. L. Multimetallic cooperativity in activation of dinitrogen at iron-potassium sites. Chem. Sci. 2014, 5, 267–274. 10.1039/C3SC52487F. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bellows S. M.; Arnet N. A.; Gurubasavaraj P. M.; Brennessel W. W.; Bill E.; Cundari T. R.; Holland P. L. The Mechanism of N–N Double Bond Cleavage by an Iron(II) Hydride Complex. J. Am. Chem. Soc. 2016, 138, 12112–12123. 10.1021/jacs.6b04654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Becke A. D. Density functional calculations of molecular bond energies. J. Chem. Phys. 1986, 84, 4524–4529. 10.1063/1.450025. [DOI] [Google Scholar]; b Perdew J. P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. B 1986, 33, 8822–8824. 10.1103/PhysRevB.33.8822. [DOI] [PubMed] [Google Scholar]
- a Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]; b Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- van Wüllen C. Molecular density functional calculations in the regular relativistic approximation: Method, application to coinage metal diatomics, hydrides, fluorides and chlorides, and comparison with first-order relativistic calculations. J. Chem. Phys. 1998, 109, 392–399. 10.1063/1.476576. [DOI] [Google Scholar]
- a Pantazis D. A.; Chen X.-Y.; Landis C. R.; Neese F. All-Electron Scalar Relativistic Basis Sets for Third-Row Transition Metal Atoms. J. Chem. Theory Comput. 2008, 4, 908–919. 10.1021/ct800047t. [DOI] [PubMed] [Google Scholar]; b Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- a Becke A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]; b Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]; c Vosko S. H.; Wilk L.; Nusair M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: a critical analysis. Can. J. Phys. 1980, 58, 1200–1211. 10.1139/p80-159. [DOI] [Google Scholar]; d Stephens P. J.; Devlin F. J.; Chabalowski C. F.; Frisch M. J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. 10.1021/j100096a001. [DOI] [Google Scholar]
- Klamt A.; Schuurmann G. COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc., Perkin Trans. 2 1993, 799–805. 10.1039/P29930000799. [DOI] [Google Scholar]
- a Zhang Y.; Oldfield E. An Investigation of the Unusual 57Fe Mössbauer Quadrupole Splittings and Isomer Shifts in 2 and 3-Coordinate Fe(II) Complexes. J. Phys. Chem. B 2003, 107, 7180–7188. 10.1021/jp030177q. [DOI] [Google Scholar]; b Zhang Y.; Mao J.; Godbout N.; Oldfield E. Mössbauer Quadrupole Splittings and Electronic Structure in Heme Proteins and Model Systems: A Density Functional Theory Investigation. J. Am. Chem. Soc. 2002, 124, 13921–13930. 10.1021/ja020298o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.