

Abstract
Sunitinib (SU) is a small molecule that inhibits the receptor tyrosine kinase (RTK) signaling pathway, and has been clinically used to treat advanced renal cell carcinoma (RCC). However, SU is not always effective as RCC is a highly chemoresistant type of cancer. One of the factors that confer chemoresistance to RCC is a hypoxic condition. Lack of oxygen activates hypoxia-inducible factor (HIF) protein, which is followed by the upregulation of growth factors, including vascular endothelial growth factor and activation of the RTK signaling pathway. In this context, histone deacetylase inhibitors (HDACIs) are considered prominent combined agents for SU as they downregulate the expression of HIFs. Therefore, the present study aimed to investigate the effectiveness of combined treatment with SU and sodium butyrate (NaBu), an HDACI. Long-term exposure to these agents exerted a stronger growth inhibitory effect in RCC cell lines compared with single treatment groups. Furthermore, combined treatment suppressed HIF-2α protein, which was induced under hypoxic conditions. In addition, this combination sustained the activity of the RTK signaling pathway to the level of intact cells, although a single treatment with SU or NaBu was demonstrated to increase this activity. Overall, it is suggested that the combination of SU and NaBu is effective for overcoming drug resistance in RCC.
Keywords: renal cell carcinoma, sunitinib, sodium butyrate, combination therapy, drug resistance, hypoxia-inducible factor, receptor tyrosine kinase signaling
Introduction
Renal cell carcinoma (RCC) accounts for 2–3% of all cancer types (1). Approximately 1/3 of patients exhibit metastatic disease at presentation and metastatic RCC (mRCC) is highly resistant to chemotherapy (2). Thus, the prognosis of mRCC is poor, with a 5-year survival rate of <10% (3). Previously, immunotherapy using interleukin-2 and interferon (IFN)-α was the standard treatment against mRCC, with a response rate of 10–20% (3). Recently, an improved understanding of cancer biology has enabled the development of molecularly targeted agents. Vascular endothelial growth factor (VEGF) and mammalian target of rapamycin (mTOR) signaling pathways have proven to be involved in the pathogenesis of mRCC, leading to the development of tyrosine kinase inhibitors (TKIs) and mTOR inhibitors (4). Sunitinib (SU) is a multi-targeted TKI that inhibits signaling by VEGF receptors (VEGFRs) 1–3, platelet-derived growth factor receptors (PDGFRs) α and β, and KIT proto-oncogene receptor tyrosine kinase (c-Kit) (5). In a previous phase III study, progression-free survival time was longer and the response rate was higher in patients with mRCC who received SU compared with that in patients who received IFN-α (6), and SU is currently the first-line agent against mRCC. However, SU treatment is not always successful due to frequent resistance and severe side effects (7). Combination therapy is one of the strategies used to solve these problems. The combinations of SU and IFN-α (8) or an mTOR inhibitor (9) have been investigated for the treatment of mRCC; both resulted in failure due to dose-limiting toxicity. Therefore, the establishment of a novel combination strategy for mRCC treatment is warranted.
In order to identify a prominent combination agent for SU, the effects of SU with sodium butyrate (NaBu) was investigated in the present study. NaBu is a short-chain fatty acid, which is present in the human gut at a concentration of 2–10 mM (10). In the 1970s, NaBu was revealed to induce the differentiation of leukemic cells, which was dependent on histone deacetylase (HDAC) inhibition (11). Furthermore, NaBu was identified to exhibit anti-tumor activity (12). On the basis of these observations, clinical trials on NaBu have been performed on a few patients with acute leukemia. However, minimal efficacy was observed due to the rapid metabolism of NaBu and weak HDAC inhibitory activity in vivo (13). Therefore, it is primarily used as a study tool to elucidate the mechanism underlying the effects of HDAC inhibitors (HDACIs) and to identify a potential enhancer of the anticancer effects of existing agents. The effects of HDACIs are generally pleiotropic, as HDACs modulate the acetylation status of histones and non-histone proteins (14). For instance, NaBu inhibits the activity of AKT serine/threonine kinase (Akt) and extracellular signal regulated kinase (ERK), which are downstream proteins of the receptor tyrosine kinase (RTK) signaling pathway (15). Additionally, NaBu has been reported to suppress the transcriptional activity of hypoxia inducible factor (HIF)-α, which is a major transcription factor of VEGF and PDGF (16). Thus, it is expected that NaBu could enhance the anticancer effect of SU, due to its potential for inhibiting the RTK signaling pathway. This study was designed to investigate the efficacy of combination therapy with SU and NaBu in RCC cells.
Materials and methods
Reagents
All reagents were purchased from Sigma-Aldrich (Merck KGaA, Darmstadt, Germany), unless otherwise indicated. SU was dissolved in dimethyl sulfoxide (DMSO; Wako Pure Chemical Industries, Ltd., Osaka, Japan) at a concentration of 10 mM and stored at −20°C. NaBu was dissolved in medium (McCoy's 5A medium or Dulbecco's modified Eagle's medium; DMEM) prior to use. MTT (Dojindo Molecular Technologies, Inc., Kumamoto, Japan) was dissolved in distilled water to achieve a concentration of 5 mg/ml and stored at −20°C.
Cell culture
Three human RCC cell lines, Caki-1 (American Type Culture Collection, Manassas, VA, USA), ACHN and 786-O (provided by Dr Tomohiro Yano, Toyo University, Tokyo, Japan), were used. Caki-1 cells were grown in McCoy's 5A medium (Sigma-Aldrich; Merck KGaA) supplemented with 10% fetal bovine serum (FBS; Equitech-Bio, Kerrville, TX, USA), 100 U/ml penicillin, 100 µg/ml streptomycin and 0.22 g/l L-glutamine (all Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA). ACHN and 786-O cells were grown in DMEM (Wako Pure Chemical Industries, Ltd.) supplemented with FBS and the aforementioned antibiotics. All cells were incubated at 37°C in 5% CO2. For hypoxic conditions, an AnaeroPack™ system (Mitsubishi Gas Chemical Company, Inc., Tokyo, Japan) was used.
Cell viability assay
A total of 2.0×103 Caki-1 cells, and 8.0×102 ACHN and 786-O cells were seeded into a 96-well plate. Following 24 h of incubation, the cells were treated with SU (1.1–3.3 µM), NaBu (1.3–2.0 mM) or both (final concentrations listed in Fig. 1A), followed by culturing for 12 h, 24 h, 2 days, 3 days, 5 days, and 7 days at 37°C. Control cells were treated with 0.1% DMSO. Drug concentrations were determined by the IC50 values for 48 h single treatment of SU or NaBu, detected in preliminary assays (data not presented). On days 1, 3, 5 and 7, 0.25 mg/ml MTT was added to each well and the plate was incubated for 1 h, at 37°C in 5% CO2. The supernatant was removed and DMSO was added to each well. The absorbance at a wavelength of 540 nm and a reference wavelength of 650 nm was read with a Multiskan JX microplate reader (Thermo Labsystems, Santa Rosa, CA, USA). Cell viability (%) was calculated as follows: [Optical density (OD) of the treated wells]/(OD of the control cells)x100.
Figure 1.

Effects of the combined treatment with SU and NaBu on the growth of human renal cancer cell lines. (A) Cells were treated with the indicated concentrations of SU, NaBu or both for 7 days and cell viability was determined using an MTT assay. *P<0.05, **P<0.01, ***P<0.001, vs. the DMSO-treated control group using the Tukey-Kramer's test. +P<0.05, ‡P<0.01 significant differences among the indicated groups following the Tukey-Kramer's test. (B) Effects of the combined treatment with SU and NaBu on the HIF-α target; VEGF expression was detected using reverse transcription-quantitative polymerase chain reaction analysis in Caki-1 cells over the course of 48 h. All data are presented as the mean ± standard deviation following three independent experiments. SU, sunitinib; NaBu, sodium butyrate; cont, control; VEGF, vascular endothelial growth factor; HIF-α, hypoxia-inducible factor-α.
Reverse transcription-quantitative polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from each cell line using RNAzol® RT reagent (Cosmo Bio Co., Ltd., Tokyo, Japan) according to the manufacturer's protocol. cDNA was synthesized in a 20 µl reaction mixture containing 500 ng of total RNA using ReverTra Ace® qPCR RT Master mix (Toyobo Co., Ltd., Osaka, Japan). qPCR was performed using 2 µl cDNA (500 ng of each original RNA) with the StepOne™ Real-Time PCR System (Applied Biosystems; Thermo Fisher Scientific, Inc.) and SYBR Premix Ex Taq™ (Takara Bio Inc., Otsu, Japan), according to the manufacturer's protocol. The thermocycling conditions maintained were as follows: 95°C for 10 sec, followed by 40 cycles at 95°C for 5 sec and 60°C for 31 sec. The expression of VEGF was determined from the threshold cycle values and were normalized to the internal standard gene, GAPDH, using a standard curve based method, according to Larionov et al (17). The primer sequences used in the present study are described in Table I. Each experiment had set duplicate samples, and final data presented is the average of 3 independent experiments.
Table I.
Primers used in reverse transcription-quantitative polymerase chain reaction analysis.
| Target gene | Sequence |
|---|---|
| VEGF (395 bp) | |
| Forward | 5′-AACTTTCTGCTGTCTTGG-3′ |
| Reverse | 5′-TTTGGTCTGCATTCACAT-3′ |
| GAPDH (180 bp) | |
| Forward | 5′-CCAACGTGTCAGTGGTGGAC-3′ |
| Reverse | 5′-CAGCGTCAAAGGTGGAGGAG-3′ |
VEGF, vascular endothelial growth factor.
Western blot analysis
A total of 2×105 Caki-1 and ACHN cells were seeded into 60-mm dishes. Following 24 h of incubation at 37°C, the cells were treated with SU, NaBu or both for each indicated period. Control cells were treated with 0.1% DMSO. Subsequently, cells were collected through scraping and dissolved in ice-cold lysis buffer [50 mM Tris (pH 7.4), 150 mM sodium chloride, 1% Triton X, 10 mM β-glycerophosphate, 1 mM sodium orthovanadate, 1 mM ethylenediaminetetraacetic acid, 1 mM phenylmethane sulfonyl fluoride and 1% protease inhibitor cocktail]. Protein concentration was determined using a DC protein assay kit (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Proteins (20 µg/lane) were electrophoresed using 7.5–15% SDS-PAGE. SDS was purchased from Wako Pure Chemical Industries, Ltd. and 30% acrylamide solution was purchased from Bio-Rad Laboratories, Inc. The separated proteins were transferred to a polyvinylidene difluoride membrane (Atto Corporation, Tokyo, Japan). Then, the membranes were blocked with 5% skim milk (Megmilk Snow Brand Co., Ltd., Tokyo, Japan) in TBS-Tween-20 (13.7 mM sodium chloride, 25 mM Tris and 0.05% Tween-20; Wako Pure Chemical Industries, Ltd.) for 1 h at room temperature. Subsequently, the membranes were incubated with primary antibodies overnight at 4°C, followed by incubation with secondary horseradish peroxidase (HRP)-conjugated antibodies (Sigma-Aldrich; Merck KGaA) for 1 h at room temperature. Antibody information is indicated in Table II. The detection was accomplished using an Immobilon™ Western Chemiluminescent HRP Substrate (Merck KGaA) and a Luminescent Image Analyzer LAS-1000 plus (Fujifilm, Tokyo, Japan). β-actin was used as the internal standard. Data is the average or representative of 3 independent experiments.
Table II.
Primary antibodies used in western blot or immunoprecipitation analyses.
| Target | Source | Cat. no. | Dilution | Species |
|---|---|---|---|---|
| Anti-phosphotyrosine pAb | Upstate Biotechnology, Inc. (Lake Placid, NY, USA) | 06–427 | 500 | Rabbit AH |
| PDGFR-β mAb | Santa Cruz Biotechnology, Inc. (Dallas, TX, USA) | sc-53872 | 2,000 | Mouse AH |
| p44/42 MAPK (ERK1/2) (137F5) mAb: ERK | Cell Signaling Technologies, Inc. (Danvers, MA, USA) | 4695 | 2,000 | Rabbit AH |
| Phopho-p44/42 MAPK (ERK1/2) | Cell Signaling Technologies, Inc. | 9101 | 2,000 | Rabbit AH |
| (Thr202/Tyr204) mAb: p-ERK Akt pAb | Rockland, Inc. (Gilbertsville, PA, USA) | 100-401–401 | 2,000 | Rabbit AH |
| Phospho-Akt (Ser473) mAb: p-Akt | Rockland, Inc. | 200-301-268 | 2,000 | Mouse AH |
| HIF-1α (28b) mAb | Santa Cruz Biotechnology, Inc. | sc-13515 | 2,000 | Mouse AH |
| HIF-2α pAb | Novus Biologicals, LLC (Littleton, CO, USA) | NB100-122 | 2,000 | Rabbit AH |
| VEGF (147) pAb | Santa Cruz Biotechnology, Inc. | sc-507 | 1,000 | Rabbit AH |
| β-actin mAb | Sigma-Aldrich; Merck KGaA (Darmstadt, Germany) | A5441 | 10,000 | Mouse AH |
mAb, monoclonal antibody; pAb, polyclonal antibody; AH, anti-human; VEGF, vascular endothelial growth factor; PDGFR, platelet-derived growth factor; ERK, extracellular signal regulated kinase; phosphor/p, phosphorylated; MAPK, mitogen-activated protein kinase; HIF, hypoxia-inducible factor; AKT, AKT serine/threonine kinase.
Immunoprecipitation analysis
A total of 2×105 786-O cells were seeded into 60-mm dishes. Following treatment with 1.5 µM SU or 1.3 mM NaBu or a combination for 24 h, cells were obtained according to the aforementioned method in the western blot analysis. Immunoprecipitation analysis was perfomed according to the manufacturer's protocol (Bio-Rad Laboratories, Inc.; 161–4823JA, SureBeads™ Protein G Magnetic Beads). Briefly, total cell lysates (400 µg/ml), SureBeads™ (Bio-Rad Laboratories, Inc.) and 5 µl anti-PDGFR-β antibody (Table II) were incubated for 1 h at room temperature. Immunoprecipitates were washed three times with lysis buffer. A total of 40 µl sample buffer [250 mM Tris (pH 6.8), 40% sucrose, 20% 2-mercaptoethanol, 8% SDS (all Wako Pure Chemical Industries, Ltd.) and 0.002% bromophenol blue (ICN Biomedicals, Eschwege, Germany)] were added, and then heated at 70°C for 10 min. For western blotting, each protein sample was diluted 4 times in lysis buffer and analyzed according to the aforementioned method. To detect Tyrosine phosphorylation (P-Tyr), the membrane was incubated with a rabbit anti-human primary antibody directed against phosphotyrosine (Table II) overnight at 4°C. The detection was accomplished using an Immobilon™ Western Chemiluminescent HRP Substrate and a Luminescent Image Analyzer ImageQuant™ LAS 4000 (GE Healthcare Life Sciences, Chalfont, UK).
Statistical analysis
Statistical analyses were performed using the Dunnett's test and the Tukey-Kramer's test (SPSS, version 22, IBM SPSS, Armonk, NY, USA). All experiments were performed >3 times. Data were expressed as mean ± standard error. P<0.05 was considered to indicate a statistically significant difference.
Results
Long-term combined treatment with SU and NaBu effectively inhibits proliferation and VEGF induction in RCC cell lines
To investigate the combined effect of SU and NaBu on RCC cell proliferation, SU and NaBu were used at 50% growth inhibitory concentrations (IC50) following a single exposure to SU or NaBu, then cell viability following combination treatment and that of the single treatment were compared. The final combined concentrations for each cell line are depicted in Fig. 1A. Cell viability gradually increased in a time-dependent manner over the course of 7 days in all three RCC cell lines. Regarding Caki-1 cells, on day 5 cell viability was significantly decreased to ~30 and 50% of the DMSO control by 1.0 mM of NaBu (P<0.001), and 3.3 µM of SU (P<0.001), respectively (Fig. 1A). By day 7, cell viability had slightly increased following treatment with SU or NaBu alone, whereas the combination of these agents had significantly suppressed this increase (P<0.001 vs. control; P<0.001 between NaBu single group and combination group). In ACHN cells, 1.1 µM of SU or 2.0 mM of NaBu alone had almost no effect on proliferation. However, cell viability significantly decreased to ~50% of that of the control cells following combined treatment with SU and NaBu by day 7. The proliferation rate of 786-O cells was slow, as compared with Caki-1 or ACHN cells. Single treatment with 1.5 µM of SU or 1.3 mM of NaBu sufficiently suppressed cell growth during the experimental term, although combined treatment demonstrated the most potent growth inhibition.
SU was originally considered an anti-angiogenic agent, thus one of the targets of SU is VEGF-mediated signaling (5). VEGF is the direct target of HIF-α protein, which servers a critical role in RCC progression. Treatment using SU leads to a gradual development of resistance to SU, with a corresponding unresponsiveness to elevations in VEGF levels. In the present study, VEGF mRNA expression gradually increased in Caki-1 cells over the course of 48 h; however, this was significantly suppressed following 24 h combined treatment with NaBu and SU (Fig. 1B).
Combined treatment with SU and NaBu suppresses RTK signaling activity
The effect of SU and NaBu on RTK signaling was subsequently investigated. The expression levels of certain tyrosine receptors, including the direct targets of SU c-Kit and PDGFR-β, were detected in all the three RCC cell lines used in the present study, particularly PDGFR-β (data not presented). The phosphorylation status of PDGFR-β, as indicated by the immunoprecipitation band with p-Tyr, was activated in untreated and single treated cells. However, combined treatment with SU and NaBu resulted in a decrease in phosphorylated PDGFR-β levels, as compared with those in the untreated control cells (Fig. 2).
Figure 2.
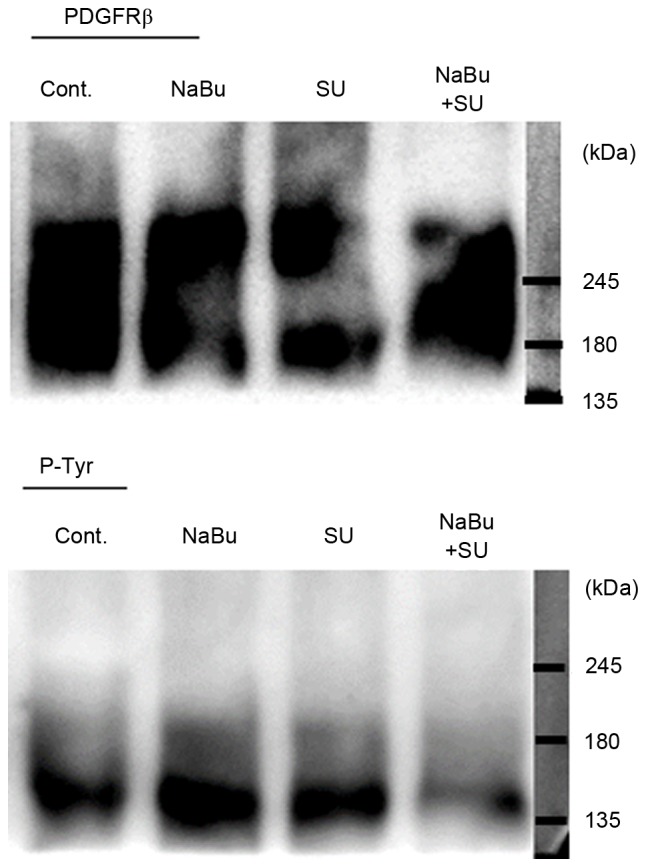
Effects of the combined treatment with SU and NaBu on the phosphorylation of PDGFR-β, as a target of RTK signaling. The 786-O cells were treated with 1.5 µM SU, 1.3 mM NaBu or both for 24 h. The cell lysates were immunoprecipitated using an anti-PDGFR-β antibody, and immunoblot analysis was performed using an anti-p-Tyr antibody. SU, sunitinib; NaBu, sodium butyrate; cont, control; PDGFR-β, platelet-derived growth factor receptor-β; p-Tyr, phosphorylated tyrosine.
The central region of solid tumors is generally exposed to hypoxic conditions, which promote tumor angiogenesis and progression by activating the expression of pro-angiogenic factors (16). An important factor in this process is HIF-α. Specifically, HIF-1α is an important transcription factor involved in acute hypoxia, whereas HIF-2α drives the cell response to chronic hypoxia (18). Therefore, the expression of HIF-α proteins, Akt, ERK and their phosphorylated/active forms in ACHN cells was investigated under normoxic and hypoxic conditions. The expression of HIF-1α increased following 4 h of hypoxic exposure, compared with the levels observed under normoxic conditions, whereas the expression levels of HIF-2α were increased following 6 h and this was maintained until 24 h following exposure (Fig. 3A). The change in the expression levels of HIF-α proteins was larger for HIF-2α compared with HIF-1α under normoxic and hypoxic conditions. The levels of HIF-2α increased 12-fold following 10 h incubation in hypoxic conditions, as compared with the levels in normoxic conditions (Fig. 3B). This increase was suppressed by the combined treatment with SU and NaBu. Furthermore, this combination suppressed the levels of phosphorylated Akt and ERK to almost the same as those present in control cells, although the phosphorylated status of these factors were also increased by a single treatment with SU or NaBu following 10 h of exposure. Notably, SU monotherapy under hypoxic conditions appeared to elevate the phosphorylation of Akt and ERK. VEGF is an essential growth factor that is directly transcribed by HIF-α (16). Under hypoxic conditions, single treatment with SU markedly decreased VEGF expression levels (to ~70% of hypoxic control), but the range of the expression levels of VEGF induced by hypoxia or NaBu treatment appeared to be small (50–70% of normoxic control) compared with that observed for other RTK signaling factors (2–5-fold compared with control; Fig. 3B).
Figure 3.

Effects of the combined treatment with SU and NaBu on the protein expression of HIFs, VEGF and downstream RTK signaling proteins Akt and ERK, and their phosphorylated variants. Intact ACHN cell culture dishes were exposed to normal oxygen tension (normoxia, 21% oxygen tension) or <0.1% oxygen tension (hypoxia) for each indicated period. (A) Representative images of three independent experiments. (B) Each column represents the mean ± standard error following three independent experiments. Cells were exposed to each drug for 10 h under normoxic or hypoxic conditions. SU, sunitinib; NaBu, sodium butyrate; cont, control; VEGF, vascular endothelial growth factor; HIF, hypoxia-inducible factor; AKT, AKT serine/threonine kinase; ERK, extracellular signal regulated kinase; p, phosphorylated.
Discussion
The present study was designed to investigate whether the novel combination therapy with SU and NaBu could overcome drug resistance in RCC cells. This combination is aimed as vertical blockage of the RTK signaling pathway. The results of the present study demonstrated that the combination treatment effectively suppressed the growth of RCC cells following long-term exposure. To confirm the involvement of the RTK signaling pathway in the combination effect, hypoxic conditions were established, which mimicked the intra-tumor environment with confirmation of increased HIF-α protein expression. Such a hypoxic adaptation should occur prior to the initiation of tumor growth. Thus, we investigated in the shortest amount of time for capturing HIF-α increase in the present study.
It was confirmed that receptors of RTK signaling, including c-Kit and PDGFRβ, were present in RCC cells used in the current study. VEGF was originally considered a specific stimulator of vascular endothelial cells, but it has been revealed to affect other cells in addition, including hematopoietic precursor cells, macrophages and hepatic sinusoid cells. At first, the effect of SU was thought to be exerted only via angiogenesis, which can induce tumor shrinkage. In a previous study, the growth of endothelial cells (HUVEC) was suppressed by the VEGF receptor tyrosine kinase inhibitor, PTK787/ZK222584, following VEGF stimulation (19). Although, VEGF has been reported to induce bladder cancer growth (20). Additionally, a VEGF antibody suppressed the growth of human cancer cells in melanoma, pancreatic cancer, cervical cancer and Kaposi sarcoma (21). The VEGFR2 antibody also suppressed leukemic cell growth (22). These reports indicate that VEGF could stimulate the growth of a cancer cell via the VEGFR autocrine loop, which is expressed on the cancer cell itself. PDGFR signaling is also a promising target for RCC (23). Activation of the c-Kit receptor has been known to induce transduction signaling, including induction of the mitogen-activated protein kinase and phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathways in RCC, which in turn leads to mast cell activation (24).
In the present study, the effects of combined treatment with SU and NaBu on RTK signaling factors were examined. Among the RTK downstream factors, ERK and Akt exist at branching points that determine the downstream direction. Cell-dependent MEK/ERK signaling and PI3K/Akt signaling pathways can collaborate with each other to maintain cell survival (25). The expression of phosphorylated ERK, the activated form of ERK, was notably variable each hour (Fig. 3A), which may be dependent on cell cycle progression. This trend was the same under normoxic and hypoxic conditions, although the peak time point was slightly shifted, whereby hypoxic exposure appeared to shift its peak earlier compared with under normoxic conditions. Akt expression was almost constant during the experimental periods in all conditions. Additionally, HIF-α protein was confirmed to be increased following hypoxic exposure compared with normoxic conditions. Hypoxic conditions are universally observed in the tumor environment and are the result of fragile development of the vascular system accompanied with a high growth rate. To survive in severe conditions, a cancer cell induces HIF-α expression, which is normally degraded by proteasomes under normoxic/aerobic conditions (26). Stable expression of HIF-α induces VEGF and PDGF transcription. Notably, the results of the present study revealed that HIF-2α induction was stronger compared with that of HIF-1α. Three HIF-α subtypes have been identified: HIF-1α, HIF-2α, and HIF-3α. Although HIF-1α is the most ubiquitous and well-studied subtype, HIF-2α is considered to serve an essential role during stable hypoxic conditions in RCC (18,27). Furthermore, it has been reported that VEGF expression is maintained through HIF-2α in RCC cells lacking HIF-1α (28).
NaBu has been reported to inhibit the activation of ERK and Akt (15); however single treatment with NaBu appeared to induce these factors compared with non-treatment control following 10 h exposure in the current study. In addition, single treatment with SU induced their activation, particularly under hypoxic conditions. This opposes the results of a previous study that demonstrated that single treatment with SU (0.5–3 µM) did not alter the expression of phosphorylated ERK and Akt in ACHN, and Caki-1 cells (29). Possible reasons for the differences in results may involve the treatment concentrations of drugs and exposure periods used. In the present study, cells were exposed to drugs for 10 h and HIF-2α protein was confirmed to be upregulated, which may be involved in the drug resistance. HIF-2α has been suggested to cause resistance to sorafenib, a RTK inhibitor, in hepatocellular carcinoma cells by activating the transforming growth factor-α/epidermal growth factor receptor signaling pathway (30). In the present study, single treatment with NaBu or SU was not observed to be effective at inhibiting RTK signaling in RCC cells under the conditions that the cells were maintained in. However, the results demonstrated that combined treatment suppressed the activation of ERK and Akt, as compared with each single treatment.
VEGF is an important protein for the induction of angiogenesis in cancerous cells and is essential for their survival (4,5,7). NaBu and SU have been known to decrease VEGF expression and signaling (5,16). However, in the present study, no significant differences were observed in the protein expression of VEGF under hypoxic exposure. A limitation of the current study was the use of a cell culture system to evaluate angiogenic capacity. Thus, further studies using in vivo or a 3D cell culture models are warranted in which the development of vessels and angiogenic factors can be detected.
In the present study, the growth rate of 786-O was dissimilar to that of the other two RCC cell lines, Caki-1 and ACHN. The latter cells demonstrated a mutually similar growth rate. This difference may be attributable to a genetic factor, including the presence of wild type von-Hippel-Lindau (VHL) protein present in Caki-1 and ACHN cells (31), or mutant VHL, which is present in 786-O cells (32). The significantly synergistic effect was only identified in Caki-1 and ACHN cells. VHL is an essential protein for the degradation of HIF-α under normoxic conditions (33). Thus, HIF-α expression remains stable in 786-O cells, even under normoxic conditions. It has been challenging to control the VHL-mutant type of cancer. However, a previous study indicated that inhibiting certain class II HDACs was effective for inducing the degradation of HIF-α in a VHL-independent manner (33). Although the growth-inhibiting effect of NaBu is small when combined with SU in 786-O cells, another HDACI, such as trichostatin A, may have a more potent inhibitory effect.
In conclusion, combination treatment with NaBu and SU has demonstrated a promising efficacy against RCC, a type of cancer known to develop SU resistance. Further studies examining whether the combination is effective in a clinical environment and studies on other HDACIs, due to the limitations of NaBu, may help to clarify whether the combination is effective for the treatment of RCC.
Acknowledgements
The authors would like to thank Dr Akihiro Hisaka (Chiba University, Chiba, Japan) for reviewing the present study. The current study was partially supported by the Grant-in-Aid for Young Scientists (B; grant nos. 24790529 and 15K19161) from the Japan Society for the Promotion of Sciences, and a research grant from the Takeda Science Foundation (2013–2015).
References
- 1.Pal SK, Hossain DM Sakib, Zhang Q, Frankel PH, Jones JO, Carmichael C, Ruel C, Lau C, Kortylewski M. Pazopanib as third-line therapy for metastatic renal cell carcinoma: Clinical efficacy and temporal analysis of cytokine profile. J Urol. 2015;193:1114–1121. doi: 10.1016/j.juro.2014.09.110. [DOI] [PubMed] [Google Scholar]
- 2.Mattei J, da Silva RD, Sehrt D, Molina WR, Kim FJ. Targeted therapy in metastatic renal carcinoma. Cancer Lett. 2014;343:156–160. doi: 10.1016/j.canlet.2013.09.038. [DOI] [PubMed] [Google Scholar]
- 3.Motzer RJ, Russo P. Systemic therapy for renal cell carcinoma. J Urol. 2000;163:408–417. doi: 10.1016/S0022-5347(05)67889-5. [DOI] [PubMed] [Google Scholar]
- 4.Mulders P. Vascular endothelial growth factor and mTOR pathways in renal cell carcinoma: Differences and synergies of two targeted mechanisms. BJU Int. 2009;104:1585–1589. doi: 10.1111/j.1464-410X.2009.08987.x. [DOI] [PubMed] [Google Scholar]
- 5.Chow LQ, Eckhardt SG. Sunitinib: From rational design to clinical efficacy. J Clin Oncol. 2007;25:884–896. doi: 10.1200/JCO.2006.06.3602. [DOI] [PubMed] [Google Scholar]
- 6.Motzer RJ, Hutson TE, Tomczak P, Michaelson MD, Bukowski RM, Rixe O, Oudard S, Negrier S, Szczylik C, Kim ST, et al. Sunitinib versus interferon alfa in metastatic renal-cell carcinoma. N Engl J Med. 2007;356:115–124. doi: 10.1056/NEJMoa065044. [DOI] [PubMed] [Google Scholar]
- 7.Domblides C, Gross-Goupil M, Quivy A, Ravaud A. Emerging antiangiogenics for renal cancer. Expert Opin Emerg Drugs. 2013;18:495–511. doi: 10.1517/14728214.2013.858697. [DOI] [PubMed] [Google Scholar]
- 8.Motzer RJ, Hudes G, Wilding G, Schwartz LH, Hariharan S, Kempin S, Fayyad R, Figlin RA. Phase I trial of sunitinib malate plus interferon-alpha for patients with metastatic renal cell carcinoma. Clin Genitourin Cancer. 2009;7:28–33. doi: 10.3816/CGC.2009.n.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel PH, Senico PL, Curiel RE, Motzer RJ. Phase I study combining treatment with temsirolimus and sunitinib malate in patients with advanced renal cell carcinoma. Clin Genitourin Cancer. 2009;7:24–27. doi: 10.3816/CGC.2009.n.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mork CN, Faller DV, Spanjaard RA. A mechanistic approach to anticancer therapy: Targeting the cell cycle with histone deacetylase inhibitors. Curr Pharm Des. 2005;11:1091–1104. doi: 10.2174/1381612053507567. [DOI] [PubMed] [Google Scholar]
- 11.Witt O, Lindemann R. HDAC inhibitors: Magic bullets, dirty drugs or just another targeted therapy. Cancer Lett. 2009;280:123–124. doi: 10.1016/j.canlet.2009.02.038. [DOI] [PubMed] [Google Scholar]
- 12.Novogrodsky A, Dvir A, Ravid A, Shkolnik T, Stenzel KH, Rubin AL, Zaizov R. Effect of polar organic compounds on leukemic cells. Butyrate-induced partial remission of acute myelogenous leukemia in a child. Cancer. 1983;51:9–14. doi: 10.1002/1097-0142(19830101)51:1<9::AID-CNCR2820510104>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 13.Patnaik A, Rowinsky EK, Villalona MA, Hammond LA, Britten CD, Siu LL, Goetz A, Felton SA, Burton S, Valone FH, Eckhardt SG. A phase I study of pivaloyloxymethyl butyrate, a prodrug of the differentiating agent butyric acid, in patients with advanced solid malignancies. Clin Cancer Res. 2002;8:2142–2148. [PubMed] [Google Scholar]
- 14.Chen S, Sang N. Histone deacetylase inhibitors: The epigenetic therapeutics that repress hypoxia-inducible factors. J Biomed Biotechnol. 2011;2011:197946. doi: 10.1155/2011/197946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jung JW, Cho SD, Ahn NS, Yang SR, Park JS, Jo EH, Hwang JW, Jung JY, Kim SH, Kang KS, Lee YS. Ras/MAP kinase pathways are involved in Ras specific apoptosis induced by sodium butyrate. Cancer Lett. 2005;225:199–206. doi: 10.1016/j.canlet.2004.11.029. [DOI] [PubMed] [Google Scholar]
- 16.Kim SH, Kim KW, Jeong JW. Inhibition of hypoxia-induced angiogenesis by sodium butyrate, a histone deacetylase inhibitor, through hypoxia-inducible factor-1alpha suppression. Oncol Rep. 2007;17:793–797. [PubMed] [Google Scholar]
- 17.Larionov A, Krause A, Miller W. A standard curve based method for relative real time PCR data processing. BMC Bioinformatics. 2005;6:62. doi: 10.1186/1471-2105-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Koh MY, Lemos R, Jr, Liu X, Powis G. The hypoxia-associated factor switches cells from HIF-1α- to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011;71:4015–4027. doi: 10.1158/0008-5472.CAN-10-4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qian DZ, Wang X, Kachhap SK, Kato Y, Wei Y, Zhang L, Atadja P, Pili R. The histone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004;64:6626–6634. doi: 10.1158/0008-5472.CAN-04-0540. [DOI] [PubMed] [Google Scholar]
- 20.Nakanishi R, Oka N, Nakatsuji H, Koizumi T, Sakaki M, Takahashi M, Fukumori T, Kanayama HO. Effect of vascular endothelial growth factor and its receptor inhibitor on proliferation and invasion in bladder cancer. Urol Int. 2009;83:98–106. doi: 10.1159/000224877. [DOI] [PubMed] [Google Scholar]
- 21.Masood R, Cai J, Zheng T, Smith DL, Hinton DR, Gill PS. Vascular endothelial growth factor (VEGF) is an autocrine growth factor for VEGF receptor-positive human tumors. Blood. 2001;98:1904–1913. doi: 10.1182/blood.V98.6.1904. [DOI] [PubMed] [Google Scholar]
- 22.Dias S, Hattori K, Zhu Z, Heissig B, Choy M, Lane W, Wu Y, Chadburn A, Hyjek E, Gill M, et al. Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Invest. 2000;106:511–521. doi: 10.1172/JCI8978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang W, Qi L, Tan M, Zhang Z, Du J, Wei X, Yao X. Effect of platelet-derived growth factor-B on renal cell carcinoma growth and progression. Urol Oncol. 2015;33:168.e17–e27. doi: 10.1016/j.urolonc.2014.12.015. [DOI] [PubMed] [Google Scholar]
- 24.Marech I, Gadaleta CD, Ranieri G. Possible prognostic and therapeutic significance of c-Kit expression, mast cell count and microvessel density in renal cell carcinoma. Int J Mol Sci. 2014;15:13060–13076. doi: 10.3390/ijms150713060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dent P. Crosstalk between ERK, AKT and cell survival. Cancer Biol Ther. 2014;15:245–246. doi: 10.4161/cbt.27541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O'Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/S0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- 27.Sowter HM, Raval RR, Moore JW, Ratcliffe PJ, Harris AL. Predominant role of hypoxia-inducible transcription factor (Hif)-1alpha versus Hif-2alpha in regulation of the transcriptional response to hypoxia. Cancer Res. 2003;63:6130–6134. [PubMed] [Google Scholar]
- 28.Shinojima T, Oya M, Takayanagi A, Mizuno R, Shimizu N, Murai M. Renal cancer cells lacking hypoxia inducible factor (HIF)-1alpha expression maintain vascular endothelial growth factor expression through HIF-2alpha. Carcinogenesis. 2007;28:529–536. doi: 10.1093/carcin/bgl143. [DOI] [PubMed] [Google Scholar]
- 29.Bai L, Yang JC, Ok JH, Mack PC, Kung HJ, Evans CP. Simultaneous targeting of Src kinase and receptor tyrosine kinase results in synergistic inhibition of renal cell carcinoma proliferation and migration. Int J Cancer. 2012;130:2693–2702. doi: 10.1002/ijc.26303. [DOI] [PubMed] [Google Scholar]
- 30.Zhao D, Zhai B, He C, Tan G, Jiang X, Pan S, Dong X, Wei Z, Ma L, Qiao H, et al. Upregulation of HIF-2α induced by sorafenib contributes to the resistance by activating the TGF-α/EGFR pathway in hepatocellular carcinoma cells. Cell Signal. 2014;26:1030–1039. doi: 10.1016/j.cellsig.2014.01.026. [DOI] [PubMed] [Google Scholar]
- 31.Tanaka T, Torigoe T, Hirohashi Y, Sato E, Honma I, Kitamura H, Masumori N, Tsukamoto T, Sato N. Hypoxia-inducible factor (HIF)-independent expression mechanism and novel function of HIF prolyl hydroxylase-3 in renal cell carcinoma. J Cancer Res Clin Oncol. 2014;140:503–513. doi: 10.1007/s00432-014-1593-7. [DOI] [PubMed] [Google Scholar]
- 32.Lieubeau-Teillet B, Rak J, Jothy S, Iliopoulos O, Kaelin W, Kerbel RS. von Hippel-Lindau gene-mediated growth suppression and induction of differentiation in renal cell carcinoma cells grown as multicellular tumor spheroids. Cancer Res. 1998;58:4957–4962. [PubMed] [Google Scholar]
- 33.Qian DZ, Kachhap SK, Collis SJ, Verheul HM, Carducci MA, Atadja P, Pili R. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res. 2006;66:8814–8821. doi: 10.1158/0008-5472.CAN-05-4598. [DOI] [PubMed] [Google Scholar]