

Summary
Background
Current treatment options for patients with relapsed or refractory (RR) lymphoma and multiple myeloma (MM) are limited, highlighting the unmet need for effective therapies in these disease settings. CUDC-907 is an oral, first-in-class, small molecule that is designed to inhibit both histone deacetylase (HDAC) and phosphoinositide 3-kinase (PI3K) enzymes, which are members of common oncogenic pathways in hematologic malignancies. This study examines CUDC-907 monotherapy in patients with RR lymphoma and MM.
Methods
This open-label, non-randomized, first-in-man, phase 1 multi-center trial enrolled adult patients with lymphoma or MM who were refractory to or relapsed after ≥2 prior regimens. CUDC-907 was orally administered in a standard 3+3 dose escalation design using three different dosing schedules which enrolled sequentially as follows: once daily (QD), then intermittent twice (BIW) or thrice weekly (TIW) that enrolled simultaneously, and finally five days on/two days off (5/2) in 21-day cycles. Dosing started at 30 mg for QD and 60 mg for other schedules, escalating in 30 mg increments. Patients continued to receive CUDC-907 until disease progression or other treatment discontinuation criteria were met. The primary objective was to determine the maximum-tolerated dose and recommended phase 2 dose (RP2D); secondary objectives were to assess the safety and tolerability, and preliminary anti-cancer activity. Results from the completed dose escalation phase are presented. Safety analyses were conducted in all patients who received at least one dose of study medication; efficacy analyses were conducted in all patients who received at least one dose of study drug and underwent at least one post-baseline response assessment. This ongoing trial is registered at ClinicalTrials.gov, number NCT01742988.
Findings
Forty-four heavily pretreated patients received CUDC-907 up to a maximum of 60 mg for the QD and 5/2 schedules, and 150 mg for the intermittent schedules in the dose escalation phase. The most common Grade ≥3 adverse events were thrombocytopenia (n=9, 20%), neutropenia (n=3, 7%), and hyperglycemia (n=3, 7%). Dose limiting toxicities (DLTs) were diarrhea and hyperglycemia; no DLTs were observed on the 5/2 schedule. Eleven of 44 patients reported serious AEs, 3 of which were considered treatment-related: epistaxis and the DLTs of diarrhea and hyperglycemia. AEs led to dose reductions in 6 patients and treatment discontinuation in 7 patients. Thirty-seven patients were evaluable for response. Five out of 9 patients with diffuse large B-cell lymphoma (DLBCL) achieved objective responses (2 complete responses [CR], 3 partial responses [PR]). Three of these objective responses (1 CR, 2PR) occurred in patients with transformed follicular (t-FL) DLBCL. Stable disease (SD) has been observed in 21 (57%) of 37 response-evaluable patients including DLBCL, Hodgkin lymphoma (HL), and MM. On the basis of the response and tolerability profile, the RP2D of CUDC-907 was determined to be 60 mg on the 5/2 schedule.
Interpretation
CUDC-907 has demonstrated tolerability and anti-tumor activity across all dosing schedules studied, including multiple objective responses and disease control in heavily pre-treated patients with DLBCL. These results support continued development of CUDC-907, alone and in combination with other therapies, for the treatment of DLBCL in refractory and relapsed settings.
Funding
Curis, Inc. with support from the Leukemia and Lymphoma Society.
Introduction
Lymphoma and multiple myeloma (MM) account for approximately 4% of all newly diagnosed cancers worldwide and are responsible for 3.7% of cancer deaths.1 Treatment options are particularly limited in the setting of multiple relapsed or refractory (RR) disease, which is characterized by diminishing likelihood, depth, and duration of disease response. Growing awareness of genetic aberrations and dysregulated oncogenic pathways has shifted therapeutic focus to molecularly targeted agents to improve disease control.
Histone deacetylase (HDAC) and phosphoinositide 3-kinase (PI3K) enzymes and their signaling pathways are validated therapeutic targets in hematologic malignancies. Four HDAC inhibitors, vorinostat (Zolinza®), belinostat (Beleodaq®), romidepsin (Istodax®), and panobinostat (Farydak®), have been approved by the U.S. Food and Drug Administration (FDA) for the treatment of RR hematologic cancers, specifically peripheral and cutaneous T-cell lymphomas (TCLs) and MM. Responses to HDAC inhibitors have also been observed in RR aggressive non-Hodgkin lymphomas (NHLs) including diffuse large B-cell lymphoma (DLBCL), Hodgkin lymphoma (HL), and acute myeloid leukemia.2 Idelalisib (Zydelig®), a small molecule inhibitor that selectively targets the PI3K p110-δ isoform, has received FDA approval for the treatment of various indolent non-Hodgkin lymphomas (iNHLs). While idelalisib has demonstrated anti-cancer activity across a range of iNHLs, significant activity has not been demonstrated in the setting of aggressive NHL.3
Emerging pre-clinical data suggest that dual targeting of HDAC and PI3K pathways may achieve synergistic anti-cancer effects.4 An HDAC inhibitor combined with a PI3K/mTOR inhibitor synergistically exhibited potent inhibition of tumor growth and prolonged survival in mouse DLBCL xenograft models, demonstrating the ability of PI3K inhibition to potentiate histone acetylation and HDAC inhibition to augment AKT dephosphorylation.5 Dual targeting is intended to overcome or thwart the emergence of resistance to molecularly targeted agents that can develop through secondary target gene mutations or compensatory activation of alternative pathways, so called “oncogenic switch.” A promising strategy for mitigating such acquired drug resistance is through simultaneous inhibition of multiple molecular pathways, using either several agents in combination or a single agent that concurrently blocks multiple targets and/or pathways.6
CUDC-907 is a first-in-class, rationally-designed small molecule that dually inhibits HDAC (class I and II) and PI3K (class I α, β, and δ) enzymes.4 This unique combination targets multiple oncogenic signaling pathways critical for cell proliferation, survival, and migration.2,3 CUDC-907 has been shown to inhibit activation of PI3K/AKT, JAK/STAT, and MAPK signaling pathways, in addition to decreasing MYC protein levels, in both solid tumor and hematologic cancer cell lines.4,7 The anti-tumor growth and pro-apoptotic activity of CUDC-907 has also been shown to be more potent than single-target HDAC or PI3K inhibitors, as demonstrated in a variety of cultured and implanted cancer cell lines representing B- and T-cell lymphomas, leukemias, and MM.7
In this phase I trial of oral CUDC-907 monotherapy, we report overall safety and preliminary efficacy results from the dose finding cohort in the setting of RR lymphoma and MM.
Methods
Study Design and Participants
A standard 3+3 trial design was used to enroll adult patients with RR lymphoma or MM across 4 US centers into sequential dose escalation cohorts. Patients were eligible if they were ≥18 years of age with histologically confirmed lymphoma or MM that was refractory to or relapsed after ≥2 prior regimens. Eligible patients had a life expectancy of >3 months, measurable or evaluable disease, an Eastern Cooperative Oncology Group (ECOG) score of ≤2, and adequate hematologic, renal, and hepatic function (absolute neutrophil count ≥1,000/μL; platelets ≥75,000/μL; creatinine ≤1.5× upper limit of normal (ULN), ≤ 2x ULN for MM patients; total bilirubin ≤1.5x ULN; AST/ALT ≤2.5x ULN). Patients with active central nervous system involvement, gastrointestinal conditions interfering with study drug absorption or tolerance, ongoing diarrhea, uncontrolled or severe cardiovascular disease, human immunodeficiency virus, and hepatitis B or C infection were excluded. Other exclusionary criteria were serious infection in the previous 14 days, graft versus host disease in the previous 3 months, and second primary malignancy in the previous 2 years (except for curatively treated solid tumors at low risk for recurrence). Patients receiving ongoing immunosuppressive therapy or those who received systemic anti-cancer therapy within 3 weeks of study entry were also excluded. Patients were assessed at baseline and at subsequent evaluations according to Revised Response Criteria for Malignant Lymphoma or the International Uniform Response Criteria for Multiple Myeloma, as appropriate for their specific tumor type.
This trial was approved by the institutional review boards of all participating centers and was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation guidelines for Good Clinical Practice. All patients provided written informed consent for participation.
Procedures
Patients received CUDC-907 capsules (Pharmatek Laboratories, Inc., San Diego, USA) orally, within 30 minutes of meals, in 21-day cycles according to one of three dosing schedules: once daily (QD), intermittent twice (BIW) or thrice weekly (TIW) that enrolled simultaneously, and five days on/two days off (5/2). A cycle duration of 21 days was chosen to allow flexibility for combination dosing with standards of care that are often dosed at this frequency. Cohorts of 3 patients received CUDC-907 starting at 30 mg for the QD schedule and 60 mg for the intermittent and 5/2 schedules. Doses escalated in 30 mg increments up to 60 mg for the QD schedule and 150 mg for the intermittent schedules. The first patient in each dose cohort was required to complete 7 days of treatment prior to enrollment of more patients into that cohort. If a DLT occurred in 1 of 3 patients in a given cohort, an additional three patients were treated at the same dose. If only 1 patient in the expanded cohort experienced a DLT, the next higher dose-level could open to enrollment. If >1 patient in a cohort experienced a DLT, then further enrollment into that cohort would cease and the previous dose-level would be explored as the maximum-tolerated dose (MTD). MTD was defined as the highest dose studied at which a DLT occurred in <2 of 6 patients during Cycle 1.
A DLT was defined as any of the following treatment-related adverse events (AEs) occurring during Cycle 1: non-hematological ≥ Grade 3 AE (excluding Grade 3 nausea or vomiting in subjects not treated with optimal antiemetic therapy), any AE resulting in a dose delay ≥7 days, Grade 4 neutropenia lasting ≥7 days or ≥Grade 3 with fever >101.3°F (38.5°C) or infection, and Grade 4 thrombocytopenia ≥7 days or ≥Grade 3 with significant bleeding. In the absence of DLTs, each patient was treated for a minimum of 21 days (1 cycle). There was no pre-determined duration of treatment and patients could continue to receive ongoing treatment until disease progression or other treatment discontinuation criteria were met (i.e., unmanageable toxicity, withdrawal of consent, physician decision, major protocol violation, or non-compliance).
To assess the primary objective, a Safety Review Committee that included the Principal Investigators and medical monitor reviewed available safety and laboratory data for all cohorts to determine whether the MTD had been reached and to decide upon further enrollment, dose escalation, cohort expansion, or discontinuation of or enrollment into a particular dose schedule.
Dose delays and modifications were allowed to ameliorate toxicities. Treatment could be held up to 7 days and restarted when retreatment criteria were met, or for longer at the investigator’s discretion with sponsor approval. Intra-patient dose-escalation was not permitted. Patients who experienced a Grade 3 DLT were either discontinued from treatment or reduced to the next lower dose and those who experienced a Grade 4 DLT were permanently discontinued from treatment.
Safety evaluations were conducted at screening, throughout the trial, and at follow-up 30 days after the last dose of CUDC-907. Evaluations included physical examination, electrocardiogram (ECG), clinical laboratory testing, and vital sign assessments. The incidence and severity of AEs were assessed according to the National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE v4.03).
Patients were re-staged according to standard response criteria as appropriate for their specific tumor type, Revised Response Criteria for Malignant Lymphoma or the International Uniform Response Criteria for Multiple Myeloma, during the last week of Cycles 2, 4, 6, then every four cycles thereafter, and at the end of treatment.8, 9 CT/MRI and/or PET scans were obtained at baseline and at each response assessment visit described previously. Patients with progressive disease were discontinued from treatment while patients deriving clinical benefit could continue to receive CUDC-907.
Plasma and tissue samples were variously obtained pre-dose and at serial time points after the first dose to characterize the pharmacokinetic (PK) profile of CUDC-907 and its major metabolites, as well as biomarkers related to HDAC and PI3K inhibition (acetylated histone H3 and phosphorylated AKT, respectively).
Outcomes
Primary objectives were to determine the MTD and recommended phase 2 dose (RP2D) of CUDC-907. Secondary objectives were assessments of safety, tolerability, and preliminary anti-tumor activity according to standard response criteria.
Statistical analysis
The size of the dose escalation cohort is based on the standard 3+3 dose escalation design used in oncology trials and no formal sample size estimation was performed. The safety population includes all patients from the dose escalation portion of the trial who received ≥1 dose of CUDC-907. The DLT-evaluable population, used for determination of the MTD, consisted of patients who received at least 66% of Cycle 1 doses without modification. Patients who experienced a DLT in Cycle 1 were included in the DLT-evaluable population regardless of dose modification. The response-evaluable population included all patients who received at least one dose of study drug and completed at least one post-baseline disease assessment. The dose expansion portion is currently ongoing and results for those patients will be reported separately. All efficacy and safety data were summarized using descriptive statistics by SAS Version 9. Data cut-off is 27 July 2015. This trial is registered with ClinicalTrials.gov, number NCT01742988.
Role of the funding source
This trial was funded by Curis, Inc with support from the Leukemia and Lymphoma Society. Curis, Inc. designed the trial and interpreted the data in collaboration with the study investigators. The data were collected and analyzed by investigators and site staff, and were reviewed and reported by the Sponsor. The authors of this report include those who are employed by the Sponsor. All authors had full access to the raw dataset upon request and attest to the accuracy of this report. The corresponding author had full access to all of the data and final responsibility for submission for publication. None of the authors are employed by the NIH or have received NIH funding related to the trial.
Results
Between 23 January 2013 and 27 July 2015, 44 heavily pretreated (median of 5 regimens) patients, including those who previously underwent autologous and/or allogeneic stem cell transplant (SCT), received CUDC-907 up to a maximum of 60 mg for the QD and 5/2 schedules and 150 mg for the intermittent schedules. In this completed dose escalation phase of the trial, 10 patients were enrolled into the QD dosing cohort, 12 into BIW, and 15 into TIW. Seven patients received the 60 mg 5/2 dose. Table 1 presents baseline patient demographics. A total of 4/44 (9%) MM patients and 40/44 (91%) lymphoma patients were enrolled. Major lymphoma histologies consisted of DLBCL, including transformed follicular lymphoma (tFL/DLBCL, high grade and composite low-high grade disease per local pathology report) and HL. Other lymphomas included T-cell, lymphoplasmacytic, small lymphocytic, mantle cell, follicular, Burkitt, gray-zone, and marginal zone lymphomas. As of 27 July 2015, 7 out of 44 total patients (16%) remained on active treatment. Thirty-seven patients discontinued study drug due to progressive disease (PD) or clinical signs of PD (n=24, 55%), physician decision (n=5, 11%), AEs (n=7, 16%), or withdrawal of consent (n=1, 2%). One patient with DLBCL discontinued treatment in order to undergo autologous SCT after achieving complete response (CR) in Cycle 2. Although the median duration of treatment for all patients was 2.1 months (interquartile range [IQR]: 1.2 to 5.7+ months) as of data cut-off, durable disease stabilization was observed across all histologies. Maximum durations of treatment ranged from 12 (HL) and 12.6 (tFL/DLBCL) months to 27.3 (DLBCL) and 30.5 (MM) months, all of which were ongoing as of the data cut-off date.
Table 1.
Safety population patient characteristics: dose escalation
| Baseline Demographics & Disposition | N=44 |
|---|---|
| Male, n (%) | 32 (72) |
| Caucasian, n (%) | 35 (80) |
| Age, median years (range) | 63 (22 – 83) |
| ≥65 years, n (%) | 18 (41) |
| Histology, n (%) | |
| DLBCL | 12 (27) |
| t-FL/DLBCL* | 7 (16) |
| HL | 12 (27) |
| Classical HL | 11 (25) |
| MM | 4 (9) |
| Other lymphoma* | 16 (36) |
| Years since diagnosis, median (range) | 5 (<1 – 43) |
| Stage, n (%) | |
| I – II | 6 (14) |
| III – IV | 33 (75) |
| Unknown | 5 (11) |
| Prior treatments, n (%) | |
| No. prior regimens, median (range) | 5 (2-10) |
| HDAC inhibitor | 6 (14) |
| PI3K inhibitor | 3 (7) |
| Rituximab | 30 (68) |
| Radiation therapy | 10 (23) |
| Stem cell transplant | 18 (41) |
| Autologous | 16 (36) |
| Allogeneic | 3 (7) |
High grade and composite low-high grade disease per local pathology report
Includes lymphoplasmacytic (n=3), small lymphocytic (n=3), mantle cell (n=3), follicular (n=2), T-cell (n=2), marginal zone (n=1), Burkitt (n=1), and gray-zone (n=1)
DLBCL – diffuse large B-cell lymphoma, t-FL/DLBCL – transformed follicular lymphoma/diffuse large B-cell lymphoma, HL – Hodgkin lymphoma, MM – multiple myeloma.
Four DLTs occurred in 3 patients at 60 mg QD (Grade 4 hyperglycemia and Grade 3 diarrhea), 150 mg BIW (Grade 3 hyperglycemia), and 150 mg TIW (Grade 3 diarrhea). The DLTs of Grade 4 and Grade 3 hyperglycemia occurred in a patient with insulin-dependent diabetes mellitus who discontinued treatment and a patient with history of glucose intolerance who remained on treatment with appropriate management of blood glucose, respectively. DLTs only occurred on the highest dose tested of the QD, BIW, and TIW schedules. No DLTs occurred on the 5/2 dosing schedule, which was subsequently determined to be the RP2D.
Adverse events (AEs) were reported in 39 (89%) of 44 patients with events of Grade ≥ 3 occurring in 19 (43%) of 44 patients. Table 2 describes AEs reported in >10% of patients and any Grade ≥ 3 events, that were generally Grade 1–2 and reversible with standard therapeutic interventions (e.g. loperamide upon onset of diarrhea), dose holds, or dose reductions. The most common AEs were diarrhea (n=25, 57%), fatigue (n=16, 36%), nausea (n=11, 25%) and thrombocytopenia (n=11, 25%). Treatment related-AEs were reported in 32 (73%) of 44 patients; the most common Grade ≥ 3 treatment-related AEs included thrombocytopenia (n=8, 18%), neutropenia (n=3, 7%), hyperglycemia (n=3, 7%), and diarrhea (n=2, 5%). Eleven (25%) of 44 patients reported serious AEs (SAEs), 3 of which were considered treatment-related: epistaxis and the DLTs of diarrhea and hyperglycemia. AEs led to dose reductions in 6 (14%) patients and treatment discontinuation in 7 (16%) patients. Although dose reductions occurred across all dosing schedules, treatment discontinuation due to AEs only occurred on the QD and intermittent schedules. There were 3 on-study deaths due to sepsis, pleural effusion, and worsening lymphoma, none of which were considered treatment-related. The overall AE profile for the 7 patients who received the 60 mg 5/2 dose was similar to that of the broader safety population.
Table 2.
Adverse events in ≥10% of patients and any grade ≥3 events (safety population, n=44*)
| Patients n, (%) | Grade 1-2 | Grade 3 | Grade 4 | Grade 5 | Total |
|---|---|---|---|---|---|
| Diarrhea | 23 (52) | 2 (5) | 0 | 0 | 25 (57) |
| Fatigue | 16 (36) | 0 | 0 | 0 | 16 (36) |
| Nausea | 11 (25) | 0 | 0 | 0 | 11 (25) |
| Thrombocytopenia | 2 (5) | 7 (16) | 2 (5) | 0 | 11 (25) |
| Constipation | 5 (11) | 0 | 0 | 0 | 5 (11) |
| Cough | 5 (11) | 0 | 0 | 0 | 5 (11) |
| Neutropenia | 2 (5) | 1 (2) | 2 (5) | 0 | 5 (11) |
| Rash | 5 (11) | 0 | 0 | 0 | 5 (11) |
| Upper respiratory infection | 5 (11) | 0 | 0 | 0 | 5 (11) |
| Hypomagnesaemia | 3 (7) | 1 (2) | 0 | 0 | 4 (9) |
| Hyperglycemia | 0 | 1 (2) | 2 (5) | 0 | 3 (7) |
| Pleural effusion | 0 | 2 (5) | 0 | 0 | 2 (5) |
| Pneumonia | 0 | 2 (5) | 0 | 0 | 2 (5) |
| Hypercalcemia | 0 | 0 | 1 (2) | 0 | 1 (2) |
| Lymphoma | 0 | 0 | 0 | 1 (2) | 1 (2) |
| Sepsis | 0 | 0 | 0 | 1 (2) | 1 (2) |
| Acute renal failure | 0 | 1 (2) | 0 | 0 | 1 (2) |
| Anemia | 0 | 1 (2) | 0 | 0 | 1 (2) |
| Increased blood alkaline phosphate | 0 | 1 (2) | 0 | 0 | 1 (2) |
| Cardiac tamponade | 0 | 1 (2) | 0 | 0 | 1 (2) |
| Dehydration | 0 | 1 (2) | 0 | 0 | 1 (2) |
| Epistaxis | 0 | 1 (2) | 0 | 0 | 1 (2) |
| Failure to thrive | 0 | 1 (2) | 0 | 0 | 1 (2) |
| Hyperuricemia | 0 | 1 (2) | 0 | 0 | 1 (2) |
| Hypokalemia | 0 | 1 (2) | 0 | 0 | 1 (2) |
| Lymph node pain | 0 | 1 (2) | 0 | 0 | 1 (2) |
| Musculoskeletal pain | 0 | 1 (2) | 0 | 0 | 1 (2) |
| Urosepsis | 0 | 1 (2) | 0 | 0 | 1 (2) |
| Vascular access complications | 0 | 1 (2) | 0 | 0 | 1 (2) |
Safety population includes any patient in the escalation phase who received at least 1 dose of CUDC-907 prior to data cut-off
In the dose escalation cohort, 7 patients discontinued treatment during Cycle 1 prior to undergoing a post-baseline assessment and were ineligible for efficacy analysis. Four of these seven patients discontinued treatment in Cycle 1 due to AEs: one patient experienced Grade 4 hypercalcemia due to PD; one patient experienced Grade 5 infection, hypotension, sepsis, and death that were deemed unrelated to treatment; one patient experienced fatigue, neutropenia, dizziness and a GI fistula deemed unrelated to study drug, and one patient experienced two DLTs consisting of Grade 4 hyperglycemia and Grade 3 diarrhea. Three of the seven patients discontinued treatment in Cycle 1 due to physician decision, PD, or withdrawal of consent.
Among 37 dose escalation patients evaluable for disease response, 5 objective responses were observed, all in patients with DLBCL, including 3 patients with t-FL/DLBCL. Table 3 shows the best treatment response by histology. Figure 1 shows the largest post-baseline tumor size reduction in response-evaluable patients with lymphoma, demonstrating anti-tumor activity across multiple histologies. Complete responses (CR) occurred in 1 patient with DLBCL in Cycle 2 (60 mg 5/2) and another patient with t-FL/DLBCL in Cycle 12 (150 mg TIW). Partial responses (PR) occurred in 1 patient with DLBCL in Cycle 4 (60 mg QD) and 2 patients with t-FL/DLBCL in Cycles 4 and 26 (150 mg BIW and 30 mg QD, respectively). The median duration of response with ongoing therapy is 4.4+ months (IQR 3.5 – 9.3), with 2 responses ongoing at the time of data cutoff. Stable disease (SD) in response-evaluable patients lasting a median of 2.9+ months (IQR 1.4-6.1) has been observed in 21 patients including DLBCL (1.7 months), HL (median 5.8+ months, IQR 5.3-7.2), and MM (median 16.3+ months, IQR 9.9-22.7). The longest duration of SD is 29.1+ months in a patient with low secretory MM. It is uncertain whether prior anti-cancer therapy received within 4 weeks of enrollment may have contributed to disease stabilization observed in this or other patients with MM (minimum 3 week wash-out period prior to treatment with CUDC-907, n=4).
Table 3.
Assessment summary for response-evaluable dose escalation patients (n=37)
| Indication | N | Best Response, N (%) | Median Treatment Duration, months (interquartile range) | |||
|---|---|---|---|---|---|---|
| CR | PR | SD | PD | |||
| All DLBCL* | 9 | 2 (22) | 3 (33) | 1 (11) | 3 (33) | 2.4 (1.7 – 5.8+) |
| ➢ t-FL/DLBCL | 5 | 1 (20) | 2 (40) | 1 (20) | 1(20) | 5.6 (2.9 – 5.8+) |
| HL | 10 | – | – | 8 (80) | 2 (20) | 5.8 (3.5 – 7.7+) |
| MM | 4 | – | – | 2 (50) | 2 (50) | 3.2 (1.9 – 10.9+) |
| Other lymphoma** | 14 | – | – | 10 (71) | 4 (29) | 2.1 (1.3–2.7+) |
| Total | 37 | 2 (5) | 3 (8) | 21 (57) | 11 (30) | 2.7 (1.4 – 5.9+) |
Includes t-FL/DLBCL and DLBCL
Includes lymphoplasmacytic (n=3), small lymphocytic (n=3), mantle cell (n=3), follicular (n=2), gray-zone (n=1), marginal zone (n=1) and T-cell (n=1).
+ = ongoing as of 27 July 2015
Figure 1. Maximum target lesion decrease in lymphomas: response-evaluable dose escalation patients (n=33).
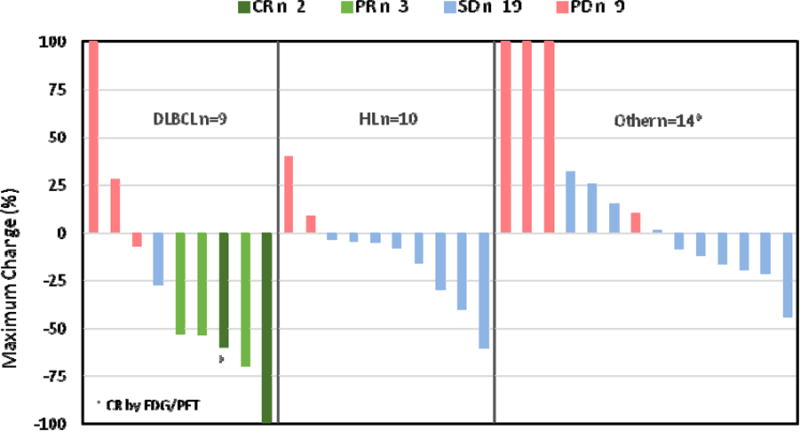
*Lymphoplasmacytic (n=3), small lymphocytic (n=3), mantle cell (n=3), follicular (n=2), gray-zone (n=1), marginal zone (n=1) and T-cell (n=1)
Plasma PK of CUDC-907 and major metabolites (including the inactive M1 metabolite and the active M2 metabolite that retains limited PI3K inhibitory activity) were determined for each dose level and schedule based on samples obtained on the 1st and 15th days of Cycle 1. Dose-related increases in plasma exposure were observed and showed considerable inter-patient variability (data not shown). Cycle 1 Day 15 plasma concentrations of CUDC-907 and metabolites from the 60 mg 5/2 dosing cohort (n=6) shown in Figure 2 indicate low parent drug and metabolite levels following two days off treatment. CUDC-907 concentrations (69.5 ng/g) in a tumor sample obtained 4 hours post-dose on Cycle 1 Day 19 from one patient enrolled into the 60 mg 5/2 expansion cohort demonstrate drug distribution to tumor tissue (data not shown), consistent with preclinical observations in rodent tissue distribution studies.
Figure 2. CUDC-907 and metabolite concentrations in plasma, 60 mg 5/2 dose.
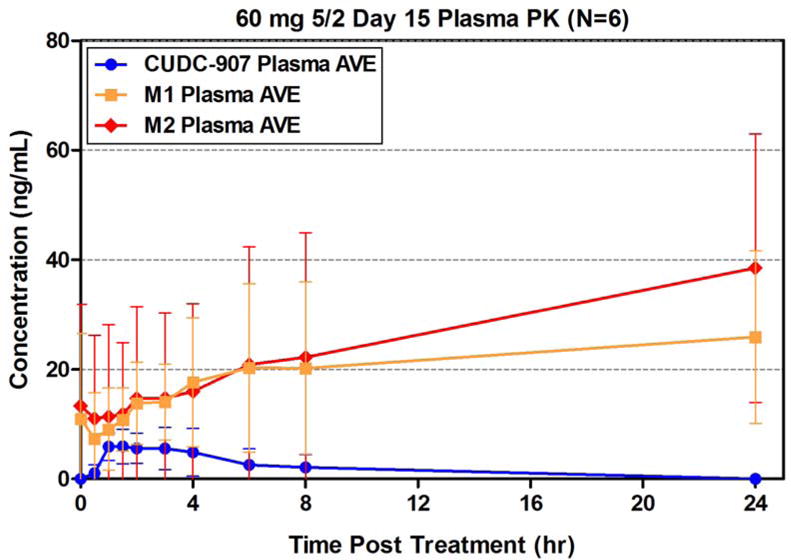
AVE – average, M1 – inactive metabolite, M2 – active metabolite. Plasma obtained from 6 patients in the expansion phase of the trial. Error bars refer to standard deviation.
Peripheral blood mononuclear cell (PBMC) western blot analysis of the 60 mg 5/2 dosing cohort (n=3) demonstrated the pharmacodynamic effects of CUDC-907 on selected biomarkers during Cycle 1 (Figure 3). Post-treatment accumulation of acetylated histone H3 indicated HDAC blockade, while decreases in AKT phosphorylation indicated PI3K pathway inhibition. These results further support selection of 60 mg 5/2 as the RP2D for future testing of CUDC-907.
Figure 3. Biomarkers of CUDC-907 activity in peripheral blood, 60 mg 5/2 dose.
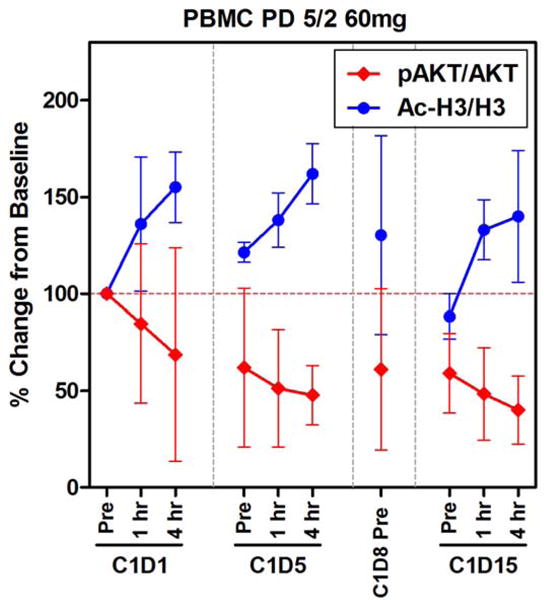
Ac-H3 – acetylated histone H3, pAKT – phosphorylated AKT, C – cycle, D – day. PBMC samples obtained from 3 patients in the expansion phase of the trial. Error bars refer to standard deviation.
Discussion
This study demonstrates that CUDC-907 monotherapy is tolerated at doses that can achieve durable disease control in heavily pre-treated patients with RR DLBCL, MM, HL, and other lymphomas. A total of four DLTs consisting of diarrhea and hyperglycemia were reported on the highest dose tested on QD, BIW, and TIW schedules; no DLT occurred on the 5/2 schedule, the RP2D. Diarrhea is a well-recognized side effect of HDAC and PI3K inhibitors and hyperglycemia is a known toxicity of HDAC inhibitors as well as an on-target toxicity consistent with inhibition of the PI3K/AKT/mTOR pathway.10–14 Both are predictable and manageable toxicities of CUDC-907. Overall, AEs were self-limiting and typically consisted of Grade 1-2 diarrhea, fatigue, nausea, and thrombocytopenia. The majority of AEs leading to treatment discontinuation were unrelated to study medication. Dose reductions due to AEs experienced by 6 patients across all dosing schedules did not appear to compromise disease control. Although the overall safety profile of CUDC-907 is consistent with those of current FDA-approved HDAC and PI3K inhibitors (i.e., gastrointestinal, hematological, etc.), serious adverse reactions associated with those drugs, such as colitis, infections, hepatotoxicity, and cardiac toxicities, have not been reported to date.10–14 Ongoing follow-up will enable longer term monitoring for emergent toxicities in patients treated with CUDC-907.
Anti-tumor activity was observed across all dosing schedules in 5 patients with DLBCL, including 3 patients with t-FL/DLBCL, with confirmed objective responses of 2 CRs and 3 PRs in this subset of 9 heavily pre-treated, response-evaluable patients (median age 72, range 41-77). While 1 patient achieved CR (5/2 schedule) by the time of Cycle 2 restaging, objective responses have typically occurred after Cycle 4, suggesting a pattern of gradual response to CUDC-907 treatment. Data from the 60 mg 5/2 cohort confirm a favorable PK profile and demonstrate therapeutic concentrations of CUDC-907 in tumoral tissue. Additionally, the inhibitory activities of CUDC-907 against HDAC and PI3K pathways were confirmed in the 60 mg 5/2 cohort as measured by changes in acetylated histone H3 and phosphorylated AKT levels in PBMCs. Although limited to 7 patients, the toxicity profile of 60 mg 5/2 was favorable and did not lead to any treatment discontinuations due to AEs, supporting the tolerability of this dose schedule. In particular, dose free-intervals allowed exposure breaks, as reflected in PK data, and are hypothesized to allow recovery of cytopenias and other toxicities. Overall, these data support 5/2 dosing as an appropriate schedule that can be delivered with manageable toxicity over lengthy periods while still achieving sufficient tumoral exposure for target inhibition. On the basis of confirmed objective responses, a tolerable safety profile, and relatively early onset of tumoral regression, CUDC-907 60 mg 5/2 was determined to be the RP2D. The dose expansion portion of this trial is currently ongoing at the RP2D, evaluating monotherapy in RR DLBCL, MM, HL and TCL, as well as combination therapy with rituximab in RR DLBCL.
Despite improvements in survival rates in the rituximab era, approximately one-third of patients with DLBCL experience RR disease with diminishing prospects for disease control.15 Successful management of RR DLBCL hinges on tumoral sensitivity to, as well as the patient’s ability to tolerate, salvage therapy and eligibility for subsequent SCT. Even when considering the limitations of cross-trial comparisons and variability introduced by small patient numbers, monotherapy salvage regimens for RR DLBCL rarely achieve response rates greater than 50%.16–21 The reported activity of single agent HDAC or PI3K inhibitors in this setting is even lower, with ORRs ≤20%.16, 22–26
Interestingly, preliminary responses on our study appeared to cluster in patients diagnosed with t-FL/DLBCL, with these patients experiencing some of the longest treatment durations on the study (median 5.6 months, IQR 2.9-5.8+) as compared to patients with de novo DLBCL (median 1.6 months, range 1.7-8.2+). Compared to patients with de novo DLBCL, those with t-FL/DLBCL had received more prior lines of therapy (median 4 vs 2), consistent with a longer duration of disease (median 5 vs 1.2 years since diagnosis). Generally, the demographic profiles of both groups, including age range (median age: t-FL/DLBCL, 72 years; DLBCL, 64 years), were comparable.
CUDC-907 is a rationally-designed novel compound that combines HDAC and PI3K inhibitor functionality in a single scaffold. The rationale for developing a dual HDAC/PI3K inhibitor is based on increasing recognition that interfering with a single target and/or pathway may not abrogate the malignant phenotype of most tumors, including DLBCL. Additionally, an extensive body of research indicates that HDAC inhibitors are most useful in combination with other targeted or cytotoxic agents, and synergistic activity with PI3K inhibition has been reported.4, 27 The ability of HDAC and PI3K inhibition to potentiate each other’s activity in DLBCL presents a promising novel strategy.5 In the setting of relapsed disease, there is substantial need to augment responses with tolerable regimens that achieve disease control and possibly enable patients to proceed to high-dose therapy and autologous SCT.28 Salvage therapy options are increasingly important for patients refractory to front-line rituximab who have a lower likelihood of response to rituximab-based salvage therapy and a correspondingly lower 3-year event-free survival of 21% and overall survival.29, 30 An oral dual HDAC/PI3K inhibitor that circumvents potentially dose limiting toxicities of established agents while achieving sufficient disease control warrants further exploration in the setting of hematologic cancers as mono – or combination therapy.
To confirm, extend, and better describe CUDC-907’s activity in the setting of RR disease, we are currently enrolling patients with DLBCL on the 5/2 schedule. Furthermore, we plan to correlate clinical responses to the molecular characteristics of the tumors, including cell of origin and mutational status, in order to identify subgroups most likely to derive the greatest benefit from this treatment. Older age and advanced disease, among other factors, are associated with worse prognosis and an increased likelihood of harboring adverse mutational aberrations, such as MYC and BCL-2.29 Additionally, CUDC-907 is being co-administered with rituximab to determine whether the combination augments the clinical activity of CUDC-907 in patients with RR DLBCL while maintaining the acceptable safety profile achieved with the RP2D monotherapy.
Durable objective responses achieved in patients with RR DLBCL demonstrate CUDC-907’s potential to address the unmet clinical need for effective therapy in this aggressive disease. Generally, dual HDAC/PI3K inhibition with CUDC-907 has been shown to be well tolerated, with a favorable toxicity profile comparable to, or slightly better than, that of FDA-approved HDAC and PI3K single-target agents. In aggregate, these findings indicate that further testing of CUDC-907 is warranted in the setting of aggressive lymphomas such as RR DLBCL and t-FL/DLBCL.
Acknowledgments
The authors thank all the patients and their families, as well as the clinical sites participating in this trial. This trial was sponsored by Curis, Inc with financial support from the Leukemia and Lymphoma Society.
Footnotes
Declaration of Interests
YA: research funding (Curis); JGB: none; MRP: none; IF: none; JFG: none; SSN: none; KK: none; AC: none; AA: none; MC, LG, JW, AM, JLV: employment (Curis); YO: none
Contributors
AY, JGB, MRP, KK, JW, JLV, and YO contributed to the study design and protocol development. AY, JGB, MRP, IF, JFG, SSN, KRK, ARC, AA, MC, and YO contributed to patient recruitment and data collection. AY, JGB, MRP, KRK, JLV, LG, JW, AM, JLV, and YO contributed to data analysis and interpretation. LG, JW, and JLV contributed to literature searches. AY, JGB, LG, JW, JLV, and YO contributed to the writing of this manuscript. LG, JW, and AM contributed to figures and tables. JW and AM contributed to sample analysis and description of bioananalytical procedures. All authors have reviewed drafts of the manuscript and have approved the final version.
References
- 1.Ferlay J, Soerjomataram I, Ervik M, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide. Lyon, France: International Agency for Research on Cancer; 2013. (IARC CancerBase No. 11). http://globocan.iarc.fr (accessed 07 Aug, 2015) [Google Scholar]
- 2.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;124(1):30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akinleye A, Avvaru P, Purqan M, Song Y, Liu D. Phosphatidylinositol 3-kinase (PI3K) inhibitors as cancer therapeutics. J Hematol Oncol. 2013;6(1):88. doi: 10.1186/1756-8722-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qian C, Lai CJ, Bao R, et al. Cancer network disruption by a single molecule inhibitor targeting both histone deacetylase activity and phosphatidylinositol 3-kinase signaling. Clin Cancer Res. 2012;18(15):4104–13. doi: 10.1158/1078-0432.CCR-12-0055. [DOI] [PubMed] [Google Scholar]
- 5.Rahmani M, Aust MM, Benson EC, Wallace L, Friedberg J, Grant S. PI3K/mTOR inhibition markedly potentiates HDAC inhibitor activity in NHL cells through BIM-and MCL-1–dependent mechanisms in vitro and in vivo. Clin Cancer Res. 2014;20(18):4849–60. doi: 10.1158/1078-0432.CCR-14-0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lackner MR, Wilson TR, Settleman J. Mechanisms of acquired resistance to targeted cancer therapies. Future Oncol. 2012;8(8):999–1014. doi: 10.2217/fon.12.86. [DOI] [PubMed] [Google Scholar]
- 7.Bao R, Wang D, Qu H, et al. Antitumor activity of CUDC-907, a dual PI3K and HDAC inhibitor, in hematological cancer models. Cancer Res. 2012;72(8 Supplement):3744. [Google Scholar]
- 8.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007;25(5):579–586. doi: 10.1200/JCO.2006.09.2403. [DOI] [PubMed] [Google Scholar]
- 9.Durie BG, Harousseau JL, Miguel JS, et al. International uniform response criteria for multiple myeloma. Leukemia. 2006;20:1467–1473. doi: 10.1038/sj.leu.2404284. [DOI] [PubMed] [Google Scholar]
- 10.Beleodaq [package insert] Irvine, CA: Spectrum Pharmaceuticals, Inc; 2014. [Google Scholar]
- 11.Farydak [package insert] East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2015. [Google Scholar]
- 12.Istodax [package insert] Summit, NJ: Celgene Corporation; 2014. [Google Scholar]
- 13.Zolinza [package insert] Whitehouse Station, NJ: Merck & Co., Inc; 2006. [Google Scholar]
- 14.Zydelig [package insert] Foster City, CA: Gilead Sciences, Inc; 2014. [Google Scholar]
- 15.Friedberg JW. Relapsed/refractory diffuse large B-cell lymphoma. ASH Education Book. 2011;2011(1):498–505. doi: 10.1182/asheducation-2011.1.498. [DOI] [PubMed] [Google Scholar]
- 16.Mondello P, Younes A. Emerging drugs for diffuse large B-cell lymphoma. Expert Rev Anticancer Ther. 2015;15(4):439–51. doi: 10.1586/14737140.2015.1009042. [DOI] [PubMed] [Google Scholar]
- 17.Ribrag V, Soria JC, Reyderman L, et al. Phase 1 first-in-human study of the enhancer of zeste-homolog 2 (EZH2) histome methyl transferase inhibitor E7438 as a single agent in patients with advanced solid tumors or B cell lymphoma. Eur J Cancer. 2014;50:197. [Google Scholar]
- 18.Palanca-Wessels MC, Salles GA, Czuczman MS, et al. Final results of a phase 1 study of the anti-CD79b antibody-drug conjugate DCDS4501A in relapsed or refractory (R/R) B-cell non-Hodgkin lymphoma (NHL) Blood. 2013;122(21):4400. [Google Scholar]
- 19.Kuruvilla J, Byrd JC, Flynn J, et al. The oral selective inhibitor of nuclear export (SINE) selinexor (KPT-330) demonstrates broad and durable clinical activity in relapsed/refractory non Hodgkin’s lymphoma. Blood. 2014;124(21):396. [Google Scholar]
- 20.Herait P, Domret H, Thieblemont C, et al. BET-Bromodomain (BRD) inhibitor OTX015: Final results of the dose-finding part of a phase I study in hematologic malignancies. Ann Oncol. 2015;26(suppl 2):ii10–ii11. [Google Scholar]
- 21.Friedberg JW, Mahadevan D, Jung J, et al. Phase 2 trial of alisertib (MLN8237), an investigational, potent inhibitor of aurora A kinase (AAK), in patients (pts) with aggressive B-and T-cell non-Hodgkin lymphoma (NHL) Blood. 2011;118(21):95. [Google Scholar]
- 22.Zaja F, Volpetti S, Chiappella A, et al. Panobinostat as salvage treatment for patients with relapsed/refractory diffuse large B-cell lymphoma not eligible to high dose therapy: a phase II study of the Fondazione Italiana Linfomi (FIL) Blood. 2014;124(21):1755. [Google Scholar]
- 23.Crump M, Andreadis C, Assouline SE, et al. A phase II study of single agent mocetinostat, an oral isotype-selective histone deacetylase (HDAC) inhibitor, in patients with diffuse large cell B-cell (DLBCL) and follicular (FL) lymphomas. J Clin Oncol. 2013;31(15):8535. [Google Scholar]
- 24.Younes A, Salles G, Bociek RG, et al. An open-label phase II study of buparlisib (BKM120) in patients with relapsed and refractory diffuse large B-cell lymphoma, mantle cell lymphoma or follicular lymphoma. Blood. 2014;124(21):1718. [Google Scholar]
- 25.Persky DO, Bernstein SH, Goldman BH, et al. A phase II study of PXD101 (belinostat) in relapsed and refractory aggressive B-cell lymphomas (rel/ref ABCL): SWOG S0520. J Clin Oncol. 2012;30(15):e18536. [Google Scholar]
- 26.Crump M, Coiffier B, Jacobsen ED, et al. Phase II trial of oral vorinostat (suberoylanilide hydroxamic acid) in relapsed diffuse large-B-cell lymphoma. Ann Oncology. 2008;19(5):964–9. doi: 10.1093/annonc/mdn031. [DOI] [PubMed] [Google Scholar]
- 27.Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs. 2010;19(9):1049–1066. doi: 10.1517/13543784.2010.510514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gisselbrecht G. Use of rituximab in diffuse large B-cell lymphoma in the salvage setting. Br J Haematol. 2008;143(5):607–12. doi: 10.1111/j.1365-2141.2008.07383.x. [DOI] [PubMed] [Google Scholar]
- 29.Hu S, Xu-Monette ZY, Tzankov A, et al. MYC/BCL2 protein coexpression contributes to the inferior survival of activated B-cell subtype of diffuse large B-cell lymphoma and demonstrates high-risk gene expression signatures: a report from The International DLBCL Rituximab-CHOP Consortium Program. Blood. 2013;121(20):4021–31. doi: 10.1182/blood-2012-10-460063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elstrom RL, Martin P, Ostrow K, et al. Response to second-line therapy defines the potential for cure in patients with recurrent diffuse large B-cell lymphoma: Implications for the development of novel therapeutic strategies. Clin Lymphoma Myeloma Leuk. 2010;10(3):192–6. doi: 10.3816/CLML.2010.n.030. [DOI] [PubMed] [Google Scholar]