

Abstract
Objective
Defective autophagy in macrophages leads to pathologic processes that contribute to atherosclerosis, including impaired cholesterol metabolism and defective efferocytosis. Autophagy promotes the degradation of cytoplasmic components in lysosomes and plays a key role in the catabolism of stored lipids to maintain cellular homeostasis. miR-33 is a post-transcriptional regulator of genes involved in cholesterol homeostasis, yet the complete mechanisms by which miR-33 controls lipid metabolism are unknown. We investigated whether miR-33 targeting of autophagy contributes to its regulation of cholesterol homeostasis and atherogenesis.
Approach and Results
Using CARS microscopy we show that miR-33 drives lipid droplet accumulation in macrophages, suggesting decreased lipolysis. Inhibition of neutral and lysosomal hydrolysis pathways revealed that miR-33 reduced cholesterol mobilization by a lysosomal-dependent mechanism, implicating repression autophagy. Indeed, we show that miR-33 targets key autophagy regulators and effectors in macrophages to reduce lipid droplet catabolism, an essential process to generate free cholesterol for efflux. Notably, miR-33 regulation of autophagy lies upstream of its known effects on ABCA1-dependent cholesterol efflux, as miR-33 inhibitors fail to increase efflux upon genetic or chemical inhibition of autophagy. Furthermore, we find that miR-33 inhibits apoptotic cell clearance via an autophagy-dependent mechanism. Macrophages treated with anti-miR-33 show increased efferocytosis, lysosomal biogenesis and degradation of apoptotic material. Finally, we show that treating atherosclerotic Ldlr−/− mice with anti-miR-33 restores defective autophagy in macrophage foam cells and plaques, and promotes apoptotic cell clearance to reduce plaque necrosis.
Conclusions
Collectively, these data provide insight into the mechanisms by which miR-33 regulates cellular cholesterol homeostasis and atherosclerosis.
Keywords: Autophagy, miR-33, Atherosclerosis, Cholesterol Efflux, Lipid Homeostasis, Macrophage
Subject codes: Vascular Biology, Metabolism, Lipids and Cholesterol
Graphical abstract
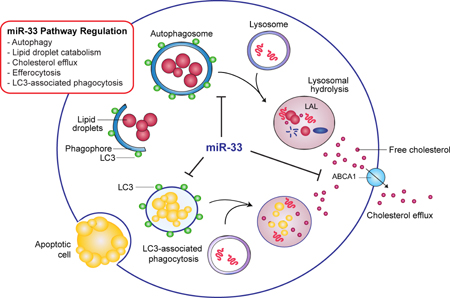
INTRODUCTION
In atherosclerosis, lipid-laden macrophage foam cells accumulate in the artery wall, where they fuel chronic inflammation that promotes plaque formation1. Autophagy plays a key role in the maintenance of cellular lipid homeostasis, and its dysregulation results in pathophysiological processes, including atherosclerosis2, 3. Autophagy of cytoplasmic lipid droplets or “lipophagy” catabolizes stored triglycerides, and promotes fatty acid oxidation to maintain cellular energy homeostasis4. It also protects against the detrimental effects of cellular cholesterol accumulation in macrophages by delivering excess cholesterol, stored as cholesterol esters in lipid droplets, to lysosomes where they are hydrolyzed by lysosomal acid lipase to free cholesterol and effluxed from the cell5. In addition, autophagy and lysosomal degradation contribute to the response to efferocytosis of apoptotic cells6, 7, which presents a lipid-loading stress on the engulfing cell. Recent studies have demonstrated that autophagy becomes progressively impaired in atherosclerosis8, and compromised autophagy in macrophages has been shown to account for a number of pro-atherosclerotic processes including impaired cholesterol metabolism5, 9, defective efferocytosis10, and activation of the inflammasome8. These studies suggest that inducing autophagy may promote regression of atherosclerotic plaques, yet this remains to be directly tested.
An improved understanding of the mechanisms regulating autophagy in macrophages may identify methods to modulate autophagy function and/or restore homeostasis in the case of autophagy dysfunction in atherosclerosis. Recent studies have shown that microRNAs (miRNAs) regulate autophagy through post-transcriptional repression of autophagy related genes (Atg) or upstream effectors (BECN1, mTOR, ULK1)11–13. miRNAs target mRNAs for translational repression or degradation by binding to complementary regions in the 3′ untranslated region of mRNAs14. Many miRNAs are highly conserved among eukaryotic species, and appear to have evolved as fine-tuners of gene expression that can simultaneously repress multiple, related genes in a biological pathway15. As such, miRNA modulation using either miRNA inhibitors or mimics is being actively investigated as a means to therapeutically target pathways in disease. However, our understanding of how miRNAs regulate autophagy in pathological settings, such as atherosclerosis, remains incomplete.
Studies from our group and others have identified miR-33 as a key regulator of metabolic gene pathways involved in cholesterol and fatty acid homeostasis14. In humans, miR-33a and b are intronic miRNAs that are co-expressed with their host genes, SREBF2 and SREBF1, which code for transcription factors that regulate cholesterol and fatty acid synthesis/uptake16–18. Thus, transcription of SREBF2 and SREBF1 also results in expression of miR-33a and b, which cooperate with their host genes to balance cellular lipid levels by repressing genes that oppose SREBP-regulated pathways, such those involved in cholesterol efflux (ABCA1, ABCG1)16–18 and fatty acid oxidation (HADHB, CROT, CPT1A, PRKAA1)19–21. Mice have only one copy of miR-33 located within Srebf2, and like its human counterpart it is co-regulated with its host gene16–18. In particular, miR-33 is a potent inhibitor of the cholesterol transporter ABCA1, which contributes to reverse cholesterol transport by mediating both cellular cholesterol efflux and the genesis of high density lipoproteins (HDL)16–18, 20, 22, 23. Inhibition of miR-33 has been shown to increase plasma levels of HDL in mice16–18 and primates24, 25, and to reduce plaque size and inflammation in mouse models of atherosclerosis26–29. Recently, we showed that miR-33 expression is induced by the intracellular pathogen Mycobacterium tuberculosis30, which survives within macrophages by evading autophagy-mediated degradation and promoting the accumulation of lipid bodies that serve as a nutrient source for the bacterium. The induction of miR-33 by M. tuberculosis results in the inhibition of integrated pathways involved in autophagy, lysosomal function and fatty acid oxidation by targeting key effectors of autophagy (Atg5, Atg12, Map1lc3b, Prkaa1, Lamp1) and repressing transcriptional regulators of autophagy and lysosome biogenesis (FOXO3 and TFEB)30, which in turn, enables intracellular survival of the bacterium30. These findings have reinforced the notion that miR-33 orchestrates gene pathway regulation as a means to fine-tune cellular metabolism. As autophagy regulates the catabolism of lipid droplet-stored cholesterol esters5 in addition to mediating bacterial delivery to the lysosome (xenophagy), we hypothesized that miR-33 may restrict the mobilization of cholesterol destined for ABCA1-mediated efflux by repressing autophagy.
Here we show that miR-33 reduces lipid droplet catabolism in macrophage foam cells by repressing key autophagy effectors (Atg5, Atg12, Map1lc3b, Prkaa1, Lamp1) and transcriptional regulators (FOXO3, TFEB). miR-33 silencing promotes lipid droplet mobilization in macrophage foam cells, a process that is upstream of ABCA1-dependent cholesterol efflux. Notably, the ability of miR-33 inhibitors to enhance cholesterol efflux are lost in macrophages deficient in autophagy, despite derepression of ABCA1 expression, highlighting the important contribution of this pathway. Finally, we show that miR-33 inhibition in atherosclerotic mice restores defective autophagy in the aorta and in macrophages of atherosclerotic plaques. Together, these results provide a better understanding of the mechanisms by which miR-33 regulates cellular cholesterol metabolism and identify miR-33 inhibition as a therapeutic means to enhance autophagy in atherosclerosis.
MATERIALS AND METHODS
Materials and Methods are available in the online-only Data Supplement.
RESULTS
miR-33 regulates lipid droplet abundance via lysosomal cholesterol efflux
Autophagy plays a critical role in the delivery of lipid droplets to the lysosome, where lipid droplet-associated neutral lipids are catabolized. To investigate whether miR-33 affects this process, we used coherent anti-Stokes Roman scattering (CARS) microscopy to perform label-free imaging of lipid droplets in macrophages. Peritoneal macrophages transfected with miR-33 and loaded with acetylated low density lipoprotein (acLDL) for 24 h showed increased lipid droplet burden and volume compared to macrophage transfected with control mimic (Figure 1A), suggesting that miR-33 represses lipid droplet catabolism. Macrophage autophagy delivers lipid droplets to the lysosome where stored cholesterol esters are hydrolyzed by lysosomal acid lipase to free cholesterol5, which can then be effluxed from the cell. This pathway can be distinguished from the actions of cytoplasmic neutral cholesteryl ester hydrolases using inhibitors that disrupt lysosomal (chloroquine) or neutral (paraoxon) lipolysis. Thus, to determine whether miR-33 regulation of autophagy contributes to its ability to repress cholesterol efflux, we tranfected peritoneal macrophages with control, miR-33 mimic or anti-miR-33 oligonucleotides and measured cholesterol efflux via ABCA1 in the presence or absence of chloroquine and paraoxon. miR-33 overexpression reduced cholesterol efflux to ApoA1 in peritoneal macrophages treated with vehicle or paraoxon, whereas this effect was abolished in those treated with chloroquine (Fig. 1B), indicating a requirement for lysosomal function. Furthermore, miR-33 inhibition increased cholesterol efflux in mouse peritoneal macrophages treated with vehicle or paraoxon (Fig. 1C), but not chloroquine, suggesting that miR-33 regulates cholesterol availability for efflux by the autophagy pathway.
Figure 1. miR-33 regulates lipid droplet catabolism.
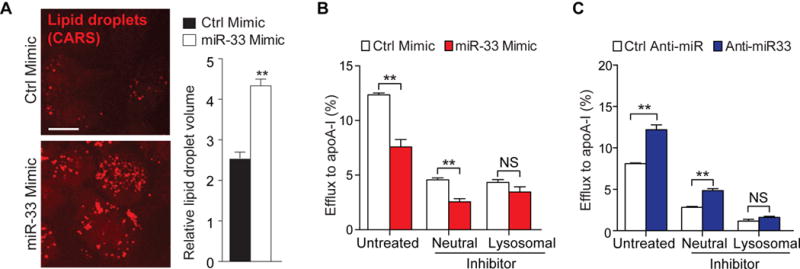
A) Coherent anti-Stokes Raman scattering (CARS) multiphoton imaging of cellular lipid droplets in peritoneal macrophages treated with miR-33 and control mimics and treated with acLDL for 24h. Quantification of lipid droplet volume by voxel analysis is shown at right. Scale bar = 10 μm. B) Cellular efflux of 3H-cholesterol to apolipoprotein A-I (apoA-I, 50μg/mL) in peritoneal macrophages treated with control or miR-33 mimics, in the presence or absence of the neutral lipolysis inhibtor paraoxon (Neutral) or the lysosomal inhibitor chloroquine (Lysosomal). C) Cellular efflux of 3H-cholesterol to apoA-I as in (B), in peritoneal macrophages treated with control or anti-miR-33 inhibitors.. *P < 0.05, **P < 0.01 compared to control treatment. NS, not significant.
miR-33 regulates lipid droplet clearance in vivo via autophagy
Recent studies from our group showed that in M. tuberculosis infected macrophages, miR-33 reduces autophagy-mediated bacterial killing by repressing autophagy effector genes (Atg5, Lamp1, Prkaa1) and AMPK-activated transcription factors (Foxo3, Tcfeb)30. To test whether miR-33 represses autophagy in atherogenic macrophages, we used a model of in vivo foam cell formation, in which we injected hypercholesterolemic Apoe−/− mice with thioglycollate i.p. to elicit the recruitment of peritoneal macrophages and treated mice with control or anti-miR-33 oligonucleotides for 72 h (Figure 2A). Macrophages isolated from anti-miR-33 treated mice showed higher mRNA levels of the miR-33 target genes Atg5, Lamp1, Prkaa1 and Abca1, but not control genes (Apoe, Cd68), compared to macrophages from mice treated with control anti-miR (Figure 2B), thus confirming repression of these genes by endogenous miR-33 in macrophage foam cells. In addition, macrophages from anti-miR-33-treated mice showed higher mRNA levels of Foxo3 and Tcfeb, as well as mRNAs for autophagy-related genes that they regulate (FOXO3: Atg4b, Atg12, Map1lc3b; TFEB: Ctsb, Lipa and Uvrag), compared to macrophages from control anti-miR treated mice (Figure 2C). Consistent with the collective increase in expression of these autophagy genes, western blotting revealed an increase in protein levels of the autophagosome marker LC3 (microtubule-associated protein 1A/1B light chain B) and reduced levels of the chaperone protein p62/SQSTM1 (which is degraded by autophagy31) in macrophages isolated from mice treated with anti-miR-33 compared to control anti-miR (Figure 2D). Immunofluorescent staining for the lipid droplet marker adipophilin showed that macrophages isolated from control anti-miR treated Apoe−/− mice contained abundant adipophilin-positive cytoplasmic neutral lipid droplets, consistent with foam cells, and this was greatly reduced in macrophages from anti-miR-33-treated mice (Figure 2E). Notably, the decrease in lipid droplets in macrophages from anti-miR-33-treated mice was associated with an increase in punctate LC3 staining, consistent with an increase in autophagosomes, and co-localization of LC3 and BODIPY+ puntae indicative of autophagic degradation of lipid droplets (Figure 2F).
Figure 2. Inhibition of endogenous miR-33 promotes autophagy in macrophage foam cells in vivo.
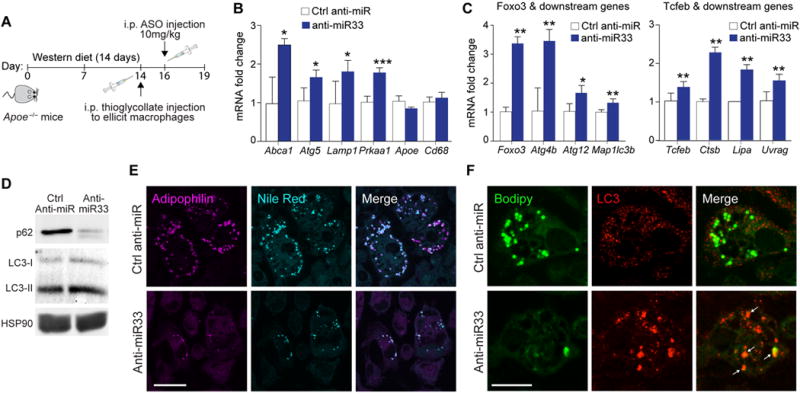
A) Schematic diagram showing the experimental outline for antisense oligonucleotide treatment of male Apoe−/− mice on a Western diet. B-C) qPCR of (B) miR-33 target genes or non-miR-33 targets, as well as (C) Foxo3, Tcfeb and their downstream transcriptional targets in macrophage foam cells isolated hyperlipidemic Apoe−/− mice treated with anti-miR-33 or control anti-miR. D) Western blotting of lysates from peritoneal macrophages isolated from Apoe−/− mice treated with anti-miR-33 or control anti-miR to detect LC3-I (cytosolic), LC3-II (autophagosomal) or the autophagy-chaperone protein p62. HSP90 is shown as an internal control. E) Fluorescence microscopy imaging of Adipophilin and neutral lipid (Nile Red-positive) droplets in peritoneal macrophages isolated from hyperlipidemic Apoe−/− mice treated with anti-miR-33 or control anti-miR. Scale bar = 25μm. F) Fluorescence microscopy imaging of LC3 and neutral lipid (BODIPY-positive) droplets in peritoneal macrophages isolated from hyperlipidemic Apoe−/− mice treated with anti-miR-33 or control anti-miR. Scale bar = 25μm. Data are the mean ± s.e.m. of 3 mice. *P<0.05, **P<0.005.
Regulation of cholesterol homeostasis by miR-33 is dependant on autophagy
To test whether miR-33 regulation of autophagy is essential for its effects on cholesterol efflux, we performed experiments in Atg5−/− autophagy-deficient macrophages. Compared to wild type BMDMs, cholesterol efflux to apoA1 is reduced by approximately 40% in Atg5−/− BMDMs, which represents the contribution of autophagy to the generation of free cholesterol for efflux, with the balance generated by neutral cholesterol ester hydrolysis. Transfection of wild type BMDMs with miR-33 reduced cholesterol efflux to apoA1 compared to control mimic treated cells, whereas miR-33 overexpression failed to decrease cholesterol efflux in Atg5−/− BMDMs (Figure 3A). Correspondingly, transfection of wild type BMDMs with anti-miR-33 increased cholesterol efflux to apoA1 (Figure 3B) and promoted the dissipation of cytoplasmic BODIPY+ lipid droplets (Figure 3C, D), whereas these effects were absent in similarly treated Atg5−/− BMDMs. Notably, wild type and Atg5−/− BMDMs transfected with anti-miR-33 showed equivalent increases in Abca1 mRNA (Figure 3E), indicating that miR-33 regulation of autophagy acts upstream of ABCA1 to control cholesterol availability for efflux.
Figure 3. miR-33 regulation of cholesterol efflux requires autophagy.
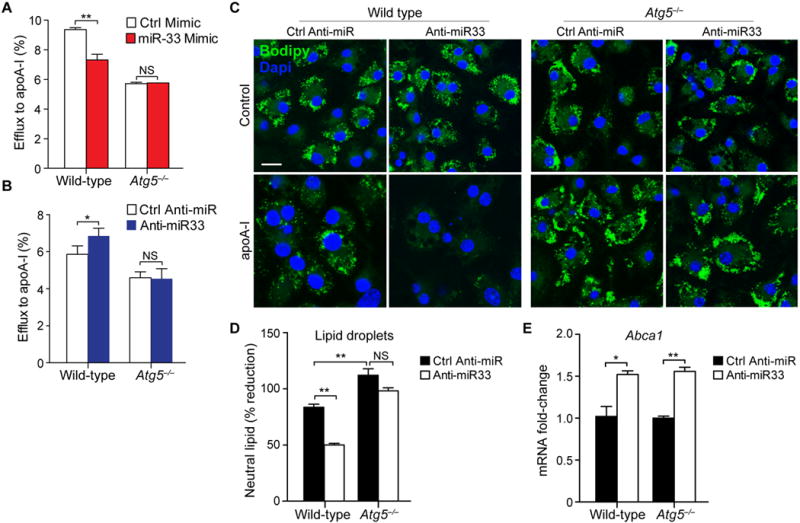
A, B) Cellular efflux of 3H-cholesterol to apoA-I in wild type and Atg5−/− macrophages treated with miR-33 and control (A) mimics or inhibitors (B). C) Immunofluorescence imaging of Bodipy-stained lipid droplets in Ctrl anti-miR or anti-miR-33 treated wild type and Atg5−/− macrophages loaded with acetylated LDL. Cells were imaged prior to (control) or after incubation with apoA-I for 4h to promote cholesterol efflux. Scale bar, 25μm. D) Quantification of neutral lipid content of cells shown in (C). E) qRT-PCR of Abca1 mRNA in cells shown in (C). *P<0.05, **P<0.005. NS, not significant.
Antagonism of miR-33 promotes autophagy in atherosclerotic plaques
Recent studies indicate that autophagy becomes impaired in atherosclerosis, and its dysfunction contributes to vascular inflammation and plaque progression8, 10. To investigate whether miR-33 inhibition could restore autophagy in atherosclerotic mice, we fed Ldlr−/− mice a western diet for 14 weeks to establish atherosclerotic plaques, and then treated mice with control anti-miR or anti-miR-33 for 4 weeks on a chow diet (Figure 4A). Using this model, we previously reported that anti-miR-33 treatment reduced atherosclerotic plaque size by 35%, compared to control anti-miR27. To evaluate autophagy in these mice, we first measured p62 protein as this chaperone protein has been shown to accumulate in the aorta with atherosclerosis progression8. Aortic lysates from control anti-miR treated Ldlr−/− mice showed high levels of p62 protein, consistent with defective autophagy (Figure 4B). Notably, treatment with anti-miR-33 reduced aortic p62 protein levels by approximately 40% (Figure 4B). Similarly, immunofluorescent staining for p62 in cross-sections of the aortic root showed markedly less p62 in CD68+ plaque macrophages of anti-miR-33 treated mice compared to control anti-miR treated mice (Figure 4C, D). Consistent with the reduced accumulation of p62 in anti-miR-33 treated mice, we observed an increase in LC3 staining in plaques from these mice, compared to control anti-miR treated mice (Figure 5A). Whereas CD68+ plaque macrophages from control anti-miR treated mice showed little LC3 staining, we observed abundant LC3 staining in CD68+ macrophages in plaques of anti-miR-33 treated mice (Figure 5A, B), which combined with our observation of reduced p62 levels suggests elevated autophagy flux in plaques of anti-miR33 treated mice. Interestingly, we also observed some LC3 staining in cells that did not express CD68, which are likely of smooth muscle cell origin (Figure 5A, B). Taken together, these data indicate that inhibition of endogenous miR-33 in vivo restores autophagy function in atherosclerotic plaques.
Figure 4. miR-33 inhibition restores defective autophagy in atherosclerosis.
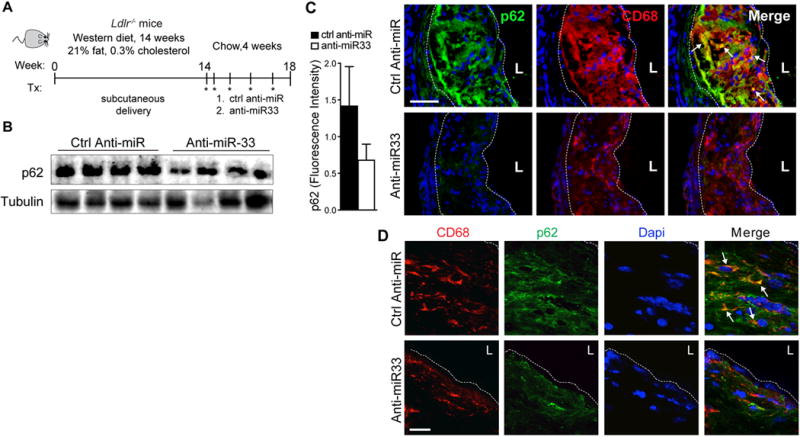
A) Schematic diagram showing the treatment regimen of male Ldlr−/− mice on a Western diet. B) Western blotting of the autophagy chaperone protein p62 in aortic lysates of Ldlr−/− mice (n=4) that were fed a western diet for 14 weeks and then treated for 4 weeks with anti-miR-33 or control anti-miR. C) Representative immunofluorescence staining of CD68 (red) and p62 (green) and their colocalization (yellow) in atherosclerotic plaques of Ldlr−/− mice described in (A). At left, quantification of p62 fluorescence intensity in plaques is shown (n=7 mice / group). D) Confocal microscopy of p62 (green) and CD68 (red), and their colocalization (merge), in aortic sinus atherosclerotic plaques of Ldlr−/− mice undergoing atherosclerosis regression. Dashed lines indicate plaque borders. L= lumen. Scale bar = 100μm.
Figure 5. miR-33 inhibition increases LC3 expression in atherosclerotic plaques.
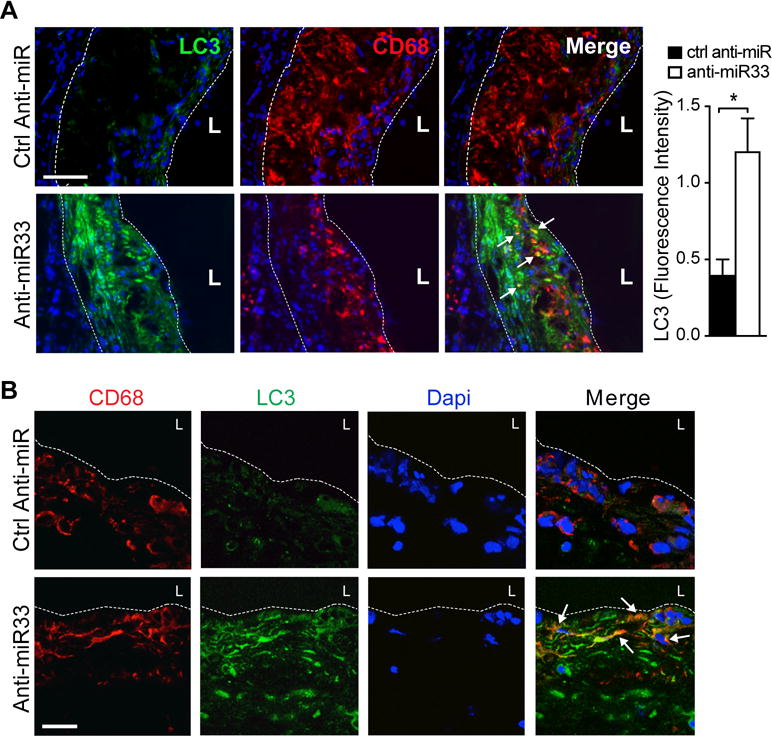
A) Representative immunofluorescence staining of CD68 (red) and LC3 (green) and their colocalization (yellow) in atherosclerotic plaques of Ldlr−/− mice described in (A). At right, quantification of LC3 fluorescence intensity in plaques is shown (n=7 mice / group). B) Confocal microscopy of LC3 (green) and CD68 (red), and their colocalization (merge), in aortic sinus atherosclerotic plaques of Ldlr−/− mice undergoing atherosclerosis regression. Dashed lines indicate plaque borders. L= lumen. Scale bar = 100μm.
miR-33 regulates efferocytosis in atherosclerosis
In addition to improvement in markers of autophagy, we observed that plaques from anti-miR-33 treated mice had reduced necrotic area compared to plaques from control anti-miR treated mice (Figure 6A). As plaque necrotic core formation is thought to result from defective clearance of apoptotic cells, or efferocytosis6, and autophagy can promote efferocytosis by regulating the efficient recognition of apoptotic cells and degradation of phagocytosed material10, 32–34, we investigated whether miR-33 alters these processes. To test the impact of endogenous miR-33 on efferocytosis, we transfected BMDMs with anti-miR-33 or control anti-miR and measured their uptake of fluorescently-labeled apoptotic Jurkat cells. Inhibition of miR-33 increased internalization of apoptotic cells by BMDMs by 30% compared to control anti-miR treatment (Figure 6B). Inhibition of miR-33 also increased efferocytotic capacity, as evidenced by a greater number of apoptotic cells per macrophage in anti-miR-33 compared to control anti-miR treated BMDMs (Figure 6C). Notably, these effects were dependent on ATG5, as miR-33 inhibition failed to increase efferocytosis in Atg5−/− macrophages (Figure 6D). As we observed an increase in TFEB upon miR-33 inhibition, we postulated that this may increase lysosome biogenesis and thus, the degradative capacity of anti-miR-33 treated macrophages. Indeed, we observed an increase in LAMP1-positive lysosomes in anti-miR-33 compared to control anti-miR treated BMDMs (Figure 6E). Consistent with an increase in lysosomal function, anti-miR-33 treated macrophages incubated with apoptotic cells contained a greater number of small diffuse apoptotic bodies and fewer intact intracellular apoptotic cells than control anti-miR treated BMDMs (Figure 6F). Together, these data suggest that by enhancing autophagy and lysosomal biogenesis, miR-33 inhibition increases macrophage apoptotic cell clearance and digestion of apoptotic cargo, which likely contribute to its beneficial effects on atherosclerosis.
Figure 6. miR-33 regulates efferocytosis in atherosclerosis.
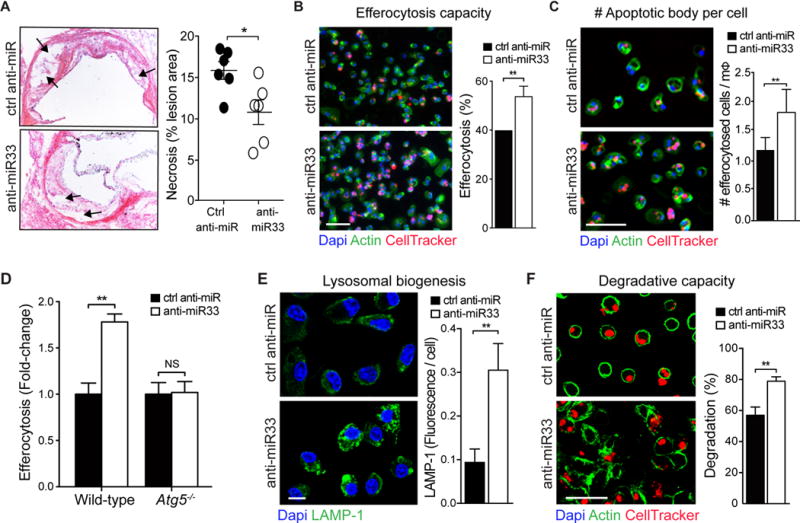
A) Quantification of necrotic area (depicted by arrows) in the aortic roots of ctrl anti-miR or anti-miR-33 treated mice, with representative images of the aortic root lesions stained with H&E presented at left of graphs. n=6 mice/group. Original magnification, ×10. B-D and F) Apoptotic Jurkat cells were added onto peritoneal macrophages treated with ctrl anti-miR or anti-miR-33, and efferocytosis was assayed and expressed as the percent of cells having efferocytosed (B), the number of ingested apoptotic cells per macrophage (mΦ) (C) or the number of cells containing cellular debris (F). In (D), the ability of miR-33 inhibition to regulate efferocytosis in autophagy-deficient Atg5−/− macrophages was measured as compared to wild-type. E) Fluorescence microscopy of lysosomal-associated membrane protein 1 (LAMP-1) in macrophages treated with anti-miR-33 or control anti-miR. (B, C, E, F) Scale bar = 50μm. *P < 0.05, **P < 0.01 compared to control treatment. NS, not significant.
DISCUSSION
In this study, we show that miR-33 has potent effects on autophagy and lysosomal function that reinforce its targeting of cholesterol efflux and reverse cholesterol transport gene pathways to mediate cholesterol homeostasis. Previous studies by our group and others established miR-33 as a potent inhibitor of cholesterol efflux and reverse cholesterol transport18, 27 through its targeting of key genes involved in cholesterol transport (Abca1 and Abcg116–18), biliary excretion (Atp8b1 and Abcb1135) and intracellular trafficking (Npc118 and Osbpl636). We now show that by targeting key autophagy regulators and effectors in macrophages, miR-33 reduces the catabolism of lipid droplets by lysosomes, a process that generates free cholesterol for efflux. Notably, miR-33 regulation of lipophagy lies upstream of its effects on ABCA1-dependent cholesterol efflux as miR-33 inhibitors fail to increase cholesterol efflux to apoA1 in the presence of genetic or chemical inhibitors of autophagy, despite increases in macrophage ABCA1 expression. These findings suggest that miR-33 primarily restricts the generation of free cholesterol via the acid lipolysis pathway, but does not affect neutral cholesterol ester hydrolysis. Together, these findings underscore the critical contribution of miR-33 regulation of autophagy to its effects on cellular lipid metabolism. They also highlight the importance of miRNAs as orchestrators of pathway control that can have profound effects on biological processes by targeting multiples genes within related pathways.
Recent studies in mouse models of atherosclerosis have shown that macrophage autophagy becomes defective with disease progression, resulting in accelerated atherogenesis8, 10. These studies suggest that mechanisms that increase autophagy in plaque macrophages may restore defective lipid metabolism and reduce inflammation, leading to plaque regression. We previously showed that miR-33 expression is increased in human atherosclerotic plaques, and that miR-33 expression is particularly abundant in pro-inflammatory M1 macrophages - the predominant macrophage phenotype in progressing plaques - as compared to pro-resolving M2 macrophages26, raising the possibility that miR-33 may contribute to autophagy inhibition in atherosclerosis. Indeed, we show herein that treating hypercholesterolemic mice with miR-33 inhibitors increases autophagosome formation and reduces lipid droplet burden in macrophage foam cells, indicating repression of these processes by miR-33 in vivo. Furthermore, treating atherosclerotic Ldlr−/− mice with miR-33 inhibitors for 4 weeks reduced p62 accumulation in the aorta and in plaque macrophages, and also increased LC3 levels in plaques, consistent with improved autophagy. These findings suggest that miR-33 inhibition may be a promising strategy to reverse autophagy dysfunction in atherosclerosis. Importantly, inhibition of miR-33 not only increases expression of its direct target genes in the autophagy pathway (mouse: Atg5, Lamp1, Prkaa1; human: ATG5, ATG12, MAP1LC3B,PRKAA1, LAMP1)30, but also promotes AMPK-dependent activation of the FOXO3 and TFEB transcription factors30, which sustains autophagy via an autoregulatory feedfoward loop31, 37. Although our study does not discriminate the contribution of miR-33’s effects on transcriptional regulation of autophagy from its repression of its direct target genes in the autophagy pathway, it highlights the roles of miRNAs as pathway regulators, which can have profound effects on biological processes by targeting multiple genes within a related pathway.
By regulating AMPK-dependent activation of FOXO3 and TFEB, miR-33 may have broad effects on lysosomal trafficking and function that reinforce its targeting of key autophagy effector genes. We recently showed that miR-33 inhibition increases TFEB protein levels and its translocation to the nucleus30, where activated TFEB promotes the expression of a network of genes involved in lysosome and autophagosome biogenesis and function38, 39. We show herein that treatment of Ldlr−/− mice with anti-miR-33 increases expression of TFEB and its transcriptional targets Ctsb (cathepsin B), Lipa (lysosomal acid lipase) and Uvrag (UV radiation resistance-associated gene) in macrophage foam cells. TFEB overexpression in atherosclerotic macrophages has been reported to enhance prodegradative responses and rescue macrophage defects seen with atherogenic lipid loading, including lysosomal dysfunction, inflammasome activation, and polyubiquitinated protein aggregation40. Thus, the potential of miR-33 inhibitors to simultaneously increase expression of genes in the autophagy pathway and activation of TFEB represents a powerful approach to enhance macrophage function in atherosclerosis.
Stimulation of the autophagolysosomal pathway in macrophages through miR-33 inhibition is likely to promote multiple protective functions that limit plaque progression, including enhancing lipid droplet catabolism and ABCA1-dependent efflux, reducing inflammasome activation, and promoting the efferocytosis of apoptotic cells8, 10, 41. Indeed, we previously showed that plaques from atherosclerotic Ldlr−/− mice treated with miR-33 inhibitors for 4 weeks have reduced plaque size, lipid content and inflammation compared to plaques from control anti-miR treated mice26, 27. Although these beneficial effects of miR-33 inhibition were initially attributed to increased levels of plasma HDL cholesterol and macrophage reverse cholesterol transport27, our current data reveals a previously unappreciated effect on plaque macrophage autophagy that would increase both cholesterol mobilization for efflux and apoptotic cell clearance. Efferocytosis becomes impaired in atherosclerotic plaques, and if not cleared, apoptotic cells undergo secondary necrosis, leading to the formation of necrotic cores that threaten plaque stability42. We find that inhibition of miR-33 in vitro promotes macrophage efferocytosis, lysosomal biogenesis and degradation of apoptotic material – processes that would stimulate tissue repair and plaque regression. Given that ATG5-deficient macrophages are capable of phagocytosing apoptotic cells at similar rates to that of wild-type7, 34, the increase in ATG5-mediated efferocytosis in anti-miR33 treated macrophages relative to wild type may be attributable to an enhanced degradative capacity of these cells and/or increased LC3. Indeed, LC3-associated phagocytosis (LAP) was recently shown to be activated in response to engulfment of apoptotic cells, and to promote degradation of the endocytosed material33, 34. This process is distinct from the classical autophagy pathway, but requires ATG533. Consistent with an improvement in efferocytosis with anti-miR33 treatment, we find that atherosclerotic Ldlr−/− mice treated with miR-33 inhibitors have reduced plaque necrotic area compared to control anti-miR treated mice, and similar findings were also observed in diabetic Ldlr−/− mice treated with anti-miR-3343. These beneficial effects related to anti-miR-33-dependent derepression of the autophagy pathway are likely to work in concert with anti-miR-33’s other known anti-atherosclerotic mechanisms (e.g., increasing HDL, reverse cholesterol transport, mitochondrial biogenesis44, fatty acid oxidation20, 23, and M2 macrophage polarization26).
Collectively, our data provide insight into the mechanisms by which miR-33 regulates cellular cholesterol homeostasis, and how its expression during atherogenesis may contribute to disease progression. Mir33 (MIR33A in humans) is embedded within an intron of the Srebf2 gene, and is transcriptionally upregulated with its host gene under conditions of low sterol. Whereas the SREBP2 protein acts to increase expression of genes involved in cholesterol synthesis and uptake (eg. Ldlr, Hmgcs, Hmgcr), miR-33 inhibits gene pathways involved in cholesterol ester hydrolysis and free cholesterol export from the cell, including intracellular trafficking and cholesterol efflux, and as described herein, autophagy and lysosomal function. Osborne and colleagues demonstrated that SREBP-2 activates autophagy genes during sterol depletion, and suggested that SREBP-2 activation provides the cell with lipids from three sources: de novo biosynthesis, lipoprotein uptake, and lipid droplet mobilization through lipophagy45. Conversely, we find that miR-33, which is co-transcribed with SREBP-2, can restrict cholesterol mobilization from lipid droplets by inhibiting autophagy. It is thus possible that co-regulation of miR-33 along with SREBP-2 could provide a negative feedback regulation of this transcription factor and allow for fine-tuning of the autophagy pathway, although the precise interplay of SREBP-2 and miR-33 to regulate autophagy remains to be fully elucidated. This miR-33/SREBP2 circuit provides an integrated system to boost cellular cholesterol levels during times of cholesterol demand. However, miR-33 expression in atherosclerotic macrophages may contribute to pathological processes, such as reducing autophagy and cholesterol efflux, which contribute to foam cell dysfunction. Although the mechansims that drive miR-33 expression in atheroma are not yet known44, we recently reported that miR-33 is induced by M. tuberculosis through an NFkB-dependent mechanism30 raising the possibility that endogenous atherosclerotic ligands that activate NFkB may contribute. Strategies that inhibit this expression of miR-33 in plaque macrophages would provide a powerful approach to restore macrophage function in atherosclerosis by increasing interrelated pathways involved in autophagy, lysosomal function, cholesterol trafficking and immunometabolism.
Supplementary Material
HIGHLIGHTS.
By repressing key autophagy effectors and transcriptional activators (FOXO3, TFEB), miR-33 reduces autophagy and lysosomal function in macrophages.
miR-33 targeting of autophagy acts upstream of its known target ABCA1, to restrict lipid droplet catabolism and generation of free cholesterol for efflux. miR-33 inhibitors fail to increase cholesterol efflux in the absence of autophagy, despite upregulation of the ABCA1 cholesterol transporter.
miR-33 inhibition in macrophages increases autophagy, lipid droplet catabolism, efferocytosis and apoptotic cell digestion.
Treatment of atherosclerotic Ldlr−/− mice with miR-33 inhibitors for 4 weeks reverses macrophage autophagy dysfunction in atherosclerotic plaques.
Acknowledgments
We thank Regulus Therapeutics for providing the anti-miR-33 and control oligonucleotides. Sources of funding: Support for this work came from the National Institutes of Health (R01HL119047 to K.J.M. and K.J.R.; R01HL129433 and R35HL135799-01 to K.J.M. and E.A.F.; 1 R01 HL106019 NIH/NHLBI to I.T.), the American Heart Association (15POST25090199 to M.O.; 13POST14490016 and K99/R00HL125667 to B.R.), K.J.R. is supported by Canadian Institutes of Health Research (MOP130365 and MSH130157).
NONSTANDARD ABBREVIATIONS AND ACRONYMS
- AcLDL
Acetylated low-density lipoprotein
- APOA1
Apolipoprotein A-I
- ABCA1
ATP-Binding cassette transporter A1
- ABCG1
ATP-binding cassette transporter G1
- AMPK
5′ AMP-activated protein kinase
- BMDM
Bone marrow derived macrophages
- CARS
Coherent anti-stokes raman scattering
- FOXO3
Forkhead box O3
- HDL
High density lipoprotein
- LAL
Lysosomal acid lipase
- LC3
Microtubule-associated protein 1A/1B light chain B
- miRNA
microRNA
- miR-33
miRNA-33
- p62
Sequestosome-1 or Ubiquitin-binding protein p62
- RCT
reverse cholesterol transport
- TFEB
Transcription factor EB
Footnotes
Disclosures: KJM and KJR have a patent on the use of miR-33 inhibitors to treat inflammation.
References
- 1.Moore KJ, Sheedy FJ, Fisher EA. Macrophages in atherosclerosis: A dynamic balance. Nature reviews Immunology. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ouimet M. Autophagy in obesity and atherosclerosis: Interrelationships between cholesterol homeostasis, lipoprotein metabolism and autophagy in macrophages and other systems. Biochimica et biophysica acta. 2013;1831:1124–1133. doi: 10.1016/j.bbalip.2013.03.007. [DOI] [PubMed] [Google Scholar]
- 3.Singh R, Cuervo AM. Autophagy in the cellular energetic balance. Cell metabolism. 2011;13:495–504. doi: 10.1016/j.cmet.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell metabolism. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nature reviews Immunology. 2010;10:36–46. doi: 10.1038/nri2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zang L, Xu Q, Ye Y, Li X, Liu Y, Tashiro S, Onodera S, Ikejima T. Autophagy enhanced phagocytosis of apoptotic cells by oridonin-treated human histocytic lymphoma u937 cells. Archives of biochemistry and biophysics. 2012;518:31–41. doi: 10.1016/j.abb.2011.11.019. [DOI] [PubMed] [Google Scholar]
- 8.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell metabolism. 2012;15:534–544. doi: 10.1016/j.cmet.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinet P, Ritchey B, Smith JD. Physiological difference in autophagic flux in macrophages from 2 mouse strains regulates cholesterol ester metabolism. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:903–910. doi: 10.1161/ATVBAHA.112.301041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell metabolism. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rayner KJ, Moore KJ. Microrna control of high-density lipoprotein metabolism and function. Circulation research. 2014;114:183–192. doi: 10.1161/CIRCRESAHA.114.300645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gupta SK, Thum T. Non-coding rnas as orchestrators of autophagic processes. Journal of molecular and cellular cardiology. 2016;95:26–30. doi: 10.1016/j.yjmcc.2015.11.012. [DOI] [PubMed] [Google Scholar]
- 13.Zhai H, Fesler A, Ju J. Microrna: A third dimension in autophagy. Cell cycle (Georgetown, Tex) 2013;12:246–250. doi: 10.4161/cc.23273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feinberg MW, Moore KJ. Microrna regulation of atherosclerosis. Circulation research. 2016;118:703–720. doi: 10.1161/CIRCRESAHA.115.306300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bartel DP. Micrornas: Target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marquart TJ, Allen RM, Ory DS, Baldan A. Mir-33 links srebp-2 induction to repression of sterol transporters. Proc Natl Acad Sci U S A. 2010;107:12228–12232. doi: 10.1073/pnas.1005191107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Najafi-Shoushtari SH, Kristo F, Li Y, Shioda T, Cohen DE, Gerszten RE, Naar AM. Microrna-33 and the srebp host genes cooperate to control cholesterol homeostasis. Science. 2010;328:1566–1569. doi: 10.1126/science.1189123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rayner KJ, Suarez Y, Davalos A, Parathath S, Fitzgerald ML, Tamehiro N, Fisher EA, Moore KJ, Fernandez-Hernando C. Mir-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramirez CM, Davalos A, Goedeke L, Salerno AG, Warrier N, Cirera-Salinas D, Suarez Y, Fernandez-Hernando C. Microrna-758 regulates cholesterol efflux through posttranscriptional repression of atp-binding cassette transporter a1. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:2707–2714. doi: 10.1161/ATVBAHA.111.232066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerin I, Clerbaux LA, Haumont O, Lanthier N, Das AK, Burant CF, Leclercq IA, MacDougald OA, Bommer GT. Expression of mir-33 from an srebp2 intron inhibits cholesterol export and fatty acid oxidation. J Biol Chem. 2010;285:33652–33661. doi: 10.1074/jbc.M110.152090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rottiers V, Najafi-Shoushtari SH, Kristo F, Gurumurthy S, Zhong L, Li Y, Cohen DE, Gerszten RE, Bardeesy N, Mostoslavsky R, Naar AM. Micrornas in metabolism and metabolic diseases. Cold Spring Harbor symposia on quantitative biology. 2011;76:225–233. doi: 10.1101/sqb.2011.76.011049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horie T, Ono K, Horiguchi M, Nishi H, Nakamura T, Nagao K, Kinoshita M, Kuwabara Y, Marusawa H, Iwanaga Y, Hasegawa K, Yokode M, Kimura T, Kita T. Microrna-33 encoded by an intron of sterol regulatory element-binding protein 2 (srebp2) regulates hdl in vivo. Proc Natl Acad Sci U S A. 2010;107:17321–17326. doi: 10.1073/pnas.1008499107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davalos A, Goedeke L, Smibert P, et al. Mir-33a/b contribute to the regulation of fatty acid metabolism and insulin signaling. Proc Natl Acad Sci U S A. 2011;108:9232–9237. doi: 10.1073/pnas.1102281108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rayner K, et al. Inhibition of mir-33a/b in non-human primates raises plasma hdl and lowers vldl triglycerides. Nature. 2011;478:404–409. doi: 10.1038/nature10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rottiers V, Obad S, Petri A, et al. Pharmacological inhibition of a microrna family in nonhuman primates by a seed-targeting 8-mer antimir. Science translational medicine. 2013;5:212ra162. doi: 10.1126/scitranslmed.3006840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ouimet M, Ediriweera HN, Gundra UM, et al. Microrna-33-dependent regulation of macrophage metabolism directs immune cell polarization in atherosclerosis. J Clin Invest. 2015;125:4334–4348. doi: 10.1172/JCI81676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rayner KJ, Sheedy FJ, Esau CC, Hussain FN, Temel RE, Parathath S, van Gils JM, Rayner AJ, Chang AN, Suarez Y, Fernandez-Hernando C, Fisher EA, Moore KJ. Antagonism of mir-33 in mice promotes reverse cholesterol transport and regression of atherosclerosis. J Clin Invest. 2011;121:2921–2931. doi: 10.1172/JCI57275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Horie T, Baba O, Kuwabara Y, et al. Microrna-33 deficiency reduces the progression of atherosclerotic plaque in apoe−/− mice. Journal of the American Heart Association. 2012;1:e003376. doi: 10.1161/JAHA.112.003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rotllan N, Ramirez CM, Aryal B, Esau CC, Fernandez-Hernando C. Therapeutic silencing of microrna-33 inhibits the progression of atherosclerosis in ldlr−/− mice–brief report. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:1973–1977. doi: 10.1161/ATVBAHA.113.301732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ouimet M, Koster S, Sakowski E, et al. Mycobacterium tuberculosis induces the mir-33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol. 2016;17:677–686. doi: 10.1038/ni.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klionsky DJ, Abdalla FC, Abeliovich H, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012;8:445–544. doi: 10.4161/auto.19496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zang L, He H, Ye Y, Liu W, Fan S, Tashiro S, Onodera S, Ikejima T. Nitric oxide augments oridonin-induced efferocytosis by human histocytic lymphoma u937 cells via autophagy and the nf-kappab-cox-2-il-1beta pathway. Free radical research. 2012;46:1207–1219. doi: 10.3109/10715762.2012.700515. [DOI] [PubMed] [Google Scholar]
- 33.Lai SC, Devenish RJ. Lc3-associated phagocytosis (lap): Connections with host autophagy. Cells. 2012;1:396–408. doi: 10.3390/cells1030396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, Green DR. Microtubule-associated protein 1 light chain 3 alpha (lc3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A. 2011;108:17396–17401. doi: 10.1073/pnas.1113421108. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 35.Allen RM, Marquart TJ, Albert CJ, Suchy FJ, Wang DQ, Ananthanarayanan M, Ford DA, Baldan A. Mir-33 controls the expression of biliary transporters, and mediates statin- and diet-induced hepatotoxicity. EMBO Mol Med. 2012;4:882–895. doi: 10.1002/emmm.201201228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ouimet M, Hennessy EJ, van Solingen C, et al. Mirna targeting of oxysterol-binding protein-like 6 regulates cholesterol trafficking and efflux. Arteriosclerosis, thrombosis, and vascular biology. 2016;36:942–951. doi: 10.1161/ATVBAHA.116.307282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sanchez AM, Candau RB, Csibi A, Pagano AF, Raibon A, Bernardi H. The role of amp-activated protein kinase in the coordination of skeletal muscle turnover and energy homeostasis. Am J Physiol Cell Physiol. 2012;303:C475–485. doi: 10.1152/ajpcell.00125.2012. [DOI] [PubMed] [Google Scholar]
- 38.Palmieri M, Impey S, Kang H, di Ronza A, Pelz C, Sardiello M, Ballabio A. Characterization of the clear network reveals an integrated control of cellular clearance pathways. Human molecular genetics. 2011;20:3852–3866. doi: 10.1093/hmg/ddr306. [DOI] [PubMed] [Google Scholar]
- 39.Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P, Sardiello M, Rubinsztein DC, Ballabio A. Tfeb links autophagy to lysosomal biogenesis. Science. 2011;332:1429–1433. doi: 10.1126/science.1204592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Emanuel R, Sergin I, Bhattacharya S, Turner JN, Epelman S, Settembre C, Diwan A, Ballabio A, Razani B. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid-induced lysosomal dysfunction and downstream sequelae. Arteriosclerosis, thrombosis, and vascular biology. 2014;34:1942–1952. doi: 10.1161/ATVBAHA.114.303342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Le Guezennec X, Brichkina A, Huang YF, Kostromina E, Han W, Bulavin DV. Wip1-dependent regulation of autophagy, obesity, and atherosclerosis. Cell metabolism. 2012;16:68–80. doi: 10.1016/j.cmet.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 42.Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Distel E, Barrett TJ, Chung K, Girgis NM, Parathath S, Essau CC, Murphy AJ, Moore KJ, Fisher EA. Mir33 inhibition overcomes deleterious effects of diabetes mellitus on atherosclerosis plaque regression in mice. Circulation research. 2014;115:759–769. doi: 10.1161/CIRCRESAHA.115.304164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karunakaran D, Thrush AB, Nguyen MA, et al. Macrophage mitochondrial energy status regulates cholesterol efflux and is enhanced by anti-mir33 in atherosclerosis. Circulation research. 2015;117:266–278. doi: 10.1161/CIRCRESAHA.117.305624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Seo YK, Jeon TI, Chong HK, Biesinger J, Xie X, Osborne TF. Genome-wide localization of srebp-2 in hepatic chromatin predicts a role in autophagy. Cell metabolism. 2011;13:367–375. doi: 10.1016/j.cmet.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.