

Abstract
Transforming growth factor-β1 (TGF-β1), secreted by main components of tumor microenvironment, is considered to be closely associated with cancer development and chemoresistance. The present study aimed to analyze the effects and mechanisms underlying TGF-β1-induced chemoresistance to oxaliplatin (LOH) in human colorectal cancer (CRC) cell lines. The cytotoxic effects of LOH subsequent to TGF-β1 treatment were assessed in three CRC cell lines by MTT assay. In addition, epithelial to mesenchymal transition (EMT), DNA damage and apoptosis assays were performed to evaluate the mechanisms of drug resistance in vitro. It was revealed that an exposure of CRC cells to TGF-β1 induced EMT. This was followed by a decrease in the levels of DNA damage and LOH-induced apoptotic cell death at certain TGF-β1 concentrations compared with untreated cells, which was responsible for LOH resistance. TGF-β1 leads to resistance to LOH in CRC cells, primarily through EMT. These data not only provide insight into the understanding of the chemoresistant mechanisms, but also may guide the clinical applications of reducing EMT to enhance the sensitivity to chemotherapy, by targeting TGF-β1.
Keywords: TGF-β1, chemoresistance, epithelial to mesenchymal transition, colorectal cancer, oxaliplatin
Introduction
Colorectal cancer (CRC) is one of the most prevalent and fatal types of disease worldwide (1). Although significant progress has been made in diagnostic techniques, chemotherapy and surgery, ~70–80% of patients with CRC will experience recurrence or metastasis, resulting from the dormancy and spread of chemotherapy-resistant cells and the majority of patients with metastatic CRC (mCRC) will succumb to the disease eventually (2,3). However, the mechanisms of chemoresistance in CRC have not been fully clarified.
Current evidence reveals that the complexity of the tumor microenvironment (TME) serves a critical role in altering the biological behavior of cancer cells, including resistance to conventional chemotherapy (4). TME is a dynamic and complicated system that consists of cancer-associated fibroblasts, endothelial cells and immune cells, and exhibits high levels of cytokines, growth factors and hormones (5,6). Previously, numerous studies indicated that a small number of cells and components may release transforming growth factor-β1 (TGF-β), and TGF-β results in an alteration in chemosensitivity in certain situations in the TME (4,7–14).
TGF-β signaling pathways are considered necessary for cancer progression (7). The main types of TGF-β, TGF-β1, TGF-β2 and TGF-β3, regulate cancer cell proliferation, migration, differentiation, apoptosis and chemosensitivity in TME (15). Of the three ligands, TGF-β1 is the most extensively studied isoform (16). TGF-β1 gives rise to drug resistance via the promotion of EMT, cancer stem cell-like properties, participating in crosstalk with interleukin 6, regulating DNA mismatch repair system and inducing cell cycle arrest and autophagy in several cancers (8–14). By contrast, blocking the TGF-β1 pathway may enhance the efficacy of chemotherapy under certain circumstances (13). These data have ensured that TGF-β1 is one of the most important types of components that affect chemoresistance in TME.
In previous decades, due to the use of standard chemotherapy containing oxaliplatin (LOH), the response rates, progression-free and overall survival have been significantly improved in patients with CRC (17,18). At present, combinations of chemotherapy containing LOH continue to exhibit powerful efficacy in the treatment of CRC (19). However, the alteration in sensitivity of CRC cells to LOH under the administration of TGF-β1 has not been previously demonstrated.
Thus, the aims of the present study were to assess whether TGF-β1 contributes to LOH chemosensitivity in cultured CRC cell lines, and to explore the mechanisms by which TGF-β1 is responsible for these effects.
Materials and methods
Cell culture and reagents
The human CRC HCT-116, CL187 and SW620 cell lines were all purchased from the ATCC (American Type Culture Collection). HCT-116 cells were cultured in McCoy's 5A medium (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). CL187 cells were cultured in high glucose Dulbecco's Modified Eagle Medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and SW620 cells were cultured in Leibovitz-15 (Gibco; Thermo Fisher Scientific, Inc.). The cell lines were cultured with 10% heat-inactivated fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.), 50 U/ml penicillin and 50 µg/ml streptomycin at 37°C supplied with 5% CO2. Human recombinant TGF-β1 (rh TGF-β1) was purchased from PeproTech, Inc. (Rocky Hill, NJ, USA); LOH was obtained from HengRui Therapeutics, Inc., (Princeton, NJ, USA). LOH was dissolved with 5% glucose at 10 mM prior to dilution to the indicated concentrations with culture medium prior to use. RhTGF-β1 was diluted according to the manufacturer's protocol.
Cell viability and proliferation assays
Cell viability was measured by MTT assay. CRC cells were plated at a density of 3×103 per well in 96-well plates. RhTGF-β1 was added at the final concentrations of 0, 3 or 5 ng/ml, or 0–20 ng/ml after 24 h. At 72 h subsequent to rhTGF-β1 treatment, CRC cells were then exposed to increasing doses of LOH for an additional 72 h, to detect LOH resistance. Then, 4 or 7 days subsequent to plating, 0.5 mg/ml MTT solution was added to the medium for 4 h incubation at 37°C, and then 100 µl dimethyl sulfoxide was added to each well. The absorbance at 570 nm was determined with a scanning multi-well spectrophotometer (Tecan US, Inc., Morrisville, NC, USA). Each assay was performed in triplicate.
Western blot analysis
HCT-116 cells were treated with rhTGF-β1 at a gradient of concentrations [1, 3, 5, 10 and 20 ng/ml], simultaneously, a mock and TGF-β1 antibody (Santa Cruz Biotechnology, Inc., Dallas, TX, USA) served as controls. Western blot analysis was performed as described previously (20). Cells were harvested at 3 days subsequent to treatment and lysed in a radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime Institute of Biotechnology, Shanghai, China) containing phenylmethylsulfonyl fluoride and leupeptin for 30 min at room temperature (RT). The soluble protein concentration was determined by bicinchoninic acid (BCA) protein assay (Boster, Wuhan, China). Protein (80 µg) was electrophoresed in SDS-PAGE and transferred to polyvinylidene fluoride membranes (EMD Millipore, Billerica, MA, USA). Primary monoclonal antibody to E-cadherin (catalog no. 20874-1-AP; dilution, 1:1,000; ProteinTech Group, Inc., Chicago, IL, USA), N-cadherin (catalog no. 610920; dilution, 1:1,000; BD Transduction Laboratories, New Jersey, USA), Snail (catalog no. #3895; dilution, 1:1,000; Cell Signaling Technology, Massachusetts, USA) and GAPDH (catalog no. G8795; dilution, 1:10,000; Sigma-Aldrich; Merck KGaA) were incubated with polyvinylidene fluoride membranes overnight at 4°C. Binding of the primary antibody was detected by horse anti-mouse immunoglobulin (catalog no. 7076; dilution, 1:2,000; Cell Signaling Technology, Inc., Danvers, MA, USA) or goat anti-rabbit immunoglobulin (catalog no. 7077; dilution, 1:2,000; Cell Signaling Technology, Inc.) conjugated to horseradish peroxidase, which was incubated for 1 h at room temperature. Electrochemiluminescent reagents were used to test the signals, according to the manufacturer's instructions (TransGen Biotech, Inc. Beijing, China). ImageJ software (version Java 1.6.0_20; National Institutes of Health, Bethesda, MD, USA) was used to analyze the gray value of each band.
Cell immunofluorescence staining
HCT-116 cells were seeded on 8-well chamber slides (BD-falcon; BD Biosciences, Franklin Lakes, NJ, USA) at ~1×104 per well. A Treatment of rhTGF-β1 at the increasing concentrations of 0, 1, 3, 5, 10 and 20 ng/ml were given for 72 h after the cells were adherent. TGF-β1 antibody-treated cells were used as a control. The cells were washed with Dulbecco's phosphate-buffered saline (D-PBS) 3 times and then fixed in 4% paraformaldehyde for 15 min at RT. Then, 1% Triton 100 in D-PBS buffer was allowed to permeate the cells at RT for 30 min. Subsequent to washing 3 times, the cells were blocked with 5% normal goat serum (Vector Laboratories, Inc., Burlingame, CA, USA) in 1% bovine serum albumin (BSA)/D-PBS buffer at RT for 1 h, and then incubation with the primary antibodies rabbit anti-E-cadherin (catalog no. 20874-1-AP; dilution, 1:1,000, ProteinTech Group, Inc.) and mouse anti-N-cadherin (catalog no. 610920; dilution, 1:1,000, BD Transduction Laboratories) overnight at 4°C. Subsequent to the 3 washes, the cells were incubated with the Alexa Fluor 594 (catalog no. A-11012; Invitrogen; Thermo Fisher Scientific, Inc.) and FITC-labeled secondary antibody (catalog no. bs-0926G-FITC; Bioss, Beijing, China) for 1 h at RT, respectively. Finally, nuclei were stained with Prolong Gold Antifade reagent with DAPI (catalog no. P-36931; Invitrogen; Thermo Fisher Scientific, Inc.). Images of the stained cells were captured with a fluorescent microscope (Olympus Corporation, Tokyo, Japan) at equal exposure time.
Flow cytometric analyses for apoptosis
A total of 5×104 cells per well were plated in 6-well plates. Then, 24 h after plating, the cells were treated for 3 days with rhTGF-β1 at doses of 0, 3 and 5 ng/ml, in addition to LOH treatment at increasing concentrations from 0 to 30 µM for 3 days. Apoptotic cell death was detected using an Annexin V-fluorescein isothiocyanate/propidium iodide (FITC/PI) double staining kit (Dojindo Molecular Technologies, Kumamoto, Japan) according to the manufacturer's protocol. The cells were washed twice with PBS and re-suspended cells in 1X Annexin-V binding buffer at a concentration of 1×106 cells/ml. Annexin V-FITC and PI were added separately to 5 µl cell suspensions and incubated for 30 min in the dark. The samples were analyzed by FACScan flow cytometer (BD Biosciences, San Jose, CA, USA). Data were analyzed by using Flow Jo software (version 7.6, Treestar).
Alkaline comet assay
The preparation samples for comet assays (Trevigen, Gaithersburg, MD, USA) were the same as the aforementioned Annexin V-FITC/PI double staining. Cells were combined with molten LM Agarose and 50 µl agarose/cells was immediately spread over a CometSlide™. The slides were cooled at 4°C in the dark for 10 min and then the slides were immersed in Lysis Solution overnight at 4°C. Subsequent to lysis, the cells were subjected to unwinding, electrophoresis and drying and staining with SYBR-Green I (Dingguo Biotechnology Co., Beijing, China). Subsequently, the cells were observed using fluorescent microscopy (Olympus Corporation). One hundred cells were analyzed for each group using Comet Score software (version 1.5, TriTek Solutions, Inc. Rancho Santa Margarita, CA, USA). The tail moment and the amount of DNA in the tail were manually scored by an examiner blinded to the treatment group. Experiments were repeated in triplicate.
Statistical analysis
Data were analyzed using JMP version 11.0 software (SAS Institute Co., Ltd., Shanghai, China). Data were described and statistically tested if they were normally distributed. Data are shown as the mean ± standard error of the mean or standard deviation from at least three independent experiments. The chemoresistance induced by TGF-β1 were compared and analyzed by one-way analysis of variance. Other data were performed with un-paired Student's t-test and nonparametric Wilcoxon test to evaluate differences among the experimental groups. P<0.05 was considered to indicate a statistically significant difference.
Results
TGF-β1 promotes chemoresistance to LOH in CRC cells
To examine whether CRC cells exhibit chemoresistance to LOH subsequent to treatment with TGF-β1, the effect of LOH on TGF-β1 treatment in all three cell lines was examined using an MTT assay. As is evident in Fig. 1A, along with the increasing concentration of LOH, CRC cells showed different levels of accumulative effects of death. By contrast, CRC cells that were pretreated with TGF-β1 demonstrated significant levels of resistance to LOH even at the highest dose of 50 µM LOH. However, no differences in viability were observed between groups treated with 5 ng/ml TGF-1 plus a serial concentration of LOH and 3 ng/ml TGF-1 plus a serial concentration of LOH. These results strongly support the initial hypothesis. These data indicate that the change in chemosensitivity to LOH of the HCT-116 cell line was the most significant among the three CRC cell lines. For the purposes of explaining the reasons for TGF-β1-treated CRC cells exhibiting chemoresistance to LOH, the colon cancer cell line HCT-116 was selected as the subject for the subsequent experiments.
Figure 1.

LOH sensitivity, morphological and vitality changes subsequent to TGF-β1 treatment. (A) All three colorectal cancer cell lines were treated with increasing concentrations of LOH for 3 days subsequent incubation with TGF-β1 for 3 days. (B) Images of TGF-β1-treated HCT-116 cells were captured after 3 and 30 days (magnification, ×200). There were no significant changes to the morphology of HCT-116 cells. (C) Viability of cells treated with TGF-β1 for 3 days was also assessed using MTT assay. The graph shows the mean ± standard deviation. *P<0.05; #, no significant differences among different groups; OD, optical density; TGF-β1, transforming growth factor β1; LOH, oxaliplatin.
No obvious morphological or changes in viability in TGF-β1-treated HCT-116 cells
Morphological observations and viability tests of TGF-β1-treated HCT-116 cells were performed. Following treatment with TGF-β1 for three days, cells with EMT phenotype (characterized by elongated morphology) were not observed, even after the 30 days of TGF-β1 treatment (Fig. 1B). Additionally, cell viability was tested using MTT assays. Similar results were revealed in that there were no significant differences among each group with the TGF-β1 concentration from 0 to 20 ng/ml observed (Fig. 1C). In conclusion, the data in Fig. 1B and C indicate that TGF-β1 does not significantly affect the morphological changes and changes to the viability of HCT-116 cells. Therefore, TGF-β1-induced chemoresistance is not associated with cell viability and cell proliferation.
TGF-β1 induces EMT in HCT-116 cells
To detect the EMT phenotype, cell immunofluorescent staining and western blot analyses were performed to examine the expression of indicators associated with EMT. It was demonstrated that with the increasing concentration of TGF-β1, HCT-116 upregulated the mesenchymal marker of neural cadherin (N-cadherin), and downregulated the epithelial marker epithelial cadherin (E-cadherin), despite the lack of typical morphology of EMT observed under the microscope. Concurrently, the level of transcription factor zinc finger protein SNAI1 (Snail), associated with EMT, also increased (Fig. 2A and B). Additionally, immunofluorescent staining was performed to evaluate EMT. Consistent with the result of western blot analyses, Fig. 2C shows that treating cells with 3 and 5 ng/ml TGF-β1 was a feasible method to inducing EMT. However, the EMT assays suggested that no significantly higher levels of N-cadherin and Snail, and lower levels of E-cadherin expression were observed among cells treated by 3–20 ng/ml TGF-β1. These data demonstrate that TGF-β1-induced EMT remains present in colon cancer HCT-116 cells. Therefore, the attenuation of the chemotherapeutic effects of LOH on HCT-116 cells may be due to TGF-β1-induced EMT.
Figure 2.

TGF-β1 promotes EMT in HCT-116 cells, which were treated with different doses of TGF-β1 for 3 days. (A and B) Western blotting of EMT markers expression levels. TGF-β1 treatment reduced the expression level of E-cadherin and promoted the expression of N-cadherin and Snail. (C) Cell immunofluorescence was performed to detect the EMT markers N-cadherin (green) and E-cadherin (red). TGF-β1 treatment reduced the expressions of E-cadherin and promoted the expression of N-cadherin. The graph shows the mean ± standard deviation. *P<0.05; **P<0.01; #P>0.05; E-cadherin/E-ca, epithelial cadherin; N-cadherin/N-ca, neural cadherin; Snail, Zinc finger protein SNAI1; EMT, epithelial mesenchymal transition; TGF-β1; transforming growth factor β1, Ab, antibody.
TGF-β1 attenuates LOH-induced DNA damage and apoptotic death in HCT-116 cells
Platinum compounds exert their anticancer effects usually through DNA damage. LOH may induce single- and double-stranded DNA breaks. Therefore, alkaline comet assays were performed to explore the levels of DNA damage in TGF-β1-induced chemoresistance to LOH. The cells were treated with TGF-β1 and LOH as aforementioned. Representative images of comet tails for each group are shown in Fig. 3C. The percentage of total DNA of cells found in the tail which reflects migrating DNA fragments from the nucleoid is shown in Fig. 3B. In addition, tail moment is an additional method used to measure the level of DNA damage, which combine the amount of DNA in the tail with the distance of migration (Fig. 3A). These collective data indicate that TGF-β1 induces HCT-116 cells to resist LOH through the attenuation of DNA damage. As DNA damage usually leads to the induction of apoptosis, the effects of TGF-β1 on apoptosis induced by LOH in HCT-116 cells were additionally characterized. Annexin/PI double staining was used to detect cell apoptosis. As is evident in Fig. 4, there was a marked trend that demonstrated that 3 and 5 ng/ml TGF-β1 exerted more powerful anti-apoptotic effects compared with untreated cells. Taken together, the data from the present study revealed that the resistance to LOH subsequent to TGF-β1 treatment on HCT-116 cells is due to attenuated DNA damage and ensuing anti-apoptotic effects caused by EMT.
Figure 3.
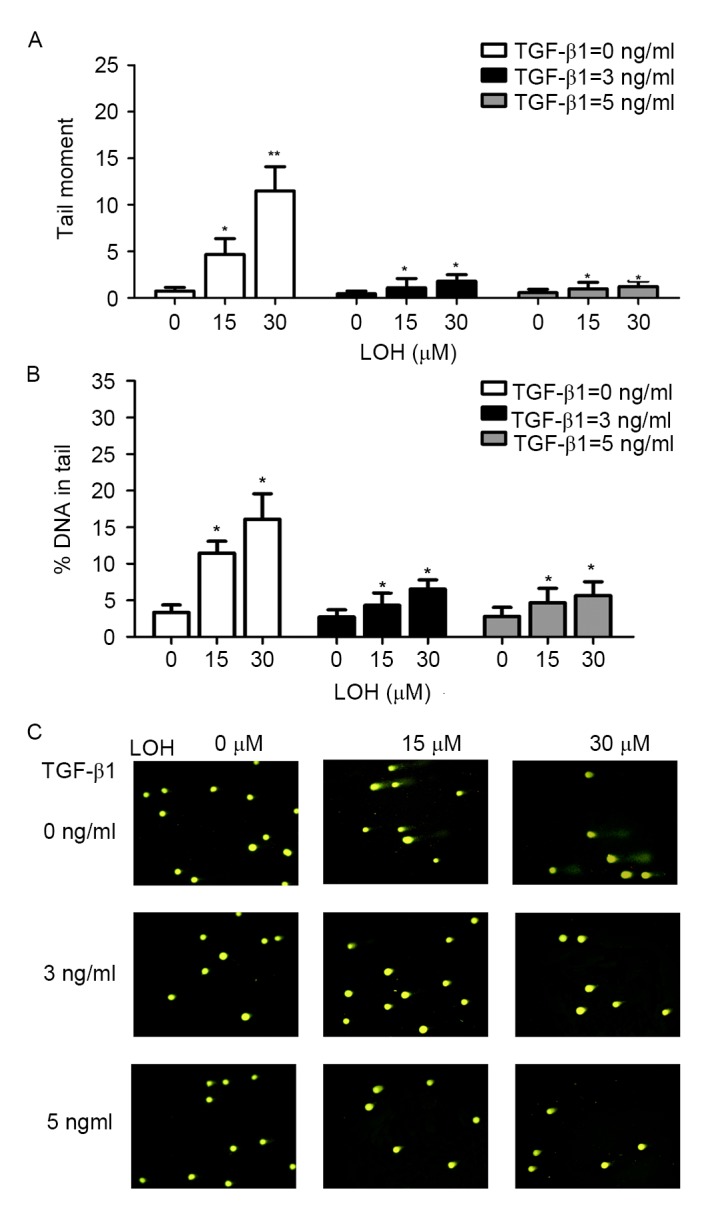
TGF-β1 attenuates induced DNA damage of LOH in HCT-116 cells. LOH-induced DNA damage was evaluated by comet assay in HCT-116 cells. The cells were treated as aforementioned. (A and B) Tail moment and percentage of DNA in the tails of each treatment group was calculated. The cells, which were treated with 3 and 5 ng/ml TGF-β1, exhibited LOH-induced attenuated DNA damage. Data is presented as the mean ± standard deviation. (C) Representative images of comet tail. Magnification, ×200. *P<0.05; **P<0.01; TGF-β1, transforming growth factor β1; LOH, oxaliplatin.
Figure 4.

TGF-β1 neutralizes the effect of LOH-induced apoptosis in HCT-116 cells. The Annexin-V-propidium iodide assay was used to measure the percentage of apoptosis by flow cytometry. Treatment with 3 and 5 ng/ml TGF-β1 for 3 days in addition to 3 days of LOH treatment led to anti-apoptotic effects. TGF-β1, transforming growth factor β1; LOH, oxaliplatin; FITC, fluorescein isothiocyanate.
Discussion
Oxaliplatin is widely used as a first-line chemotherapeutic agent globally in patients with mCRC (21). However, the mechanisms underlying LOH resistance are modulated by multiple factors, and have not been fully understood until now.
Extensive data suggest that the TME serves a crucial role in generating and promoting chemoresistance (4,8,9,22,23). TGF-β is a universal cytokine in the TME, which may be produced by cancer-associated fibroblasts, cancer-associated macrophages, mesenchymal stem cells, platelets and even cancer cells. TGF-β may not only maintain homeostasis in healthy cells, but also regulate the proliferation, migration, differentiation, invasion, metastasis and drug resistance of cancer cells via the Smad and/or non-Smad pathways (7,15,24). A number of studies suggested that TGF-β resulted in chemoresistance through several mechanisms. Among the chemoresistant effects caused by TGF-β, EMT was identified most frequently (8–14,25). EMT is considered a common process induced by TGF-β1, in which cancer cells lose epithelial features and acquire a mesenchymal phenotype with enhanced malignant behaviors such as invasion and metastasis, and chemoresistance in numerous cancer cell types (26–30).
There are no studies investigating the chemoresistant effect of TGF-β1-induced EMTs on CRC cells in the published literature. In the present study, the CRC cell lines used exhibited chemoresistance to LOH subsequent to TGF-β1 treatment. EMT, attenuated DNA damage and anti-apoptotic effects were demonstrated in the detailed experiments using TGF-β1-treated CRC cells, which may account for the TGF-β1-induced LOH resistance in CRC. Additionally, it was revealed that the LOH cytotoxicity assay and the DNA damage and apoptotic assays agreed with the phenotypical changes of EMT and cells demonstrated significant changes with the treatment of 3 and 5 ng/ml TGF-β1 compared with their untreated counterparts in the CRC cell lines. These data reveal that 3 and 5 ng/ml TGF-β1 are appropriate concentrations for producing chemoresistance effects in the CRC cell line. In the present study, CRC cells treated with >5 ng/ml TGF-β1 did not exhibit significant changes in cell viabilities and EMT phenotypes, and these results suggested that the subsequent experiments should be performed with concentrations of TGF-β1 treatment <5 ng/ml. The final data indicated that CRC cells demonstrated no significant differences between the groups treated by 3 and 5 ng/ml TGF-β1 in the MTT, EMT, comet and apoptotic assays.
The promotion of resistance to platinum-based chemotherapy by EMT has been demonstrated previously (25). However, LOH is a third-generation platinum compound that varies its mechanisms of resistance from its former associated platinum compounds. A number of previous studies revealed that numerous genes, proteins and transcription factors are involved in sensitivity of oxaliplatin treatment (19,31–36). Nevertheless, to the best of our knowledge, a small number of studies have addressed the association between the aforementioned LOH chemoresistance mechanisms and the biological functions of the TGF-β1 signaling pathways except for phosphoinositide-3-kinase (PI3K)/protein kinase B signaling. Additionally, TGF-β in the TME is a potent, multi-functional cytokine that exerts either tumor-suppressing or tumor-promoting activities, which depends on the specific TME, cancer stage, cancer type and chemotherapeutic agents present (30,37). These data indicate that the mechanisms underlying TGF-β1-induced chemoresistance to LOH remain incompletely characterized. Additional studies should focus on the TGF-β1 pathway associated with LOH resistance. In addition, except for EMT assays performed in the present study, other methods should be studied due to the complexity of LOH resistance caused by TGF-β1, which may include changing of CSC properties, crosstalk with other cytokines, regulating DNA mismatch repair system, modulating cell cycle arrest and autophagy.
In conclusion, the present study demonstrated that 3 and 5 ng/ml TGF-β1 induced EMT efficiently, which resulted in the attenuation of DNA damage and anti-apoptotic effects in CRC cells. These changes account for LOH resistance. Additional studies should focus on blocking the TGF-β pathway for re-sensitizing patients with CRC to LOH.
Acknowledgements
The authors would like to thank Miss Miao Guo and Dr Yanan Xu (Immunology and Allergy laboratory, The First Affiliated Hospital of Jinzhou Medical University) for excellent technical assistance with the flow cytometry-based analysis. The present study was supported by General Program of the National Natural Science Foundation of China (grant no., 81472460), The President Foundation of Jinzhou Medical University and The Leading Project on Colorectal Cancer of Jinzhou Medical University.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Zakaria S, Donohue JH, Que FG, Farnell MB, Schleck CD, Ilstrup DM, Nagorney DM. Hepatic resection for colorectal metastases: Value for risk scoring systems? Ann Surg. 2007;246:183–191. doi: 10.1097/SLA.0b013e3180603039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nautiyal J, Kanwar SS, Yu Y, Majumdar AP. Combination of dasatinib and curcumin eliminates chemo-resistant colon cancer cells. J Mol Signal. 2011;6:7. doi: 10.1186/1750-2187-6-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deep G, Agarwal R. Targeting tumor microenvironment with silibinin: Promise and potential for a translational cancer chemopreventive strategy. Curr Cancer Drug Targets. 2013;13:486–499. doi: 10.2174/15680096113139990041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013;501:346–354. doi: 10.1038/nature12626. [DOI] [PubMed] [Google Scholar]
- 7.Pickup M, Novitskiy S, Moses HL. The roles of TGFβ in the tumour microenvironment. Nat Rev Cancer. 2013;13:788–799. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asiedu MK, Ingle JN, Behrens MD, Radisky DC, Knutson KL. TGFbeta/TNF(alpha)-mediated epithelial-mesenchymal transition generates breast cancer stem cells with a claudin-low phenotype. Cancer Res. 2011;71:4707–4719. doi: 10.1158/0008-5472.CAN-10-4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L, Li R, Zhao QD, Yang Y, Lu ZH, Wei LX. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014;352:160–168. doi: 10.1016/j.canlet.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 10.Mallini P, Lennard T, Kirby J, Meeson A. Epithelial-to-mesenchymal transition: What is the impact on breast cancer stem cells and drug resistance. Cancer Treat Rev. 2014;40:341–348. doi: 10.1016/j.ctrv.2013.09.008. [DOI] [PubMed] [Google Scholar]
- 11.Yamada D, Kobayashi S, Wada H, Kawamoto K, Marubashi S, Eguchi H, Ishii H, Nagano H, Doki Y, Mori M. Role of crosstalk between interleukin-6 and transforming growth factor-beta 1 in epithelial-mesenchymal transition and chemoresistance in biliary tract cancer. Eur J Cancer. 2013;49:1725–1740. doi: 10.1016/j.ejca.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Yu Y, Wang Y, Ren X, Tsuyada A, Li A, Liu LJ, Wang SE. Context-dependent bidirectional regulation of the MutS homolog 2 by transforming growth factor β contributes to chemoresistance in breast cancer cells. Mol Cancer Res. 2010;8:1633–1642. doi: 10.1158/1541-7786.MCR-10-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabe Y, Shi YX, Zeng Z, Jin L, Shikami M, Hatanaka Y, Miida T, Hsu FJ, Andreeff M, Konopleva M. TGF-β-neutralizing antibody 1D11 enhances cytarabine-induced apoptosis in AML cells in the bone marrow microenvironment. PLoS One. 2013;8:e62785. doi: 10.1371/journal.pone.0062785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang H, Wu H, Zheng J, Yu P, Xu L, Jiang P, Gao J, Wang H, Zhang Y. Transforming growth factor β1 signal is crucial for dedifferentiation of cancer cells to cancer stem cells in osteosarcoma. Stem Cells. 2013;31:433–446. doi: 10.1002/stem.1298. [DOI] [PubMed] [Google Scholar]
- 15.Ma S, Qu W, Mao L, Zhu Z, Jia L, Zhao L, Zheng X. Antitumor effects of oncolytic adenovirus armed with Drosophila melanogaster deoxyribonucleoside kinase in colorectal cancer. Oncol Rep. 2012;27:1443–1450. doi: 10.3892/or.2012.1665. [DOI] [PubMed] [Google Scholar]
- 16.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 17.Lievre A, Samalin E, Mitry E, Assenat E, Boyer-Gestin C, Lepère C, Bachet JB, Portales F, Vaillant JN, Ychou M, Rougier P. Bevacizumab plus FOLFIRI or FOLFOX in chemotherapy-refractory patients with metastatic colorectal cancer: A retrospective study. BMC Cancer. 2009;9:347. doi: 10.1186/1471-2407-9-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Gramont A, Figer A, Seymour M, Homerin M, Hmissi A, Cassidy J, Boni C, Cortes-Funes H, Cervantes A, Freyer G, et al. Leucovorin and fluorouracil with or without oxaliplatin as first-line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18:2938–2947. doi: 10.1200/JCO.2000.18.16.2938. [DOI] [PubMed] [Google Scholar]
- 19.Seetharam R, Sood A, Goel S. Oxaliplatin: Pre-clinical perspectives on the mechanisms of action, response and resistance. Ecancermedicalscience. 2009;3:153. doi: 10.3332/ecancer.2009.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu Z, Mao L, Zhao L, Sun Z, Wang Z, Xu H, Zheng X. Synergistic therapeutic effect in gastric cancer cells produced by oncolytic adenovirus encoding Drosophila melanogaster deoxyribonucleoside kinase. Cancer Biol Ther. 2011;11:874–882. doi: 10.4161/cbt.11.10.15182. [DOI] [PubMed] [Google Scholar]
- 21.Cassidy J, Clarke S, Díaz-Rubio E, Scheithauer W, Figer A, Wong R, Koski S, Rittweger K, Gilberg F, Saltz L. XELOX vs FOLFOX-4 as first-line therapy for metastatic colorectal cancer: NO16966 updated results. Br J Cancer. 2011;105:58–64. doi: 10.1038/bjc.2011.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rebucci M, Michiels C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem Pharmacol. 2013;85:1219–1226. doi: 10.1016/j.bcp.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 23.Iseri OD, Kars MD, Arpaci F, Atalay C, Pak I, Gündüz U. Drug resistant MCF-7 cells exhibit epithelial-mesenchymal transition gene expression pattern. Biomed Pharmacother. 2011;65:40–45. doi: 10.1016/j.biopha.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 24.Flavell RA, Sanjabi S, Wrzesinski SH, Licona-Limon P. The polarization of immune cells in the tumour environment by TGFbeta. Nat Rev Immunol. 2010;10:554–567. doi: 10.1038/nri2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Marchini S, Fruscio R, Clivio L, Beltrame L, Porcu L, Nerini I Fuso, Cavalieri D, Chiorino G, Cattoretti G, Mangioni C, et al. Resistance to platinum-based chemotherapy is associated with epithelial to mesenchymal transition in epithelial ovarian cancer. Eur J Cancer. 2013;49:520–530. doi: 10.1016/j.ejca.2012.06.026. [DOI] [PubMed] [Google Scholar]
- 26.Thiery JP. Epithelial-mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–454. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 27.Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 28.Bierie B, Moses HL. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 29.Akhurst RJ. TGF beta signaling in health and disease. Nat Genet. 2004;36:790–792. doi: 10.1038/ng0904-1024a. [DOI] [PubMed] [Google Scholar]
- 30.Wendt MK, Tian M, Schiemann WP. Deconstructing the mechanisms and consequences of TGF-β-induced EMT during cancer progression. Cell Tissue Res. 2012;347:85–101. doi: 10.1007/s00441-011-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aaltonen LA, Peltomäki P, Leach FS, Sistonen P, Pylkkänen L, Mecklin JP, Järvinen H, Powell SM, Jen J, Hamilton SR, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993;260:812–816. doi: 10.1126/science.8484121. [DOI] [PubMed] [Google Scholar]
- 32.Gnoni A, Russo A, Silvestris N, Maiello E, Vacca A, Marech I, Numico G, Paradiso A, Lorusso V, Azzariti A. Pharmacokinetic and metabolism determinants of fluoropyrimidines and oxaliplatin activity in treatment of colorectal patients. Curr Drug Metab. 2011;12:918–931. doi: 10.2174/138920011798062300. [DOI] [PubMed] [Google Scholar]
- 33.Kashiwagi E, Izumi H, Yasuniwa Y, Baba R, Doi Y, Kidani A, Arao T, Nishio K, Naito S, Kohno K. Enhanced expression of nuclear factor I/B in oxaliplatin-resistant human cancer cell lines. Cancer Sci. 2011;102:382–386. doi: 10.1111/j.1349-7006.2010.01784.x. [DOI] [PubMed] [Google Scholar]
- 34.Kieran D, Woods I, Villunger A, Strasser A, Prehn JH. Deletion of the BH3-only protein puma protects motoneurons from ER stress-induced apoptosis and delays motoneuron loss in ALS mice; Proc Natl Acad Sci USA; 2007; pp. 20606–20611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moser C, Lang SA, Kainz S, Gaumann A, Fichtner-Feigl S, Koehl GE, Schlitt HJ, Geissler EK, Stoeltzing O. Blocking heat shock protein-90 inhibits the invasive properties and hepatic growth of human colon cancer cells and improves the efficacy of oxaliplatin in p53-deficient colon cancer tumors in vivo. Mol Cancer Ther. 2007;6:2868–2878. doi: 10.1158/1535-7163.MCT-07-0410. [DOI] [PubMed] [Google Scholar]
- 36.Xiu P, Dong X, Dong X, Xu Z, Zhu H, Liu F, Wei Z, Zhai B, Kanwar JR, Jiang H, et al. Secretory clusterin contributes to oxaliplatin resistance by activating Akt pathway in hepatocellular carcinoma. Cancer Sci. 2013;104:375–382. doi: 10.1111/cas.12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gatza CE, Holtzhausen A, Kirkbride KC, Morton A, Gatza ML, Datto MB, Blobe GC. Type III TGF-β receptor enhances colon cancer cell migration and anchorage-independent growth. Neoplasia. 2011;13:758–770. doi: 10.1593/neo.11528. [DOI] [PMC free article] [PubMed] [Google Scholar]