

Abstract
In this report, we present a new path to the control of quantum dot surface chemistry that can lead to a better understanding of nano-scale interfaces and the development of improved photocatalysts. Control of the synthetic methodology leads to QDs that are concomitantly ligated by crystal-bound organics at the surface anion sites and small X-type ligands on the surface cation sites.
Semiconducting nanocrystals, also known as quantum dots (QDs), are a hybrid material consisting of an inorganic semiconductor core and capping ligands.1 The capping ligands are vital to the colloidal stability of the QDs, and the interface between the semiconductor core and the ligands has a significant effect on the opto-electronic properties of the QDs through the passivation or creation of surface trap states. Understanding and controlling the interface between the capping ligands and the core facilitates the targeted implementation of QDs in catalytic, biomedical, and electronic applications.2
The colloidal methods used to synthesize the QDs are typically performed in organic solvents yielding a layer of native capping ligands with polar head groups and long alkyl chains. These long-chain ligands coordinate to the particle surfaces, decreasing the surface energy of the QD during synthesis, and allowing for control of size, crystalline structure, shape, and colloidal stability.3,4 Typically, organic capping ligands passivate the surface atoms by coordination through either a dative bond (L-type) or an anionic ligand (X-type) to surface metal cations.2,5,6 The long alkyl chains of the organic capping ligands prevent solubility of the particles in polar media and are insulating to charge transfer. This ligand corona needs to be removed, exchanged, or modified for applications in photovoltaics, photocatalysis, and biological systems to impart solubility in different media, improve charge transfer, or allow for functionalization.
Recently, we characterized a novel binding mode for organic capping ligands on QDs that we termed crystal-bound ligands, where the thiol ligands occupy high coordination sites,7 results in increased ligand stability such as resistance to ligand exchange and photooxidation,8 and therefore is a promising new ligand type for many applications.
Crystal-bound ligands inherently passivate the surface sulfur sites with alkyl groups. Passivation of anion sites has been studied previously with cationic organometallic complexes such as cadmium oleate. The anionic organic tail is lost to maintain charge balance when small, X-type, anionic ligands are introduced to passivate the surface metal sites of the quantum dot.9 In contrast, passivation of the anion sites with crystal-bound ligands should allow for simultaneous passivation of the surface metal sites with small, X-type, anionic ligands10 without the removal of the alkyl chains, but has yet to be experimentally demonstrated. Similarly, metal chalcogenide–thiol clusters show highly bridged thiols,11 which are reminiscent of the high coordination number of the crystal-bound binding mode. Due to the shape of the clusters, exposed cations only occur at the apices and can be passivated by other ligands. In quasi-spherical QD, we expect a greater diversity in surfaces and a much greater mixture of cation and anion exposure.
It is not known how this dual-mode passivation, and in particular, the crystal-bound component operates to modify the opto-electronic properties of quantum dots. In this paper, we demonstrate the role that the dual-mode crystal-bound and X-type passivation of QDs can play on the optical properties. Furthermore, we show the potential for improving the catalytic function of QDs through this controlled dual-mode coordination by comparing them to QDs with a similar surface-bound capping mode in a photocatalytic reaction.
Crystal-bound ligands occur on QDs when a primary thiol is used as the sulfur source in the synthesis of a metal sulfide. The metal–thiolate complex decomposes producing the nanocrystal.12 At lower temperatures, the final layer of organics is not thermally cleaved.13 Previously, we demonstrated crystal-bound ligands on Cu2S and CuInS2 nanocrystals.7 Here we extended this work to the synthesis of a ZnS shell around CdSe with crystal-bound ligands. CdSe seeds were synthesized14 (d = 2.8 ± 0.2 nm, n = 125, QY = 26%) and shelled by ZnS (d = 3.2 ± 0.2 nm, n = 127, QY = 50%)15 following literature procedures. To synthesize a secondary shell with crystal-bound ligands, the premade core/shells were injected with dodecanethiol (DDT) into a solution of Zn(OAc)2 in oleic acid and trioctylamine at 150 1C for 1 h.16 The resulting QDs increased in size to 3.9 ± 0.3 nm n = 133 and the quantum yield to 80% (Fig. 1). We considered the possibility that the differences in behavior (vide infra) are a result of differing surface densities of ligands resulting from the differing synthetic routes. However, the ligand density was determined by 1H NMR and was found to be similar for the crystal-bound DDT QDs (3.9 ligands per nm2) and the CdSe@ZnS seeds that had undergone a room temperature ligand exchange to DDT (3.6 ligands per nm2). The peak of the S 2p XPS spectra of QDs with crystal-bound thiols is shifted towards a lower binding energy than QDs with surface-bound thiols (ESI†). The decreased high binding energy component for QDs with crystal-bound thiols is consistent with a lack of low binding-energy surface thiolates, as we reported previously for crystal-bound ligands on Cu2S.7
Fig. 1.
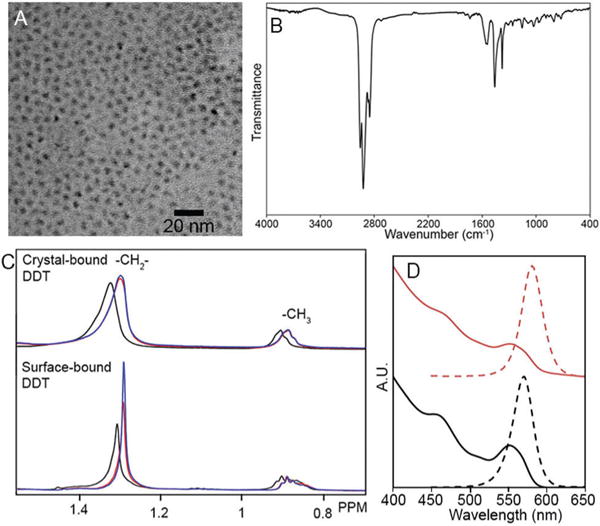
TEM of CdSe@ZnS core/shell nanocrystals with crystal-bound DDT (A) and IR (B). 1H NMR of CdSe@ZnS with crystal-bound (top) and surface-bound (bottom) DDT in D8-THF, initial (black), after the addition of base (red) and after 10 s of sonication (blue) (C). Absorbance and PL of CdSe@ZnS core@shell nanocrystals with crystal-bound DDT of CdSe (red) and after (black) (D).
IR spectroscopy of the QDs capped with crystal-bound DDT shows vibrational modes at 1560 cm−1 and 1379 cm−1, which can be assigned to X-type coordination of acetate to the surface Zn sites (Fig. 2B).17 While oleic acid is also part of the synthesis, the absence of signals due to the double bond by IR and NMR suggest that acetate causes the observed C–O stretching modes rather than oleate (ESI†).
Fig. 2.
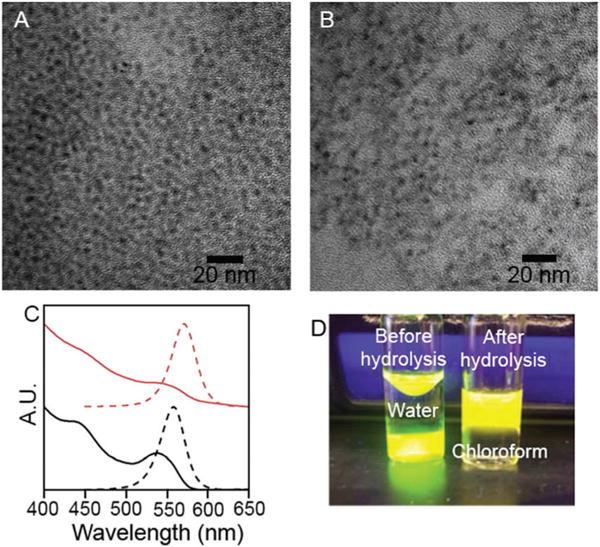
TEM of CdSe@ZnS with crystal-bound D3MP before (A) and after (B) hydrolysis. Absorbance and PL spectra of CdSe@ZnS with crystal-bound D3MP (black) and MPA (red) (C). D3MP capped CdSe@ZnS before and after hydrolysis (D).
1H NMR was used to show increased stability of the organic chain for QDs with crystal-bound ligands over QDs with surface-bound ligands when undergoing ligand exchange at the cation sites. The as-synthesized QDs with crystal-bound and surface-bound DDT were suspended in THF and exposed to aqueous base to facilitate a ligand exchange at the cation sites to X-type ligand, OH−.18 No additional free thiol ligands were added. Quadripolar effects of the metal atoms of the QD quench the signals of protons close to the QD,19 therefore an increase in the –CH2– peak is evidence of an increase in the free ligand concentration as thiol ligands are removed from the QD surfaces (Fig. 2C). Such an increase in free ligand was observed for the particles capped with surface-bound DDT, indicating the exchange of X-type dodecanethiolate by OH−. No change in solubility was observed for the crystal-bound case, indicating the crystal-bound ligands are more robustly attached to the particle surfaces.
Despite the 1H NMR evidence that CdSe@ZnScapped with crystal-bound DDT did not undergo loss of the organic ligands, the QDs underwent a near immediate quench and red-shift in the photo-luminescence (PL) upon exposure to base (Fig. 2D). This observation hinted that the crystal-bound ligand binding mode provides new optoelectronic properties; however, further study of this phenomenon and comparison to the surface-bound ligand binding mode became challenging because the QDs with surface bound thiols quickly precipitated from solution after the base exposure. As a result, we changed to our system to study the effects of our surface chemistry in aqueous phase catalysis, compared to traditional surface-bound ligands.
To allow for phase transfer of the crystal-bound capped CdSe@ZnS into an aqueous environment, dodecyl-3-mercaptopropionate (D3MP) was used in place of DDT in the synthesis.7 D3MP replaces DDT in the crystal-bound shell synthesis yielding similar particles as the DDT sample, where the particle size increased to 3.9 ± 0.3 nm n = 121 as did the quantum yield to 83%. A base hydrolysis was used to cleave the ester rendering the QDs water soluble (ESI†). After the hydrolysis the particles maintained their size and shape (3.9 ± 0.3 nm n = 146). The PL was shifted to lower energies and quenched to 6% (Fig. 2).
When the size of the CdSe core was changed, so too was the magnitude of the fluorescence red shift. Larger CdSe cores resulted in a smaller shift (4.06 nm, −0.025 eV) and smaller cores resulted in a greater shift (2.65 nm, −0.068 eV) (ESI†). In contrast, when native ligands on the CdSe@ZnS were exchanged to give surface-bound mercaptopropionic acid (MPA), no significant shift was observed in the fluorescence spectrum (ESI†). Two non-mutually-exclusive hypotheses emerged regarding atomistic origin of the red shift in the QD PL spectrum for crystal-bound thiols upon hydrolysis. The first hypothesis is that the addition of base alters the coordination of the surface Zn from acetate to hydroxide, as small anions are known to charge balance the surface of QDs.10 This change in surface cation chemistry alters the electron wavefunction since the conduction band is dominated by the orbitals of the cations.20 The second possibility is that the hydrolysis of the ester causes a shift in the band gap. Crystal-bound thiols are part of the anion sublattice of the QD, which dominates the orbital make-up of the valence band in CdSe and ZnS.20 As a result, the hole wavefunction may be perturbed by energetic subtleties between ester and carboxylate groups on the crystal-bound ligands.21
Density functional theory (DFT) was used to investigate the change in the QD band gap for different surface terminations providing an atomistic origin of the red shift upon hydrolysis. The calculations were performed for slab models of the (110) surface, which is the lowest energy surface of zinc blende ZnS and likely to dominate faceting of the cubic crystal-structured QDs.22 HR TEM of D3MP capped core–shell QDs before and after hydrolysis showed lattice fringing consistent with CdSe throughout, indicating ZnS is strained to match the CdSe lattice. Therefore, the calculations used CdSe lattice parameters for CdSe for the strained ZnS shell (ESI†).
Two sets of slab models were constructed to simulate quantum confinement of two QD sizes. The thicker slab model consists of 11 layers of CdSe and 2 layers of ZnS (one on each side), emulating the larger nanoparticles. The thinner slab model consists of 5 layers of CdSe and 2 layers of ZnS, emulating the smaller QDs. The confinement energies (Econfinement) from the models are shown in Table 1, along with the experimental data showing that the two are within reasonable agreement. Details of calculations are in ESI.†
Table 1.
Experimental and computational redshifts
| Experiment
| |||
|---|---|---|---|
| Econfinement (eV) | Eshift (eV) | ||
| Large QDs (4.06 nm) | 0.372 | −0.025 | |
| Medium QDs (3.04 nm) | 0.432 | −0.036 | |
| Small QDs (2.65 nm) | 0.523 | −0.068 | |
|
DFT (Fig. 3) | |||
| Model A
|
Model B
|
Model C
|
|
| Econfinement (eV) | Eshift (eV) | Eshift (eV) | |
|
| |||
| Thick slab | 0.259 | 0.013 | −0.042 |
| Thin slab | 0.616 | 0.003 | −0.164 |
Fig. 3A–C shows the three types of surface terminations of the CdSe@ZnSconsidered in the calculations. Model A represents the state of QD surface before hydrolysis, when the Zn site is terminated by acetate. Models B and C represent two possible models of the surface after hydrolysis. In Model B, hydrolysis only happens at the ester ligand leaving crystal-bound MPA, while in Model C the hydrolysis occurs on both the ester and the Zn site to give Zn–OH cation termination.
Fig. 3.
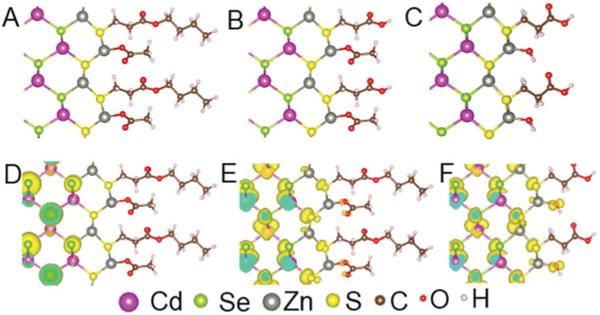
Models of the CdSe@ZnS core shell surface used in calculations (A–C). (D) The hole wavefunction (VBM) in Model A for thick slab of CdSe/ZnS core shell. (E) The electron wavefunction (CBM). (F) The electron wavefunction (CBM) of Model C.
The calculated shift (Eshift) in the band gap of the CdSe@ZnSupon hydrolysis of the dual-mode crystal-bound/surface-bound capping ligands are shown in Table 1. The hydrolysis of D3MP ligand in Model B introduces very small increase in the band gap of 13 meV for the thicker slab and 3 meV for the thin slab. Therefore, the large experimentally observed redshift cannot be explained by hydrolysis of the D3MP ligands alone. Instead, additional surface contributions must be considered. Combining the hydrolysis of D3MP ligand with a change of Zn termination from Zn–acetate to Zn–OH results in a reduction of the band gaps: −42 meV for the thicker slab and −164 meV for the thinner slab. Both results are of the same order of magnitude as the experimental data (−25 to −68 meV for large and small QDs, respectively). The small discrepancies between the theoretical and experimental redshifts are likely due to the use of slab models that are confined in 1-dimension to simulate the confinement energy of nanoparticles that are confined in 3-dimensions, an oversimplification of the exposed ZnS crystallographic surfaces, and tensile strain on the ZnS shell.
The DFT calculations show that the band gap in the dual mode capping is altered mostly by the change in Zn termination from Zn–acetate to Zn–OH and not by the hydrolysis of the ester of crystal-bound D3MP. The calculations show strong confinement of the hole wavefunction by the ZnS layer (Fig. 3D), but the electron wavefunction penetrates the ZnS shell and contains contributions from the absorbent on the Zn site (Fig. 3E). Therefore, the conduction band minimum (CBM) is sensitive to the change of the Zn site passivation. Indeed, after the change in Zn passivation from acetate to hydroxide there is an increased contribution in the electron wavefunction from the hydroxide (Fig. 3D and F). The greater extension of the electron wavefunction in the Zn–OH surface indicates that the quantum confinement effect on electron is weaker than in the Zn–acetate surface and thus the band gap is smaller, consistent with the observed redshift after hydrolysis. It should be noted that although one would expect good confinement of electrons in CdSe@ZnS core–shell structure23 from the bulk band alignment, the ZnS layer is highly strained on CdSe,24 which alters the band positions in ZnS by lowering the conduction band.
Red shifts in the band gap of QDs have been associated with the extension of the exciton wavefunction into organo-sulfur ligands such as dithiocarbamates and thiols.21 However, our calculations suggest that for the crystal-bound thiols, the organic ligands bound to the anion sublattice are electronically benign and therefore solely provide colloidal stability. It is the change in inorganic surface cation ligand that causes the modification of the band gap. These calculations explain our initial observation that QDs with crystal-bound DDT undergo shifts in the fluorescence spectrum upon exposure to base without loss of the organic ligands (Fig. 1).
The changes in fluorescence and colloidal stability of the QDs with surface-bound DDT after exposure to base suggests that hydroxide removes surface-bound DDT to expose cation sites and change their termination to Zn–OH. The removal of the aliphatic ligands harms the colloidal stability in organic solvents. In contrast, the organic component in the crystal-bound surface allows for organic–inorganic capping. The organic ligand corona is not disturbed by the hydroxide treatment changing the cation termination to Zn–OH and the colloidal stability is maintained.
This dual-mode ligand structure has important implications for QDs used in applications where charge transport and transfer is crucial, such as solution-based water reduction catalysts that rely on QD photosensitizers.25 The organic crystal-bound ligands provide solubility of the QDs in desired media and enhanced ligand stability under light exposure.7 The exposed cation sites can be used to modify the electronic properties as evident by the increased leakage of the electron wavefunction to the surface upon cation OH− termination in the DFT calculations (Fig. 3F). While the DFT calculations do not show the production of mid-gap states upon surface hydroxylation, the increased contribution of the surface atoms to the wavefunction causes the exciton to be more prone to collisional recombination. In addition to the observed fluorescence quench, the QDs are correspondingly more efficient at electron transfer to solution species, aiding in photocatalysis.
As a demonstration of the improved charge transfer possible for this dual-mode crystal-bound and X-type ligand QD capping over more standard surface-bound ligand systems, the photodegradation of methylene blue in aqueous solution was perfomed.26 The change in the absorbance of the dye was monitored in the presence of CdSe@ZnS nanocrystals capped with traditional surface-bound MPA and CdSe@ZnS nanocrystals capped with crystal-bound MPA and –OH anion terminating (Fig. 4). After 1 h of irradiation by visible light, the hybrid ligand capped QDs facilitated a 40% degradation of the methylene blue, whereas the QDs with traditional surface-bound organic passivation provided only 17% degradation. The improved photocatalytic degradation of methylene blue shows the dual passivation results in improved photoinduced charge transfer to solution based species.
Fig. 4.
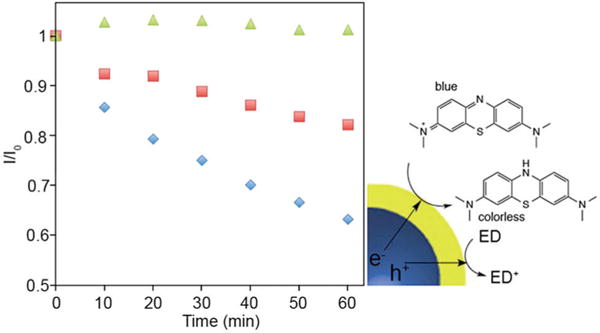
The change in absorbance of methylene blue at 666 nm with CdSe@ZnS photocatalysts with crystal-bound ligands (blue), surface bound ligands (red) and no QDs (green).
In this dual-mode capping system, the organic capping component controls the colloidal stability. Crystal-bound thiols, which terminate the anion sites, are not displaced under base exposure. The use of the ester containing D3MP as a thiol ligand provides a route to modify the solubility of particles in polar and nonpolar media as well as provides a chemical handle for later surface functionalization. In essence, the organic ligand corona is robust and can be tailored to give solubility in water or organic solvents. Concomitantly, the inorganic component modifies the surface cation chemistry and influences the electronic properties in CdSe@ZnS quantum dots, improving catalytic activity.
Supplementary Material
Acknowledgments
We acknowledge funding from the NSF-1253105 and NSF EPS-1004083. MJT thanks the Mitchum E. Warren Fellowship. AMF thanks the NIH MARC U-STAR program supported in part by grant 2T34GM007663 from the NIH. SC thanks the EDGE Program. XS and STP would like to acknowledge support from U.S. DOE grant DE-FG02-09ER46554 and by the McMinn Endowment at Vanderbilt University. This research used computational resources at NERSC, a User Facility supported by the Office of Science of the U.S. DOE under Contract No. DE-AC02-05CH11231 and HPC facilities at University of the Memphis.
Footnotes
Electronic supplementary information (ESI) available: Additional synthetic details, characterization of materials, and discussion of ligand stability.
Notes and references
- 1.Yin Y, Alivisatos AP. Nature. 2005;437:664–670. doi: 10.1038/nature04165. [DOI] [PubMed] [Google Scholar]
- 2.Owen JS, Park J, Trudeau PE, Alivisatos AP. J Am Chem Soc. 2008;130:12279–12281. doi: 10.1021/ja804414f. [DOI] [PubMed] [Google Scholar]
- 3.Green M. J Mater Chem. 2010;20:5797–5809. [Google Scholar]
- 4.Scher EC, Manna L, Alivisatos AP. Philos Trans R Soc, A. 2003;361:241–255. doi: 10.1098/rsta.2002.1126. [DOI] [PubMed] [Google Scholar]
- 5.Owen J. Science. 2015;347:615–616. doi: 10.1126/science.1259924. [DOI] [PubMed] [Google Scholar]
- 6.Anderson NC, Owen JS. Chem Mater. 2013;25:69–76. [Google Scholar]
- 7.Turo MJ, Macdonald JE. ACS Nano. 2014;8:10205–10213. doi: 10.1021/nn5032164. [DOI] [PubMed] [Google Scholar]
- 8.Aldana J, Wang YA, Peng XG. J Am Chem Soc. 2001;123:8844–8850. doi: 10.1021/ja016424q. [DOI] [PubMed] [Google Scholar]
- 9.Ip AH, Thon SM, Hoogland S, Voznyy O, Zhitomirsky D, Debnath R, Levina L, Rollny LR, Carey GH, Fischer A, Kemp KW, Kramer IJ, Ning ZJ, Labelle AJ, Chou KW, Amassian A, Sargent EH. Nat Nanotechnol. 2012;7:577–582. doi: 10.1038/nnano.2012.127. [DOI] [PubMed] [Google Scholar]
- 10.Zherebetskyy D, Scheele M, Zhang YJ, Bronstein N, Thompson C, Britt D, Salmeron M, Alivisatos P, Wang LW. Science. 2014;344:1380–1384. doi: 10.1126/science.1252727. [DOI] [PubMed] [Google Scholar]
- 11.Herron N, Calabrese JC, Farneth WE, Wang Y. Science. 1993;259:1426–1428. doi: 10.1126/science.259.5100.1426. [DOI] [PubMed] [Google Scholar]
- 12.Choi SH, An K, Kim EG, Yu JH, Kim JH, Hyeon T. Adv Funct Mater. 2009;19:1645–1649. [Google Scholar]
- 13.Chen O, Zhao J, Chauhan VP, Cui J, Wong C, Harris DK, Wei H, Han HS, Fukumura D, Jain RK, Bawendi MG. Nat Mater. 2013;12:445–451. doi: 10.1038/nmat3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen O, Chen X, Yang YA, Lynch J, Wu HM, Zhuang JQ, Cao YC. Angew Chem, Int Ed. 2008;47:8638–8641. doi: 10.1002/anie.200804266. [DOI] [PubMed] [Google Scholar]
- 15.Dethlefsen JR, Dossing A. Nano Lett. 2011;11:1964–1969. doi: 10.1021/nl200211n. [DOI] [PubMed] [Google Scholar]
- 16.Jun S, Jang E. Angew Chem, Int Ed. 2013;52:679–682. doi: 10.1002/anie.201206333. [DOI] [PubMed] [Google Scholar]
- 17.Dhas NA, Zaban A, Gedanken A. Chem Mater. 1999;11:806–813. [Google Scholar]
- 18.Nag A, Kovalenko MV, Lee JS, Liu WY, Spokoyny B, Talapin DV. J Am Chem Soc. 2011;133:10612–10620. doi: 10.1021/ja2029415. [DOI] [PubMed] [Google Scholar]
- 19.Hens Z, Martins JC. Chem Mater. 2013;25:1211–1221. [Google Scholar]
- 20.Alivisatos AP. Science. 1996;271:933–937. [Google Scholar]
- 21.Frederick MT, Weiss EA. ACS Nano. 2010;4:3195–3200. doi: 10.1021/nn1007435. [DOI] [PubMed] [Google Scholar]
- 22.Hamad S, Cristol S, Callow CRA. J Phys Chem B. 2002;106:11002–11008. [Google Scholar]
- 23.Reiss P, Protiere M, Li L. Small. 2009;5:154–168. doi: 10.1002/smll.200800841. [DOI] [PubMed] [Google Scholar]
- 24.Wei SH, Zunger A. Appl Phys Lett. 1998;72:2011–2013. [Google Scholar]
- 25.Han ZJ, Qiu F, Eisenberg R, Holland PL, Krauss TD. Science. 2012;338:1321–1324. doi: 10.1126/science.1227775. [DOI] [PubMed] [Google Scholar]
- 26.Costi R, Saunders AE, Elmalem E, Salant A, Banin U. Nano Lett. 2008;8:637–641. doi: 10.1021/nl0730514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.