

Abstract
Platelet-derived growth factor receptor (PDGFR)-β is an important tyrosine kinase and its downregulation has been reported to alter the radiosensitivity of glioma cells, although the underlying mechanism is unclear. In order to investigate the effect of PDGFR-β on the radiosensitivity of glioblastoma, the present study transfected C6 glioma cells with a PDGFR-β-specific small interfering (si)RNA expression plasmid, and downregulation of the expression of PDGFR-β in C6 glioma cells was confirmed by western blotting and immunohistochemical analysis. Clone formation assays and xenograft growth curves demonstrated that PDGFR-β-siRNA enhanced the radiosensitivity of C6 glioma cells in vitro and in vivo. Furthermore, MTT and xenograft growth curves demonstrated that PDGFR-β-siRNA inhibited the proliferation of C6 glioma cells in vitro and in vivo, and terminal deoxynucleotidyl transferase dUTP nick end-labeling and immunohistochemical analyses demonstrated that PDGFR-β-siRNA induced apoptosis and inhibited the expression of Ki-67, cyclin B1 and vascular endothelial growth factor in C6 glioma cell xenografts. Taken together, these results suggested that PDGFR-β may be used as a target for the radiosensitization of glioblastoma.
Keywords: glioblastoma, platelet-derived growth factor receptor-β, radiosensitization, RNA interference
Introduction
Glioblastoma multiforme (GBM) is the most common intracranial malignancy (1), accounting for 15.6% of all primary brain tumors and 45.2% of primary malignant brain tumors (1). At present, the standard treatment of GBM involves surgery combined with postoperative synchronous chemoradiotherapy and adjuvant chemotherapy (2). Despite improvements in minimally invasive surgical techniques and the use of intensity modulated conformal radiotherapy (RT) and temozolomide chemotherapy, the 5-year overall survival rate of GBM is still only 9.8% (3). Postoperative RT is the most important adjuvant therapy for GBM as it can reduce the likelihood of tumor recurrence (4). However, RT is not always ideal because of poor glioma cell radiosensitivity and an insufficient blood and oxygen supply to the postoperative tumor area. In addition, the brain is a dose-limiting organ for radiation, such that it is difficult to enhance the local control of a tumor by increasing the radiation dose. Therefore, methods to augment the radiosensitivity of GBM have emerged as an important research topic. Furthermore, to improve the outcomes of patients following traditional surgery and chemoradiotherapy, studies investigating strategies to improve the survival rate of GBM by targeted molecular therapy are urgently required.
Platelet-derived growth factor (PDGF) was first identified in fibroblasts and smooth muscle cells and was considered a powerful pro-mitotic agent (5,6) as it may be physically separated from human platelets (7). The PDGF receptor (PDGFR) is a type of tyrosine kinase receptor. PDGF and PDGFR have both been shown to be overexpressed in glial tumor cell lines and tumor surgical samples, and their increased expression has been correlated with a higher tumor grade. In previous studies, analysis of the expression levels of PDGF-A and PDGF-B, as well as their receptor PDGFR-β, in glioma cells was performed through in situ hybridization and immunohistochemical staining (8–10). Binding of PDGF to PDGFR-β activates an intracellular signal transduction pathway involving a cascade of mitogen-activated protein kinase proteins and resulting in the proliferation of malignant cells (11). In addition, PDGFR-β has been closely related to tumor angiogenesis. During this process, new extravascular blood vessels are covered by vascular smooth muscle cells and adventitial cells, and the development of these cells requires the activation of PDGFR. PDGF is also chemotactic and causes these cells to concentrate around the neovascular networks in order to stimulate their growth and promote tumor angiogenesis. Furthermore, PDGF is able to induce the transcription and secretion of vascular endothelial growth factor (VEGF), thereby promoting tumor angiogenesis indirectly (12).
At present, a variety of clinical PDGFR tyrosine kinase inhibitors are used to treat glioma. Imatinib has been shown to enhance the radiosensitivity and chemosensitivity of gliomas, inhibit glioma cell clone formation and arrest glioma cells in the G0/G1 and G2/M phases (13,14). Holdhoff et al (15) observed glioma cell lines while administering imatinib combined with RT, and observed that imatinib was able to radiosensitize the cells and inhibit tyrosine phosphorylation of numerous intracellular proteins in a dose-dependent manner. Although it is known that PDGFR-β is the key tyrosine kinase in the alteration of the radiosensitivity of glioma cells (16), the downstream pathways of such reactions require further elucidation. Imatinib is a phenylaminopyrimidine derivative that selectively inhibits several receptor tyrosine kinases thought to play a role in tumor proliferation and progression. These include the oncogenic BCR-ABL fusion protein found in leukemia cells, PDGFR and KIT, which is the oncogenic product of the stem cell factor receptor gene c-Kit. Other non-receptor tyrosine kinases, serine/threonine kinases and growth factor receptors, including epidermal growth factor receptor (EGFR) and fibroblast growth factor receptor, are at least two orders of magnitude more resistant to imatinib than BCR-ABL and PDGFR (17). These findings suggested that the radiosensitizing effect of imatinib on gliomas may involve the inhibition of multiple receptor tyrosine kinases, including PDGFR and other non-receptor tyrosine kinases.
In order to investigate the effect of PDGFR-β on the radiosensitivity of glioma, the present study used RNA interference (RNAi) technology to silence PDGFR-β expression in C6 glioma cells, and observed the proliferation, cell cycle distribution and apoptosis of C6 glioma cells in vivo and in vitro. Furthermore, the present study evaluated the possibility of combining RNAi technology with RT to enhance the radiosensitivity of gliomas, as well as the underlying molecular mechanisms.
Materials and methods
Ethics statement
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of Central South University (Changsha, China). The protocol was approved by the Committee on the Ethics of Animal Experiments of Central South University (permit no. 2014033-2). Animals were sacrificed by sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
Cells and cell culture
C6 glioma cells were purchased from the cell center of Xiangya Medical College, Central South University. The cells were divided into three groups, as follows: i) Untreated C6 glioma cells were designated the control group (CON); ii) C6 glioma cells transfected with PDGFR-β-small interfering (si)RNA vector were designated the knockdown group (KD); and iii) C6 glioma cells transfected with the empty vector were designated the negative control group (NC). CON, NC and KD cells were cultured in Dulbecco's modified Eagle's medium (Hyclone; GE Healthcare Life Sciences, Logan, UT, USA) supplemented with 10% heat-inactivated fetal bovine serum (Hyclone; GE Healthcare Life Sciences), 50 U/ml penicillin and 50 mg/ml streptomycin in a humidified atmosphere containing 5% CO2 at 37°C.
Construction and transfection of the RNAi-PDGFR-β vector
The 19-nucleotide siRNA targeting the rat PDGFR-β gene with a high gene-silencing efficacy (sense, caG GTG GTG TTT GAG GCT TAT; antisense, ATA AGC CTC AAA CAC CAC CTG) and the negative control siRNA (sense, TTC TCC GAA CGT GTC ACG T; antisense, ACG TGA CAC GTT CGG AGA A) were chemically synthesized by Shanghai GeneChem Co., Ltd. (Shanghai, China). The short hairpin (sh)RNA corresponding to the 6-nucleotide loop sequence (CTC GAG) of the PDGFR-β-siRNA, and its negative control containing the 9-nucleotide loop sequence (TTC AAG AGA) flanked by sense and antisense siRNA sequences, were synthesized by polymerase chain reaction (PCR) as previously described (18). The PDGFR-β-shRNA was inserted immediately into the shRNA expression system of the pU6-vshRNA-UBI-GFP vector (Shanghai GeneChem Co., Ltd.) to generate an RNAi-PDGFR-β vector, in which PDGFR-β shRNA was driven by the LV promoter and green fluorescent protein (GFP) was the reporter gene. All generated plasmids were identified by PCR and sequencing. Detailed vector maps and sequence information are available on request.
Transfection
For transfection, C6 glioma cells were seeded into 6-well plates at a concentration of 5×104 cells/well in a humidified incubator containing 5% CO2 at 37°C. Upon reaching 15% confluence, the cells were transfected with 10-µl diluents containing 4×107 RNAi-PDGFR-β vector or negative control vector, and were added to 890 µl enhancing solution (REVG0002; Shanghai GeneChem Co., Ltd.) and 100-µl polybrene (5 µl/ml) at a multiplicity of infection of 2. If there were no obvious signs of cell toxicity, the medium was changed after 24 h, and the cells were incubated for 2–3 days. GFP reporter gene expression was observed by fluorescence microscopy; a fluorescence rate of >80% suggested transfection success.
Western blot analysis
CON, NC and KD cells were homogenized in lysis buffer [50 mM Tris (pH 7.5), 1% Nonidet P-40, 150 mM NaCl, 2 mM EGTA, 1 mM Na3VO4, 100 mM NaF, 10 mM Na4P2O7, 1 mM phenylmethylsulfonyl fluoride, 10 g/ml aprotinin and 10 g/ml leupeptin]. Protein lysates were centrifuged at 14,000 × g for 10 min at 4°C, after which the total protein concentration was determined using the BCA protein assay (Beijing Dingguo Changsheng Biotechnology, Co., Ltd., Beijing, China). Cell lysates containing 50 µg protein were separated by 8% SDS-PAGE and transferred onto polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA, USA). The membranes were blocked with 5% (wt/vol) skimmed milk, and then probed with rabbit anti-rat PDGFR-β (dilution 1:10,000; sc-358943; Santa Cruz Biotechnology, Inc., Dallas, TX, USA) and β-actin (dilution 1:10,000; AP0060; Bioworld Technology, Inc., St. Louis Park, MN, USA) monoclonal antibodies overnight at 4°C. The membranes were then washed three times [with 50 mM Tris (pH 7.5), 0.5% Tween-20, 150 mM NaCl and 2 mM EGTA] and incubated with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (dilution 1:10,000; A0208; Beyotime Institute Biotechnology, Haimen, China) for 1 h at room temperature, followed by visualization using enhanced chemiluminescence (ECL) Western blotting detection reagents (Beyotime Institute Biotechnology). Protein band intensities were measured using ImageJ v1.45 software (National Institutes of Health, Bethesda, MD, USA) and normalized using the β-actin band intensity as the internal standard of the total protein load.
Cell viability assay
CON, NC and KD cells were plated onto 96-well plates at a density of 1×104/well, and the cell viability was determined using MTT assays after 12, 24, 48 and 72 h. Briefly, 10 µl MTT (Beijing Dingguo Changsheng Biotechnology, Co., Ltd.) stock solution (5 mg/ml) was added to each well, and the plates were incubated at 37°C for 4 h. Subsequently, 150 µl DMSO was added to each well. The plate was shaken on a rotary platform at room temperature for 10 min, after which the optical density was measured at 570 nm. Data are expressed as a percentage of the control. Experiments were performed in triplicate and repeated at least three times.
Colony formation assay
CON, NC and KD cells were plated onto 6-well plates at a density of 1×103 cells/well and cultured in 5% CO2 at 37°C for 12 days. The cells were exposed to fractionated 6-MV X-rays (0, 2, 4, 6 or 8 Gy doses) at a dose rate of 2.0 Gy/min using a Siemens PRIMUS™ Linear Accelerator (Siemens Ltd., Malvern, PA, USA). Colonies were stained with 0.005% crystal violet in methanol. Colony formation (≥50 cells) was counted under an inverted Olympus microscope (Olympus Corporation, Tokyo, Japan). Experiments were performed in triplicate and repeated thrice.
Cell cycle analysis
C6 glioma cells were divided into the CON+RT group, NC+RT group (NC+RT) and KD+RT group. The cells were treated with a radiation dose of 3 Gy at a dose rate of 2 Gy/min using a 6-MV X-ray. Cells were trypsinized, washed with cold PBS and re-suspended in PBS. Subsequently, the cells were fixed in 70% ethanol for 30 min, after which 5 µl propidium iodide (final concentration, 250 ng/ml; BD Biosciences, Franklin Lakes, NJ, USA) was added to the mixture. Flow cytometry was performed to detect and analyze changes in the cell cycle distribution using a FACScan flow cytometer (BD Biosciences).
Xenografts and experimental design
A total of 50 4-to-6-week-old female nude mice were purchased from the Hunan Slack King of Laboratory Animal Co., Ltd., (Changsha, China). All mice were housed in the Unit for Laboratory Animal Medicine, Xiangya Hospital (Changsha, China), with a 12-h light/dark cycle, free access to water and a standard mouse diet. The mice were divided into five groups of 10 mice each, as follows: i) CON group, ii) NC group, iii) KD group, iv) RT group and vi) KD+RT group. Cells (1×106) were injected subcutaneously into the left side of the axillary rear position of the mice. The CON and RT groups were injected with C6 cells only, the NC group with C6 cells and empty vector, and the KD and KD+RT groups with C6 cells and the RNAi-PDGFR-β vector. When the tumor volume had reached an average size of 150 mm3, the mice in the RT and KD+RT groups were irradiated with a dose of 10-Gy radiation. During radiation treatment, the entire mouse body was covered with lead sheets, with the exception of the tumor area, so that only the tumor was exposed to the radiation. After 2 days, four mice from each group were sacrificed. Subcutaneous tumors were removed and fixed in buffered formaldehyde for hematoxylin-eosin (H&E) staining, immunohistochemical analysis and terminal deoxynucleotidyl transferase dUTP nick end-labeling (TUNEL) assays. Subcutaneous tumor growth of the remaining six mice in each group was measured every 3 days using vernier calipers. Tumor volume was calculated according to the equation: Volume (mm3) = 1 / 2 × (Rmax × Rmin2), where R is the tumor diameter. Mice were sacrificed after 28 days.
H&E staining and immunohistochemistry
Formalin-fixed paraffin-embedded tumors were cut into 4-µm thick sections for H&E and immunohistochemical analysis. H&E was performed using standard histological techniques. Immunohistochemical analysis was conducted in accordance with the manufacturer's protocol. Briefly, the tissue sections were incubated with one of the following antibodies: Rabbit anti-mouse PDGFR-β polyclonal antibody (dilution 1:100; sc-358943; Santa Cruz Biotechnology, Inc.), rabbit anti-mouse Ki-67 monoclonal antibody (dilution 1:400; ab16667; Abcam, Cambridge, UK), rabbit anti-mouse cyclin B1 polyclonal antibody (dilution 1:100; sc-752; Santa Cruz Biotechnology, Inc.) and rabbit anti-mouse VEGF polyclonal antibody (dilution 1:400; ab51745; Abcam). Cyclin B1 and Ki-67 were expressed in the nuclei of tumor cells, PDGFR-β was expressed in the cytomembrane and VEGF was expressed in the cytoplasm. Positively stained cells were counted in five randomly selected fields.
Cell apoptosis by TUNEL
To detect apoptotic cells in subcutaneous tumors, TUNEL assays were performed using a commercial apoptosis detection kit (KGA 105; Nanjing KeyGen Biotech Co., Ltd., Nanjing, China), according to the manufacturer's protocol. TUNEL-positive cells displayed compaction or segregation of the nuclear chromatin, or nuclei that were broken up into discrete fragments. Positively stained cells were counted in five representative fields. The apoptosis index was calculated according to the following formula: Apoptosis index (%) = number of TUNEL-positive cells / total cell count × 100.
Statistical analysis
The experimental data were analyzed using SPSS version 17.0 (SPSS, Inc., Chicago, IL, USA) and quantitative data are presented as the mean ± standard deviation. Two groups were compared using the Student's t-test and multiple groups were compared using one-way analysis of variance followed by post-hoc tests such as the least significant difference test. The results of the TUNEL and immunohistochemical assays were compared using the χ2 test. P<0.05 was considered to indicate a statistically significant difference.
Results
PDGFR-β-shRNA downregulates PDGFR-β expression in C6 glioma cells
As shown in Fig. 1, western blotting was used to assess the expression levels of PDGFR-β in the CON, NC and KD group C6 cells. The density ratio of PDGFR-β/β-actin was 0.6298±0.555 in the Con group, 0.6125±0.696 in the NC group and 0.1020±0.0126 in the KD group. There were significant differences in the protein expression levels of PDGFR-β between the KD group and the CON and NC groups (P=0.0001). These results suggest that RNAi-PDGFR-β significantly inhibits PDGFR-β expression in glioma cells.
Figure 1.

Effect of PDGFR-β-specific short hairpin RNA on PDGFR-β expression in C6 glioma cells. Whole-cell lysates were prepared and subjected to western blotting. Data were obtained from three independent experiments and are shown as the mean ± standard deviation. *P<0.05 vs. CON or NC. PDGFR-β, platelet-derived growth factor receptor-β; CON, control C6 cells; NC, negative control C6 cells; KD, PDGFR-β-knockdown C6 cells.
PDGFR-β-specific shRNA enhances the radiosensitivity of C6 glioma cells in vitro
In order to investigate the radiosensitivity of the KD cells, a clonogenic survival assay was performed (Fig. 2) and quantified by determination of the radiobiological parameters (Table I). Clone formation assays indicated that the mean lethal dose (D0) of radiation in the KD+RT group was decreased from 2.391 to 1.091 Gy compared with the RT group, and the radiosensitization ratio was 2.192. These results suggest that RNAi-PDGFR-β specifically increases the level of radiation-induced C6 glioma cell death and enhances the radiosensitivity of C6 glioma cells in vitro.
Figure 2.

PDGFR-β-specific short hairpin RNA enhances the radiosensitivity of C6 glioma cells. CON, NC and KD cells were irradiated with 0, 2, 4, 6 or 8 Gy radiation. Clone formation assays demonstrated the dose-dependent reduction of the survival fraction of C6 cells. CON, control C6 cells; NC, negative control C6 cells; KD, PDGFR-β-knockdown C6 cells; PDGFR-β, platelet-derived growth factor receptor-β.
Table I.
Radiobiological parameters of NC and KD C6 glioma cells combined with radiotherapy.
| Group | K | N | D0 | Dq | SER (KD vs. CON) | SER (KD vs. NC) | R2 |
|---|---|---|---|---|---|---|---|
| CON | 0.4183 | 2.543 | 2.391 | 1.805 | – | – | 0.988 |
| NC | 0.4968 | 2.920 | 2.013 | 1.771 | – | – | 0.988 |
| KD | 0.9164 | 5.616 | 1.091 | 1.813 | 2.192 | 1.845 | 1.000 |
CON, control C6 cells; NC, negative control C6 cells; KD, PDGFR-β-knockdown C6 cells. D0, mean lethal dose; SER, radiosensitization ratio; N, extrapolation number; Dq, quasithreshold dose.
PDGFR-β-specific shRNA enhances the radiosensitivity of C6 glioma cell xenografts
To further examine the effects of RNAi-PDGFR-β, in vivo experiments with C6 cell xenograft tumors were conducted. An in vivo radiation dose of 10 Gy was used to simplify the assessment of the effects following treatment. No significant differences were observed in the tumor volumes between the CON and NC groups; however, the tumor volume of the KD group was significantly smaller compared with the CON and NC groups after 28 days (P=0.02; Fig. 3). The tumor volumes in the RT and KD+RT groups were significantly smaller compared with those in the CON, NC and KD groups at 28 days (P=0.0001; Fig. 3). In addition, the tumor volume in the KD+RT group was significantly smaller than the RT group at 28 days (P=0.0001; Fig. 3). These results suggest that RNAi-PDGFR-β specifically enhances the radiosensitivity of C6 glioma cell xenografts in vivo.
Figure 3.
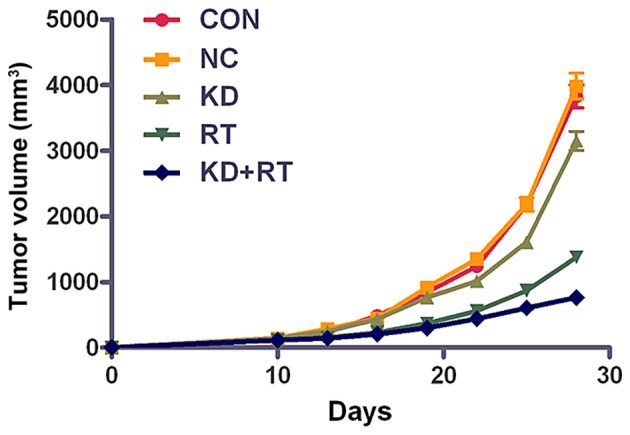
In vivo effects of PDGFR-β-specific short hairpin RNA in a C6 glioma cell xenograft model. No significant difference in tumor volume was observed between the CON and NC groups (P>0.05), while the tumor volume in the KD group was significantly reduced compared with the CON and NC groups at 28 days (P<0.01). A significant reduction in tumor volume was observed in the RT and KD+RT groups at 28 days (P<0.01), as compared with the CON, NC and KD groups. The tumor volume of the KD+RT group was significantly decreased compared with the RT group at 28 days (P<0.01). Data are presented as the mean ± standard deviation. CON, mice injected with control C6 cells; NC, mice injected with negative control C6 cells; KD, mice injected with PDGFR-β-knockdown C6 cells; RT, radiotherapy; PDGFR-β, platelet-derived growth factor receptor-β.
PDGFR-β-specific shRNA reduces the viability of C6 glioma cells
To investigate the effect of PDGFR-β-specific shRNA on C6 glioma cell viability, MTT assays were performed. The results demonstrated that the KD cells had a significantly reduced viability compared with the CON and NC cells at 12 (all P=0.04), 24 (P=0.04 vs. CON; P=0.03 vs. NC), 48 (P=0.03 vs. CON; P=0.04 vs. NC) and 72 h (all P=0.04) (Fig. 4).
Figure 4.

Effect of PDGFR-β-specific short hairpin RNA on the variability of C6 glioma cells. The viability of the CON, NC and KD cells was determined using MTT assays. The viability of the KD cells was decreased compared with the CON and NC cells from 12–72 h. *P<0.05 vs the CON cells. Data were obtained from three independent experiments and are shown as the mean ± standard deviation. CON, control C6 cells; NC, negative control C6 cells; KD, PDGFR-β-knockdown C6 cells; PDGFR-β, platelet-derived growth factor receptor-β.
PDGFR-β silencing causes cell cycle arrest in the G1 and G2/M phases
Cell cycle profiles were determined by flow cytometry. The fractions of KD+RT group cells in the G1 and G2/M phases were significantly increased by 9.47 and 5.88%, respectively, compared with those in the CON+RT group (G1 phase, P=0.02; G2/M phase, P=0.00; Fig. 5). Furthermore, the fraction of KD+RT group cells in the S phase was significantly decreased by 15.34% compared with the CON and NC groups (all P=0.001; Fig. 5). These results suggest that the knockdown of PDGFR-β expression enhances radiation-induced cell cycle arrest in the G1 and G2/M phases.
Figure 5.
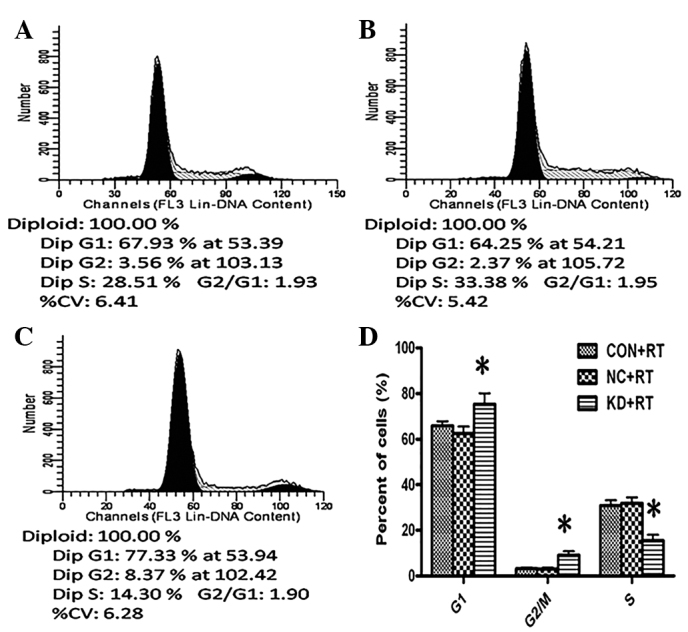
Cell cycle distributions of the (A) CON, (B) NC and (C) KD cells following RT were determined by flow cytometry. (D) Quantitative analysis of cell cycle distributions. For the KD+RT cells the S phase fraction was reduced and more cells were arrested in the G1 and G2/M phases. *P<0.05 vs. the CON+RT cells. Data were obtained from three independent experiments and are shown as the mean ± standard deviation. CON, control C6 cells; NC, negative control C6 cells; KD, platelet-derived growth factor receptor-β-knockdown C6 cells; RT, radiotherapy.
Immunohistochemical analysis of xenograft sections for PDGFR-β expression
PDGFR-β was expressed in all groups examined, although the degree of expression varied widely among the groups. Immunohistochemical analysis showed that the expression of PDGFR-β in the KD and RT groups was significantly reduced compared with the CON and NC groups (KD vs. CON and NC, P=0.01; RT vs. CON and NC, P=0.03; Fig. 6 and Table II). Furthermore, the expression of PDGFR-β in the KD+RT group was significantly lower compared with the KD and RT groups (all P=0.001; Fig. 6 and Table II).
Figure 6.

TUNEL assay and expression of PDGFR-β, Ki-67, VEGF and cyclin B1 in C6 cell xenografts using immunohistochemistry (magnification, ×400). (A) Radiation and PDGFR-β-specific shRNA induced cell apoptosis in C6 cell xenografts. Positive apoptotic cells were identified as brown, with a brownish/black nuclear stain. (B) The expression of PDGFR-β in C6 cells in vivo. Positive PDGFR-β expression was identified by a brown cytoplasm/membrane stain. (C) The expression of Ki-67 in C6 cells in vivo. Positive Ki-67 expression was identified by a brown nuclear stain. (D) The expression of cyclin B1 in C6 cells in vivo. Positive cyclin B1 expression was identified by a brown nuclear stain. (E) The expression of VEGF in C6 cells in vivo. Positive VEGF expression was identified as a brown cytoplasmic stain. TUNEL, terminal deoxynucleotidyl transferase dUTP nick end-labeling; PDGFR-β, platelet-derived growth factor receptor-β; VEGF, vascular endothelial growth factor; CON, control C6 cells; NC, negative control C6 cells; KD, PDGFR-β-knockdown C6 cells; RT, radiotherapy.
Table II.
AI and expression of PDGFR-β, Ki-67, cyclin B1 and VEGF in C6 cell xenografts.
| Group | AI (%) | PDGFR-β (%) | Ki-67 (%) | Cyclin B1 (%) | VEGF (%) |
|---|---|---|---|---|---|
| CON | 2.77 | 53.28 | 81.75 | 55.38 | 55.33 |
| NC | 3.02 | 54.62 | 78.88 | 65.27 | 54.78 |
| KD | 5.12 | 30.83 | 77.89 | 54.25 | 46.36 |
| RT | 28.4 | 37.38 | 57.23 | 35.45 | 59.24 |
| KD+RT | 48.2a | 17.33a | 41.38a | 27.23a | 25.43 |
| χ2 | 112.52 | 42.33 | 57.64 | 26.81 | 28.87 |
P<0.0001 vs. the KD group. AI, apoptosis index; CON, control C6 cells; NC, negative control C6 cells; KD, PDGFR-β-knockdown C6 cells; PDGFR-β, platelet-derived growth factor receptor-β; VEGF, vascular endothelial growth factor.
Immunohistochemical analysis of xenograft sections for Ki-67 and cyclin B1 expression
Immunohistochemical analysis indicated that the expression of Ki-67 in the KD group was marginally lower than its expression level in the CON group, although the difference was not significant (P=0.29). The expression of Ki-67 in the RT and KD+RT groups was significantly lower compared with the CON, NC and KD groups (P<0.0001), and the expression of Ki-67 in the KD+RT group was significantly lower compared with that in the RT group (P<0.0001). PDGFR-β-specific shRNA did not affect the expression of cyclin B1 in C6 cell xenografts, although the expression of cyclin B1 was reduced in the RT group (P=0.26 vs. CON). The expression of cyclin B1 in the RT and KD+RT groups was lower compared with the CON, NC and KD groups (all P=0.0003). The expression of cyclin B1 in the KD+RT group was significantly lower than that in the RT group (P=0.0002; Fig. 6 and Table II).
TUNEL analysis of cell apoptosis in C6 glioma cell xenografts
TUNEL analysis showed that there were no significant differences in the apoptosis indexes of C6 glioma cell xenografts among the CON, NC and KD groups (P=0.06). The apoptosis indexes of the RT and KD+RT groups were significantly higher than those of the CON, NC and KD groups (all P<0.0001). The apoptosis index of the KD+RT group was significantly higher than that of the RT group (P=0.01; Fig. 6 and Table II.). These results suggest that radiation and PDGFR-β-specific shRNA induces cell apoptosis in C6 glioma cell xenografts.
Immunohistochemical analysis of xenograft sections for VEGF
Immunohistochemical analysis of C6 cell xenograft tissues demonstrated exposure of C6 cell xenografts to RT resulted in a slight increase in VEGF expression (P=0.24), and suggested there was a radiation-induced increase in VEGF protein levels. The expression of VEGF in the KD group was lower compared with the CON and NC groups (all P=0.01). The expression of VEGF in the KD+RT group was significantly lower compared with the RT group (P=0.01). This finding indicates that, when KD cells are irradiated, VEGF levels are reduced as compared with the irradiation alone control. Therefore, PDGFR-β-specific shRNA reduced the radiation-stimulated increase in VEGF protein expression levels.
Discussion
Targeted molecular therapy is a novel therapeutic strategy for GBM (18). In previous studies, targeting of PDGFR using selected inhibitors achieved some success in the treatment of gliomas. However, the molecular mechanisms remain unclear. The present study demonstrated that PDGFR-β-specific shRNA enhanced the radiosensitivity of C6 glioma cells in vivo and in vitro, and that the molecular mechanisms involved promoting radiation-induced arrest of C6 glioma cells in the G1 and G2/M phases and reducing the expression levels of Ki-67, cyclin B1 and VEGF. These results suggested that PDGFR-β may be used a target for enhancement of the radiosensitivity of GBM.
Generally, it is thought that the G2 and M phases are the most sensitive to radiation therapy, followed by the G1 phase, while the S phase is the least sensitive to RT (19). Radiation is able to cause M phase delay, as well as cell cycle arrest in the G0/1 and G2/M phases (20). Cyclin B1, which is closely associated with mitosis, is not expressed in normal brain tissue or is expressed at extremely low levels; however, it has been shown to be overexpressed in glial tumors and its upregulation was correlated with a higher tumor grade (21). Cyclin B1 plays an important role in accelerating cell cycle progression and regulating the transformation of the G2/M phase. To date, studies have reported that excessive cyclin B1 expression is able to cause radiation resistance, while decreased cyclin B1 expression is able to enhance G2/M phase arrest and increase radiosensitivity (22,23). Ren et al (13) reported that the PDGFR inhibitor, imatinib, was able to inhibit glioma cell colony formation and to cause the arrest of glioma cells in the G0/G1 and G2/M phases, with a concomitant reduction of the number of cells in the S phase. The present study demonstrated that PDGFR-β-specific shRNA caused C6 cells to arrest in the G0/G1 and G2/M phases. Furthermore, the positive expression rates of cyclin B1 in the RT and KD+RT groups were significantly reduced compared with the other groups. In addition, the expression levels of cyclin B1 in the KD+RT group were reduced to a greater extent compared with the RT group. These results suggested that PDGFR-β-targeted therapy combined with RT was able to reduce the expression of cyclin B1, thereby blocking cells in the G2/M period and increasing the radiosensitivity of the cells.
Apoptosis is regulated by the programmed cell death gene and is the main form of tumor cell death induced by radiation therapy (24). The present study demonstrated that the apoptosis index of the KD+RT group was higher than the other three groups, and that this group had the most TUNEL-positive cells. These results indicated that inhibition of PDGFR-β expression by RNAi combined with RT was able to enhance apoptosis compared with RT alone, suggesting that RNAi of PDGFR-β might enhance the tumor radiosensitivity of C6 gliomas.
Ki-67 is a nuclear antigen associated with cell proliferation. The expression levels of Ki-67 have been correlated with glioma grading and prognosis (25). Lafuente et al (26) reported that the expression of Ki-67 was correlated with the expression of PDGFR-β, suggesting that PDGFR-β overexpression may promote cell proliferation. In the present study, expression of Ki-67 was inhibited in the RT and KD+RT groups, and the lowest number of positive tumor nuclei was found in the combined treatment group. These results indicated that PDGFR-β-specific shRNA combined with RT may inhibit cell proliferation, leading to a greater antitumor effect than RT alone.
Previous studies have shown that anti-angiogenesis strategies combined with RT are able to increase the radiation sensitivity and improve the efficacy of RT (27,28). GBM is a typical tumor that is rich in blood vessels, and its processes of development, invasion and metastasis depend on angiogenesis, which is closely related to PDGF and VEGF signaling pathways (29). When tumor cells release PDGF-activated PDGFR-β, it is able to induce endothelial cells and smooth muscle cells to proliferate and migrate, stimulating the formation of new blood vessels, which has a direct role in tumor angiogenesis. In addition, abnormal expression of PDGF stimulates VEGF secretion via autocrine and paracrine signaling, which indirectly promotes tumor angiogenesis (30). VEGF is the most effective factor in promoting vascular growth, as it induces endothelial cell proliferation and the formation of blood vessels, and also increases the permeability of blood vessels. In a previous study, upregulation of VEGF resulted in the resistance of tumors to radiation (27). PDGFR-β is highly expressed in vascular endothelial cells and around microvasculature in glioma, and it is closely related to the formation of blood vessels (31). In the present study, as compared with the control and empty vector groups, reduced expression of VEGF was seen in the KD and KD+RT groups, although the expression of VEGF was increased in the RT group. PDGFR-β-specific shRNA combined with RT was able to reduce the expression of VEGF, and thus affect tumor angiogenesis, while radiation alone induced increased expression of VEGF. Gorski et al (32) reported that the level of VEGF expression increased following RT, which is consistent with our immunohistochemical results.
The present study demonstrated that PDGFR-β-specific shRNA inhibits the proliferation of C6 cells in vitro and in vivo. RT combined with RNAi-PDGFR-β displayed synergistic antitumor effects on C6 glioma cells in vitro and in vivo. The molecular mechanism of the radiosensitizing effect of RNAi-PDGFR-β is involved in cell cycle arrest, cell apoptosis and anti-angiogensis. A single therapeutic target is often not effective; thus, multiple target treatments should be adopted. Omuro (33) reported that the results of single-target drugs targeting EGFR, VEGFR and PDGFR in malignant gliomas (glioblastomas and anaplastic forms of astrocytomas, oligodendrogliomas and oligoastrocytomas) have been disappointing. Multi-targeting drugs or combinations of two or more single-targeting drugs should to be used to overcome the resistance of malignant glioma.
In conclusion, the present study demonstrated that PDGFR-β-specific shRNA downregulated the expression of PDGFR-β at the protein level and enhanced the radiosensitivity of C6 cells in vivo and in vitro. The molecular mechanism of the enhancement of radiosensitivity by RNAi-PDGFR-β may involve cell apoptosis, cell cycle arrest and anti-angiogenesis. Therefore, PDGFR-β may be an appropriate target for selectively enhancing the radiosensitivity of glioblastoma.
Acknowledgements
The present study was supported by the Hunan Province Science and Technology Program (grant no. 2011SK3223) and the Hunan Provincial Natural Science Foundation of China (grant no. 2012JJ5043).
References
- 1.Ostrom QT, Gittleman H, Farah P, Ondracek A, Chen Y, Wolinsky Y, Stroup NE, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006–2010. Neuro Oncol. 2013;15(Suppl 2):ii1–ii56. doi: 10.1093/neuonc/not151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, et al. European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10:459–466. doi: 10.1016/S1470-2045(09)70025-7. [DOI] [PubMed] [Google Scholar]
- 3.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 4.Nakagawa K, Aoki Y, Fujimaki T, Tago M, Terahara A, Karasawa K, Sakata K, Sasaki Y, Matsutani M, Akanuma A. High-dose conformal radiotherapy influenced the pattern of failure but did not improve survival in glioblastoma multiforme. Int J Radiat Oncol Biol Phys. 1998;40:1141–1149. doi: 10.1016/S0360-3016(97)00911-5. [DOI] [PubMed] [Google Scholar]
- 5.Kohler N, Lipton A. Platelets as a source of fibroblast growth-promoting activity. Exp Cell Res. 1974;87:297–301. doi: 10.1016/0014-4827(74)90484-4. [DOI] [PubMed] [Google Scholar]
- 6.Ross R, Glomset J, Kariya B, Harker L. A platelet-dependent serum factor that stimulates the proliferation of arterial smooth muscle cells in vitro; Proc Natl Acad Sci USA; 1974; pp. 1207–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heldin CH, Westermark B, Wasteson A. Platelet-derived growth factor: Purification and partial characterization; Proc Natl Acad Sci USA; 1979; pp. 3722–3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hermansson M, Nistér M, Betsholtz C, Heldin CH, Westermark B, Funa K. Endothelial cell hyperplasia in human glioblastoma: Coexpression of mRNA for platelet-derived growth factor (PDGF) B chain and PDGF receptor suggests autocrine growth stimulation; Proc Natl Acad Sci USA; 1988; pp. 7748–7752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maxwell M, Naber SP, Wolfe HJ, Galanopoulos T, Hedley-Whyte ET, Black PM, Antoniades HN. Coexpression of platelet-derived growth factor (PDGF) and PDGF-receptor genes by primary human astrocytomas may contribute to their development and maintenance. J Clin Invest. 1990;86:131–140. doi: 10.1172/JCI114675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Plate KH, Breier G, Farrell CL, Risau W. Platelet-derived growth factor receptor-beta is induced during tumor development and upregulated during tumor progression in endothelial cells in human gliomas. Lab Invest. 1992;67:529–534. [PubMed] [Google Scholar]
- 11.Chen D, Li Y, Mei Y, Geng W, Yang J, Hong Q, Feng Z, Cai G, Zhu H, Shi S, et al. miR-34a regulates mesangial cell proliferation via the PDGFR-β/Ras-MAPK signaling pathway. Cell Mol Life Sci. 2014;71:4027–4042. doi: 10.1007/s00018-014-1599-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nazarenko I, Hede SM, He X, Hedrén A, Thompson J, Lindström MS, Nistér M. PDGF and PDGF receptors in glioma. Ups J Med Sci. 2012;117:99–112. doi: 10.3109/03009734.2012.665097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ren H, Tan X, Dong Y, Giese A, Chou TC, Rainov N, Yang B. Differential effect of imatinib and synergism of combination treatment with chemotherapeutic agents in malignant glioma cells. Basic Clin Pharmacol Toxicol. 2009;104:241–252. doi: 10.1111/j.1742-7843.2008.00371.x. [DOI] [PubMed] [Google Scholar]
- 14.Russell JS, Brady K, Burgan WE, Cerra MA, Oswald KA, Camphausen K, Tofilon PJ. Gleevec-mediated inhibition of Rad51 expression and enhancement of tumor cell radiosensitivity. Cancer Res. 2003;63:7377–7383. [PubMed] [Google Scholar]
- 15.Holdhoff M, Kreuzer KA, Appelt C, Scholz R, Na IK, Hildebrandt B, Riess H, Jordan A, Schmidt CA, van Etten RA, et al. Imatinib mesylate radiosensitizes human glioblastoma cells through inhibition of platelet-derived growth factor receptor. Blood Cells Mol Dis. 2005;34:181–185. doi: 10.1016/j.bcmd.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 16.Ranza E, Bertolotti A, Facoetti A, Mariotti L, Pasi F, Ottolenghi A, Nano R. Influence of imatinib mesylate on radiosensitivity of astrocytoma cells. Anticancer Res. 2009;29:4575–4578. [PubMed] [Google Scholar]
- 17.Savage DG, Antman KH. Imatinib mesylate-a new oral targeted therapy. N Engl J Med. 2002;346:683–693. doi: 10.1056/NEJMra013339. [DOI] [PubMed] [Google Scholar]
- 18.Chen SW, Zhang XR, Wang CZ, Chen WZ, Xie WF, Chen YX. RNA interference targeting the platelet-derived growth factor receptor beta subunit ameliorates experimental hepatic fibrosis in rats. Liver Int. 2008 doi: 10.1111/j.1478-3231.2008.01759.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reardon DA, Desjardins A, Vredenburgh JJ, Herndon JE, II, Coan A, Gururangan S, Peters KB, McLendon R, Sathornsumetee S, Rich JN, et al. Phase II study of Gleevec plus hydroxyurea in adults with progressive or recurrent low-grade glioma. Cancer. 2012;118:4759–67. doi: 10.1002/cncr.26541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Biade S, Stobbe CC, Chapman JD. The intrinsic radiosensitivity of some human tumor cells throughout their cell cycles. Radiat Res. 1997;147:416–421. doi: 10.2307/3579497. [DOI] [PubMed] [Google Scholar]
- 21.Chen H, Huang Q, Dong J, Zhai DZ, Wang AD, Lan Q. Overexpression of CDC2/CyclinB1 in gliomas, and CDC2 depletion inhibits proliferation of human glioma cells in vitro and in vivo. BMC Cancer. 2008;8:29. doi: 10.1186/1471-2407-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He J, Li J, Ye C, Zhou L, Zhu J, Wang J, Mizota A, Furusawa Y, Zhou G. Cell cycle suspension: A novel process lurking in G2 arrest. Cell Cycle. 2011;10:1468–1476. doi: 10.4161/cc.10.9.15510. [DOI] [PubMed] [Google Scholar]
- 23.Maity A, Hwang A, Janss A, Phillips P, McKenna WG, Muschel RJ. Delayed cyclin B1 expression during the G2 arrest following DNA damage. Oncogene. 1996;13:1647–1657. [PubMed] [Google Scholar]
- 24.Silva MF, Khokhar AR, Qureshi MZ, Farooqi AA. Ionizing radiations induce apoptosis in TRAIL resistant cancer cells: in vivo and in vitro analysis. Asian Pac J Cancer Prev. 2014;15:1905–1907. doi: 10.7314/APJCP.2014.15.5.1905. [DOI] [PubMed] [Google Scholar]
- 25.Takano S, Ishikawa E, Sakamoto N, Matsuda M, Akutsu H, Noguchi M, Kato Y, Yamamoto T, Matsumura A. Immunohistochemistry on IDH 1/2, ATRX, p53 and Ki-67 substitute molecular genetic testing and predict patient prognosis in grade III adult diffuse gliomas. Brain Tumor Pathol. 2016;33:107–116. doi: 10.1007/s10014-016-0260-x. [DOI] [PubMed] [Google Scholar]
- 26.Lafuente JV, Adán B, Alkiza K, Garibi JM, Rossi M, Cruz-Sánchez FF. Expression of vascular endothelial growth factor (VEGF) and platelet-derived growth factor receptor-beta (PDGFR-beta) in human gliomas. J Mol Neurosci. 1999;13:177–185. doi: 10.1385/JMN:13:1-2:177. [DOI] [PubMed] [Google Scholar]
- 27.Fenton BM, Paoni SF, Ding I. Pathophysiological effects of vascular endothelial growth factor receptor-2-blocking antibody plus fractionated radiotherapy on murine mammary tumors. Cancer Res. 2004;64:5712–5719. doi: 10.1158/0008-5472.CAN-04-0434. [DOI] [PubMed] [Google Scholar]
- 28.Shannon AM, Williams KJ. Antiangiogenics and radiotherapy. J Pharm Pharmacol. 2008;60:1029–1036. doi: 10.1211/jpp.60.8.0009. [DOI] [PubMed] [Google Scholar]
- 29.Jain RK. Normalization of tumor vasculature: An emerging concept in antiangiogenic therapy. Science. 2005;307:58–62. doi: 10.1126/science.1104819. [DOI] [PubMed] [Google Scholar]
- 30.Cohen-Jonathan Moyal E. Angiogenic inhibitors and radiotherapy: From the concept to the clinical trial. Cancer Radiother. 2009;13:562–567. doi: 10.1016/j.canrad.2009.07.007. (In French) [DOI] [PubMed] [Google Scholar]
- 31.Raymond E. PDGFR inhibition in brain tumours-oft expectation fails where most it promises. Eur J Cancer. 2009;45:2236–2238. doi: 10.1016/j.ejca.2009.05.031. [DOI] [PubMed] [Google Scholar]
- 32.Gorski DH, Beckett MA, Jaskowiak NT, Calvin DP, Mauceri HJ, Salloum RM, Seetharam S, Koons A, Hari DM, Kufe DW, Weichselbaum RR. Blockage of the vascular endothelial growth factor stress response increases the antitumor effects of ionizing radiation. Cancer Res. 1999;59:3374–3378. [PubMed] [Google Scholar]
- 33.Omuro AM. Exploring multi-targeting strategies for the treatment of gliomas. Curr Opin Investig Drugs. 2008;9:1287–1295. [PubMed] [Google Scholar]