

Abstract
TNF family members and their receptors contribute to increased gene expression for inflammatory processes and intracellular cascades leading to programmed cell death, both via activation of NF-κB. TNF receptor (TNFR)-associated factors (TRAFs) are cytoplasmic adaptor proteins binding to various receptors of the TNFR family. In an attempt to delineate the role of individual TRAFs, we compared NF-κB activation by CD40wt and CD40 mutants with different TRAF recruitment patterns. Recognized only recently, NF-κB signaling occurs at least via two different pathways. Each pathway results in nuclear translocation of two different Reldimers, the canonical p50/RelA and the noncanonical p52/RelB. Here, we show that via TRAF6, CD40 mediates only the activation of the canonical NF-κB pathway. Via TRAF2/5, CD40 activates both the canonical and the noncanonical NF-κB pathways. We observed that TRAF3 specifically blocked the NF-κB activation via TRAF2/5. This inhibitory effect of TRAF3 depends on the presence of an intact zinc finger domain. Paradoxically, suppression of TRAF2/5-mediated NF-κB activation by TRAF3 resulted in enhanced transcriptional activity of TRAF6-mediated canonical NF-κB emanating from CD40. We also observed that 12 TNFR family members (p75TNFR, LTβR, RANK, HVEM, CD40, CD30, CD27, 4-1BB, GITR, BCMA, OX40, and TACI) are each capable of activating the alternative NF-κB pathway and conclude that TRAF3 serves as a negative regulator of this pathway for all tested receptors.
Keywords: TNF receptor superfamily, Rel family, NF-κB-inducing kinase
CD40 belongs to a subgroup of TNF receptor (TNFR) superfamily (TNFRSF) members that do not have a death domain and trigger intracellular signaling events via direct interaction with TNFR-associated factors (TRAFs). Signals through these TNFRSFs are essential for the development and coordinated function of lymphoid tissues, the induction of inflammatory responses, and tissue homeostasis in bone and mammary glands (reviewed in ref. 1). They may also provide necessary signals for the development of hair follicles and sweat-gland formation. The severe disease states resulting from loss-of-function mutations in genes that code for this group of receptors, such as X-linked hyper IgM syndrome (CD40-CD40L), anhidrotic ectodermal dysplasia (EDA/EDAR/XEDAR), or familial expansile osteolysis (RANK/RANKL), illustrate the functional importance of these molecules (1).
TRAFs were initially discovered through their ability to bind to the p75TNFR and were classified as a gene family on the basis of a conserved domain at the C terminus (2). This domain was termed TRAF domain and characterizes all six known TRAFs. The TRAF domain enables TRAFs to interact with various receptors, including the members of TNFRSF and also with multiple intracellular signaling molecules (reviewed in ref. 3). The TRAF proteins thus represent the molecular link between several signaling pathways and the TNFRSFs. They also act as the branching points between pathways mediating as contradicting functions as apoptosis and gene activation, i.e., via NF-κB or AP1 (4). The N-terminal two-thirds of all known TRAFs consists of several zinc-binding motifs that are believed to mediate the activation of downstream signaling molecules such as NF-κB-inducing kinase (NIK) (5) or c-Jun N-terminal kinase (6). TRAF1 that lacks zinc-binding motifs is the only exception. The initial characterization of TRAF functions was based on experiments with dominant negative mutants. These studies indicated that TRAF2, -5, and -6 were important for the activation of NF-κB (7–10). Gene inactivation studies confirmed these observations. The knockout of the TRAF2 gene not only resulted in premature death and increased sensitivity for TNF-mediated cell death but also in a reduced NF-κB activation via CD40 and the p55TNFR (11). Similarly, the disruption of the TRAF5 gene resulted in reduced NF-κB signaling by CD27 and CD40 (12). However, the inactivation of the TRAF2 or TRAF5 gene did not completely block NF-κB signaling of the TNFRSFs. Similar observations were made in TRAF6 knockout mice (13, 14). Taken together, these studies suggested that TRAFs act in concert and are able to supplement each other's function. The possibility that TNFRSF members mediate NF-κB activation in a TRAF-independent fashion was never completely ruled out.
Recent work in the NF-κB field revealed the existence of two NF-κB-signaling pathways that mediate the nuclear translocation of two different NF-κB binding activities, derived from the precursor proteins NF-κB1 (p105) and NF-κB2 (p100) (reviewed in refs. 15 and 16). These pathways were termed “canonical” and “noncanonical” NF-κB pathway and result in the nuclear appearance of p50/RelA or p52/RelB complexes with different consequences for NF-κB-dependent gene expression. Gene-inactivation studies suggest that the genetic program influenced by the canonical NF-κB pathway codes for inflammation and protection from cell death, whereas the noncanonical NF-κB pathway seems to control genes that are essential for the correct and coordinated development of lymphatic tissues (16).
The goal of this study was to obtain a better understanding of the interplay between the TRAFs in the activation of the two known NF-κB-signaling pathways. CD40, one of the best studied TNFRSF members was used as the model system to study the effect of mutations that interfere with TRAF recruitment on NF-κB signaling. A systematic analysis of the NF-κB components found in the nucleus after CD40 activation revealed that TRAF6 recruitment activates only the canonical NF-κB pathway, whereas TRAF2/5 recruitment leads to the activation of canonical and noncanonical NF-κB. It is shown that TRAF3 regulates the signal intensity of these two pathways. TRAF2/5-mediated NF-κB translocation into the nucleus was inhibited in TRAF3high cells. TRAF6-dependent NF-κB translocation was not affected; however, its transcriptional activity was enhanced. Suppressing the expression of TRAF2, TRAF5, or NIK with siRNA reconfirmed the findings with the CD40 mutants and supported the notion that both TRAF2/5-mediated NF-κB-signaling events, canonical and noncanonical, depend at least to some degree on the function of NIK. As shown here with 12 TRAF-binding TNFRSFs, the regulatory role of TRAF3 is not restricted to CD40.
Materials and Methods
Cell Lines. The human embryonic kidney cell line 293T was kept in Dulbecco's MEM Glutamax I medium (GIBCO). All media were supplemented with 5% heat-inactivated FCS (Biochrom), 100 units/ml penicillin, 0.1 mg/ml streptomycin, and 1 mM sodium pyruvate (GIBCO). For transient transfection experiments, the medium also contained 20 mM Hepes.
cDNA Cloning of CD40, CD40 Mutants, and CD40 Receptor Hybrid Mutants. The human CD40 cDNA was cloned as described in ref. 17. The CD40 mutants used in this study where generated with the QuikChange Site-Directed Mutagenesis kit (Stratagene).
PCR-directed mutagenesis was used for the construction of the CD40 receptor hybrids. A unique MluI site was created at the extracellular to cytoplasmic domain border of the CD40 cDNA and at the corresponding site of the TNFRSF cDNAs. The site was used to exchange the region encoding for the CD40-transmembrane and -cytoplasmic domains with the corresponding TNFRSF domains of interest. The TNFRSF cDNAs were either cloned by RT-PCR with the published sequence information or obtained as I.M.A.G.E. clones (http://image.llnl.gov/image/html/idistributors.shtml). All cloning steps were done under the strict consideration that the encoded amino acid sequences were identical to the corresponding wild-type receptors. For expression, all constructs were inserted in pEF-BOS (18).
The human CD40L cDNA was obtained from American Type Culture Collection (ATCC 79814), inserted into pEF-BOS, and stably overexpressed in BHK cells (CD40L+ cells).
The human TRAF2 cDNA was cloned as described in ref. 19. All other human TRAF cDNAs were cloned by RT-PCR with the published sequence information. The cDNAs coding for the deletion mutants ΔTRAF389–567 and ΔTRAF3324–567 were generated by restriction. The ΔTRAF3 constructs were expressed in pcDNA3.1His (Invitrogen), and the TRAF5 cDNA was expressed in pCMVTag3 (Invitrogen). All other TRAF constructs were inserted in pcDNA3.
The NF-κB reporter plasmid pIC277-ΔAP2 containing the two NF-κB sites from the ICAM-1 promoter was kindly provided by Judith Johnson (Institute for Immunology, Munich University, Munich).
All cDNA constructs used in this study were reconfirmed by sequencing before use.
NF-κB Reporter Assay. Subconfluent 293T cells were transfected in 96-well microtiter plates with a plasmid DNA mixture containing 6 ng of TRAF3 or empty control vector, 10 ng of receptor construct, and 180 ng of pIC277ΔAP2 per well, using the Ca phosphate precipitation method (20). Eighteen hours after transfection, formaldehyde-fixed CD40L+ cells were added for 3 h at an effector-to-target ratio of 2:1. The cells were then lysed in a buffer containing 25 mM glycylglycin (pH 7.8), 10 mM MgSO4, 0.5% Triton X-100, and 1% BSA. Fifty microliters of each lysate was immediately assayed for luciferase activity by using a substrate buffer containing 25 mM glycylglycin (pH 7.8), 60 mM DTT, 10 mM MgSO4, 1 mM ATP, and 35 μM Luciferin (PJK Industrievertretungen Handel). The plates were read in a Victor multilabel counter (PerkinElmer). All results were normalized on the basis of receptor expression as determined by a CD40 ELISA with the mAb Ro1 (21) and a polyclonal rabbit anti-CD40 (17) and HPLC-purified sCD40 as a standard.
Measurement of TRAF Recruitment. Subconfluent 293T cells were transfected in 3.5-cm dishes with 1 μg of expression plasmid coding for the receptor of interest and 4 μg of a TRAF expression plasmid as described above. The cells were detached 26 h after transfection, washed once in ice-cold PBS, and detergent-extracted with PBS containing a protease inhibitor mixture (0.16 mM Pefabloc, 105 units of aprotinin), 1% Triton X-100, and 0.1% Na Azide (lysis buffer). The lysates were serially diluted in lysis buffer containing 1% BSA and then reacted overnight with ELISA microtiter plates (Greiner, Nurtingen, Germany) coated with the anti-CD40 mAb Ro1 (21). TRAFs bound to the studied signaling domains were detected with rabbit anti-TRAF2 (sc-7187, Santa Cruz Biotechnology), rabbit anti-TRAF3 (Pharmingen), or rabbit anti-TRAF5 (sc-7220, Santa Cruz Biotechnology). The assay was developed using peroxidase-labeled anti-rabbit Ig (Dianova, Hamburg, Germany). A standard extract arbitrarily defined as 1,000 units/ml for each CD40/TRAF combination was used to eliminate assay-to-assay variations. All results were normalized on the basis of CD40 expression as determined by ELISA in the same lysates. Possible effects of variations in TRAF expression on the results of this assay were ruled out in pilot experiments. These experiments demonstrated that the TRAF recruitment could be reliably and reproducibly determined when the TRAF and CD40 expression plasmids were used at a ratio of 4:1 or above (data not shown).
Western Blot for NF-κB Proteins. Subconfluent 293T cells were transfected with 180 ng of plasmid encoding CD40, CD40 mutants, or receptor hybrid constructs together with TRAF3 or a control plasmid as described above. Nuclear extracts were prepared 26 h later as described in ref. 22. Samples of the nuclear extract or the cytoplasmic fraction were run on SDS/PAGE under reducing conditions and electrophoretically transferred onto poly(vinylidene difluoride) membranes. The membranes were blocked with 2.5% skim milk in PBS and briefly rinsed with PBS/Tween. The blots were incubated overnight with the appropriate antibody for NF-κB monomers. Mouse anti-p52 (Upstate Biotechnology Technology, Lake Placid, NY), rabbit anti-p50, mouse anti-RelA, and rabbit anti-RelB (sc-7178, sc-8008, and sc-226, respectively, from Santa Cruz Biotechnology) were used. Equal loading was reconfirmed with the monoclonal mouse anti-actin Ab (AC15, Sigma). The blots were developed using peroxidase-labeled secondary antibodies and the chemiluminescence-based SuperSignal West Pico kit (Pierce). For reexposure with a second anti-NF-κB antibody, the blots were briefly treated with H2O2.
Suppression of TRAF2/5 and NIK with siRNA. Subconfluent 293T cells were transfected with 12.5 μg of siRNA plasmid in 90-mm dishes as described above. A mixture of 1161-NIK and 1511-NIK in pSuper (OligoEngine) was used for suppression of NIK expression (23). TRAF2 and -5 expression was inhibited with siRNA constructs containing the nucleotide sequences 5′-GTGTCGAGTCCCTTGCAGA and 5′-CATTGCATCTGGCTGTCCC in pSuppressor (Imgenex). Empty vectors served as controls. The cells were detached 48 h (for NIK 24 h) after transfection, counted and seeded into six-well plates (TPP, Trasadingen, Switzerland) at a density of 5 × 105 cells per well. Three hours later, the cells were transfected again as described above.
Results
TRAF3 Inhibits NF-κB Activation of a CD40 Mutant Devoid of TRAF6 Binding. CD40 recruits all known TRAFs except TRAF4 despite a relatively short signaling domain (24), complicating the analysis of TRAF functions in a given signal transduction pathway. In an attempt to circumvent this problem, we systematically analyzed the signaling of CD40 mutants with isolated TRAF recruitment defects. As described by others (24), the mutation of E in position 235 results in loss of TRAF6 binding (Fig. 1). TRAF2, -3, and -5 bind to overlapping regions that all include the motif PxQxT254 (24). Therefore, it appeared logical that a single mutation of T254 to A (T254A) eliminates the binding of all three TRAFs (24). A more careful analysis, however, showed that this mutation only reduces TRAF2/5 recruitment and has no effect on TRAF3 binding (Fig. 1) (25). Reduction of TRAF2/3/5 binding to CD40 to background levels was seen only when the PxQxT254 and two QE motifs at amino acid positions 263 and 274 were mutated to Ala or when all three motifs were deleted (TAQEQE and 1–245 in Fig. 1).
Fig. 1.

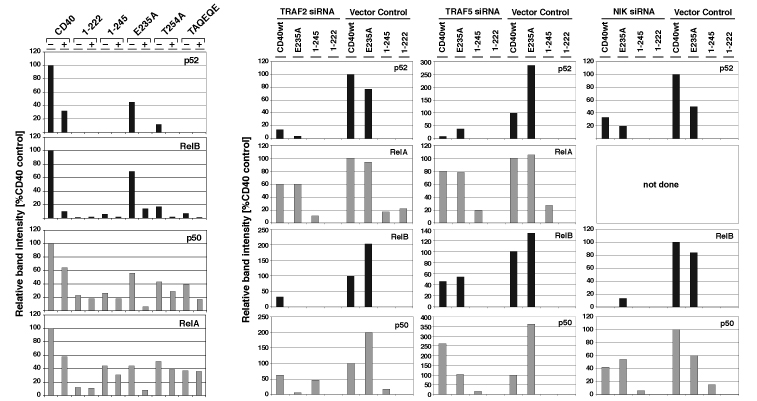
NF-κB activation and TRAF recruitment by CD40 and CD40 mutants. (A) Schematic presentation of the TRAF interaction sites in the CD40-signaling domain. Shown are the interactions sites of TRAF6 and TRAF2/3/5 as well as the mutations relevant in this study. (B) CD40-mediated activation of NF-κB ceases only when the TRAF6, the TRAF2/3/5, and two further QE motifs in the CD40 C terminus are mutated or deleted (correlation with TRAF recruitment). All assays were done as described in Materials and Methods. Induction of NF-κB activity was determined after 3 h of stimulation with CD40L+ cells. The results were normalized on the basis of receptor expression. TRAF recruitment is expressed as the percentage of the CD40wt control (arbitrarily assumed to be 100%).
Paradoxically, the TRAF recruitment data did not correlate with the NF-κB-signaling activity of these CD40 mutants in reporter gene assays. Isolated recruitment defects of TRAF6 (E235A) or TRAF2/3/5 (1–245) had no apparent effect on the CD40's ability to activate NF-κB-dependent transcription (Fig. 1B). A significant reduction of the NF-κB signaling was seen only when the binding of all four TRAFs was eliminated (EATAQEQE and 1–245 in Fig. 1). This suggested that CD40 can compensate for the isolated loss of TRAF6- or TRAF2/5-mediated NF-κB activation.
Next, we systematically tested NF-κB signaling of CD40 and its mutants in cells overexpressing one TRAF of interest. In these experiments, we observed that TRAF3 overexpression suppresses the NF-κB-signaling activity of E235A (Fig. 2), the CD40 mutant that binds only TRAF2, -3, and -5. No such effect was seen with CD40wt. CD40 mutants that bound only TRAF6 activated NF-κB in TRAF3high cells 1.5- to 2-fold more potently than CD40wt (1–245 in Fig. 2). Inhibition of CD40 and the p75TNFR-mediated NF-κB activation by TRAF3 had been observed in earlier studies (7). However, the effect had been attributed to binding competition between TRAF2, -3, and -5 for their receptor binding site (26–28). Structure function studies of TRAF3 demonstrated that deletion of the zinc-binding motifs does not affect its competence for receptor interaction (29, 30). Thus, it should be possible to use zinc-finger-less TRAF3 mutants to discern between effects that result from active TRAF3 signaling or from displacement of TRAF2/5 from the common CD40-binding site. As shown in Fig. 2, two such TRAF3 mutants, ΔTRAF389–567 and ΔTRAF3324–567, had no affect on the NF-κB-signaling activity of CD40 or its mutants. Thus, we concluded that TRAF3 acts as a molecular switch between two NF-κB-signaling pathways originating from CD40, the first triggered via TRAF6 and the second via TRAF2 or -5. This TRAF3 function was apparently not mediated by replacement of TRAF2/5 from their receptor binding site.
Fig. 2.

TRAF3 inhibits NF-κB signaling of CD40 mutants with TRAF6 recruitment defects (dependence on zinc finger motifs). CD40wt or CD40 mutants were transfected into 293T cells together with TRAF3, ΔTRAF389–567, or ΔTRAF3324–567 expression plasmids and a NF-κB reporter plasmid. Eighteen hours later the cells were stimulated for 3 h with CD40L+ cells, then lysed and Luciferase activity was determined. All results were normalized on the basis of CD40 expression. Shown are the average values of duplicate determinations of one representative experiment of four.
TRAF3 Inhibits TRAF2/5-Mediated Activation of the Noncanonical NF-κB Pathway. Recent studies demonstrated that the activation of NF-κB occurs through two different signaling pathways (reviewed in ref. 16). The first or canonical pathway operates via the kinase IKKβ and results in the degradation of IkBα and subsequently in the nuclear translocation of p50/RelA heteromers. It essentially requires the protein IKKγ (NEMO). The second or noncanonical pathway is activated by IKKα, operates independently from IKKγ, and results in the degradation of NF-κB2 (p100) and the nuclear translocation of p52/RelB. Coope et al. (31) demonstrated recently that CD40 activates both the canonical and the noncanonical NF-κB pathway. This study also shows that the T254A mutation reduces CD40 signaling for the noncanonical NF-κB pathway. In view of these data, we performed a detailed analysis of the NF-κB proteins translocated into the nucleus in response to CD40wt and CD40 mutants. Mutation or deletion of the TRAF2/3/5-binding site in CD40 as in T254A, 1–245, or TAQEQE reduced or completely eliminated the nuclear translocation of the p52/RelB complex (Fig. 3A; see also Fig. 6, which is published as supporting information on the PNAS web site). This observation correlated with reduced or absent processing of p100 to p52 in the cytoplasm (Fig. 3A, compare nuclear p52 with cytoplasmic p52). Inactivation of the TRAF6-binding site, on the other hand, led only to a slight reduction of CD40-mediated nuclear translocation of p52 (E235A in Figs. 3 and 6) or CD40 induced p100 processing. The translocation of p50/RelA heteromers appeared to be mediated by both TRAF6 and TRAF2/5. Neither the loss of TRAF6 binding (E235A in Fig. 3 A and B) nor the loss of TRAF2/5 binding completely abolished CD40-mediated translocation of p50/RelA complexes into the nucleus (1–245 or TAQEQE in Fig. 3).
Fig. 3.

TRAF3 inhibits TRAF2/5- but not TRAF6-mediated NF-κB activation. Subconfluent 293T cells were transfected with CD40 or CD40 mutant expression plasmids together with TRAF3wt (A), ΔTRAF3324–567 (B), or a control plasmid (A and B). The cells were harvested 26 h after transfection, and nuclear extracts were prepared as described in ref. 22. Five micrograms of protein of each extract was analyzed by Western blot. The p100 degradation was analyzed in the cytoplasmic fraction of the same experiment. CD40 or CD40mut expression was also determined in the cytoplasmic fractions and is shown as the percentage of the control experiment with CD40wt. Shown is one representative result of four experiments.
Studying CD40-mediated NF-κB signaling in TRAF3high cells revealed that TRAF3 inhibits the ability of CD40 to trigger the nuclear translocation of p52/RelB complexes. The CD40wt signal was significantly reduced, and complete suppression was found with the CD40 mutant E235A that binds only TRAF2, -3, and -5 (Fig. 3, blot 1). Notably, TRAF3 blocked E235A-mediated translocation of both p50/RelA and p52/RelB heteromers. The data show that TRAF2/5 recruitment results in the activation of both canonical and noncanonical NF-κB and that TRAF3 acts as a negative regulatory element in this pathway. TRAF6, in contrast, serves CD40 exclusively for the activation of the canonical pathway.
The specificity of this TRAF3 effect on the signaling pathway originating from the TRAF2/5-binding motif could be demonstrated with 1–245. This CD40 mutant binds only TRAF6 and was unable to trigger the degradation of p100 in the cytoplasm (compare nuclear and cytoplasmic p52 in Fig. 3A) or the nuclear translocation of p52/RelB (Fig. 3). However, 1–245 still mediated the nuclear translocation of p50/RelA, although at clearly reduced levels (Fig. 3). This NF-κB signal was not affected by changes in the cellular TRAF3 expression level (Fig. 3) showing that TRAF3 does not suppress TRAF6-mediated NF-κB translocation.
TRAF2, TRAF5, and NIK Contribute to CD40-Mediated Activation of the Canonical and Noncanonical NF-κB. In the next set of experiments, the siRNA approach was used to test the effect of reduced TRAF2, TRAF5, or NIK expression on CD40-mediated NF-κB activation. As expected from the studies with CD40 mutants, the ability of CD40wt or E235A to signal for nuclear translocation of p52/RelB complexes was completely blocked or clearly reduced in TRAF2, TRAF5, and NIK targeted cells (Figs. 4 and 6). This finding demonstrates that all three proteins contribute significantly to CD40-mediated activation of noncanonical NF-κB. CD40-induced nuclear translocation of p50/RelA was less affected by the decreased TRAF2/5 expression, thus reconfirming that CD40 signals the activation of canonical NF-κB also via TRAF6 and that this occurs independently from TRAF2/5. The observation that CD401–245-induced p50/RelA was not reduced in TRAF2- or TRAF5-targeted cells also supported this notion. The nuclear translocation of p50/RelA triggered by this mutant appeared even slightly increased in these cells. Similar consequences for the activation of canonical NF-κB were seen in NIK-targeted cells. CD40- and E235A-mediated p50 activation was reduced but not completely blocked, and the ability of CD401–245 to trigger this signaling event was unaffected (Fig. 4 Bottom).
Fig. 4.

Targeting of TRAF2, TRAF5, or NIK expression with siRNA reduces CD40-mediated activation of the noncanonical NF-κB pathway. Subconfluent 293T cells were transfected in a two-step procedure with the indicated siRNA constructs and with CD40 or the indicated CD40 mutants. Nuclear extracts were prepared and analyzed as described in Fig. 3.
These results further support the view that CD40 uses two signaling options for NF-κB activation, one via TRAF6, resulting in the activation of canonical NF-κB, and the other via TRAF2/5/NIK, leading to activation of canonical and noncanonical NF-κB. Because of limitations in the transfection efficiency, it was impossible to completely eliminate the expression of the targeted proteins. Thus, the question whether TRAF2/5-mediated NF-κB signaling operates entirely via NIK ultimately cannot be answered.
TRAF3 Is Recruited by the Majority of the TRAF-Binding TNFR SF Members and Regulates Their Ability to Activate the Noncanonical NF-κB. Gene-inactivation experiments show that TRAF3 is important for the survival of an adult organism (32). Therefore, we were interested in determining whether the regulatory function of TRAF3 on NF-κB signaling was restricted to CD40. Because our experiments showed that TRAF3 blocked only TRAF2/5-mediated NF-κB, we compared the binding of TRAF2, -3, and -5 to 12 different TNFRSF members. All receptors tested were able to bind TRAF3, although their binding capacity differed vastly, ranging from 20-fold more (RANK) to 20-fold less (OX40) than CD40 (Fig. 5A). All receptors tested also bound TRAF2 and -5, again with large differences in the binding capacity, RANK displaying the highest and OX40 the lowest binding capacity (Fig. 5A).
Fig. 5.

TRAF3 inhibits noncanonical NF-κB activation by TRAF-binding TNFRSF members. (A) TRAF2, -3, and -5 recruitment to TNFRSF members. TRAF recruitment to TNFRSF members was determined by coexpression of the indicated receptor and the TRAF of interest in 293T cells. TRAF-binding was quantified by ELISA as described. TRAF-binding to CD40 was used as a reference. The receptor expression in each individual experiment was determined and used to normalize the recruitment data. Shown is the data and standard deviation of three independent experiments. (B) TRAF3 inhibits the activation of noncanonical NF-κB through TNFRSF members. Shown is the nuclear translocation of p52 in response to the indicated TNFRSF members in 293T cells expressing TRAF3 at low versus high levels. Transfections were done as described above. Receptor expression was adjusted in pilot experiments to intermediate p52 signal intensity. The amount of receptor-expression plasmid used in the experiment and the resulting receptor expression are shown. Receptor expression was determined in the cytoplasmic fractions of the nuclear extracts used for the detection of nuclear p52.
Consistent with the TRAF recruitment data, all 12 receptors also activated the noncanonical NF-κB pathway as judged by nuclear translocation of p52. The signal capacity for the activation of the noncanonical NF-κB pathway correlated roughly with the recruitment data for TRAF5. RANK delivered the most potent signal and a receptor expression of 8,000 receptors per cell was sufficient to achieve p52 translocation at detectable levels. Similar signal intensity was found for the LTβR. For CD30, a 5-fold higher receptor expression was required. A further 5- to 10-fold increase in receptor expression was needed to obtain detectable p52 translocation into the nucleus with the p75TNFR, BCMA, HVEM, CD40, TACI, and 4-1BB. The highest receptor expression of ≈106 per cell was needed for CD27, GITR, and OX40. Next, we tested the signaling for noncanonical NF-κB in TRAF3high cells. As expected, in TRAF3high cells the noncanonical NF-κB signal was completely suppressed or reduced to very low levels as in the case of RANK.
Discussion
In this study, we used a set of CD40 mutants with defined TRAF recruitment defects to find out how TRAF2, -3, -5, and -6 cooperate for the activation of NF-κB. The results substantiated and extended previous observations showing that CD40 activates both the canonical and the noncanonical NF-κB pathway (31) and revealed a previously uncharacterized TRAF3 function. Our data demonstrate that CD40 uses TRAF6 to activate exclusively the canonical NF-κB pathway, whereas TRAF2/5 recruitment mediates both the activation of canonical and noncanonical NF-κB. TRAF3 inhibits the TRAF2/5-mediated NF-κB signal and enhances the transcriptional activity of TRAF6-mediated NF-κB. Two zinc-finger-less TRAF3 mutants did not have this activity, although they were reported to be fully competent for CD40 or LTβR interaction (29, 30). Competition of TRAF3 with TRAF2/5 for the common binding site in CD40 can thus not be the molecular basis for the TRAF3 activity described here. We could further show that this TRAF3 function is not restricted to CD40. All 12 TRAF-binding receptors of the TNFRSF tested here, including RANK, CD30, the LTβR, BCMA, HVEM, the p75TNFR, TACI, CD40, CD27, GITR, 4-1BB, and OX40, activate the noncanonical NF-κB pathway. TRAF3 blocked or down-regulated this signal in all 12 cases.
The ability of CD40 to activate the transcription factor NF-κB was first observed >10 years ago (33). With the isolation of the TRAF proteins it became clear that TRAF2, -5, and -6 participate in this signaling function (7–9). However, despite exhaustive structure function analysis it is still unclear how the three TRAF molecules cooperate to generate the NF-κB signal induced by CD40 (24, 34–38). Destruction of the TRAF6- or TRAF2/3/5 binding sites had limited or no effect on TNFRSF-mediated NF-κB signaling and it was speculated that CD40 also uses TRAF-independent mechanisms to activate this transcription factor (24, 39). With the discovery of the noncanonical NF-κB pathway, it became clear that NF-κB activation had to be reanalyzed. Coope et al. (31) were the first to show that CD40 activates both NF-κB-signaling pathways. Their study also provided the first evidence that TRAF2 recruitment and function are important for CD40-mediated activation of noncanonical NF-κB. The data presented here substantiate and extend these observations. Two CD40 mutants that recruit only TRAF6 (1–245 and TAQEQE) were completely unable to activate the noncanonical NF-κB pathway. A CD40 mutant that signals only via TRAF2, -3, and -5, on the other hand, activated the noncanonical and, unexpectedly, also the canonical NF-κB pathway. Comparing these mutants in reporter gene assays, in turn, reconfirmed previous observations. The loss of TRAF6- or TRAF2/3/5 binding had no significant effect on CD40's ability to activate the transcription of an NF-κB reporter gene. However, the data shown here do not support the possibility of TRAF-independent NF-κB-signaling mechanisms. The translocation of NF-κB heteromers into the nucleus clearly depends on intact TRAF binding sites, and their destruction results in the expected loss of function with respect to NF-κB signaling. Antagonism between TRAF6- and TRAF2/5-mediated NF-κB signals would also explain why isolated TRAF recruitment defects do not correlate with NF-κB reporter gene assays. Observations made with the LTβR support such a possibility. Thus, it was shown that IKKα inactivation results in increased NF-κB signaling of the LTβR via the canonical pathway (40). A similar observation was made in this study. TRAF3-induced blockade of TRAF2/5-mediated signaling via the noncanonical NF-κB pathway resulted in the increased response of an NF-κB-driven reporter gene to the classical NF-κB induced by TRAF6. The molecular basis of this phenomenon is unclear, and further studies will be needed to understand it. An informative approach, in this respect, would be the use of NF-κB reporter genes that respond specifically to p50/RelA or p52/RelB.
With the demonstration that TRAF2 mediates NF-κB activation, it was also shown that TRAF3 may act as a negative regulator of NF-κB activation (7). Since then, several studies reported similar effects of TRAF3 on the NF-κB signaling of the LTβR (41), OX40 (42), BAFF (43), LMP (26, 27), and CD40 (44). Until it was demonstrated that TRAF2 and TRAF3 do not interfere with each other's CD40 binding (39), this TRAF3 function was attributed to competition between TRAF3 and TRAF2/5 for their common receptor-binding site (7, 26, 27, 43, 44). A common problem of these studies is that they did not account for the existence of the two NF-κB-signaling pathways and measured only molecular events that characterize the canonical NF-κB pathway. As shown here, the suppressive effect of TRAF3 on CD40wt-mediated NF-κB signaling cannot be picked up in reporter-gene assays. Only the detailed analysis of the NF-κB heteromers in the nucleus after CD40 activation demonstrated that the NF-κB signal originating from TRAF2/5 is the target of the regulatory activity of TRAF3. Using a CD40 mutant that cannot signal via TRAF6 allowed similar conclusions. With the help of this mutant, it was also possible to demonstrate that this TRAF3 activity requires intact zinc-binding motifs and cannot be the result of binding competition between TRAF2/5 and TRAF3. This notion is also supported by a study by Takaori-Kondo et al. (42), who first suggested that the zinc-binding motifs in TRAF3 were needed for the inhibition of OX40-mediated NF-κB activation.
TRAF2/5-mediated NF-κB signals essentially require the kinase NIK (23). NIK is thus the most likely molecular target for the NF-κB suppressive effect of TRAF3. Indeed, it has been shown that TRAF3 binds to NIK (6). A very recent study provides evidence that this interaction may result in the ubiquitination and subsequent degradation of NIK (45). However, observations made by other colleagues do not support this mechanism of TRAF3 function. Takaori-Kondo et al. (42) show that TRAF3 mutants that lack the NIK interaction site still suppress the NF-κB activation via OX40. They also suggest that TRAF2 is the most likely molecular target of TRAF3. The observation that TRAF3-deficiency in B cells has opposing effects on the NF-κB signaling of CD40 and LMP (44) is also inconsistent with the model of TRAF3 action suggested by Liao et al. (45). The observation by Ardila-Osorio et al. (46) that TRAF degradation may differ in different cellular environments adds to the confusion. Further studies will be needed to resolve the controversial observations and explain the mechanism by which TRAF3 inhibits TRAF2/5-mediated NF-κB signaling.
In view of the lethal outcome of the TRAF3 gene inactivation, it was important to consider whether the TRAF3 function described here is of wider importance for the signaling of the TNFRSF. As shown here, this is indeed the case. Our findings support a model where it is the function of TRAF3 to block the alternative NF-κB pathway and shift the NF-κB complexes translocated into the nucleus in response to TNFRSFs toward a p50/RelA constellation. Whether the loss of a factor that serves this purpose may have lethal consequences cannot be said at the moment, and it also remains to be seen whether our in vitro observations will be reconfirmed in vivo. However, considering the importance of NF-κB for survival, it is not unlikely that the inability to generate the appropriate NF-κB activity at the appropriate time point may have lethal consequences, particularly when an entire receptor family is affected.
Supplementary Material
Acknowledgments
This work was supported by Deutsche Forschungsgemeinschaft Grant SFB455 and a grant from Wissenschaftliches Herausgebergremium der Münchener Medizinische Wochenschrift. J.H. is a fellow of the Boehringer Ingelheim Fonds. S.P. is a fellow of the Hanns-Seidl-Stiftung.
Abbreviations: NIK, NF-κB-inducing kinase; TNFR, TNF receptor; TNFRSF, TNFR superfamily; TRAF, TNFR-associated factor.
References
- 1.Locksley, R. M., Killeen, N. & Lenardo, M. J. (2001) Cell 104, 487–501. [DOI] [PubMed] [Google Scholar]
- 2.Rothe, M., Wong, S. C., Henzel, W. J. & Goeddel, D. V. (1994) Cell 78, 681–692. [DOI] [PubMed] [Google Scholar]
- 3.Dempsey, P. W., Doyle, S. E., He, J. Q. & Cheng, G. (2003) Cytokine Growth Factor Rev. 14, 193–209. [DOI] [PubMed] [Google Scholar]
- 4.Hsu, H., Shu, H.-B., Pan, M.-G. & Goeddel, D. V. (1996) Cell 84, 299–308. [DOI] [PubMed] [Google Scholar]
- 5.Malinin, N. L., Boldin, M. P., Kovalenko, A. V. & Wallach, D. (1997) Nature 385, 540–544. [DOI] [PubMed] [Google Scholar]
- 6.Song, H. Y., Regnier, C. H., Kirschning, C. J., Goeddel, D. V. & Rothe, M. (1997) Proc. Natl. Acad. Sci. USA 94, 9792–9796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rothe, M., Sarma, V., Dixit, V. M. & Goeddel, D. V. (1995) Science 269, 1424–1427. [DOI] [PubMed] [Google Scholar]
- 8.Ishida, T. K., Tojo, T., Aoki, T., Kobayashi, N., Ohishi, T., Watanabe, T., Yamamoto, T. & Inoue, J. (1996) Proc. Natl. Acad. Sci. USA 93, 9437–9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ishida, T., Mizushima, S., Azuma, S., Kobayashi, N., Tojo, T., Suzuki, K., Aizawa, S., Watanabe, T., Mosialos, G., Kieff, E., et al. (1996) J. Biol. Chem. 271, 28745–28748. [DOI] [PubMed] [Google Scholar]
- 10.Cao, Z., Xiong, J., Takeuchi, M., Kurama, T. & Goeddel, D. V. (1996) Nature 383, 443–446. [DOI] [PubMed] [Google Scholar]
- 11.Yeh, W. C., Shahinian, A., Speiser, D., Kraunus, J., Billia, F., Wakeham, A., de la Pompa, J. L., Ferrick, D., Hum, B., Iscove, N., et al. (1997) Immunity 7, 715–725. [DOI] [PubMed] [Google Scholar]
- 12.Nakano, H., Sakon, S., Koseki, H., Takemori, T., Tada, K., Matsumoto, M., Munechika, E., Sakai, T., Shirasawa, T., Akiba, H., et al. (1999) Proc. Natl. Acad. Sci. USA 96, 9803–9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lomaga, M. A., Yeh, W. C., Sarosi, I., Duncan, G. S., Furlonger, C., Ho, A., Morony, S., Capparelli, C., Van, G., Kaufman, S., et al. (1999) Genes Dev. 13, 1015–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Naito, A., Azuma, S., Tanaka, S., Miyazaki, T., Takaki, S., Takatsu, K., Nakao, K., Nakamura, K., Katsuki, M., Yamamoto, T. & Inoue, J. (1999) Genes Cells 4, 353–362. [DOI] [PubMed] [Google Scholar]
- 15.Pomerantz, J. L. & Baltimore, D. (2002) Mol. Cell 10, 693–695. [DOI] [PubMed] [Google Scholar]
- 16.Bonizzi, G. & Karin, M. (2004) Trends Immunol. 25, 280–288. [DOI] [PubMed] [Google Scholar]
- 17.Hess, S., Kurrle, R., Lauffer, L., Riethmüller, G. & Engelmann, H. (1995) Eur. J. Immunol. 25, 80–86. [DOI] [PubMed] [Google Scholar]
- 18.Mizushima, S. & Nagata, S. (1990) Nucleic Acids Res. 18, 5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mullinax, R., Sorge, J., Hess, S. & Engelmann, H. (1996) Strategies 9, 81–83. [Google Scholar]
- 20.Kingston, R. E., Chen, C. A., Okayama, H. & Rose, J. K. (1993) in Current Protocols in Molecular Biology, eds. Ausubel, F. M., Brent, R., Kingston, R. E., Moore, D. D., Seidman, J. G., Smith, J. A. & Struhl, K. (Wiley, Boston), Vol. 2, pp. 9.1–9.1.11. [Google Scholar]
- 21.Schwabe, R. F., Hess, S., Johnson, J. P. & Engelmann, H. (1997) Hybridoma 16, 217–226. [DOI] [PubMed] [Google Scholar]
- 22.Fessele, S., Boehlk, S., Mojaat, A., Miyamoto, N. G., Werner, T., Nelson, E. L., Schlondorff, D. & Nelson, P. J. (2001) FASEB J. 15, 577–579. [DOI] [PubMed] [Google Scholar]
- 23.Ramakrishnan, P., Wang, W. & Wallach, D. (2004) Immunity 21, 477–489. [DOI] [PubMed] [Google Scholar]
- 24.Pullen, S. S., Dang, T. T., Crute, J. J. & Kehry, M. R. (1999) J. Biol. Chem. 274, 14246–14254. [DOI] [PubMed] [Google Scholar]
- 25.Haxhinasto, S. A., Hostager, B. S. & Bishop, G. A. (2002) J. Immunol. 169, 1145–1149. [DOI] [PubMed] [Google Scholar]
- 26.Devergne, O., Hatzivassiliou, E., Izumi, K. M., Kaye, K. M., Kleijnen, M. F., Kieff, E. & Mosialos, G. (1996) Mol. Cell. Biol. 16, 7098–7108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miller, W. E., Mosialos, G., Kieff, E. & Raab Traub, N. (1997) J. Virol. 71, 586–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hostager, B. S. & Bishop, G. A. (1999) J. Immunol. 162, 6307–6311. [PubMed] [Google Scholar]
- 29.Cheng, G., Cleary, A. M., Ye, Z.-s., Hong, D. I., Lederman, S. & Baltimore, D. (1995) Science 267, 1494–1498. [DOI] [PubMed] [Google Scholar]
- 30.Force, W. R., Cheung, T. C. & Ware, C. F. (1997) J. Biol. Chem. 272, 30835–30840. [DOI] [PubMed] [Google Scholar]
- 31.Coope, H. J., Atkinson, P. G., Huhse, B., Belich, M., Janzen, J., Holman, M. J., Klaus, G. G., Johnston, L. H. & Ley, S. C. (2002) EMBO J. 21, 5375–5385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu, Y., Cheng, G. & Baltimore, D. (1996) Immunity 5, 407–415. [DOI] [PubMed] [Google Scholar]
- 33.Berberich, I., Shu, G. L. & Clark, E. A. (1994) J. Immunol. 153, 4357–4366. [PubMed] [Google Scholar]
- 34.Pullen, S. S., Miller, H. G., Everdeen, D. S., Dang, T. T., Crute, J. J. & Kehry, M. R. (1998) Biochemistry 37, 11836–11845. [DOI] [PubMed] [Google Scholar]
- 35.Pullen, S. S., Labadia, M. E., Ingraham, R. H., McWhirter, S. M., Everdeen, D. S., Alber, T., Crute, J. J. & Kehry, M. R. (1999) Biochemistry 38, 10168–10177. [DOI] [PubMed] [Google Scholar]
- 36.Park, Y. C., Burkitt, V., Villa, A. R., Tong, L. & Wu, H. (1999) Nature 398, 533–538. [DOI] [PubMed] [Google Scholar]
- 37.McWhirter, S. M., Pullen, S. S., Holton, J. M., Crute, J. J., Kehry, M. R. & Alber, T. (1999) Proc. Natl. Acad. Sci. USA 96, 8408–8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ni, C. Z., Welsh, K., Leo, E., Chiou, C. K., Wu, H., Reed, J. C. & Ely, K. R. (2000) Proc. Natl. Acad. Sci. USA 97, 10395–10399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leo, E., Welsh, K., Matsuzawa, S., Zapata, J. M., Kitada, S., Mitchell, R. S., Ely, K. R. & Reed, J. C. (1999) J. Biol. Chem. 274, 22414–22422. [DOI] [PubMed] [Google Scholar]
- 40.Dejardin, E., Droin, N. M., Delhase, M., Haas, E., Cao, Y., Makris, C., Li, Z. W., Karin, M., Ware, C. F. & Green, D. R. (2002) Immunity 17, 525–535. [DOI] [PubMed] [Google Scholar]
- 41.Force, W. R., Glass, A. A., Benedict, C. A., Cheung, T. C., Lama, J. & Ware, C. F. (2000) J. Biol. Chem. 275, 11121–11129. [DOI] [PubMed] [Google Scholar]
- 42.Takaori-Kondo, A., Hori, T., Fukunaga, K., Morita, R., Kawamata, S. & Uchiyama, T. (2000) Biochem. Biophys. Res. Commun. 272, 856–863. [DOI] [PubMed] [Google Scholar]
- 43.Xu, L. G. & Shu, H. B. (2002) J. Immunol. 169, 6883–6889. [DOI] [PubMed] [Google Scholar]
- 44.Xie, P., Hostager, B. S. & Bishop, G. A. (2004) J. Exp. Med. 199, 661–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liao, G., Zhang, M., Harhaj, E. W. & Sun, S. C. (2004) J. Biol. Chem. 279, 26243–26250. [DOI] [PubMed] [Google Scholar]
- 46.Ardila-Osorio, H., Pioche-Durieu, C., Puvion-Dutilleul, F., Clausse, B., Wiels, J., Miller, W., Raab-Traub, N. & Busson, P. (2005) Int. J. Cancer 113, 267–275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}