

Abstract
Dilated cardiomyopathy (DCM) is associated with extensive pathological cardiac remodeling and involves numerous changes in the protein expression profile of the extracellular matrix of the heart. We obtained seven human, end-stage, failing hearts with DCM (DCM-failing) and nine human, nonfailing donor hearts and compared their extracellular matrix protein profiles. We first showed that the DCM-failing hearts had indeed undergone extensive remodeling of the left ventricle myocardium relative to nonfailing hearts. We then isolated the extracellular matrix from a subset of these hearts and performed a proteomic analysis on the isolated matrices. We found that the levels of 26 structural proteins were altered in the DCM-failing isolated cardiac extracellular matrix compared to nonfailing isolated cardiac extracellular matrix. Overall, most of the extracellular matrix proteins showed reduced levels in the DCM-failing hearts, while all of the contractile proteins showed increased levels. There was a mixture of increased and decreased levels of cytoskeletal and nuclear transport proteins. Using immunoprobing, we verified that collagen IV (α2 and α6 isoforms), zyxin, and myomesin protein levels were reduced in the DCM-failing hearts. We expect that these data will add to the understanding of the pathology associated with heart failure with DCM.
Keywords: Dilated cardiomyopathy, Heart failure, Extracellular matrix, Proteomics
1. Introduction
Heart failure is a complex, end-stage disease that culminates from any of a number of primary cardiovascular diseases, such as hypertension, coronary artery disease, and myocardial infarction, to list a few. In the United States, 5.7 million people are living with heart failure. A grim 50% of those diagnosed with heart failure will die within 5 years of diagnosis [1]. As part of the compensatory phase in the pathway toward worsening heart failure, the heart undergoes a process of pathological remodeling. The heart attempts to compensate for reduced cardiac output by enlarging the chambers of the heart in dilated cardiomyopathy (DCM) or thickening the ventricle walls in hypertrophic cardiomyopathy. While these compensatory mechanisms initially increase cardiac output, they ultimately lead to reduced cardiac function and development or worsening of heart failure.
The pathological remodeling process in the heart often involves changes in the expression of specific extracellular matrix (ECM) proteins, upsetting a delicate balance normally maintained in healthy myocardium and the development of fibrosis [2,3]. It is conventionally known that the healthy cardiac ECM is composed of approximately 85% collagen I, 10% collagen III, 5% additional collagen isoforms, and other matrix proteins such as laminin and fibronectin [4–6]. Active and resolved pathological remodeling of the heart promotes changes to the relative amounts of the various matrix proteins as well as expression of wound-specific matricellular proteins, such as tenascin C, not normally expressed in the adult heart [7,8]. These changes in the ECM have a significant impact on the heart during pathological cardiac remodeling because the ECM of the heart plays a vital role in its structure and function. The ECM of the heart accounts for 2%–4% of its volume [2,3]. Cardiomyocytes are anchored to the ECM through focal adhesions. This connection stabilizes cardiomyocytes during contraction as well as transmits the contractile force throughout the myocardium, which supports coordinated contraction [9]. Thus, when the ECM composition is altered during pathological remodeling, it has the potential to significantly impact the heart’s structure and function on many levels.
Cardiac fibroblasts reside within the cardiac ECM. These fibroblasts are responsible for the homeostatic maintenance of the ECM and interact with cardiomyocytes through paracrine signaling [10,11]. Cardiac fibroblasts also drive the process of ECM remodeling. Histologically, DCM is most often characterized by interstitial fibrosis [12]. In this type of fibrosis, the ECM surrounding cardiomyocytes expands. This is in contrast with replacement fibrosis commonly observed in myocardial infarcts, where cardiomyocytes have been lost and are replaced by a fibrotic scar.
We were interested in characterizing the ECM proteins expressed in failing hearts with DCM, focusing our efforts on decellularized, isolated ECM. We chose to study isolated ECM in order to focus very specifically on changes in the matrix and not changes that occur in the various cells that may reside in the heart (such as cardiomyocytes, fibroblasts, endothelial cells, smooth muscle cells, and infiltrating inflammatory cells). We intend to use this information to assist in analyzing the interaction between cardiac cells and the surrounding ECM, but we believe that this information may be more broadly useful. To our knowledge, there has not been a global, quantitative, proteomic analysis performed specifically on decellularized, isolated cardiac ECM from human patients with end-stage heart failure [New York Heart Association (NYHA) class III or IV] and DCM. There have been previously published studies on the “matrisome” in mouse hearts [13] and in a pig model of ischemia–reperfusion injury [14], whole-tissue proteomic studies in human hearts with inflammatory DCM and NYHA class II or III heart failure [15], and a comparative study of human hearts with DCM and ischemic cardiomyopathy [16]. The human heart studies used cardiac tissue with cellular components included in the analysis, and the animal studies were not focused on DCM and, obviously, not in humans. We believe that a proteomic analysis of decellularized, isolated ECM would assist in better understanding the pathology of DCM, focusing specifically on the contribution of the remodeled matrix. Moreover, we feel that it is important to assess human clinical cases. This additional understanding of the pathology in DCM could lead to better diagnostic criteria, identification of novel treatments to prevent or reverse pathological remodeling, and more reliable measures of successful reverse remodeling. Rather than reassessing the protein changes in high-abundance ECM proteins such as collagen I and collagen III, we instead chose to focus this proteomic study on lower-abundance proteins to expand our knowledge of ECM changes that occur in DCM-associated heart failure.
To perform this study, we assessed human hearts from end-stage heart failure patients with DCM. We compared them to nonfailing human hearts without diagnosed heart failure (although many had established cardiovascular disease risks and possible heart disease, listed in the Supplemental Table. We assessed the extent of remodeling and the protein profile in the ECM isolated from a subset of these hearts. We report here the identification of 26 structural proteins with significant differences in their levels between DCM-failing and nonfailing hearts.
2. Materials and methods
2.1. Procurement of human heart tissue
The studies on human heart tissue were performed under the guidelines of the Declaration of Helsinki, with oversight by the Institutional Review Boards at The Ohio State University (protocol no. 2012H0197) and University of New Mexico Health Sciences Center (protocol no. 13-358). Failing heart samples were obtained from patients who were diagnosed with heart failure who were receiving a heart transplant at The Ohio State University Wexner Medical Center. Nonfailing hearts were obtained in collaboration with the Lifeline of Ohio Organ Procurement program from organ donors without diagnosed heart failure whose hearts were not suitable for transplanting. These donors died from causes other than heart failure. Of note, the nonfailing donor hearts were not necessarily devoid of heart disease, and all donors possessed varying degrees of cardiovascular disease risks (Supplemental Table). In one case (sample 352041), the donor had an ejection fraction of only 25% and would likely have been diagnosed with heart failure before death in different circumstances. This donor died from a cerebral vascular accident/intracerebral hemorrhage. It is not feasible to obtain 100% completely healthy “control” human hearts devoid of any sort of cardiovascular disease for research purposes since such hearts are almost exclusively utilized for cardiac transplantation. With the exception of the abovementioned donor heart, the nonfailing hearts have ejection fractions in the normal range. All have anatomical characteristics (left ventricular wall thickness, left/right ventricular volume ratios, and atrial dimensions) that were well within the normal range for a healthy heart (data not shown). Additionally, ex vivo contractile studies performed on isolated trabecular muscles from several of the nonfailing donor hearts confirmed that contractile function was normal. That is, they possessed positive force–frequency relationship and a robust response to beta-adrenergic stimulation [17]. Finally, studies on overall histopathology and specific protein levels and localization support that the nonfailing hearts show significantly reduced pathology compared to failing hearts [18].
Hearts were removed from the patients/donors and immediately submersed in ice-cold cardioplegic solution containing 110 mM NaCl, 16 mM KCl, 16 mM MgCl2, 10 mM NaHCO3, and 0.5 mM CaCl2. Wet weights of the hearts were taken along with documentation of gross anatomical features. The hearts were transported to the laboratory in the same cold cardioplegic solution and processed immediately. Tissue was frozen in Optimal Cutting Temperature (O.C.T.) medium on liquid nitrogen-cooled isopentane, or flash-frozen in cryogenic tubes in liquid nitrogen, and stored at −80°C or below until used for this study.
2.2. Histological staining
Slides were prepared from tissue frozen in blocks of O.C.T. cut into 8-μm sections. The slides were stored at −80°C or below until staining. After incubating the slides in histological stains (specified below), the slides were cleared twice with HistoChoice (Sigma H2779) and then mounted with Cytoseal XYL mounting medium (Richard Allan Scientific 8313-4). Images were taken on either an Olympus IX-83 microscope with an Olympus DP73 digital camera, or an Olympus BX40 microscope with an Olympus DP70 camera. The images were analyzed using Olympus CellSens version 1.13 software.
2.2.1. Masson’s trichrome
The slides were fixed with Bouin’s fixative (71% picric acid, 24% formaldehyde, 5% acetic acid) and then stained with a kit according to the manufacturer’s instructions (Sigma HT15).
2.2.2. Hematoxylin and eosin (H&E)
The slides were fixed for 30 s with 100% ethanol, washed with tap water, stained with Harris hematoxylin (Sigma HHS32), washed with tap water, stained with eosin Y (Sigma HT110132), washed with tap water, and then dehydrated with 70%–90%–100% ethanol.
2.2.3. Picrosirius red
The slides were incubated in xylene for 10 min, hydrated with 100%–96%–80%–70% ethanol, and then washed with distilled water. The slides were stained with 0.1% Sirius red (Direct Red 80, Sigma 365548) for 60 min, rinsed with distilled water, and then stained with Weigert’s iron hematoxylin (Sigma HT1079) for 8 min. The slides were washed with tap water for 5 min, treated with 0.01 M HCl for 2 min, and then rinsed with tap water. Slides were dehydrated with 70%–80%–96%–100% ethanol.
2.3. Immunofluorescence
Slides with 8-μm cardiac sections (as above) were thawed and equilibrated in potassium phosphate-buffered saline (KPBS; 164.4 mM K2HPO4, 35.6 mM KH2PO4, 320 mM NaCl). Gelatin (1% in KPBS) was used as a block to nonspecific antibody binding. The slides were washed with 0.02% gelatin in KPBS (KPBSG) before being probed overnight at 4°C with primary antibodies. The antibodies were diluted as follows in KPBSG: mouse collagen I (Sigma C2456, lot 113M4774) 1:400 and rabbit collagen III (Abcam ab7778, lot GR145848-8) 1:400. After three washes in KPBSG, the slides were then probed with the following secondary antibodies for 1–2 h at room temperature at a 1:400 dilution in KPBSG: goat antimouse AlexaFluor 488 (Life Technologies A11013, lot 1495793), goat antirabbit AlexaFluor 555 (Life Technologies A21429, lot 1010124), or goat antirabbit AlexaFluor 488 (Life Technologies A11034, lot 1423009). Slides were mounted with ProLong Diamond with 4′, 6-diamidino-2-phenylindole (Life Technologies P36962) and cured for several hours to overnight. Images were taken on an Olympus IX-71 microscope with an Olympus BP72 camera and analyzed using Olympus CellSens version 1.13 software.
2.4. ECM isolation
Flash-frozen human heart tissue was thawed and minced. Decellularization water [10 ml; 2.5 mM ethylenediaminetetraacetic acid (EDTA), protease inhibitor cocktail (Sigma S8820), 100 U penicillin–100 μg/ml streptomycin (Sigma P0781), and 50 μg/ml gentamycin (Sigma G1272) in distilled, deionized water] was added to approximately 2–5 g of heart tissue. After 30-min incubation on a rotator, the decellularization water was discarded and 10 ml of decellularization buffer (2.5 mM EDTA, 1% sodium dodecyl sulfate, and, as above, protease inhibitor cocktail, penicillin–streptomycin and gentamycin) was added. After 6 h on the rotator with decellularization buffer, the buffer was removed and fresh buffer was added. The decellularization buffer was exchanged every 8–12 h for 5 days. At this time, decellularization was complete as evidenced by white, translucent tissues. Tissues were then incubated in decellularization water for 24 h on the rotator. The water was then exchanged for fresh decellularization water for 3×5-min washes. There was complete removal of cellular material as evidenced by H&E and Masson’s trichrome staining (Fig. 2). The tissue was lyophilized and stored at −20°C for a few days prior to mass spectrometric analysis.
Fig. 2.
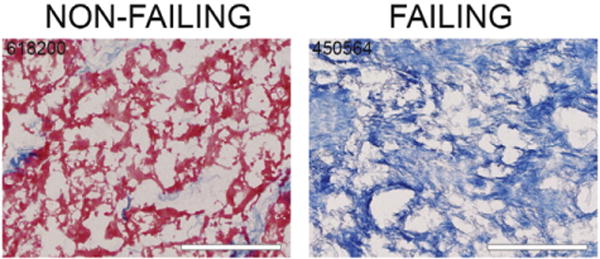
Decellularization of cardiac ECM. The cellular material was removed from the left ventricle midmyocardial tissue of human hearts. Trichrome staining shows the removal of cellular material leaving behind an isolated ECM. Scale bars=100 μm. Sample numbers used in these representative images are indicated.
2.5. Mass spectrometry and proteomic analysis
2.5.1. Equipment
An Acquity ultra performance liquid chromatography (UPLC) M-Class (Water’s Corp., Milford, MA, USA) was used to perform two-dimensional (2D) LC separations. The first-dimension chromatographic separations were performed on a BEH C18 column (Water’s Corp., 186007471) with the following specifications: 5 mm×300 μm, 3-μm particle size, and 130-Å pore size. A 2D Symmetry C18 column size (Water’s Corp., 186007497) with the following specifications was used for desalting: 20 mm×180 μm, 5-μm particle size, and 100-Å pore size. The second-dimension separations were performed on a PepMap C18 column (Thermo Scientific ES800) with the following specifications: 15 cm×75 μm, 30-μm particle size, and 100-Å pore size.
2.5.2. 2D LC
For the first dimension of 2D LC analysis, 12 μg of digest was fractionated by basic (pH 10) reverse-phase high-performance liquid chromatography (HPLC) on a Water’s BEH C18 column using a Water’s Acquity UPLC M-Class system. Solvent A was composed of 20 mmol/l ammonium formate (pH 10), and solvent B was 100% acetonitrile. Peptides were eluted from the first-dimension fraction column in 12 successive fractions using acetonitrile at 8.0%, 10.7%, 12.4%, 13.7%, 14.9%, 16.0%, 17.2%, 18.4%, 19.7%, 21.4%, 24.1%, and 50.0%. Each eluted fraction was then trapped, desalted, and separated using Thermo EasySpray PepMap 100 analytical column. Solvent A was 100% H2O with 0.1% formic acid, and solvent B was 100% acetonitrile with 0.1% formic acid. The flow rate for the analytical column was 500 μl/min. The gradient was 0 to 3 min, 0% solvent B; 3 to 45 min, 45% solvent B; and 45 to 47 min, 45%–85% solvent B.
2.5.3. Orbitrap fusion
Capillary liquid chromatography–nanospray tandem mass spectrometry (Capillary-LC/MS/MS) of protein identification was performed on a Thermo Scientific Orbitrap Fusion Mass Spectrometer equipped with EASY-Spray Sources operated in positive ion mode. Samples were separated on an EASY-Spray nanocolumn (PepMap RSLC, C18 3 μ 100 A, 75 μm×150 mm, Thermo Scientific) using a 2D rapid-separation LC HPLC system from Thermo Scientific. Each sample was injected into the μ-Precolumn Cartridge (Thermo Scientific) and desalted with 0.1% formic acid in water for 5 min. The injector port was then switched to inject, and the peptides were eluted off of the trap onto the column. Mobile phase A was 0.1% formic acid in water, and acetonitrile (with 0.1% formic acid) was used as mobile phase B. Flow rate was set at 300 nl/min. Typically, mobile phase B was increased from 2% to 35% in 30 min and then increased from 35% to 55% in 5 min and again from 55% to 90% in 5 min and then kept at 90% for another 2 min before being brought back quickly to 2% in 1 min. The column was equilibrated at 2% of mobile phase B (or 98% A) for 20 min before the next sample injection. MS/MS data were acquired with a spray voltage of 1.7 kV, and a capillary temperature of 275°C is used. The scan sequence of the mass spectrometer was based on the preview mode data-dependent TopSpeed method: the analysis was programmed for a full scan recorded between m/z 400 and 1600 and an MS/MS scan to generate product ion spectra to determine amino acid sequence in consecutive scans starting from the most abundant peaks in the spectrum in the next 3 s. To achieve high-mass-accuracy MS determination, the full scan was performed at Fourier transfer mode, and the resolution was set at 120,000. The AGC Target ion number for Fourier transfer full scan was set at 2×105 ions, maximum ion injection time was set at 50 ms, and microscan number was set at 1. Multiple-stage mass spectrometry (MSn) was performed using ion trap mode to ensure the highest signal intensity of MSn spectra using both collision-induced dissociation (CID) (for 2+ and 3+ charges) and electron transfer dissociation (ETD) (for 4+ −6 + charges) methods. The AGC Target ion number for ion trap MSn scan was set at 1000 ions, maximum ion injection time was set at 100 ms, and microscan number was set at 1. The CID fragmentation energy was set to 35%. Dynamic exclusion is enabled with a repeat count of 1 within 60 s and a low mass width and high mass width of 10 ppm.
Sequence information from the MS/MS data was processed by converting the .raw files into a merged file (.mgf) using an in-house program, RAW2MZXML_n_MGF_batch (merge.pl, a Perl script). Isotope distributions for the precursor ions of the MS/MS spectra were deconvoluted to obtain the charge states and monoisotopic m/z values of the precursor ions during the data conversion. The resulting .mgf files were searched using Mascot Daemon by Matrix Science version 2.3.2 (Boston, MA, USA), and the database was searched against the most recent SwissProt or National Center for Biotechnology Information databases. The mass accuracy of the precursor ions was set to 10 ppm for data acquired on Fusion. However, sometimes, MASCOT peak detection chooses the 13C peak rather than the monoisotopic 12C peak, especially for peptides with a large m/z. Thus, the number of 13C peaks was set at 1 to get a match to an accidental pick of 13C peak while still using the tight mass tolerance to increase search efficiency. The fragment mass tolerance was set to 0.5 Da. Considered variable modifications were oxidation (Met), deamidation (N and Q), and carbamidomethylation (Cys). Four missed cleavages for the enzyme were permitted. A decoy database was also searched to determine the false discovery rate (FDR), and peptides were filtered according to the FDR. The significance threshold was set at P<.05, and bold red peptides are required for valid peptide identification. Only proteins identified with a minimal of two peptides as well as <1% FDR are accepted for quantitation. Label-free quantitation was performed using the spectral count approach, in which the relative protein quantitation is measured by comparing the number of MS/MS spectra identified from the same protein in each of the multiple LC/MS/MS datasets. Scaffold version 4.3.4 (Proteome Software, Inc., Portland, OR, USA) was used for data analysis.
2.6. Immunoblotting
Left ventricle myocardial samples were decellularized as above under the section “ECM isolation.” The individual nonfailing or DCM-failing isolated ECM were pooled together. Using a ceramic mortar and pestle cooled with liquid nitrogen, the ECM was ground into a fine powder. ECM solubilization buffer [500 μl; 4 M urea, 20% glycerol, 3.8% sodium dodecyl sulfate, 75 mM Tris–HCl pH 6.8, 100 mM 2,4-dithiothreitol, 1× protease inhibitor cocktail (Sigma S8820)] was added to 35 mg of ECM powder. The samples were incubated at 4°C for ~2 h; samples were vortexed every 10 min. Another 500 μl of solubilization buffer was added to make the mixture less viscous. Insoluble pieces of ECM were removed by using a small steel mesh strainer. Equal volumes of 4× Laemmli (Bio-Rad 161-0747) with 10% beta-mercaptoethanol were added to the ECM solution at a ratio of one part ECM sample to three parts 4× Laemmli.
Individual, nondecellularized left ventricle myocardial samples were homogenized using a ceramic mortar and pestle cooled with liquid nitrogen, in which the left ventricular tissue was ground into a fine powder. Solubilization buffer [500 μl; 4 M urea, 20% glycerol, 3.8% sodium dodecyl sulfate, 75 mM Tris–HCl, 1× protease inhibitor cocktail (Sigma S8820)] was added to each sample; samples were incubated for 1 h at 4°C and were vortexed every 15 min. Samples were centrifuged at ~3200×g for 10 min at 4°C. The supernatant was removed and used for analysis. Equal amounts of individual nonfailing or DCM-failing supernatant were pooled together. Equal volumes of 4× Laemmli (Bio-Rad 161-0747) with 10% beta-mercaptoethanol were added to the homogenate at a ratio of one part homogenate sample to three parts 4× Laemmli.
Protein concentrations were measured using a Qubit 2.0 Fluorometer (Life Technologies/Thermo Fisher Scientific Q32866) and Qubit Protein Assay Kit (Thermo Fisher Scientific Q33212). Total protein at 7 μg was loaded per well after boiling the samples for 2 min.
For immunoblot analysis, proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) with 4%–20% Criterion TGX Stain-Free Precast gels (Bio-Rad 5678094) run at 120 V. Proteins were transferred electrophoretically to nitrocellulose membranes (Bio-Rad 1704159) using the “high-molecular weight” preset protocol in the Trans-Blot Turbo Transfer System (Bio-Rad 1704155, 25 V for 10 min). Proteins on the blot were then transiently stained with Ponceau S dye, rinsed well with distilled water, and then imaged with ProteinSimple FluorChem R system (ProteinSimple, San Jose, CA, USA). The blots were stored in Tris-buffered saline plus Tween-20 (TBST) overnight at 4°C.
The blots were blocked with 5% nonfat milk in TBST for 2 h, and then the membrane sections were rinsed 3×5 min in TBST. The whole left-ventricle-plus-ECM blots were blocked for 2 h at room temperature followed by overnight probing at 4°C with rabbit collagen I (Abcam ab2921) diluted 1:1000 in 1% normal goat serum (NGS) in TBST or rabbit fibronectin (Abcam ab2413) diluted 1:1000 in 1% NGS in TBST. The blots were incubated with the secondary antibody solution containing horseradish-peroxidase-conjugated donkey antirabbit IgG (Millipore AP182P) diluted 1:10,000 or 1:20,000 and Strep-Tactin HRP Conjugate (Bio-Rad 161-0381) in TBST with 1% normal goat serum. The ECM-only blots were blocked for 2 h at room temperature and then probed at 4°C overnight with rabbit collagen IV α2 antibody (Abcam ab69782) diluted 1:500 in TBST with 1% normal goat serum (1% NGS in TBST), goat collagen IV α6 antibody (Abcam ab99212) diluted 1:100 in TBST with 1% bovine serum albumin (1% BSA in TBST), rabbit zyxin antibody (Abcam ab109316) diluted 1:5000 in 1% NGS in TBST, and rabbit myomesin (Abcam ab201228) diluted 1:10,000 in 1% NGS in TBST. The blots were then washed 3×5 min in TBST. The blots were incubated for 2 h at room temperature with the secondary antibody solution (1% NGS or 1% BSA in TBST) containing 1:20,000 and Strep-Tactin HRP Conjugate (Bio-Rad 161-0381) and either horseradish-peroxidase-conjugated donkey antirabbit IgG (Millipore AP182P) diluted 1:10,000 or horseradish-peroxidase-conjugated donkey antigoat IgG (Abcam ab6885) diluted 1:10,000. All blots were then rinsed 3×5 min in TBST, developed with ECL 2 Western Blotting Substrate (Pierce 80196), and then imaged with the ProteinSimple FluorChem R system (ProteinSimple, San Jose, CA, USA) using the chemiluminescence application.
2.7. Statistics
Student’s t tests were used to assess statistically significant differences.
3. Results
3.1. Clinical and demographic data from patients and donors
We analyzed the left ventricular midmyocardium samples from seven patients with end-stage heart failure (DCM-failing) and nine organ donors without diagnosed heart failure whose hearts were deemed not suitable for transplant (nonfailing). Table 1 describes demographic and clinical data from these patients and donors. There was a trend toward nonfailing donors being of younger age (44±15 years versus 51±15 years in DCM-failing patients), but this difference was not statistically significant. Additionally, most of the DCM-failing samples were from males (71% male), while the nonfailing samples were mostly from females (33% male), but this parameter was also not significantly different between the groups. We acknowledge, however, that one limitation of this study may be that we are analyzing ECM differences between slightly older males (DCM-failing) and younger females (nonfailing). Most of the samples were from Caucasians in both groups.
Table 1.
Clinical data, demographic data, and fibrosis of the heart samples
| Sample number | Age (years) | Sex | Ethnic descent | Heart weight (g) | % Ejection fraction | NYHA class/ACC grade or no. of CVD risk factors | % Fibrosis | Remodeling score |
|---|---|---|---|---|---|---|---|---|
| DCM-failing | ||||||||
| 303122 | 51 | F | Caucasian | 508 | <20 | IV/D | 7.5 | 2 |
| 278931 | 64 | M | Caucasian | 678 | 40 | III/D | 19.1 | 2.5 |
| 906620 | 65 | M | Caucasian | 543 | <20 | IV/D | 13.3 | 3 |
| 733252 | 61 | M | African | 340 | <20 | IV/D | 18.7 | 2 |
| 450564 | 30 | M | Caucasian | 635 | 20 | IV/D | 14.3 | 2.5 |
| 896123 | 30 | M | Caucasian | 791 | 5 | IV/D | 16.5 | ND |
| 522421 | 56 | F | Caucasian | 470 | 10–15 | III/D | 1.2 | ND |
| Mean ± S.D. | 51±15 | – | – | 566±148 | 20±4 | – | 12.9±6.5 | 2.4±0.4 |
| Median | 56 | – | – | 543 | 19 | – | 14.3 | 2.5 |
| Percentage in group | – | 71% (5/7) M | 86% (6/7) Caucasian | – | 86% (6/7) with LVEF ≤35% | 29% (2/7) class III, 71% (5/7) class IV | 71% (5/7) have >10% fibrosis | – |
| Nonfailing | ||||||||
| 168021 | 62 | F | Caucasian | 482 | ND | 10 | 5.5 | 1.5 |
| 352041 | 28 | F | African | 446 | 25 | 3 | 6.6 | 2 |
| 474083 | 41 | F | African | ND | 55 | 6 | 4.7 | 1.5 |
| 488240 | 51 | F | Caucasian | ND | ND | 9 | 6.3 | 1 |
| 618200 | 58 | F | Caucasian | 636 | ND | 10 | 4.7 | 1 |
| 685884 | 36 | M | Caucasian | 896 | 60 | 3 | 9.1 | 3 |
| 947202 | 34 | M | Caucasian | 499 | ND | 4 | 2.8 | 0 |
| 845013 | 26 | M | Caucasian | 497 | ND | 2 | 5.3 | ND |
| 947200 | 63 | F | Caucasian | 608 | ND | 2 | 0.4 | ND |
| Mean ± S.D. | 44±15 | – | – | 581±155 | 47±11 | 5 | 5.0±2.4 | 1.4±0.9 |
| Median | 41 | – | – | 499 | 55 | 4 | 5.3 | 1.5 |
| Percentage in group | – | 33% (3/9) M | 78% (7/9) Caucasian | – | 33% (1/3) with LVEF ≤35% | – | 0% (0/9) have >10% fibrosis | – |
| Failing versus nonfailing | ||||||||
| P value | .39 | .13 | .69 | .86 | .017 | – | .0045 | .056 |
Demographic and clinical data were taken from the patients’ and donors’ medical records. The percent fibrosis was determined using Masson’s-trichrome-stained slides and Olympus CellSens imaging software. The remodeling score was determined visually using a combination of Masson’s-trichrome-, picrosirius-red-, and H&E-stained slides. A score of 0 indicates no fibrosis, while 3 indicates extensive fibrosis. Student’s t tests were used to determine statistically significant differences between the two groups.
Key: CVD = cardiovascular disease, ND = not determined, S.D. = standard deviation, NYHA = New York Heart Association, ACC = American College of Cardiology.
Heart weights (wet) were similar between the DCM-failing patients and nonfailing donors. We only obtained left ventricular ejection fraction (LVEF) data from three of the nine nonfailing donors, but this measure of cardiac function was still significantly higher in the nonfailing donors (47%±11%) compared to the DCM-failing patients (20%±4%). Eighty-six percent of the DCM-failing patients met the functional definition of heart failure with LVEFs at or below 35%. Please note that for the LVEF values listed as <20% and 10%–15%, we assumed the highest possible value (19 or 15, respectively) when performing our statistical analysis. Thus, the actual differences between the groups could be even greater. All of the DCM-failure patients were in end-stage disease, with New York Heart Association (NYHA) class III or IV designations and American College of Cardiology (ACC) grade D.
3.2. Fibrosis in failing hearts with DCM
We verified that the failing heart samples were fibrotic using Masson’s trichrome and picrosirius red stains, both of which highlight collagenous scar tissue. Additionally, we showed the prevalence of inflammation using H&E staining. Masson’s trichrome staining (Fig. 1a) showed that five of seven DCM-failing samples were fibrotic (≥10% of the total area containing blue-staining collagen-based fibrosis), while none of the nonfailing samples showed were extensively fibrotic. Moreover, the DCM-failing samples showed both interstitial and replacement fibrosis. A significantly higher percentage of the tissue in DCM-failing samples (12.9%±6.5%) was fibrotic scar tissue compared to nonfailing samples (5.0%±2.4%, P<.005).
Fig. 1.
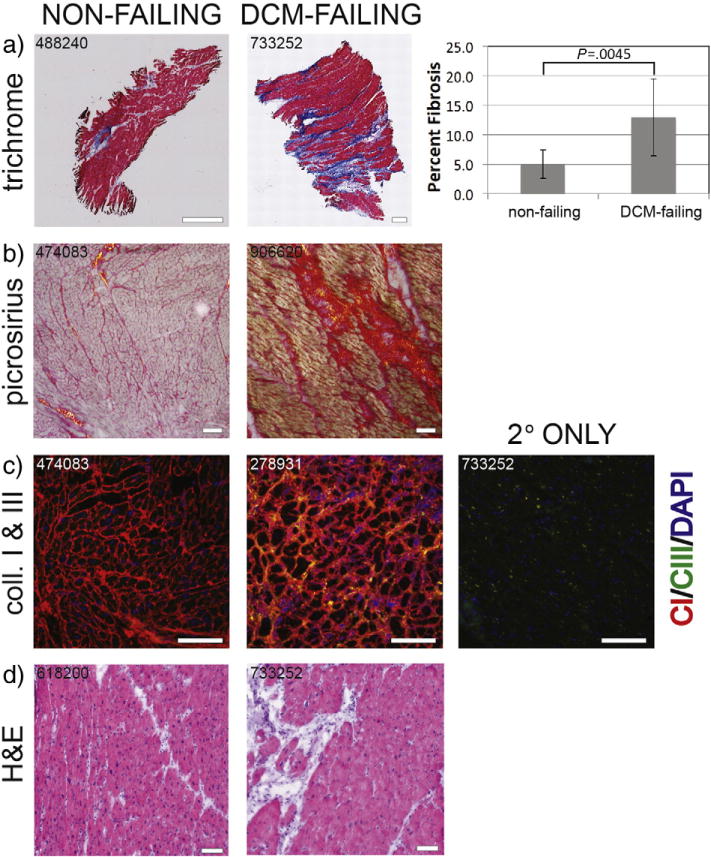
Fibrosis and collagen expression in the left ventricle myocardial tissue from human hearts. (a) Masson’s-trichrome-stained sections. Cardiomyocytes are stained in red; nuclei in brown–black; and collagenous, fibrotic scar tissue in blue. Scale bars=1 mm. Percent fibrosis was determined by percentage blue stain of total tissue area in nonfailing (n=9) and DCM-failing (n=7) tissue samples. Student’s t test was used to assess statistically significant differences between the groups. (b) Picrosirius-red-stained sections. Cardiomyocytes are pale pink, collagen-I fibrotic tissue is in red (red–orange birefringence under the microscope’s polarized light), and collagen-III fibrotic tissue is in yellow (yellow–green birefringence under the microscope’s polarized light). Scale bars=100 μm. (c) Collagen I (red) and collagen III (green) immunofluorescently labeled sections. Scale bars=100 μm. (d) H&E-stained sections. Cardiomyocytes are in a darker pink, nuclei are in purple–blue, and fibrotic scar tissue is in pale pink. Scale bars=100 μm. Sample numbers used in these representative images are indicated. Key: CI or Coll I = collagen I, CIII or Coll III = collagen III, H&E = hematoxylin and eosin.
To determine whether the scar tissue was made up of collagen I and/or collagen III fibers, we used picrosirius red staining and viewed the sections under polarized light. The collagen I fibers had an orange–red birefringence, and the collagen III fibers had a yellow–green birefringence. We determined that the scar tissue was made up of primarily collagen I (Fig. 1b). We also verified the picrosirius red staining data regarding collagen I and collagen III prevalence by immunofluorescence staining (Fig. 1c). We observed that collagen I expression was higher in DCM myocardium. H&E staining revealed that the DCM samples showed an increase in inflammatory infiltrates in the fibrotic scars (Fig. 1d).
We visually scored the extent of the remodeling using a combination of trichrome-, picrosirius-red-, and H&E-stained sections using a scale of 0 (no remodeling) to 3 (extensive remodeling). The average remodeling score for nonfailing hearts was 1.4, while the average score for failing hearts was 2.4. However, the difference just missed statistical significance. Altogether, these data show that the DCM-failing samples had undergone extensive remodeling with substantial fibrotic scar tissue. Thus, we were satisfied that the DCM-failing samples would be good representatives of ECM damage in DCM pathology and the nonfailing samples were relatively normal.
3.3. Proteomic analysis of failing hearts with DCM
To determine ECM protein levels in the ECM of DCM-failing versus nonfailing hearts, we decellularized left ventricular midmyocardial tissue to leave behind isolated ECM. Masson’s-trichrome-stained sections of isolated ECM (Fig. 2) support that all soluble and cellular materials were completely removed. We also showed that the decellularization process preserved protein integrity through immunoblotting comparing whole heart tissue and isolated ECM from both DCM-failing and nonfailing hearts, probing for collagen I and fibronectin (Fig. 3). We were able to detect collagen I and fibronectin bands in both whole heart and isolated ECM. Although we loaded 7 μg total protein per lane, based on Ponceau S total protein staining, the whole heart lanes appeared to be loaded with more total protein than isolated ECM lanes. Even so, the collagen I and fibronectin bands were stronger in the isolated ECM lanes, supporting that the ECM proteins were preserved. Additionally, the collagen I and fibronectin levels were higher in the failing samples compared to the nonfailing samples, supporting that the ECM of DCM-failing hearts was expanded relative to nonfailing hearts.
Fig. 3.
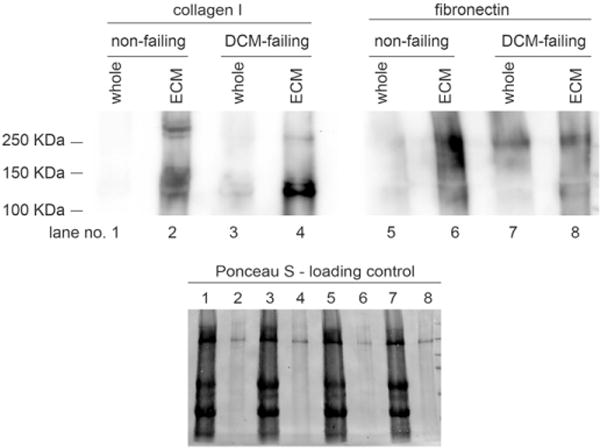
Immunoblot analysis of collagen I and fibronectin. Individual isolated ECM from each of the nonfailing (n=9) and DCM-failing (n=7) groups was pooled; the ECM was solubilized and then was subjected to SDS-PAGE and electrophoretic protein transfer onto nitrocellulose blots. Transient Ponceau S staining was used to show total protein loading levels. The blot was probed with anti-collagen-I and antifibronectin antibodies.
We then subjected three decellularized DCM-failing ECM samples (from patients 278931, 733252, 450564) and three decellularized nonfailing ECM samples (from donors 474083, 618200, 947202) to LC–MS proteomics analysis. These samples were chosen since the DCM-failing samples showed extensive fibrosis and the nonfailing samples showed very little fibrosis; we wanted to best highlight differences between highly remodeled, DCM tissue and nonremodeled, nonfailing tissue. A total of 1133 proteins were identified in this analysis. Of these, 75 showed statistically significant differences between nonfailing and DCM-failing hearts. We have listed the 26 structural proteins (ECM, cytoskeletal, and contractile proteins) in Table 2.
Table 2.
Structural proteins identified by the proteomic analysis of isolated ECM from DCM-failing and nonfailing hearts
| Protein | Uniprot code | Function | Mean spectral counts
|
P value | |
|---|---|---|---|---|---|
| Nonfailing | Failing | ||||
| Increased in failing hearts | |||||
| Hyaluronan-binding protein 2 (a.k.a. factor VII-activating protease) | HABP2 | ECM | 0 | 2 | .088 |
| Lactadherin (a.k.a. milk fat globule) | MFGM | ECM | 2 | 6 | .096 |
| Myomesin-1 | MYOM1 | Contractile | 33 | 70 | .007 |
| Myosin regulatory light chain 12A | ML12A | Contractile | 1 | 2 | .090 |
| PDZ and LIM domain protein | PDLI5 | Contractile | 45 | 76 | .093 |
| Myosin-14 | MYH14 | Contractile | 2 | 13 | .094 |
| γ-Sarcoglycan | SGCG | Cytoskeleton | 2 | 3 | .042 |
| LIM and calponin homology domain | LIMC1 | Cytoskeleton | 5 | 9 | .086 |
| FYVE and coiled-coil domain-containing protein 1 | FYCO1 | Cytoskeleton | 1 | 5 | .089 |
| Dystroglycan | DAG1 | Cytoskeleton | 0 | 5 | .100 |
| Ran binding protein 3 | RANB3 | Nuclear transport | 0 | 1 | .009 |
| Decreased in failing hearts | |||||
| Fibulin 2 | FBLN2 | ECM | 71 | 31 | .014 |
| Collagen IV α2 | CO4A2 | ECM | 135 | 72 | .039 |
| Inter-α-trypsin inhibitor heavy chain H1 | ITIH1 | ECM | 4 | 2 | .039 |
| ECM protein 1 (a.k.a. secretory component glycoprotein) | ECM1 | ECM | 8 | 4 | .052 |
| Target of Nesh-SH3 (a.k.a. ABI3BP) | TARSH | ECM | 5 | 2 | .057 |
| Integrin β-1 binding protein | ITBP2 | ECM | 1 | 0 | .063 |
| Collagen IV α6 | CO4A6 | ECM | 6 | 2 | .065 |
| von Willebrand factor A domain containing protein 1 | VWA1 | ECM | 5 | 3 | .066 |
| Collagen XV α1 | COFA1 | ECM | 13 | 6 | .092 |
| 40S ribosomal protein SA (a.k.a. 67KDa laminin receptor) | RSSA | ECM binding | 4 | 2 | .008 |
| Proteoglycan 3 | PRG3 | ECM binding | 5 | 3 | .072 |
| Tenascin | TENA | Matricellular | 17 | 2 | .067 |
| Zyxin | ZYX | Cytoskeleton | 3 | 1 | .076 |
| Coiled-coil domain-containing proteins | CCDC6 | Cytoskeleton | 3 | 0 | .079 |
| Four and a half LIM domains protein | FHL3 | Nuclear transport | 2 | 0 | .008 |
Three DCM-failing samples with extensive remodeling and three nonfailing isolated cardiac ECM samples with very little remodeling were used in the proteomic analysis. The spectral counts are the average values from the two groups. The P values were determined using Student’s t tests. Statistical significance was set at a liberal value of P<.1 since, out of necessity, a low number of samples per group were analyzed.
Eleven of the 26 proteins had significantly higher levels in the DCM-failing hearts, while 15 had reduced levels. Most of the 14 proteins that are identified specifically as ECM or matricellular proteins were reduced in the DCM-failing heart isolated ECM, with the exception of hyaluronan binding protein 2 and lactadherin. All four of the identified contractile proteins were increased. Of the six identified cytoskeletal proteins, four were increased and two were decreased. Finally, one of the nuclear transport proteins showed higher levels and one showed lower levels.
The remaining 49 proteins could not strictly be categorized as structural but may be more obviously associated with the ECM after further analysis, which is currently under way.
We verified the results of the proteomics data by immunoblotting with the ECM proteins, collagen IV (α2 and α6 isoforms), zyxin, and myomesin. These proteins’ spectral counts in DCM-failing hearts were lower, and the immunoblot data (Fig. 4) support that protein levels were indeed lower in the isolated ECM of DCM-failing hearts compared to nonfailing hearts.
Fig. 4.
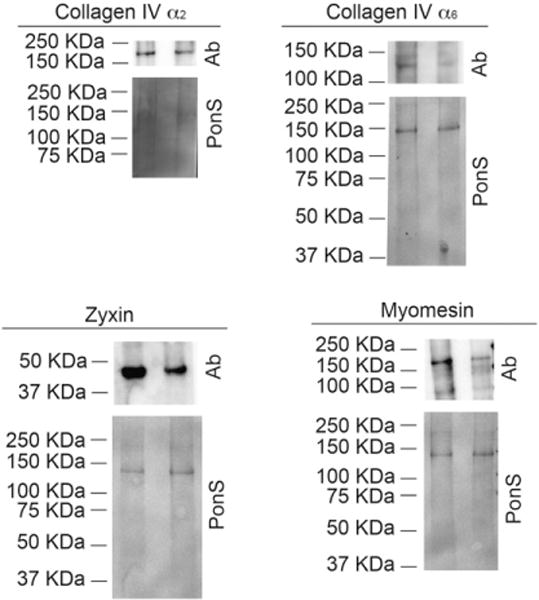
Immunoblot analysis of collagen IV α2, collagen IV α6, zyxin, and myomesin, proteins identified in the proteomics screen. Individual isolated ECM from each of the nonfailing (n=9, lanes on the left) and DCM-failing (n=7, lanes on the right) groups was pooled; the ECM was solubilized and then was subjected to SDS-PAGE and electrophoretic protein transfer onto a nitrocellulose blot. Transient Ponceau S (“PonS”) staining was used to show total protein loading levels. The blots were probed with antibodies (“Ab”) against collagen IV α2, collagen IV α6, zyxin, and myomesin.
Overall, we found that the ECM isolated from the significantly remodeled DCM failing hearts had a different pattern of structural protein levels than the isolated ECM from nonfailing hearts.
4. Discussion
We decided to conduct this proteomic analysis of the isolated ECM from failing human hearts with DCM for our studies under way on cell–ECM interactions. We believe that this information will be generally useful in understanding the pathology of remodeling in heart failure. These data may be useful in numerous applications, such as in diagnoses of the pathology of heart failure remodeling or discovery of novel treatments.
We first showed that the DCM-failing samples had characteristic remodeling; thus, we were confident that we could identify differences that arise from the remodeling process. Indeed, the DCM-failing samples had significantly more fibrosis along with increased inflammation. These are all indicative of extensive remodeling. While the DCM-failing samples showed histologically typical interstitial fibrosis throughout the multiple sections from each heart we analyzed in this and other studies [18, unpublished studies], we were surprised to see that some samples also showed replacement fibrosis. To control for inter- and intraspecimen variability to the best of our ability, we chose random pieces of tissue from each of the multiple heart samples in both groups to perform our analyses and have done so for every experiment described in this report. To ensure that we highlighted differences between highly remodeled DCM-failing ECM and relatively normal nonfailing ECM, we chose to perform further analysis on three of the most remodeled DCM-failing hearts and three of the least remodeled nonfailing hearts.
We then performed a proteomic analysis of the decellularized, isolated ECM from the three selected DCM-failing and the three selected nonfailing midmyocardial left ventricle samples. We tried to enrich for the low-abundance proteins in this analysis to capture novel changes in protein expression beyond what is known about changes in major ECM proteins such as collagens I and III, laminin, fibronectin, etc.
Interestingly, most of the ECM proteins showed reduced levels in failing hearts. This is contrary to what we expected since there is a known expansion of the ECM during the pathological remodeling process of the heart. Indeed, immunofluorescence on collagen I and collagen III (Fig. 1) and immunoblots on collagen I and fibronectin (Fig. 3) showed an increased abundance of major cardiac ECM proteins in the DCM-failing samples. Most likely, the expanded ECM is primarily made up of high-abundance proteins, like collagen I; however, high-abundance proteins were not the focus of our analysis. The only ECM proteins that were at higher levels in DCM-failing isolated ECM were hyaluronan binding protein 2 and lactadherin. To the best of our knowledge, this is the first association with hyaluronan binding protein and the heart. This protein is otherwise associated with immune modulation [19,20] and atherosclerotic plaque formation [21]. Lactadherin is a membrane glycoprotein that promotes phagocytosis of apoptotic cells. It also has implications in antimicrobial activity, wound healing, and positive regulation of cell proliferation and of cell adhesion. [22–24] The fact that two ECM-related proteins that were up-regulated in the DCM-failing hearts are implicated in immune responses and wound healing supports the notion that the failing hearts were in a phase of pathological remodeling.
Unexpectedly, only one matricellular protein was identified: tenascin (isoform not specified). We had expected a number of matricellular proteins to be identified since matricellular protein up-regulation has been implicated in active cardiac remodeling. In our study, tenascin levels were lower in the DCM-failing ECM. Perhaps because the hearts are from patients in end-stage failure, we may have captured a more resolved pathological remodeling phase, past the peak of expected matricellular protein expression [25]. Tenascin, which is not typically expressed in normal hearts, was, perhaps, higher in the nonfailing ECM because nonfailing donors had an average of five cardiovascular disease risk factors even though they were not diagnosed with heart failure prior to death. Thus, some donors may have been in early and active stages of cardiac remodeling, when tenascin levels would be higher compared to a later phase of pathological remodeling captured in the end-stage failing hearts.
We were intrigued by the fact that all of the contractile proteins were found in higher levels in the failing hearts. One possible explanation for this is that in an effort to compensate for reduced cardiac output, there was an up-regulation of the contractile apparatus to strengthen contractile force. This possibility could be further explored to deepen the understanding of the process of pathological cardiac remodeling.
Some of the proteins identified in our proteomic screen have known associations with heart disease, such as zyxin, collagen XV, four-and-a-half LIM domains protein, and RAN binding protein 3. Zyxin, found at higher levels in DCM-failing hearts in our study, is expressed at focal contacts between cardiomyocytes and the extracellular environment, and it acts as a scaffolding protein around α-actinin and Ena/vasodilator-stimulated phosphoprotein proteins. It is involved in actin polymerization [26]. Zyxin was up-regulated in a swine model of myocardial infarction treated with induced pluripotent cell-derived cardiomyocytes, endothelial cells, and smooth muscle cells [27]. Redistribution of zyxin into the nucleus of cardiomyocytes protects them from atrial natriuretic peptide-induced apoptosis [28]. Down-regulation of collagen XV leads to cardiomyopathy in a mouse model [29], and it was down-regulated in our DCM-failing heart ECM. The cytoskeletal protein four-and-a-half LIM domains protein, down-regulated in the DCM-failing heart ECM, was shown in a previous study [30] to be down-regulated in DCM at a gene expression level. Ras-related nuclear protein (RAN) binding protein 3 is a nuclear transport protein whose gene expression goes down in DCM [30]. However, protein levels in our studies was increased in the DCM-failing hearts.
At least four of the DCM-failing samples likely had idiopathic cardiomyopathy leading to extensive left ventricular dilation and pathological remodeling of the myocardium (Supporting Table). Owing to this, the fibrosis pattern was not exactly the same in every sample, with some showing replacement fibrosis in addition to the prototypical interstitial remodeling [12]. It is beyond the scope of this study to compare the proteomic profiles of the interstitial ECM versus the replacement scar ECM, but it would be an excellent follow-up to determine whether the protein content between these two distinct fibrotic presentations is similar or rather different.
Some of the alterations in ECM protein levels between DCM-failing versus nonfailing hearts might be explained by different levels of matrix turnover-modulating proteins between the groups. Therefore, we searched the proteomics data for the expression of a disintegrin and metalloproteinase (ADAMs), a disintegrin and metalloproteinase with thrombospondin motifs (ADAMTs), matrix metalloproteinases (MMPs), and tissue inhibitors of matrix metalloproteinases (TIMPs). We found that of the following matrix modulators identified, there were no significant differences between the groups (P=.15–.95): ADAMTS-1, ADAMTS-4, ADAMTS-5, MMP-12, MMP-19, MMP-23, MMP-28, and TIMP-3.
Ischemic cardiomyopathy (ICM) is also characterized by an increase in fibrotic scar tissue, but replacement fibrosis dominates the area of ischemia or infarct [12]. While a comparison between the ECM protein profiles in ICM versus DCM is outside of the scope of this study, we found it useful to compare literature reports of ICM proteomic changes to the DCM proteomics in ours and others’ studies. Roselló-Lletí et al. performed a direct comparison of human whole-heart tissue proteomics between ICM and DCM samples [16]. They observed that all of the structural proteins identified in their study were increased in ICM relative to DCM samples. There was no overlap between the proteins they identified and the proteins we identified in our proteomic analysis. Barallobre-Barreiro et al. performed a proteomic profile of the isolated ECM from a pig model of cardiac ischemia–reperfusion injury 15 and 60 days postinjury [14]. There were several proteins identified in their study that were also identified in our study: collagen IV, collagen XV, lactadherin, target of nesh-SH3, and tenascin. In their study, all of these proteins were increased in the ischemia–reperfusion model compared to controls, whereas in our study, only lactadherin was increased in DCM-failing hearts compared to nonfailing hearts. This difference might be attributed to the animal model or even the stage of pathological remodeling (2 or 4 weeks postinjury in their study; years post heart failure diagnosis in ours).
We did not assess gene expression differences in this study since we were focused on the alterations in actual protein content in DCM. However, an excellent review on DCM genomic and proteomic changes has recently been published [6]. Additionally, Raghow reviewed “omics” changes in cardiomyopathies and heart failure, and this review addresses several ECM and structural gene alterations in DCM [31]. We refer the reader to these reviews for information on gene expression changes in DCM.
Overall, we observed a reduction in ECM proteins but greater amounts of contractile proteins. The cytoskeletal and nuclear transport protein levels were variable. We believe that this study shows how pathological remodeling affects the ECM of failing hearts with DCM, focusing specifically on isolated ECM. We have studies ongoing looking at the influence of the failing heart ECM on cell phenotypes. In summary, this proteomic study highlighted several ECM-associated proteins with previously established connections to heart disease, which help support the validity of this study. At the same time, the data highlighted proteins with novel connections to the heart, which contribute a deeper understanding of the proteins expressed in the ECM of the heart and their alterations in the pathology of DCM-associated heart failure.
Supplementary Material
Acknowledgments
We thank the Lifeline of Ohio Organ Procurement program for the availability of donor hearts. We thank Indika Mallawaarachchi for assistance with statistical analysis. We thank Carlos A. Roldan for a careful review of the clinical data to assist in determining likely causes of the patients’ DCM and heart disease in the donors.
Footnotes
Funding: This work was supported by funds from the University of New Mexico, the American Heart Association (grant number 15BGIA22840012), and the National Institutes of Health (grant number ULTR001448 and FlyBase grant number R25HG007630).
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.carpath.2016.10.001.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, et al. Heart disease and stroke statistics—2016 update: a report from the American Heart Association. Circulation. 2016;133:e38–60. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 2.Segura AM, Frazier OH, Buja LM. Fibrosis and heart failure. Heart Fail Rev. 2014;19:173–85. doi: 10.1007/s10741-012-9365-4. [DOI] [PubMed] [Google Scholar]
- 3.Yabluchanskiy A, Chilton RJ, Lindsey ML. Left ventricular remodeling: one small step for the extracellular matrix will translate to a giant leap for the myocardium. Congest Heart Fail. 2013;19:E5–8. doi: 10.1111/chf.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weber KT. Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol. 1989;13:1637–52. doi: 10.1016/0735-1097(89)90360-4. [DOI] [PubMed] [Google Scholar]
- 5.Jugdutt BI. Ventricular remodeling after infarction and the extracellular collagen matrix: when is enough enough? Circulation. 2003;108:1398–403. doi: 10.1161/01.CIR.0000085658.98621.49. [DOI] [PubMed] [Google Scholar]
- 6.Louzao-Martinez L, Vink A, Harakalova M, Asselbergs FW, Verhaar MC, Cheng C. Characteristic adaptations of the extracellular matrix in dilated cardiomyopathy. Int J Cardiol. 2016;220:634–46. doi: 10.1016/j.ijcard.2016.06.253. [DOI] [PubMed] [Google Scholar]
- 7.Takawale A, Sakamuri SS, Kassiri Z. Extracellular matrix communication and turnover in cardiac physiology and pathology. Commun Phys. 2015;5:687–719. doi: 10.1002/cphy.c140045. [DOI] [PubMed] [Google Scholar]
- 8.Rienks M, Papageorgiou A-P. Novel regulators of cardiac inflammation: matricellular proteins expand their repertoire. J Mol Cell Cardiol. 2016;91:172–8. doi: 10.1016/j.yjmcc.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 9.Valiente-Alandi I, Schafer AE, Blaxall BC. Extracellular matrix-mediated cellular communication in the heart. J Mol Cell Cardiol. 2016;91:228–37. doi: 10.1016/j.yjmcc.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bang C, Antoniades C, Antonopoulos AS, Eriksson U, Franssen C, Hamdani N, et al. Intercellular communication lessons in heart failure. Eur J Heart Fail. 2015;17:1091–103. doi: 10.1002/ejhf.399. [DOI] [PubMed] [Google Scholar]
- 11.Kohl P, Camelliti P, Burton FL, Smith GL. Electrical coupling of fibroblasts and myocytes: relevance for cardiac propagation. J Electrocardiol. 2005;38:45–50. doi: 10.1016/j.jelectrocard.2005.06.096. [DOI] [PubMed] [Google Scholar]
- 12.Sheppard M. Cardiac hypertrophy, heart failure and cardiomyopathy. In: Sheppard M, editor. Practical cardiovascular pathology. 2nd. Boca Raton, FL: CRC Press; 2011. pp. 133–92. [Google Scholar]
- 13.Tian Y, Koganti T, Yao Z, Cannon P, Shah P, Pietrovito L, et al. Cardiac extracellular proteome profiling and membrane topology analysis using glycoproteomics. Proteomics Clin Appl. 2014;8:595–602. doi: 10.1002/prca.201400009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barallobre-Barreiro J, Didangelos A, Schoendube FA, Drozdov I, Yin X, Fernández-Caggiano M, et al. Proteomics analysis of cardiac extracellular matrix remodeling in a porcine model of ischemia/reperfusion injury. Circulation. 2012;125:789–802. doi: 10.1161/CIRCULATIONAHA.111.056952. [DOI] [PubMed] [Google Scholar]
- 15.Hammer E, Goritzka M, Ameling S, Darm K, Steil L, Klingel K, et al. Characterization of the human myocardial proteome in inflammatory dilated cardiomyopathy by label-free quantitative shotgun proteomics of heart biopsies. J Proteome Res. 2011;10:2161–71. doi: 10.1021/pr1008042. [DOI] [PubMed] [Google Scholar]
- 16.Roselló-LLetí E, Alonso J, Cortés R, Almenar L, Martínez-Dolz L, Sánchez-Lázaro I, et al. Cardiac protein changes in ischaemic and dilated cardiomyopathy: a proteomic study of human left ventricular tissue. J Cell Mol Med. 2012;16:2471–86. doi: 10.1111/j.1582-4934.2012.01565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milani-Nejad N, Canan BD, Elnakish MT, Davis JP, Chung JH, Federov VV, et al. The Frank–Starling mechanism involves deceleration of cross-bridge kinetics and is preserved in failing human right ventricular myocardium. Am J Physiol Heart Circ Physiol. 2015;309:H2077–86. doi: 10.1152/ajpheart.00685.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swager SA, Delfín DA, Rastogi N, Wang H, Canan BD, Federov VV, et al. Claudin-5 levels are reduced from multiple cell types in human failing hearts and are associated with mislocalization of ephrin-B1. Cardiovasc Pathol. 2015;24:160–7. doi: 10.1016/j.carpath.2014.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stephan F, Aarden LA, Zeerleder S. FSAP, a new player in inflammation? Hamostaseologie. 2012;32:51–5. doi: 10.5482/ha-1187. [DOI] [PubMed] [Google Scholar]
- 20.Bost F, Diarra-Mehrpour M, Martin JP. Inter-alpha-trypsin inhibitor proteoglycan family—a group of proteins binding and stabilizing the extracellular matrix. Eur J Biochem. 1998;252:339–46. doi: 10.1046/j.1432-1327.1998.2520339.x. [DOI] [PubMed] [Google Scholar]
- 21.Kanse SM, Parahuleva M, Muhl L, Kemkes-Matthes B, Sedding D, Preissner KT. Factor VII-activating protease (FSAP): vascular functions and role in atherosclerosis. Thromb Haemost. 2008;99:286–9. doi: 10.1160/TH07-10-0640. [DOI] [PubMed] [Google Scholar]
- 22.Newburg DS. Human milk glycoconjugates that inhibit pathogens. Curr Med Chem. 1999;6:117–27. [PubMed] [Google Scholar]
- 23.Lönnerdal B. Human milk proteins: key components for the biological activity of human milk. Adv Exp Med Biol. 2004;554:11–25. [PubMed] [Google Scholar]
- 24.Aziz M, Jacob A, Matsuda A, Wang P. Review: milk fat globule-EGF factor 8 expression, function and plausible signal transduction in resolving inflammation. Apoptosis. 2011;16:1077–86. doi: 10.1007/s10495-011-0630-0. [DOI] [PubMed] [Google Scholar]
- 25.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92:635–88. doi: 10.1152/physrev.00008.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ermolina LV, Martynova NY, Zaraisky AG. The cytoskeletal protein zyxin—a universal regulator of cell adhesion and gene expression. Russ J Bioorg Chem. 2010;36:29–37. doi: 10.1134/s1068162010010036. [DOI] [PubMed] [Google Scholar]
- 27.Chang Y-H, Ye L, Cai W, Lee Y, Guner H, Lee Y, et al. Quantitative proteomics reveals differential regulation of protein expression in recipient myocardium after trilineage cardiovascular cell transplantation. Proteomics. 2015;15:2560–7. doi: 10.1002/pmic.201500131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kato T, Muraski J, Chen Y, Tsujita Y, Wall J, Glembotski CC, et al. Atrial natriuretic peptide promotes cardiomyocyte survival by cGMP-dependent nuclear accumulation of zyxin and Akt. J Clin Invest. 2005;115:2716–30. doi: 10.1172/JCI24280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rasi K, Piuhola J, Czabanka M, Sormunen R, Ilves M, Leskinen H, et al. Collagen XV is necessary for modeling of the extracellular matrix and its deficiency predisposes to cardiomyopathy. Circ Res. 2015;107:1241–52. doi: 10.1161/CIRCRESAHA.110.222133. [DOI] [PubMed] [Google Scholar]
- 30.Molina-Navarro MM, Triviño JC, Martínez-Dolz L, Lago F, González-Juanatey JR, Portolés M, et al. Functional networks of nucleocytoplasmic transport-related genes differentiate ischemic and dilated cardiomyopathies. A new therapeutic opportunity. PLoS One. 2014;9:e104709. doi: 10.1371/journal.pone.0104709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raghow R. An ‘omics’ perspective on cardiomyopathies and heart failure. Trends Mol Med. 2016;22:813–27. doi: 10.1016/j.molmed.2016.07.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.