

Abstract
CB2 cannabinoid receptor-selective agonists are promising candidates for the treatment of pain. CB2 receptor activation inhibits acute, inflammatory, and neuropathic pain responses but does not cause central nervous system (CNS) effects, consistent with the lack of CB2 receptors in the normal CNS. To date, there has been virtually no information regarding the mechanism of CB2 receptor-mediated inhibition of pain responses. Here, we test the hypothesis that CB2 receptor activation stimulates release from keratinocytes of the endogenous opioid β-endorphin, which then acts at opioid receptors on primary afferent neurons to inhibit nociception. The antinociceptive effects of the CB2 receptor-selective agonist AM1241 were prevented in rats when naloxone or antiserum to β-endorphin was injected in the hindpaw where the noxious thermal stimulus was applied, suggesting that β-endorphin is necessary for CB2 receptor-mediated antinociception. Further, AM1241 did not inhibit nociception in μ-opioid receptor-deficient mice. Hindpaw injection of β-endorphin was sufficient to produce antinociception. AM1241 stimulated β-endorphin release from rat skin tissue and from cultured human keratinocytes. This stimulation was prevented by AM630, a CB2 cannabinoid receptor-selective antagonist and was not observed in skin from CB2 cannabinoid receptor-deficient mice. These data suggest that CB2 receptor activation stimulates release from keratinocytes of β-endorphin, which acts at local neuronal μ-opioid receptors to inhibit nociception. Supporting this possibility, CB2 immunolabeling was detected on β-endorphin-containing keratinocytes in stratum granulosum throughout the epidermis of the hindpaw. This mechanism allows for the local release of β-endorphin, where CB2 receptors are present, leading to anatomical specificity of opioid effects.
Keywords: β-endorphin, nociception, pain, keratinocyte, skin
CB2 cannabinoid receptor-selective agonists are very promising candidates for the treatment of pain. Activation of CB2 cannabinoid receptors inhibits nociception to thermal and mechanical stimuli (1, 2), thermal and tactile hypersensitivity produced by peripheral inflammation (2–4), and tactile and thermal hypersensitivity produced in a neuropathic pain model (5). Experimental findings suggesting that activation of peripheral (noncentral nervous system) CB2 receptors is necessary and sufficient to inhibit pain responses come from site-specific injections of CB2 receptor-selective agonists and antagonists (1, 3, 4). Importantly, CB2 cannabinoid receptor-selective agonists do not cause central nervous system (CNS) effects (1, 6), consistent with the inability to demonstrate the expression of CB2 receptors in the normal CNS (7–10). The lack of CNS effects is an important feature of this class of drug candidates because the efficacy of current pain therapies is frequently limited by CNS side effects. However, enthusiasm for this therapeutic approach has been tempered by the lack of information regarding the mechanism underlying the inhibition of nociceptive responses by CB2 receptor activation. CB2 cannabinoid receptors have not been found in the CNS or on peripheral neurons, suggesting that activation of CB2 receptors produces antinociception indirectly, by causing the release from nonneuronal cells of mediators that alter the responsiveness of primary afferent neurons to noxious stimuli. One type of cells that might mediate the actions of CB2 receptor-selective agonists is keratinocytes, which have been reported to express CB2 receptors (11) and to contain endogenous opioid peptides (12–14) and which are found in abundance in skin, where nociceptive stimuli have been applied when testing the antinociceptive effects of CB2 receptor-selective agonists. Therefore, we tested the hypothesis that activation of keratinocyte CB2 receptors results in the release of the endogenous opioid peptide β-endorphin, which then acts on primary afferent neurons to inhibit nociception.
Methods
Animals. All procedures were approved by the University of Arizona Animal Care and Use Committee and conform to the guidelines for the use of laboratory animals of the National Institutes of Health (publication no. 80–23, 1966). Male Sprague–Dawley rats (Harlan–Sprague–Dawley, Indianapolis) were 250–350 g at the time of testing. Mice were 20–30 g at the time of testing. Breeding pairs of mice heterozygous for the disrupted CB2 cannabinoid receptor gene (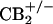 mice) (15) were kindly provided by Nancy Buckley (California State Polytechnic University, Pomona) and Andreas Zimmer (University of Bonn Medical School, Bonn). Breeding and genotyping were performed as described by Buckley et al. (15). Breeding pairs of mice heterozygous for the disrupted μ-opioid receptor gene (μ+/- mice) (16) were kindly provided by George Uhl (Molecular Neurobiology Branch, National Institute on Drug Abuse, Baltimore). Breeding and genotyping were performed as described by Sora et al. (16). Animals were maintained in a climate-controlled room on a 12-h light/dark cycle and were allowed to have food and water ad libitum.
mice) (15) were kindly provided by Nancy Buckley (California State Polytechnic University, Pomona) and Andreas Zimmer (University of Bonn Medical School, Bonn). Breeding and genotyping were performed as described by Buckley et al. (15). Breeding pairs of mice heterozygous for the disrupted μ-opioid receptor gene (μ+/- mice) (16) were kindly provided by George Uhl (Molecular Neurobiology Branch, National Institute on Drug Abuse, Baltimore). Breeding and genotyping were performed as described by Sora et al. (16). Animals were maintained in a climate-controlled room on a 12-h light/dark cycle and were allowed to have food and water ad libitum.
Drugs and Chemicals. Except where noted, chemicals were purchased from Sigma. β-Endorphin, β-endorphin antiserum, and nonimmune rabbit serum were purchased from Peninsula Laboratories. AM1241 is a CB2 receptor agonist with 70-fold selectivity for rodent CB2 receptors in vitro (5). AM630 is a CB2 receptor antagonist with 70- to 165-fold selectivity for CB2 receptors (17, 18).
Drug Administration. AM1241 was dissolved in DMSO and administered i.p. in 0.5 ml to rats and 70 μl to mice 20 min before nociceptive testing. All other drugs were dissolved in normal saline and administered s.c. to rats in the dorsal surface of the hindpaw (intrapaw) in 50 μl. Drugs were injected in the dorsal surface of the hindpaw to allow regional administration of drugs while minimizing any effects of the injection itself or of the vehicle on responses to stimuli applied to the plantar hindpaw. We had shown that injection of AM1241 in the dorsal surface of the hindpaw produced antinociceptive responses only in the same hindpaw (1). AM1241 was injected i.p., and other drugs or reagents were injected s.c. in the paw to avoid chemical interactions that might occur if both were injected s.c. in the same location. We had previously shown that the antinociceptive effects of i.p. AM1241 were prevented by intrapaw injection of the CB2 receptor antagonist AM630 (1), suggesting that AM1241 exerts its antinociceptive effects at the site of application of the nociceptive stimulus. Testing took place 20 min after drug administration.
Measurement of Thermal Withdrawal Latency. The method of Hargreaves et al. (19) was used. Animals were acclimated within Plexiglas enclosures on a clear glass plate maintained at 30°C. A radiant heat source (high-intensity projector lamp) was focused onto the plantar surface of the hind paw. When the paw was withdrawn, a motion detector halted the stimulus and a timer. A maximal cutoff of 40 sec was used to prevent tissue damage.
Measurement of β-Endorphin Release From Skin Tissue. Reagent preparation. AM1241 was dissolved in DMSO at a concentration of 2.5 μg/ul. AM1241 solution (100 μl) was then dissolved into 1 ml of Hanks' balanced salt solution (HBSS; 1.26 mM CaCl2/5.33 mM KCl/0.44 mM KH2PO4/0.5 mM MgCl2/0.41 mM MgSO4/138 mM NaCl/4 mM NaHCO3/0.3 mM Na2HPO4/5.6 mM glucose, pH 7.4), containing 1% BSA. Subsequent dilutions were made in HBSS/BSA to achieve the desired final concentration of AM1241. DMSO was added as necessary so that each sample contained an equivalent amount. The same method was used to prepare AM630.
Tissue preparation. Animals were euthanized by using 4% halothane. Skin from the plantar surface of the hindpaw was quickly collected and placed in HBSS at 37°C. A punch, 8 mm in diameter, was used to prepare skin samples of equivalent surface area. Each 8-mm skin sample was cut in half and equilibrated in HBSS for 30 min at 37°C.
Release assay. Each skin sample was placed in a 1.5-ml polypropylene tube containing 150 μl HBSS/BSA. AM1241 was added to achieve the desired final concentration. DMSO was present at a final concentration of 0.2%. Tubes containing both AM1241 + AM630 were prepared in an analogous manner. Tissue was placed in 120 μl of HBSS/BSA containing AM630. Five minutes later, 30 μl of AM1241 in HBSS/BSA was added. Each tube was incubated at 37°C for 30 min with periodic gentle agitation to improve oxygenation. The supernatant was collected and placed on ice. β-Endorphin content in the supernatant was measured immediately by using a commercially available enzyme immunoassay (Peninsula Laboratories).
β-Endorphin Release from Cultured Keratinocytes. Cultured human keratinocytes (HaCaT) cells (20) were kindly provided by N. E. Fusenig (German Cancer Research Center, Heidelberg). They were grown in 12-well plates in Iscove's modified Dulbecco's medium, supplemented with 10% FBS and penicillin-streptomycin (Invitrogen) at 37°C. Each well contained 350 μl for the release assay. AM1241 and AM630 were dissolved in DMSO and subsequently diluted in culture medium. After the addition of AM1241 and AM630 (where used), plates were incubated for 30 min. The media was collected by pipetting. β-Endorphin was measured by enzyme immunoassay.
Immunofluorescence. Hindpaw glabrous skin was removed from four male adult Sprague–Dawley rats (200–250 g), killed with an overdose of sodium pentobarbital, and perfused transcardially with 0.9% saline, followed by 4% paraformaldhyde in 0.1 M PBS at pH 7.4 and 4°C. The skin was postfixed at 4°C in the perfusion fixative for 4 h, cryoprotected in 30% sucrose in PBS, and sectioned at 14 μm on a cryostat in a plane perpendicular to the skin surface and parallel to the long axis of the foot (see Fig. 5). The sections were mounted onto alternating chrome-alum-gelatin-coated slides, air dried overnight, and processed for immunolabeling as described in detail in ref. 21 with rabbit antibody raised against an immunogen consisting of an 18-aa sequence found near the C terminus of the rat CB2 receptor (1:200; Chemicon), rabbit anti-ETRB (1:200; Alamone Labs, Jerusalem), or rabbit anti-β-endorphin (1:1,000; gift of R. G. Allen, Oregon Health and Science University, Portland). When anatomical segregation of labeling was evident in single-label preparations, double labeling was conducted by incubating in the first rabbit primary antibody, followed by the anti-rabbit Cy3, and then incubating the second rabbit primary antibody, followed by the anti-rabbit Alexa Fluor 488. The extent of any undesired crosslabeling between the second secondary antibodies and first primary antibodies or between the first secondary antibodies and second primary antibodies could be deduced from the single-label studies. Otherwise, to minimize complicating crosslabeling, the first rabbit primary antibody was labeled with Fab fragment goat anti-rabbit Cy3 (1:500; Jackson ImmunoResearch). To control for nonspecific labeling, incubations were conducted without the primary antibodies or with primary antibodies preabsorbed with their specific blocking peptide. The sections were viewed, and the images were digitally captured and processed as described in ref. 21.
Fig. 5.

Immunofluorescence labeling with antibodies against CB2, β-endorphin, and ETRB in the epidermis of glabrous skin from the hindpaw of a rat. As shown in the diagram, the sections are perpendicular to the surface and parallel to the long axis of the hindpaw near the medial border, where they pass through proximal and distal volar pads. Images in A, C, and E are taken from the site indicated by the broken line rectangle along the distal slope of the distal pad. Images in B, D, and F are from a flat site proximal to the proximal pad. The dotted lines indicate the location of the basement membrane at the epidermal-dermal border. SC, stratum corneum; SL, stratum lucidum; SG, stratum granulosum; SS, stratum spinosum; SB, stratum basalis. (Scale bar: 50 μm.) (A and B)CB2 immunolabeling is fairly uniformly distributed throughout the epidermis and is primarily limited to keratinocytes in stratum granulosum (between arrows). (C and D) β-endorphin is also fairly uniformly distributed but spans through stratum granulosum and extends well into stratum spinosum (between arrows). (E and F) ETRB labeling is restricted to the distal and proximal slopes of the volar pads (between arrows). ETRB is expressed in keratinocytes spanning stratum granulosum and extending into stratum spinosum.
Data Analysis. Differences between groups was tested by using ANOVA, followed by post hoc testing with the Student t test with Bonferroni's correction. Significance was defined as P < 0.05.
Results
The CB2 cannabinoid receptor-selective agonist AM1241 (100 μg/kg, i.p.) increased paw withdrawal latency to a thermal stimulus by 55% in rats (P < 0.05 compared with baseline values) (Fig. 1), demonstrating the production of antinociception to thermal stimuli. The vehicle (DMSO) had no effect, as observed in previous studies (1). Naloxone [10 μg, s.c. in the dorsal surface of the paw (intrapaw)] completely prevented the antinociceptive effects of AM1241 (P < 0.05 compared with AM1241 alone). Prevention of the effects of AM1241 by naloxone would be explained if AM1241 stimulated the release of endogenous opioids, and they, in turn, produced antinociceptive effects. In this regard, antiserum to β-endorphin (2 μg, intrapaw) prevented AM1241-induced antinociception (P < 0.05 compared with AM1241 alone), presumably by sequestering released β-endorphin. Nonimmune control serum had no effect.
Fig. 1.

The CB2 receptor-selective agonist AM1241 (100 μg/kg i.p.) produced antinociception to thermal stimuli. Naloxone (Nal) (10 μg, intrapaw) prevented the antinociceptive effects of AM1241, as did antiserum (AS) (2 μg, intrapaw) to β-endorphin. Nonimmune control serum (CS) (10 μg, intrapaw) had no effect. Data are expressed as mean ± SEM. n = 6 per group. *, P < 0.05 compared with baseline (BL). #, P < 0.05 compared with AM1241 alone.
To further test the role of β-endorphin in mediating the antinociception produced by AM1241, we administered AM1241 to mice lacking the gene for the μ-opioid receptor (μ-/- mice). β-Endorphin is a selective agonist at the μ-opioid receptor (22). AM1241 inhibited thermal nociception in wild-type (μ+/+) mice (Fig. 2). Paw withdrawal latency was increased by 127% at a dose of 10 mg/kg i.p. (P < 0.05 compared with baseline values). AM1241 produced significantly less antinociception in μ-opioid receptor-deficient (μ-/-) mice than in wild-type (μ+/+) mice (P < 0.05), suggesting that endogenous opioid activity at the μ-opioid receptor is necessary for CB2 receptor-mediated antinociception.
Fig. 2.

AM1241 (i.p.) produced dose-dependent antinociception in wild-type (μ+/+) mice but not in μ-opioid receptor knockout (μ-/-) mice. Data are expressed as mean ± SEM. n = 6 per group. #, P < 0.05 compared with μ+/+ mice.
Intrapaw injection of the β-endorphin peptide in rats similarly inhibited nociception to thermal stimuli. Forty micrograms increased paw withdrawal latency by 84% from 21.2 ± 0.8 sec to 39.1 ± 0.7 sec (P < 0.05). The effects of β-endorphin were completely prevented by naloxone (10 μg, intrapaw) and by antiserum to β-endorphin (2 μg, intrapaw). Paw withdrawal latency after AM1241 plus naloxone was 21 ± 2 sec, after AM1241 plus β-endorphin antiserum was 17 ± 2 sec, and after nonimmune control serum was 33 ± 3 sec. Nalaxone, β-endorphin antiserum, and nonimmune control serum had no effect on paw withdrawal latencies when administered in the absence of AM1241. These results demonstrate that β-endorphin is sufficient to produce the pattern of antinociception that follows CB2 receptor activation.
To test whether CB2 receptor activation is capable of stimulating β-endorphin release, we tested the effect of AM1241 in an in vitro β-endorphin release assay. AM1241 (10 μM) increased β-endorphin release from rat skin tissue by 93% (P < 0.05 compared with vehicle) (Fig. 3A). The CB2 receptor-selective antagonist AM630 completely prevented AM1241-stimulated β-endorphin release (P < 0.05 compared with AM1241 alone). AM630 had no effect on β-endorphin release in the absence of AM1241. AM1241 stimulated β-endorphin release from paw skin obtained from wild-type 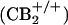 mice (Fig. 3B) but had no effect on the release from skin of CB2 receptor-deficient
mice (Fig. 3B) but had no effect on the release from skin of CB2 receptor-deficient 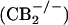 mice. These results strongly suggest that AM1241-stimulated β-endorphin release is mediated by CB2 receptors.
mice. These results strongly suggest that AM1241-stimulated β-endorphin release is mediated by CB2 receptors.
Fig. 3.

The CB2 receptor-selective agonist AM1241 stimulated β-endorphin release from glabrous paw skin. (A) Rat paw skin. The CB2 receptor antagonist AM630 (10 μM) prevented the effects of AM1241 (10 μM). AM630 had no effect on β-endorphin release in the absence of AM1241. (B) Mouse paw skin. AM1241 (10 μM) stimulated β-endorphin release from the skin of wild-type  but not from CB2 receptor-knockout
but not from CB2 receptor-knockout 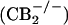 mice. Data are expressed as mean ± SEM. n = 12 per group. *, P < 0.05 compared with vehicle; #, P < 0.05 compared with 10 μM AM1241 alone.
mice. Data are expressed as mean ± SEM. n = 12 per group. *, P < 0.05 compared with vehicle; #, P < 0.05 compared with 10 μM AM1241 alone.
Similarly, AM1241 stimulated β-endorphin release from cultured human keratinocytes (HaCaT) cells (Fig. 4). AM1241 (1 μM) stimulated β-endorphin release by 146 ± 19% (P < 0.05 compared with vehicle). AM630 (1 μM) inhibited AM1241-stimulated β-endorphin release, suggesting that AM1241 stimulation of β-endorphin release is mediated by CB2 receptors. AM630 did not affect β-endorphin release in the absence of AM1241. Reverse transcription-PCR analysis has demonstrated the presence of the CB2 receptor mRNA in HaCaT cells (M.M.I., M.-C. Luo, and J.L., unpublished data).
Fig. 4.

AM1241 stimulated β-endorphin release from cultured human keratinocytes (HaCaT) cells. AM630 (1 μM) inhibited the effects of AM1241 (1 μM). AM630 had no effect in the absence of AM1241. Data are expressed as percent of release in medium alone and presented as mean ± SEM. n = 12 per group. *, P < 0.05 compared with medium alone. #, P < 0.05 compared with 1 μM AM1241.
Based on results indicating that CB2 receptors mediate β-endorphin release from keratinocytes, immunolabeling was conducted on sections of rat glabrous hindpaw skin with antibodies against CB2 receptors and β-endorphin. Labeling was also conducted with an antibody against endothelin B receptors (ETRBs), receptors that had been linked to an endothelin-mediated release of β-endorphin from keratinocytes (23). CB2 immunolabeling was intensely expressed throughout all areas of the epidermis, strictly among the uppermost layer of living keratinocytes in stratum granulosum (Figs. 5 and 6). No definitive labeling was detected when the primary antiserum was preabsorbed with blocking peptide. β-Endorphin immunolabeling was expressed on the same keratinocytes in all areas of the epidermis, such that virtually all CB2-positive keratinocytes appear to contain β-endorphin. β-Endorphin immunolabeling also continued onto deeper CB2-negative keratinocytes extending into stratum spinosum. Thus, whereas β-endorphin distribution followed the continuous pattern of CB2 distribution, β-endorphin also extended among deeper keratinocytes. In some locations, the depth of expression of both CB2 and β-endorphin was proportionately thinner than in most areas (Fig. 6A). Interestingly, ETRB labeling overlapped with CB2 but was limited to certain areas of the hindpaw, such as the flat surfaces proximal to and between the pronounced volar pads and to restricted sites on the distal and proximal slopes of the volar pads. Thus, CB2 expression is more continuous throughout the hindpaw epidermis, whereas ETRB is discontinuous. Moreover, within overlapping sites of CB2 receptor and ETRB immunolabeling, the most superficial keratinocytes in stratum granulosum expressed predominantly, if not uniquely, CB2, whereas ETRB expression also continued onto keratinocytes in the upper part of stratum spinosum. The full depth of the ETRB expression was comparable with that of β-endorphin. Given that CB2 was expressed relatively uniformly but superficially and ETRB distribution extended deeper but was discontinuous, the more uniform expression of β-endorphin extending through stratum granulosum and into stratum spinosum indicates that many β-endorphin-positive keratinocytes, especially in stratum spinosum, lack detectable CB2 or ETRB. Of immediate relevance to the hypothesis being tested, these results demonstrate that immunodectable CB2 is indeed expressed on β-endorphin-positive keratinocytes in stratum granulosum throughout the glabrous hindpaw epidermis.
Fig. 6.

Double-labeling immunofluorescence with antibodies against CB2, β-endorphin, and ETRB in the epidermis of glabrous skin from the hindpaw of a rat. The images are from sections that alternate with those in Fig. 5. Images are from locations indicted by the same letters in the schematic in Fig. 5. The dotted lines indicate the location of the basement membrane at the epidermal-dermal border. SC, stratum corneum; SL, stratum lucidum; SG, stratum granulosum; SS, stratum spinosum; SB, stratum basalis. (Scale bar, 50 μm; scale bar for insets, 25 μm.) (A and B) Anti-CB2 labeling revealed with anti-rabbit Fab conjugated with Cy3, followed by anti-β-endorphin labeling revealed with anti-rabbit Ig conjugated with Alexa Fluor 488. CB2 immunolabeling is mostly limited to stratum granulosum (red arrows), whereas β-endorphin immunolabeling spreads through stratum granulosum and into stratum spinosum (green arrows). Although fairly ubiquitously distributed throughout all areas of the epidermis, both labels can in parallel be restricted relatively superficially in some locations (*). B Inset is a 2× enlargement of the site in the small rectangle demonstrating the overlap of CB2 labeling (yellow arrows) in stratum granulosum with β-endorphin, which also extends more deeply into stratum spinosum (green arrow). (C and D) Anti-ETRB labeling revealed with anti-rabbit Fab conjugated with Cy3, followed by anti-CB2 labeling revealed with anti-rabbit Ig conjugated with Alexa Fluor 488. CB2 labeling (between green arrows) is fairly uniformly distributed in all areas of the epidermis and overlaps with the restricted sites of ETRB labeling in the volar pads and patchy areas of ETRB labeling in flat skin surfaces (red arrows and arrowheads). D Inset is a 2× enlargement of the site in the small rectangle demonstrating some superficial to deep segregation among the overlap of CB2 and ETRB labeling. The most superficial keratinocytes express primarily CB2 (green arrowhead) and the deeper ones primarily ETRB (red arrowheads). Keratinocytes in between express both (yellow arrowheads).
Discussion
The mechanism of CB2 cannabinoid receptor-mediated antinociception has not been readily explained because CB2 receptors are not normally present in the CNS or on peripheral neurons (7–9, 24). Therefore, we hypothesized that CB2 receptor activation produces antinociception indirectly by modulating the release from local cells of substances that affect the responsiveness of primary afferent neurons to noxious stimuli. Keratinocytes are very abundant in skin and have been reported to express CB2 receptors (11). Further, keratinocytes constitutively express proopiomelanocortin (23, 25), which is the precursor for a variety of peptides, including the endogenous opioid peptide β-endorphin. Therefore, we hypothesized that CB2 receptor activation produces antinociception by stimulating the release from keratinocytes of β-endorphin, which in turn produces antinociception by acting at μ-opioid receptors on primary afferent neurons. The data in this article strongly support this hypothesis.
It is also possible that other mediators, in addition to β-endorphin, might also be released from local cells after activation of CB2 receptors, contributing to the antinociceptive effects of CB2 receptor activation. However, β-endorphin release appears to play a critical role in CB2 receptor-mediated antinociception because the effects of AM1241 were completely prevented by a β-endorphin-sequestering antiserum. Release of additional mediators may explain the antiallodynic effects of AM1241 in the spinal nerve ligation model of neuropathic pain (5) in which allodynia is resistant to peripherally administered opioids (M.M.I. and T.P.M., unpublished data).
Similarly, we have not excluded the possibility that components of skin other than keratinocytes might contribute to the release of β-endorphin in response to CB2 receptor activation. Immune cells express CB2 receptors (7) and are capable of releasing endogenous opioids (26). Thus, it is possible that resident immune and inflammatory cells in skin and s.c. tissue may augment CB2 receptor-induced β-endorphin release. However, it is likely that keratinocytes are the major source of β-endorphin in skin due to their abundance compared with resident immune cells.
A significant unanswered question is the intracellular signaling pathway that couples CB2 receptor activation to β-endorphin release. Activation of CB2 cannabinoid receptors results in inhibition of adenylyl cyclase activity by a Gi/Go protein (27) and stimulates mitogen-activated protein kinase (28). Activation of a Gi protein is typically predicted to inhibit exocytosis. However, activation of some G protein-coupled receptors has been reported to result in release processes that are pertussis toxin-sensitive, suggesting that they are mediated by Gi or Gi/Go proteins (29–31). It is also possible that the ability of CB2 receptors to stimulate β-endorphin release is mediated by another class of G-proteins.
The ETRB receptor has been linked to an endothelin-mediated release of β-endorphin (23). That study also demonstrated that calcitonin gene-related peptide-containing sensory endings in the epidermis express μ-opioid receptor, which may be the site of β-endorphin-mediated antinociception. The distribution of CB2 of ETRB extended deeper than did that of CB2. The distribution was more continuous, whereas ETRB localized to specific areas. These similarities and differences in distributions support the concept that both CB2 and ETRB can mediate β-endorphin release but may act together or independently in anatomically distinct locations. Moreover, undiscovered factors may also mediate β-endorphin release from keratinocytes that lack either CB2 or ETRB.
We have demonstrated that antinociception produced by CB2 receptor-selective agonists may be mediated by stimulation of β-endorphin release from CB2-expressing cells. The β-endorphin released thus appears to act at μ-opioid receptors, probably on the terminals of primary afferent neurons, to produce peripheral antinociception. This mechanism allows for the local release of endogenous opioids limited to sites where CB2 receptors are present, thereby leading to anatomical specificity of opioid effects. In this way, CB2 receptor activation may produce peripheral antinociception without CNS side effects.
Acknowledgments
We thank Mike Pennington for the technical assistance in growing the cultured human keratinocytes, Marilyn Dockum for assistance in tissue processing, and Dr. Joseph Mazurkiewicz for providing β-endorphin antibody. This work was supported by National Institute on Drug Abuse Grant DA015866 (to T.P.M.) and U.S. Public Health Service Grant NS34692 (to F.L.R.).
Abbreviations: ETRB, endothelin B receptor; HBSS, Hanks' balanced salt solution.
References
- 1.Malan, T. P., Ibrahim, M. M., Deng, H., Liu, Q., Mata, H. P., Vanderah, T., Porreca, F. & Makriyannis, A. (2001) Pain 93, 239-245. [DOI] [PubMed] [Google Scholar]
- 2.Clayton, N., Marshall, F. H., Bountra, C. & O'Shaughnessey, C. T. (2002) Pain 96, 253-260. [DOI] [PubMed] [Google Scholar]
- 3.Quartilho, A., Mata, H. P., Ibrahim, M. M., Vanderah, T. W, Porreca, F., Makriyannis, A. & Malan, T. P. (2003) Anesthesiology 99, 955-960. [DOI] [PubMed] [Google Scholar]
- 4.Nackley, A. G, Makriyannis, A. & Hohmann, A. G. (2003) Neuroscience 119, 747-757. [DOI] [PubMed] [Google Scholar]
- 5.Ibrahim, M. M., Deng, H., Zvonok, A., Cockayne, D. A., Kwan, J., Mata, H. P., Vanderah, T. W., Lai, J., Porreca, F., Makriyannis, A. & Malan, T. P., Jr. (2003) Proc. Natl. Acad. Sci. USA 100, 10529-10533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hanus, L., Breuer, A., Tchilibon, S., Shiloah, S., Goldenberg, D., Horowitz, M., Pertwee, R. G., Ross, R. A., Mechoulam, R. & Fride, E. (1999) Proc. Natl. Acad. Sci. USA 96, 14228-14233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Munro, S., Thomas, K. L. & Abu-Shaar, M. (1993) Nature 365, 61-65. [DOI] [PubMed] [Google Scholar]
- 8.Facci, L., Toso, R. D., Romanello, S., Buriani, A., Skaper, S. D. & Leon, A. (1995) Proc. Natl. Acad. Sci. USA 92, 3376-3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schatz, A. R., Lee, M., Condie, R. B., Pulaski, J. T. & Kaminski, N. E. (1997) Toxicol. Appl. Pharmacol. 142, 278-287. [DOI] [PubMed] [Google Scholar]
- 10.Zimmer, A, Zimmer, A. M, Hohmann, A. G, Herkenham, M. & Bonner, T. I. (1999) Proc. Natl. Acad. Sci. USA 96, 5780-5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Casanova, M. L., Blazquez, C., Martinez-Palacio, J., Villanueva, C., Fernandez-Acenero, M. J., Huffman, J. W., Jorcano, J. L. & Guzman, M. (2003) J. Clin. Invest. 111, 43-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cabot, P. J., Carter, L., Gaiddon, C., Zhang, Q., Schafer, M., Loefflerm J. P. & Stein, C. (1997) J. Clin. Invest. 100, 142-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rittner, H. L., Brack, A., Machelska, H., Mousa, S. A., Bauer, M., Schafer, M. & Stein, C. (2001) Anesthesiology 95, 500-508. [DOI] [PubMed] [Google Scholar]
- 14.Kauser, S., Schallreuter, K., Thody, A., Gummer, C. & Tobin, D. (2003) J. Invest. Dermatol. 120, 1073-1080. [DOI] [PubMed] [Google Scholar]
- 15.Buckley, N. E., McCoy, K. L., Mezey, E., Bonner, T., Zimmer, A., Felder, C. C., Glass, M. & Zimmer, A. (2000) Eur. J. Pharmacol. 396, 141-149. [DOI] [PubMed] [Google Scholar]
- 16.Sora, I., Takahashi, N., Funada, M., Ujike, H., Revay, R. S., Donovan, D. M, Miner, L. L. & Uhl, G. R. (1997) Proc. Natl. Acad. Sci. USA 94, 1544-1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross, R. A, Brockie, H. C., Stevenson, L. A., Murphy, V. L., Templeton, F., Makriyannis, A. & Pertwee, R. G. (1999) Br. J. Pharmacol. 126, 665-672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hosohata, Y., Quock, R. M., Hosohata, K., Makriyannis, A., Consroe, P., Roeske, W. R. & Yamamura, H. I. (1997) Eur. J. Pharmacol. 321, R1-R3. [DOI] [PubMed] [Google Scholar]
- 19.Hargreaves, K. M., Dubner, R., Brown, F., Flores, C. & Joris, J. (1988) Pain 32, 77-88. [DOI] [PubMed] [Google Scholar]
- 20.Breitkreutz, D., Schoop, V. M., Mirancea, N., Baur, M., Stark, H. J. & Fusenig, N. E. (1998) Eur. J. Cell Biol. 75, 273-286. [DOI] [PubMed] [Google Scholar]
- 21.Paré, M., Elde, R., Mazurkiewicz, J. E., Smith, A. M. & Rice, F. L. (2001) J. Neurosci. 21, 7236-7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Helmeste, D. M. & Li, C. H. (1986) Proc. Natl. Acad. Sci. USA 83, 67-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khodorova, A., Navarro, B., Jouaville, L. S., Murphy, J. E., Rice, F. L., Mazurkiewicz, J. E., Long-Woodward, D., Stoffel, M., Strichartz, G. R., et al. (2003) Nat. Med. 9, 1055-1061. [DOI] [PubMed] [Google Scholar]
- 24.Galiegue, S., Mary, S., Marchand, J., Dussossoy, D., Carriere, D., Carayon, P., Bouaboula, M., Shire, D., Le Fur, G. & Casellas, P. (1995) Eur. J. Biochem. 232, 54-61. [DOI] [PubMed] [Google Scholar]
- 25.Wintzen, M., Yaar, M., Burbach, P. & Gilchrest, B. (1996) J. Invest. Dermatol. 106, 673-678. [DOI] [PubMed] [Google Scholar]
- 26.Stein, C., Hassan, A. H. S., Przewlocki, R., Gramsch, C., Peter, K. & Herz, A. (1990) Proc. Natl. Acad. Sci. USA 87, 5935-5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bayewitch, M., Avidor-Reiss, T., Levy, R., Barg, J., Mechoulam, R. & Vogel, Z. (1995) FEBS Lett. 375, 143-147. [DOI] [PubMed] [Google Scholar]
- 28.Ho, B. Y., Uezono, Y., Takada, S., Takase, I. & Izumi, F. (1999) Recept. Channels 6, 363-374. [PubMed] [Google Scholar]
- 29.Shefler, I., Seger, R. & Sagi-Eisenberg, R. (1999) J. Pharmacol. Exp. Ther. 289, 1654-1661. [PubMed] [Google Scholar]
- 30.Conant, K., Haughey, N., Nath, A., St. Hillaire, C., Gary, D. S., Pardo, C. A., Wahl, L. M., Bilak, M., Milward, E. & Mattson, M. P. (2002) J. Neurochem. 82, 885-893. [DOI] [PubMed] [Google Scholar]
- 31.Ferry, X., Eichwald, V., Daeffler, L. & Landry, Y. (2001) J. Immunol. 197, 4805-4813. [DOI] [PubMed] [Google Scholar]