

Abstract
We report on a 12-yr-old female and a 14-yr-old male with Seckel syndrome. The 12-yr-old female had pancytopenia, which is seen occasionally in patients with Seckel syndrome and is also a feature of Fanconi anemia, a well-recognized autosomal recessive dwarfism syndrome with chromosome instability. Chromosome breakage analysis of both of our patients also indicated chromosome instability. We suggest that there may be a subgroup of Seckel syndrome patients with chromosome instability and/or hematological problems.
Keywords: pancytopenia, chromosome instability
INTRODUCTION
Seckel [1960] described an autosomal recessive disorder with mental deficiency, microcephaly, prenatal onset of marked growth deficiency, and characteristic facial appearance (prominent beaked nose and receding chin). Over 40 patients are reported to have met the diagnostic criteria for Seckel syndrome [Majewski and Goecke, 1982; Thompson and Pembrey, 1985]. A literature review indicates that 7 patients (20%) with Seckel syndrome also had hypoplastic anemia [Upjohn, 1955; Seckel, 1960; Lilleyman, 1984; Majewski et al, 1982]. Herein we report the clinical and cytogenetic findings on 2 patients with Seckel syndrome, including one with pancytopenia.
CLINICAL REPORT
K.G. was a term white female whose birth weight and length were 1,362 g and 35.5 cm, respectively. The pregnancy and family histories were unremarkable. Consanguinity was denied. Her prominent features noted at age 4 yr were height and weight at −5 SD; microcephaly (−7 SD); prominent beaked nose; high, arched palate; micrognathia; bilateral clinodactyly of the fifth finger; small incisors; absent bone dysplasia; and moderate mental retardation (Fig. 1). She was diagnosed by several clinical geneticists as having Seckel syndrome.
Fig. 1.
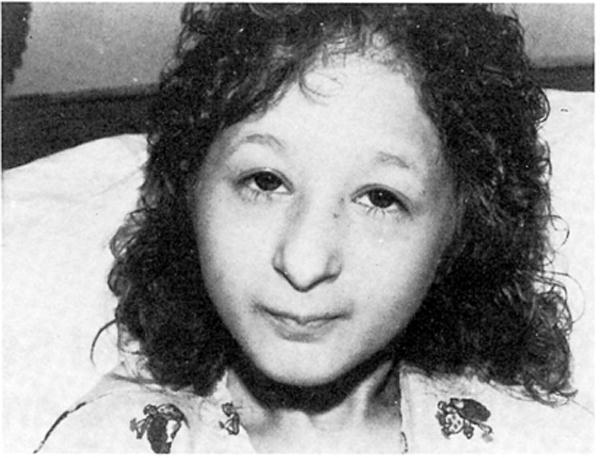
Frontal view of K.G. at 12 yr of age.
She developed anemia at 8 yr which was followed by pancytopenia. At 12 yr she weighed 16.6 kg (50th centile for 4 yr), was 109 cm tall (50th centile for 5.3 yr), and had a head circumference of 40 cm (50th centile for 3 months). After numerous transfusions a bone marrow transplant was undertaken at 12 yr but death occurred 2 wk later. Chromosome studies showed a 46,XX chromosome constitution. A 10% chromosome breakage rate in routine culture preparations was noted, which was 3 times higher than in control subjects established by the laboratory. Because of the child’s death, additional cytogenetic studies were not available.
J.J. was born to a 35-yr-old, gravida 4, para 4 woman after an 8-month uncomplicated pregnancy. His birth weight and length were 1,121 g and 38.1 cm, respectively. There were no neonatal complications although the child was hospitalized for 2 months for weight gain. Frequent respiratory infections were noted during the first 2 yr of life. Tonic-clonic seizures were noted at 2 yr and were medically treated. Psychomotor and growth retardation were noted during infancy and the bone age was considerably delayed. The family history was remarkable for another sib with severe microcephaly, growth retardation, and unusual facial appearance, who died shortly after birth. Consanguinity was denied.
On physical examination at 13 yr, his height, weight, and head circumference were 98.5 cm, 10.25 kg and 42.2 cm, respectively (all less than 3rd centile). In general he was an extremely growth-retarded, well-proportioned white male with a small, slender body habitus and reduced muscle mass. He had a narrow, triangular face with a prominent nose, small, low-set ears, and severe microcephaly (Fig. 2). Biparietal narrowing, a prominent occiput, and 5th-finger clinodactyly were also observed. He had bilateral iris colobomas, a left cataract, and a high, arched palate. He had paralysis of the right phrenic nerve, documented by nerve conduction studies, which led to surgical plication of the diaphragm at 12 yr. Cryptorchidism was also noted. The cardiovascular, abdominal, and neurological examinations were normal. His mental age was judged to be 5 yr at a chronological age of 13 yr.
Fig. 2.

Frontal view of J.J. at 10 yr of age.
EEG, EKG, echocardiogram, cervical spine X-ray, hematogram, liver enzymes, and urinary metabolic studies were all normal. A CT scan of the head showed mild cerebral dysplasia with no focal lesions. A hand x-ray showed clinodactyly of the 5th finger bilaterally and increased density of the epiphysis consistent with the diagnosis of Seckel syndrome. The bone age was 7.5 yr at a chronological age of 13 yr. Blood was obtained for chromosome analysis at 14 yr of age. There was no history of anemia at the time of examination. The child died 2 months later due to aspiration.
MATERIALS AND METHODS
Detailed chromosome breakage studies on K.G. were not available due to the patient’s death, although the routine chromosome analysis showed 10% chromosome breakage, or 3 times the control level established for the laboratory. Routine cytogenetic analysis, chromosome breakage studies with mitomycin C (MMC), folate-deficient culture conditions, and sister chromatid exchange (SCE) frequency were available on J.J.
Peripheral blood was cultured in RPMI 1640 with 20 ng/ml MMC for 48 and 96 hr at 37°C from J.J. and 12 control subjects. Approximately 100 diploid cells (≥ 44 chromosomes) at 96-hr exposure and 50 cells at 48-hr exposure were analyzed. Thirty minutes before harvest, Colcemid (0.2 μg/ml final concentration) was added. Conventional air-dried slides were prepared and stained with Giemsa. The aberrations were classified as either chromatid (breaks, gaps, and exchange figures) or chromosome (dicentrics, rings, double minutes, fragments, and markers).
Peripheral blood (0.3 ml) was cultured in 10 ml of folate-deficient Medium 199 for 96 hr at 37°C from J.J. and 12 control subjects. Colcemid (0.15 μg/ml final concentration) was added 30–45 min before harvesting. Conventional harvest methods were used and air-dried slides were stained with Giemsa. All fragile sites as well as chromosome and chromatid aberrations were recorded from 100 metaphases.
Sister chromatid exchanges were analyzed from J.J. and 12 control subjects. Peripheral blood (0.5 ml) was obtained and cultured for 72 hr in the presence of 20 μM 5-bromodeoxy-uridine. The chromosomes were stained with the FPG technique. Cells were analyzed and the presence of first, second, and third mitotic divisions was recorded. The number of SCEs from 30 analyzable metaphases was recorded and the average frequency determined. The replicative index was calculated from the percentage of cells in first, second, and third division [Watanabe and Endo, 1984].
RESULTS
Table I summarizes the MMC data from J.J. and 12 control subjects grown in 20 ng/ml MMC for 96 hr. Eighteen aberrations (2 chromosome fragments, 8 chromatid gaps, 7 chromatid breaks, and 1 triradial) were found in 100 cells analyzed from J.J. Sixty-five aberrations (6 chromosome fragments, 2 ring chromosomes, 34 chromatid gaps, 21 chromatid breaks, and 2 quadriradials) were found in 1,201 cells from the 12 control subjects. The range of total chromosome aberrations for 100 cells was 3–9 for the 12 control subjects. The average frequency and standard deviation for total chromosome and chromatid aberrations per 100 cells were 5.4 ± 1.8, for chromatid aberrations were 4.8 ± 1.4, and for chromosome aberrations were 0.5 ± 0.6 for the 12 control subjects.
TABLE I.
Percentage of Cells With Aberrations Treated With 20 ng/ml Mitomycin C for 96 Hr
| Subject group | Chromatid and chromosome aberrations | Chromatid aberrations | Chromosome aberrations |
|---|---|---|---|
| J.J. | 18 | 16 | 2 |
| Control | 5.3 | 4.6 | 0.7 |
| (N = 12) |
Table II summarizes the MMC data from J.J. and 12 control subjects grown in 20 ng/ml MMC for 48-hr exposure. Twenty aberrations (8 chromatid gaps, 10 chromatid breaks, and 2 triradials) were found in 100 cells analyzed from J.J. Twenty-nine aberrations (4 chromosome fragments, 14 chromatid gaps, and 11 chromatid breaks) were found in 600 cells from the 12 control subjects. The range of total chromosome aberrations for 50 cells was 0 to 4 for the 12 control subjects. The average frequency and standard deviation for total chromosome and chromatid aberrations per 50 cells were 2.4 ± 1.3, for chromatid aberrations 2.1 ± 1.2, and for chromosome aberrations 0.3 ± 0.5 for the 12 control subjects.
TABLE II.
Percentage of Cells With Aberrations Treated With 20 ng/ml Mitomycin C for 48 Hr
| Subject group | Chromatid and chromosome aberrations | Chromatid aberrations | Chromosome aberrations |
|---|---|---|---|
| J.J. | 20 | 20 | 0 |
| Control | 4.8 | 4.2 | 0.6 |
| (N = 12) |
The average frequency for total chromosome and chromatid aberrations per 100 cells grown in folate-deficient culture conditions was 19 for J.J. and 5 ± 4.7 for the 12 control subjects. The average frequency and standard deviation of SCEs were 15.3 ± 3.2 for J.J. and 9.1 ± 1.9 with an average range from 5.9 to 12.4 for the 12 control subjects. The average replicative indices for J.J. and the control subjects were 1.88 and 1.82, respectively.
DISCUSSION
Our 2 patients represent additional cases of Seckel syndrome. At least 8 patients with Seckel syndrome have had chromosome studies and 2 patients were found to be mosaic for sex chromosome aneuploidy (45,X/46,XX or 46,XX/47,XXX) [Majewski and Goecke, 1982]. Two sibs without hematological problems have also had chromosome breakage and SCE analyses but no abnormality was identified [Cervenka et al, 1979].
Our chromosome breakage data would suggest instability in our patients studied in 2 separate laboratories. Mitomycin C-induced chromosome breakage is a well-recognized feature of Fanconi anemia and other autosomal recessive disorders of known chromosome instability such as Bloom syndrome, another type of human dwarfism characterized by an elevation of the number of SCEs.
The question arises as to whether Seckel syndrome is heterogeneous with a distinct subgroup of patients with associated chromosome instability and bone marrow failure or hematological abnormalities. We would suggest that other patients with Seckel syndrome be evaluated for hematological disorders and/or chromosome breakage in order to determine the true incidence of these findings in this rare dwarfism syndrome.
References
- Cervenka J, Tsuchiya H, Ishiki T, Susuki M, Mori H. Seckel’s dwarfism: Analysis of chromosome breakage and sister chromatid exchanges. Am J Dis Child. 1979;133:555–556. doi: 10.1001/archpedi.1979.02130050099023. [DOI] [PubMed] [Google Scholar]
- Lilleyman JS. Constitutional hypoplastic anemia associated with familial “bird-headed” dwarfism (Seckel syndrome) Am J Pediatr Hematol Oncol. 1984;6:207–209. [PubMed] [Google Scholar]
- Majewski F, Goecke T. Studies of microcephalic primordial dwarfism I. Am J Med Genet. 1982;12:7–21. doi: 10.1002/ajmg.1320120103. [DOI] [PubMed] [Google Scholar]
- Majewski F, Ranke M, Schinzel A. Studies of microcephalic primordial dwarfism II. Am J Med Genet. 1982;12:23–35. doi: 10.1002/ajmg.1320120104. [DOI] [PubMed] [Google Scholar]
- Seckel HPG. Bird Headed Dwarfs. Basel and New York: S. Karger; 1960. [Google Scholar]
- Thompson E, Pembrey Μ. Seckel syndrome: An overdiagnosed Syndrome! J Med Genet. 1985;22:192–201. doi: 10.1136/jmg.22.3.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upjohn C. Familial dwarfism associated with microcephaly, mental retardation and anemia. Proc Soc Med. 1955;48:334–335. [PubMed] [Google Scholar]
- Watanabe T, Endo A. The SCE test as a tool for cytogenetic monitoring of human exposure to occupational and environmental mutagens. In: Tice R, Hollaender A, editors. Sister Chromatid Exchanges. New York: Plenum; 1984. pp. 946–947. [DOI] [PubMed] [Google Scholar]