

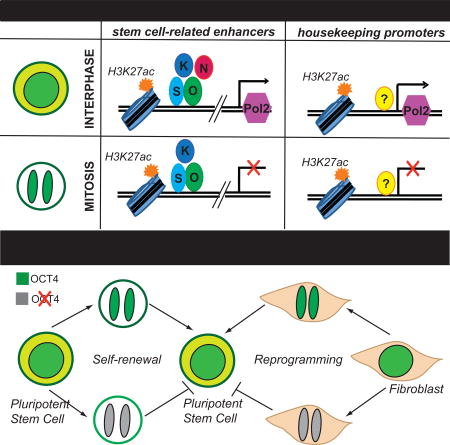
SUMMARY
During mitosis, transcription is halted and many chromatin features are lost, posing a challenge for the continuity of cell identity, particularly in fast cycling stem cells, which constantly balance self-renewal with differentiation. Here we show that, in pluripotent stem cells, certain histone marks and stem cell regulators remain associated with specific genomic regions of mitotic chromatin, a phenomenon known as mitotic bookmarking. Enhancers of stem cell-related genes are bookmarked by both H3K27ac and the master regulators OCT4, SOX2 and KLF4, while promoters of housekeeping genes retain high levels of mitotic H3K27ac in a cell-type invariant manner. Temporal degradation of OCT4 during mitotic exit compromises its ability both to maintain and induce pluripotency, suggesting that its regulatory function partly depends on its bookmarking activity. Together, our data document a widespread yet specific bookmarking by histone modifications and transcription factors promoting faithful and efficient propagation of stemness after cell division.
INTRODUCTION
Cell identity is determined by characteristic gene expression programs and chromatin landscapes, which are constantly supervised by key transcription factors (TFs), known as master regulators (Natoli, 2010; Young, 2011). This tightly controlled system is temporarily destabilized during mitosis, when dramatic molecular changes occur, such as global transcriptional shut down, alterations in the abundance of histone modifications and dissociation of most of the transcription factors and cofactors from condensed mitotic chromatin (Martinez-Balbas, 1995; Gottesfeld and Forbes, 1997; Wang and Higgins, 2013). How cell-type specific programs are faithfully restored in daughter cells constitutes a fundamental -and yet unanswered- question in biology. Studies in different somatic cell types proposed that the heritability of defined gene expression programs might rely on the mitotic persistence of either epigenetic marks (histone and DNA modifications) and/or TFs on the chromatin, a phenomenon known as mitotic bookmarking. There are examples supporting at least partial retention of specific active or repressive histone marks, which then enables rapid recruitment of the respective histone readers and writers upon G1 entry, ensuring self-perpetuation of gene expression states (Kouskouti and Talianidis, 2005; Margueron and Reinberg, 2010; Valls et al., 2005; Zaidi et al., 2010). There are also a number of studies documenting that selected TFs remain associated with mitotic chromatin, facilitating rapid reactivation of critical genes for the respective cell identity (Caravaca et al., 2013; Kadauke and Blobel, 2013; Kadauke et al., 2012; Young et al., 2007). Interestingly, previously reported bookmarking TFs are either master regulators of cell identity, such as GATA1 in hematopoietic cells (Kadauke et al., 2012), and/or “pioneer” factors - able to bind otherwise “inaccessible” nucleosomal regions (Zaret and Carroll, 2011) such as FoxA1 in liver progenitors (Caravaca et al., 2013). Collectively, these suggest that both epigenetic and TF bookmarking mechanisms contribute to the faithful propagation of cell identity after cell division. Whether similar mechanisms are also important for stem cell fate heritability remains understudied.
Pluripotent stem cells (PSCs) are endowed with the remarkable capacity to self-renew indefinitely, while preserving the potential to differentiate into all somatic cell types in response to developmental cues (Evans, 2011; Tabar and Studer, 2014). In addition, PSCs are characterized by an extremely rapid (10–12 hour) cell cycle, which lacks G0 phase and has an unusually short G1 phase (Savatier et al., 2002). This necessitates the presence of very efficient mechanisms for preserving or resetting PSC-specific transcriptional patterns. Notably, there is a well-defined network of TFs that control maintenance and acquisition of stem cell identity (Apostolou and Hochedlinger, 2013; Takahashi and Yamanaka, 2006; Young, 2011). Among them, OCT4, SOX2, ESRRB and KLF4 have been reported to also function as pioneer factors (Iwafuchi-Doi and Zaret, 2014; Soufi et al, 2012; Soufi et al., 2015), sharing critical properties with known bookmarking TFs. However, evidence for a potential bookmarking role for some of these TFs has only recently begun to emerge (Festuccia et al, 2016; Teves et al, 2016; Deluz et al, 2016). Similarly, although the functional genomic elements that control stem cell identity and the histone marks that decorate them are well characterized (Hawkins et al., 2010; Mikkelsen et al., 2007; Whyte et al., 2013), their status during mitosis remains unknown.
Here, combining biochemical and single-cell imaging approaches, we systematically investigated the extent of mitotic retention for selected histone modifications and pluripotency-associated TFs. Our analyses identified in a genome-wide scale the specificity of bookmarking and provided evidence for its biological significance in stem cell identity.
RESULTS
Specific histone modifications and pluripotency-related transcription factors are maintained on mitotic chromatin
To determine candidate bookmarking factors in PSCs, we first tested a number of histone modifications and TFs for their relative ability to remain associated with mitotic chromatin of mouse embryonic stem cells (ESCs). To do so, we performed subcellular fractionation of both asynchronous ESCs and ESCs arrested in mitosis to a high purity (typically >92%), as assayed by FACS analysis for the presence of the mitotic marker Histone 3 phospho-Serine 10 (H3Ser10p) (Tapia et al., 2006) (Figure 1A). Western blot analysis validated the high purity of the separate fractions, exhibiting complete depletion of cytoplasmic proteins, such as Tubulin and GAPDH, from nuclear and chromatin extracts and detection of Histone 3 predominantly in the chromatin fraction (Figure S1A). When we focused on the chromatin-bound fractions, we found that architectural factors, such as SMC3 and CTCF, remained associated with chromatin during mitosis, in agreement with previous studies (Burke et al., 2005; Peters et al., 2008), whereas the RNA polymerase II phospho-Ser2 (marker for transcriptional elongation) and Mediator levels were markedly decreased in accordance with a global transcriptional silencing during mitosis (Gottesfeld and Forbes, 1997) (Figures 1B and 1E). These results demonstrated that our chromatin extracts had the required purity and quality for downstream analyses.
Figure 1. Histone modifications and pluripotency-related transcription factors are largely retained on the mitotic chromatin.

A. Representative FACS plots showing the percentage of asynchronous or nocodazole-treated ESCs (after mitotic shake-off) expressing the mitotic marker H3Ser10p. B–D. Western blot analyses showing the relative levels of positive and negative controls (B), selected histone modifications (C) and transcription factors (D) on the chromatin fraction of asynchronous (A) or mitotic (M) ESCs. E. Quantitation of (B–D) gel bands using Image J. The relative levels of each protein in mitotic chromatin fractions are plotted as a percentage of the respective levels in asynchronous chromatin fractions. Error bars indicate standard deviation based on at least two independent synchronization and fractionation experiments in two different ESC lines (ESC V6.5 and ZHBTc4.1). Asterisks indicate significant downregulation when compared to H3 levels, as calculated by t-test (**p<0.01, ***p<0.001). F. Representative live-imaging photos of mitotic ESCs expressing ectopic KLF4, ESRRB, OCT4 and SOX2 fused with a GFP reporter. Overexpression of GFP alone is used as a control. The chromosomes were stained using a cell-permeable DNA dye (Vybrant Violet). G. Time-lapse images of dividing blastomeres after microinjection of KLF4-GFP mRNA into one out of two blastomeres in 2-cell stage embryos (See also Supplemental Movie 1).
We then tested the relative abundance of common histone modifications on asynchronous and mitotic chromatin. In agreement with previous findings in other cell types (Valls et al., 2005; Wang and Higgins, 2013), all tested histone methylation marks (H3K27me3, H3K9me3, H3K4me1 and H3K4me2) were highly retained during mitosis (Figures 1C and 1E). In contrast, the level of acetylated H3 was decreased, partly due to the reduction of H3K9 and H3K14 acetylation. Interestingly, H3K27 acetylation, which is a hallmark of active regulatory elements and particularly enhancers (Creyghton et al., 2010), was detected at similar levels in asynchronous and mitotic ESCs. These results show that – at the global level – some of the key regulatory histone modifications are maintained on mitotic chromatin of ESCs, suggesting that inheritance of these chromatin marks may play a role in faithful propagation of pluripotent cell identity upon cell division.
We also tested the association of known pluripotency-related TFs with mitotic chromatin. UTF1, a chromatin-associated factor with histone-like characteristics (Kooistra et al., 2009; van den Boom et al., 2007), served as a positive control showing almost identical levels in asynchronous and mitotic chromatin extracts (Figures 1D–E). In agreement with recent studies (Festuccia et al, 2016; Teves et al, 2016; Deluz et al, 2016), this assay showed that SOX2, ESRRB and OCT4 as well as KLF4 were also highly retained on mitotic chromatin (ranging between 70–100% of respective levels in asynchronous cells). By contrast, factors such as NANOG and REX1 showed markedly reduced levels in the chromatin fraction during mitosis (Figures 1D–E). Of note, the protein levels of several TFs including NANOG were reduced in total mitotic cell extracts (Figure S1B), suggesting cell-cycle dependent regulation of protein abundance. However, a clear correlation between total protein levels and chromatin retention was not observed, implying that additional mechanisms such as active dissociation from or preferential association with mitotic chromatin may therefore operate in a TF-specific manner. Taken together these results reveal that many pluripotency-related TFs remain associated with mitotic chromatin in ESCs.
To independently verify our findings at the single-cell level, we performed immunofluorescence experiments on metaphase spreads of partly synchronized ESCs after fixation with methanol:acetic acid solution (Figure S1C–D). OCT4, SOX2, KLF4 and ESRRB were detected both on interphase nuclei and on the condensed chromosomes. In contrast, RNA PolII and NANOG were excluded from the mitotic chromosomes, in concordance with our Western blot analyses. Of note, incubation with matched IgG isotypes or only secondary antibodies showed no specific signal on either interphase nuclei or condensed chromosomes. In parallel, live imaging of ESCs overexpressing GFP fusions with OCT4, SOX2, KLF4 or ESRRB (Figure S1E) confirmed a strong enrichment for each of these TFs on mitotic chromosomes, whereas overexpressed GFP protein alone was diffused throughout the cell volume (Figure 1F). Finally, introduction of a KLF4-GFP chimeric protein into 2-cell mouse embryos followed by live imaging demonstrated in vivo the ability of this protein to associate with mitotic chromosomes in dividing blastomeres (Figure 1G and Supplemental Movie 1). Together, these results confirmed at a single cell level the ability of critical stem cell regulators to remain associated with chromatin during cell division, suggesting a potential bookmarking function.
H3K27 acetylation bookmarks distinct sets of regulatory elements during mitosis
Next, we sought to determine in a genome-wide scale the extent and specificity of mitotic retention of H3K27ac, which constitutes an attractive candidate bookmarking feature given its association to active regulatory elements. ChIP-seq experiments in asynchronous and mitotically-arrested ESCs revealed highly overlapping, but distinct, genome-wide occupancy patterns (Figure 2A–C). There was a large portion of H3K27ac sites that were retained in mitotic cells (Bookmarked, B class), whereas distinct sets of H3K27ac peaks were detected only in asynchronous (A class) or only in mitotic cells (M class) (Figure 2C). Examples of ChIP-seq tracks for each category are shown in Figure 2D.
Figure 2. ChIP-seq experiments reveal distinct, but overlapping, patterns of H3K27 acetylation on asynchronous and mitotic ESCs.

A. Numbers of H3K27ac ChIP-seq peaks in asynchronous and mitotic ESCs and extent of overlap between them. B–C Averaged coverage (B) and enrichment (C) plots of H3K27ac ChIP-seq signals in mitotic and asynchronous ESCs. Bookmarked (B) H3K27ac peaks have comparable signal in both conditions, whereas (A) are preferentially-enriched in asynchronous and (M) in mitotic cells. ChIP-seq signals 2.5 kb upstream/downstream of peak centers are shown. D. Examples of H3K27ac tracks that belong in each of the A, B and M categories. E. mRNA levels of genes proximal to H3K27ac peaks that belong to each of the A, B or M categories. Published RNA-seq data from mouse ESCs (Shen et al. 2012) were used and standardized transcripts per million (TPM) were plotted. The most proximal gene to each peak was considered. Genes that were not assigned to any of the ChIP-seq peaks are shown as “Rest”. The expression levels of genes proximal to bookmarked H3K27ac sites were significantly higher compared to the other categories (p<0.001). F. Genomic distribution of each category of H3K27ac peaks relative to genes. Genome was partitioned into Proximal to TSS (including TSS and 1kb flanking regions), Gene bodies (excluding regions proximal to TSS) and Intergenic regions (rest of the genome). G–H. Top 10 Gene Ontology (GO) annotations enriched in bookmarked H3K27ac peaks that were either (G) proximal to TSS (<2.5kb) or (H) located >2.5 kb away from any TSS. I. Barplot showing the percentage of typical enhancers and super-enhancers that retain H3K27 acetylation during mitosis (Whyte et al., 2013).
Correlation of our H3K27ac ChIP-seq data with published RNA-seq data from asynchronous ESCs showed that genes proximal to B-class H3K27ac peaks displayed stronger transcriptional activity compared to the ones that lose or gain H3K27ac during mitosis (Figure 2E). When we analyzed the distribution of H3K27ac peaks that belong in each category across the genome, we noticed a clear overrepresentation of promoter-proximal peaks among B targets, while A and M targets were mostly located >2.5kb away from Transcriptional Start Sites (TSS), either within gene bodies or in intergenic regions (Figure 2F). Gene ontology analyses using GREAT (Genomic Regions Enrichment of Annotations Tool) (McLean et al., 2010) on the B-class H3K27ac peaks enriched for distinct functional annotations depending on their proximity to the TSS. Specifically, TSS-proximal peaks strongly enriched for genes involved in fundamental cellular processes, such as cell cycle progression and RNA and DNA metabolism (Figure 2G). On the other hand, distal B-class H3K27ac peaks (>2.5kb from TSS) were predominantly linked to stem cell maintenance-related genes (Figure 2H), including Nanog, Pou5f1 and Sox2. In fact, we observed that more than 50% of typical ESC-enhancers and more than 90% of the so-called super-enhancers (Whyte et al., 2013), which are linked to genes critical for stem cell identity, remained bookmarked by H3K27ac during mitosis (Figure 2I). These results, together, indicate that H3K27 acetylation during mitosis marks two distinct sets of genomic regions: (1) enhancers of stem cell-associated genes and (2) promoters of cell cycle-regulating genes.
To test whether the observed behavior of H3K27 acetylation during mitosis was characteristic of ESCs, we repeated the above analysis with published H3K27ac ChIP-seq dataset from G1E erythroblasts (Hsiung et al., 2016). G1E erythroblasts showed a similar degree of overlap between mitotic and asynchronous H3K27ac peaks as well as a conserved pattern of genomic distribution, with B-class peaks again predominantly located around promoters (not shown). Comparison of the ESC and G1E datasets revealed that a large proportion of bookmarked H3K27ac peaks were common among the different cell types (Figure S2A). GREAT analysis of the common bookmarked peaks showed a strong enrichment around promoters of genes that were mostly involved in cell cycle, RNA processing and ribosome and chromosome organization (Figures S2B–C). In contrast, ESC-specific and G1E–specific bookmarked H3K27ac peaks were mostly located away from promoters and were strongly associated with genes characteristic for each specific cell identity (Figures S2B–C). Examples are shown in Figure S2D. In agreement with these results, motif scanning analyses using either the ESC or the G1E bookmarked H3K27ac enhancer peaks enriched for binding sites recognized by the master regulators of each cell type, such as OSN (OCT4-SOX2-NANOG) and ESRRB or GATA1, respectively (Figure S2E and Supplemental Table 2). On the other hand, bookmarked promoter H3K27ac peaks in both cell types enriched for motifs for common general transcription factors involved in cell cycle regulation, including MeCP2 and E2F3. Taken together, these results propose that H3K27 acetylation is a previously unappreciated mitotic mark with dual bookmarking behavior on both promoters of cell cycle and homeostasis-related genes and enhancers of genes important for cell identity.
KLF4, SOX2 and OCT4 bookmark critical stem cell regulatory elements during mitosis
Previous studies have shown that bookmarking factors may associate with mitotic chromatin in a non-specific manner, coating the compacted chromosomes, or by specific binding at selected genomic sites (Caravaca et al., 2013; Kadauke and Blobel, 2013). To distinguish between these possibilities, we investigated the genome-wide binding patterns of the critical pluripotency regulators KLF4 (K), OCT4 (O) and SOX2 (S) (Apostolou and Hochedlinger, 2013; Young, 2011) in mitotic and asynchronous ESCs. Unbiased clustering of all ChIP-seq samples generated two distinct groups based on the TF they were targeting (KLF4 group and OCT4/SOX2 group), but failed to clearly separate mitotic from asynchronous samples, highlighting their global similarities (Figure S3A). Stringent differential analyses of the asynchronous and mitotic binding sites for each TF revealed a large number of common genomic targets showing that 25–60% of the asynchronous KOS peaks were maintained during mitosis (Bookmarked, B), whereas the rest were markedly decreased (Asynchronous-only, A) (Figures 3A and S3B). Examples of bookmarked or non-bookmarked genomic regions are shown in Figure 3B. Of note, genomic regions that were found enriched in mitotic samples (Mitotic-only, M) had overall lower and more variable signal among replicates and thus were excluded from downstream analyses.
Figure 3. ChIP-seq assays in mitotic and asynchronous ESCs identify the bookmarked genomic targets of KLF4, OCT4 and SOX2 (KOS).

A. Venn diagrams showing numbers of unique or common KOS-binding sites in asynchronous (A) and mitotic (M) mESCs. ChIP-seq peaks consistently detected in at least two of three biological replicates were used for the analyses (see Supplemental Material). The percent of asynchronous peaks that were also detected in mitotic cells (bookmarked) are reported in parentheses. B. Examples of genomic regions that retained (black outline) or lost (red outline) KOS binding during mitosis. Note that the extent of KOS retention was similar or higher compared to the previously reported bookmarking factor ESRRB (published datasets from Festuccia et al., 2016). C. Top 5 gene ontology categories (GO) enriched in KLF4 (blue), OCT4 (red) and SOX2 (green) binding sites that were either common between A and M (bookmarked) or present only in A (A-only). GOs associated with stem cell identity (bold) were overrepresented in bookmarked peaks. D. Venn diagram depicting the number of ESC-specific super-enhancers (SE) (Whyte et al., 2013) that remained bookmarked by individual TFs and combinations, highlighting the high frequency of combinatorial bookmarking. E. Barplot showing the percentage of KLF4, SOX2 or OCT4-bound typical enhancers (TE) and SE that remained bookmarked during mitosis.
In addition to the large number of KOS bookmarked genomic sites, the strength of mitotic retention (expressed as normalized ChIP-seq signal) in most of these sites was comparable to the respective signal in asynchronous ESCs (Figures 3B and S3B). This finding was surprising given that previously reported bookmarked targets by various TFs, including the recently described ESRRB and SOX2 in ESCs (Festuccia et al., 2016; Deluz et al, 2016), are often characterized by weaker binding during mitosis, suggesting either technical or biological differences between studies. To test the possibility that the strength of the detected signal in our mitotic samples was due to contamination from residual G2 cells (not more than 10% in our mitotic samples), we mixed interphase ESCs and neuronal progenitor cells (NPCs), which do not express KLF4, in a ratio of 10:90 and performed KLF4 ChIP-seq and ChIP-qPCR. Figures S3B–C confirmed that the signal from mitotic samples around bookmarked binding sites was much higher than the one from the ESC:NPC mixture, and was in fact similar to the respective signal in asynchronous ESCs. These results validated that KOS remain strongly bound on selected genomic sites during mitosis.
To investigate whether lost (A) or bookmarked (B) binding sites were linked to genes with distinct biological functions, we performed GREAT analysis. Genomic sites from which the TFs dissociated during mitosis mostly enriched for signaling, cell cycle and differentiation-associated genes. In contrast, genomic regions bookmarked by these TFs were proximal to genes involved in chromatin modification as well as stem cell maintenance and function (Figure 3C). Among them, 40–50% of typical stem cell-enhancers and more than 60% of super-enhancers (Whyte et al., 2014) remained bookmarked by one or more of the tested TFs (Figures 3D–E), suggesting an important role for maintenance of stem cell identity after cell division. Next, we performed motif analysis for each group of TF binding sites. For all three TFs, their specific DNA-binding motif was present in 90–95% of their respective bookmarked sites, suggesting that mitotic retention requires specific and direct binding (Supplementary Table 3). Of note, this percentage was only slightly lower (82–90%) among the rest of the target sites, indicating that solely the presence of the specific motif is not predictive of mitotic binding. In addition to OCT4, SOX2 and KLF4 motifs, a large number of other DNA sequence motifs scored as highly significant over background among bookmarked sites. After filtering out TFs that are minimally or not expressed in ESCs, we found a number of potentially relevant TF motifs that were preferentially enriched among the KLF4 bookmarked targets, such as sequences for E2F7, KLF3 and TFDP1 binding (Figure S3D). A distinct set of motifs was overrepresented among OCT4 and SOX2 bookmarked binding sites, including the composite OCT4/SOX2/NANOG (OSN) motif. Interestingly, all targets bookmarked by one or more of KOS had a higher frequency of ESRRB motif compared to the asynchronous- or mitotic-only binding sites. These results suggest that KOS binding during mitosis may be dependent on or facilitated by additional TFs. Comparison of our results with published ChIP-seq datasets demonstrated that the frequency of bookmarked target sites for OCT4 and SOX2 that coincide with binding of two or more additional TFs (ESRRB, NANOG, SOX2 and OCT4) was dramatically higher compared to the A-only sites (Figure S3E), further suggesting that TF synergy increases the likelihood and/or strength of binding during mitosis.
Finally, to gain insights into the interconnection between TFs and H3K27ac on mitotic bookmarking we performed enrichment analysis of the observed-over-expected overlap of bookmarked genomic regions (Figure S3F). This analysis revealed that H3K27ac-bookmarked enhancers preferentially enriched for cooperative mitotic binding by combinations of TFs, such as KOS and KS, whereas H3K27ac-bookmarked promoters preferentially enriched for KLF4 mitotic retention. These results provide evidence that distinct regulatory regions remain bookmarked by unique combinations of TFs and/or H3K27ac.
Temporal degradation of OCT4 during mitotic exit compromises its ability to maintain and induce pluripotency
Our findings support that ESC master regulators are largely retained on selective genomic regions during mitosis, suggesting a potential bookmarking function important for stem cell identity. To test this hypothesis, we focused on the master regulator OCT4 and established a system that has been previously shown to enable conditional protein degradation during mitotic exit (Kadauke et al., 2012). Specifically, we engineered a chimeric GFP-OCT4 protein (GFP-DB-OCT4 or GDO) which carries a destruction box (DB) of the mouse Cyclin B (Glotzer et al., 1991; Holloway et al., 1993), which is targeted for degradation by the Anaphase Promoting Complex (APC) during M-to-G1 transition (Figure 4A). As a control, we also generated a non-degradable version (GFP-DBmut-OCT4 or GMO) that carries a mutated DB sequence (R42A, DBmut), which has been shown to escape targeting by APC (Kadauke et al., 2012). By FACS analysis as well as live imaging, we confirmed that GMO-infected cells displayed stable GFP expression throughout the cell cycle, whereas in GDO-infected cells, GFP was rapidly lost during M-to-G1 transition, reappeared about 1h later and gradually accumulated at later stages of the cell cycle (Figure S4A–C and Supplemental Movies 2 and 3).
Figure 4. Temporal degradation of OCT4 during mitotic exit compromises its ability to maintain pluripotency.

A. Graphic illustration of our experimental strategy to test the effect of mitotic degradation of OCT4 on stem cell identity. The GFP-DB-OCT4 (GDO) chimeric protein carrying the destruction box (DB) of Cyclin B is degraded specifically during mitotic exit. In contrast, GFP-DBmut-OCT4 (GMO) carrying an R42A mutation (DBmut) remains unaffected (See also Figure S1A–S1C and Supplemental Movies 2–3). B. Quantitation of Western blot experiments (see Figure S4C) testing protein levels of selected stem cell regulators in ZHBTc4.1 cells infected with GDO or GMO, and either untreated or treated with doxycycline (dox). Gel bands quantitated by ImageJ were normalized to the uninfected no dox control for each protein, and then normalized to the level of exogenous OCT4 for each condition. Error bars indicate standard deviation of 2 technical replicates, and results from two independent experiments are shown. Asterisk indicate p-value<0.05, as calculated by t-test C. Barplot showing the quantity and quality of colonies formed by ZHBTc4.1 ESCs expressing either GMO or GDO in the presence of doxycycline. GFP positive cells were sorted and plated on feeders and colonies were scored 7–10 days later based either on their morphology or NANOG expression. The results of 3 independent experiments are summarized. T-test showed significantly (p-value<0.05) higher percentage of pluripotent-like colonies in the GMO expressing cells. Of note, uninfected ZHBTc4.1 fully differentiated in the presence of dox. D. Examples of pluripotent, partially differentiated and fully differentiated colonies based on their morphology (Bright field) and the presence of NANOG positive cells (red) as detected by immunofluorescence.
We then tested the relative ability of GDO and GMO chimeric proteins to preserve stemness when ectopically expressed in ZHBTc4.1 ESCs that rapidly silence endogenous OCT4 expression upon doxycycline treatment (Niwa et al., 2000). Western Blot analyses of total protein extracts validated that doxycycline induced efficient depletion of endogenous OCT4, while expression of the exogenous GDO and GMO chimeric proteins were not affected (Figure S4D). In agreement with previous reports, silencing of endogenous OCT4 resulted in abrupt differentiation of uninfected ZHBTc4.1 cells, as shown by the loss of critical stem cell regulators such as NANOG, SOX2 and ESRRB (Figure S4D), and by their inability to give rise to alkaline phosphatase (AP) positive colonies (Figure S4E). Although both GDO and GMO expression were able to ameliorate these phenotypes, GDO-infected ESCs were impaired in maintaining sufficient mRNA and protein expression of stem cell regulators upon endogenous OCT4 depletion (Figures 4B, S4D and S4F). In agreement, scoring of ESC colonies based either on their morphology or NANOG expression showed that GDO expressing cells formed a significantly higher percentage of partially or completely differentiated colonies upon doxycycline treatment compared to the GMO-expressing cells (Figures 4C–D). Taken together, these results suggest that temporal degradation of OCT4 during mitotic exit compromises its ability to maintain pluripotency.
We next investigated the role of OCT4 mitotic bookmarking on reprogramming somatic cells to induced pluripotent stem cells (iPSCs) by overexpressing GDO or GMO along with wild type SOX2, KLF4 and cMYC (Sommer et al., 2009) in mouse embryonic fibroblasts (MEFs) (Figure 5A and S5A). Cells expressing GDO protein were deficient in upregulating the early pluripotency marker SSEA-1 compared to the GMO-expressing ones (Figure S5B). Consistently, GDO-KSM reprogrammed MEFs generated substantially reduced numbers of iPSC colonies, as assessed by ESC-like morphology (Figure 5B), alkaline phosphatase staining and NANOG expression (Figures S5C–D), compared to the ones expressing GMO or wild type OCT4. Of note, immunofluorescence experiments on metaphase chromosome spreads from OKSM MEFs undergoing reprogramming revealed persistent binding of the ectopic OCT4, SOX2 and KLF4 factors on the condensed chromosomes, and exclusion of RNA PolII (Figure 5C). Collectively, these results demonstrate the capacity of OCT4, SOX2 and KLF4 to bind on mitotic chromatin of both ESCs and somatic cells and provide evidence that bookmarking is also important for induction of pluripotency.
Figure 5. Temporal degradation of OCT4 during mitotic exit compromises its ability to induce pluripotency.

A. Experimental strategy to test the relative ability of GDO and GMO to induce pluripotency when overexpressed together with SOX2, KLF4 and cMYC in Mouse Embryonic Fibroblasts (MEFs). Wild type OCT4 was used as a positive control. B. Quantitation of the induced pluripotent stem cell (iPSC) colonies that were generated upon reprogramming with the respective construct. The number of colonies was normalized to the infection efficiency as described in the supplemental experimental procedures. Three independent experiments are shown. C. Representative immunofluorescence photos of metaphase chromosomes from MEFs expressing OKSM after doxycycline induction. OCT4, SOX2 and KLF4 were efficiently detected on the mitotic chromosomes, whereas RNA Polymerase II (PolII) was excluded from the condensed chromosomes. Matched IgG isotypes (goat for KOS and rabbit for PolII) were used as negative controls.
DISCUSSION
Our study revealed a widespread yet specific retention of selected histone modifications and stem cell regulators on mitotic chromatin of ESCs, arguing that extensive chromatin and TF bookmarking might ensure faithful propagation of pluripotency.
Among the highly retained histone modifications that we observed, H3K27ac was particularly intriguing, since histone acetylation levels were overall decreased during mitosis, in concordance with reduced transcriptional activity. Given that H3K27ac is strongly linked to enhancer activity and cell-type specificity, we considered it an attractive bookmarking candidate for propagation of cell identity. Interestingly, analysis of H3K27ac ChIP-seq data from ESCs as well as erythroblasts revealed a bookmarking preference for both cell-type specific enhancers and housekeeping promoters. Whether the preferential mitotic retention of H3K27ac represents a residual mark of high transcriptional activity during interphase or an actual bookmarking mechanism that ensures rapid transcriptional reactivation of essential genes during mitotic exit remains to be determined. In support of the latter hypothesis, presence of H3K27ac in mitotic erythroblasts has been correlated with increased transcriptional activity during early G1 (Hsiung et al., 2016). However, more definitive studies will be required to interrogate the significance of this mark in transcriptional memory.
In addition to the H3K27ac-bookmarked targets, we also identified distinct subsets of genomic regions that lost or gained this histone mark during mitosis, suggesting that mechanisms for deposition and removal of this histone modification may be at least partially active during mitosis or mitotic entry. HDACs have been reported to play crucial roles in cell cycle progression through mitosis (Li et al., 2006; Cimini et al, 2003), resulting in decreased histone acetylation levels, and so the loss of H3K27ac peaks was not unexpected. In contrast, the de novo acetylation of distinct genomic sites during mitosis is puzzling. A potential explanation would be that these mitotic-specific enhancer elements play a role in maintaining or inducing transcriptional activity of selected genes during mitosis, a possibility that could be addressed by nascent RNA sequencing experiments. Of note, although global transcriptional shut down during mitosis is well documented, an increasing number of studies report subsets of genes that “escape” this silencing (Halley-Stott et al., 2014; Liang et al., 2015; Liu, et al., 2017; Sciortino et al., 2001).
Previous studies in somatic cells have reported that most TFs and cofactors dissociate from mitotic chromatin, except for a few bookmarking factors (Martinez-Balbaz, 1995; Kadauke and Blobel, 2013). Here, using multiple independent experimental approaches, we report that a large number of stem cell regulators, including the recently described ESRRB and SOX2 (Festuccia et al, 2016; Deluz et al; 2016), were largely retained on mitotic chromatin of ESCs. Interestingly, in contrast with these studies, our data revealed a strong, widespread-yet-specific binding of KOS during mitosis. Biological and/or technical differences, such as synchronization and immunoprecipitation efficiencies, may explain these discrepancies. Notably, our findings are in agreement with a recently proposed model that TF retention on mitotic chromatin of ESCs may be a much broader phenomenon than previously appreciated (Teves et al., 2016). Whether this phenomenon is characteristic of pluripotent stem cell chromatin or is also present in other fast-cycling progenitor or even differentiated cells remains to be tested. Global proteomics assays in various cell types could enable determination of the relative chromatin composition of mitotic and interphase cells and give a better understanding of the key players and principles that govern this time window.
Importantly, despite the widespread mitotic retention we observed, specific TFs, such as NANOG and REX1, were mostly dissociated from mitotic chromatin. The biological significance and underlying mechanisms of retaining or excluding specific TFs from mitotic chromatin warrant further investigation. Interestingly, all of the TFs that remained bound on mitotic chromatin have been reported to either have histone-like properties (UTF1) or function as pioneer factors (OCT4, SOX2, KLF4 and ESRRB) (Soufi et al., 2012; Soufi et al., 2015), suggesting that intrinsic DNA-binding properties -in addition to TF cooperativity- may determine the bookmarking potential of each protein. In agreement, our experiments showed that ectopically expressed KLF4 in blastomeres and reprogramming TFs in MEFs were able to access mitotic chromatin. However, the bookmarking capacity and specificity of defined TFs in different cellular contexts still need to be determined.
The high degree and specificity of mitotic retention of KLF4, OCT4 and SOX2 we observed in ESCs strongly supports a potential bookmarking role for faithful inheritance of stemness in daughter cells. Indeed, our functional assays confirmed that temporal degradation of OCT4 during G1 entry induced destabilization of stem cell identity and unscheduled differentiation. Although we cannot exclude that the slow recovery of OCT4 protein levels (3h until full recovery, Figure S4C) may also account for the observed phenotypes, our results highlight the importance of this time window for OCT4 to safeguard pluripotency. Moreover, we showed that OCT4 presence during mitotic exit was also critical for efficient reprogramming of somatic cells into iPSCs. Elegant experiments in the past have shown that cell division is required for successful TF-mediated reprogramming, proposing that transient silencing of the somatic-specific transcriptional program and dissociation of the lineage-specifying factors during mitosis provide a unique chance for a new cell fate to arise (Egli et al., 2008; Egli et al., 2011; Koche et al., 2011). Our findings suggest that active mitotic binding of reprogramming factors may facilitate overwriting somatic cell identity.
Mitosis is believed to represent a temporal crisis of cell identity and a window of opportunity for cell fate transitions. Multiple studies have shown that during mitotic exit and G1 entry ESCs are the most susceptible to environmental cues that induce lineage specification (Halley-Stott et al, 2012; Pauklin and Vallier, 2013; Dalton, 2015; Pauklin et al, 2016). Therefore, understanding the mechanisms that govern this critical cell-cycle window will enable the identification of molecular levers that could be used to shift the balance of self-renewal/differentiation to promote a certain lineage. Our results constitute an important step towards this direction by revealing patterns and basic principles of mitotic occupancy for critical chromatin marks and stem cell regulators.
EXPERIMENTAL PROCEDURES
See Supplemental Material for details
Cell Culture
V6.5, ZHBTc4.1 (Niwa et al., 2000) and H2B–mCherry expressing ESCs were cultured on irradiated feeder cells in KO-DMEM (Invitrogen) supplemented with Glutamax, pen-strep, nonessential amino acids, β-mercaptoethanol, 1000 U/ml LIF and 15% heat-inactivated fetal bovine serum.
Mitotic arrest and Subcellular fractionation
ESCs were passaged on gelatinized plates the day before synchronization. Nocodazole (200ug/ml) was added to the medium for 7hr prior to collection by mitotic shake-off for Western blot and ChIP-seq assays. Synchronization efficiency was determined by FACS analysis on ethanol-fixed cells stained with H3Ser10p and DAPI. Chromatin-associated extracts were prepared using a subcellular protein fractionation kit (ThermoScientific, #78840) following manufacturer’s instructions.
ChIP-seq assays
ChIP was performed as described previously (Apostolou et al, 2013). Related antibodies and primers are listed in Supplemental Tables 1 and 4. Library construction and ChIP-seq analysis are described in the Supplemental Material.
DNA constructs, infections and functional assays
Turbo GFP followed by either the Cyclin B destruction box (DB) (13–90aa) or a mutated R24A version (DBmut) was cloned in frame with Oct4 into: (i) the pHAGE-Ef1a vector to generate GDO or GMO constructs or (ii) the pHAGE-tetO-Stemcca (Sommer et al., 2009) vector to generate the GDO-KSM Fand GMO constructs and and GMO-KSM constructs. Virus production and transduction was performed as described previously (Apostolou et al, 2013).
ZHBTc4.1 ESCs either uninfected or infected with GDO or GMO were plated on gelatin without feeders in presence or absence of dox (2ug/ml) in regular ESC medium (+LIF) or in the absence of LIF (−LIF). Cells were then collected for western blot analyses, qRT-PCR or colony formation and scoring as described in Supplemental Material.
Mouse Embryonic Fibroblasts (MEFs) carrying the Rosa26-rtTA allele were infected with GDO-KSM, GMO-KSM and then reprogrammed as described before (Stadtfeld, Apostolou et al, 2012).
Statistical analyses
Two-sample two-sided t-test or Wilcoxon test were used to calculate the reported p values in Figures 1E, 4B and S4F or 2E, respectively.
Supplementary Material
Acknowledgments
We are grateful to Todd Evans, Ritu Kumar, Matthias Stadtfeld and Konrad Hochedlinger for critical reading of the manuscript. We also want to thank Ari Melnick and Anja Fischer for antibodies and Minjung Kang for help with confocal imaging. B.P. is supported by a Medical Scientist Training Program grant from the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under award number T32GM007739 to the Weill Cornell/Rockefeller/Sloan Kettering Tri-Institutional MD-PhD Program. DCG is supported by the New York Stem Cell Foundation and the Family-Friendly Postdoctoral Initiative at Weill Cornell Medicine. This work was funded by the NIH Director’s New Innovator Award (DP2DA043813), the Edward Jr Mallinckrodt Foundation and the Tri-Institutional Stem Cell Initiative by the Starr Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
All ChIP-seq data reported in this paper have been deposited to GEO under the accession number GSE92846.
AUTHOR CONTRIBUTION
EA conceived, designed and supervised the study, performed experiments and wrote the manuscript with help from all authors. YL performed all the bioinformatics analyses under the supervision of OE and EA and with input from AD. BPW and DK performed fractionation and western blot analyses. BPW performed IF on metaphase spreads. DCG performed ChIP-seq experiments and validations. BPW and JL generated the GDO and GMO constructs and performed the functional analyses for the maintenance and induction of pluripotency, respectively. DCG and KK performed live imaging of GDO and GMO expressing ESCs with guidance from PG, while NS and VG performed the microinjections and live imaging of blastomeres with guidance from AKH.
References
- Apostolou E, Hochedlinger K. Chromatin dynamics during cellular reprogramming. Nature. 2013;502:462–471. doi: 10.1038/nature12749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apostolou E, Ferrari F, et al. Genome-wide chromatin interactions of the Nanog locus in pluripotency, differentiation, and reprogramming. Cell Stem Cell. 2013;12:699–712. doi: 10.1016/j.stem.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burke LJ, Zhang R, Bartkuhn M, Tiwari VK, Tavoosidana G, Kurukuti S, Weth C, Leers J, et al. CTCF binding and higher order chromatin structure of the H19 locus are maintained in mitotic chromatin. The EMBO journal. 2005;24:3291–3300. doi: 10.1038/sj.emboj.7600793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravaca JM, Donahue G, Becker JS, He X, Vinson C, Zaret KS. Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes & development. 2013;27:251–260. doi: 10.1101/gad.206458.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimini D, Mattiuzzo M, Torosantucci L, Degrassi F. Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Molecular biology of the cell. 2003;14:3821–3833. doi: 10.1091/mbc.E03-01-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronado D, Godet M, Bourillot PY, Tapponnier Y, Bernat A, Petit M, Afanassieff M, Markossian S, Malashicheva A, Iacone R, et al. A short G1 phase is an intrinsic determinant of naive embryonic stem cell pluripotency. Stem cell research. 2013;10:118–131. doi: 10.1016/j.scr.2012.10.004. [DOI] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:21931–21936. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalton S. Linking the Cell Cycle to Cell Fate Decisions. Trends in cell biology. 2015;25:592–600. doi: 10.1016/j.tcb.2015.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deluz C, Friman ET, Strebinger D, Benke A, Raccaud M, Callegari A, Leleu M, Manley S, Suter DM. A role for mitotic bookmarking of SOX2 in pluripotency and differentiation. Genes & development. 2016;30:2538–2550. doi: 10.1101/gad.289256.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli D, Birkhoff G, Eggan K. Mediators of reprogramming: transcription factors and transitions through mitosis. Nature reviews Molecular cell biology. 2008;9:505–516. doi: 10.1038/nrm2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egli D, Chen AE, Saphier G, Ichida J, Fitzgerald C, Go KJ, Acevedo N, Patel J, et al. Reprogramming within hours following nuclear transfer into mouse but not human zygotes. Nat Commun. 2011;2:488. doi: 10.1038/ncomms1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans M. Discovering pluripotency: 30 years of mouse embryonic stem cells. Nature reviews Molecular cell biology. 2011;12:680–686. doi: 10.1038/nrm3190. [DOI] [PubMed] [Google Scholar]
- Festuccia N, Dubois A, Vandormael-Pournin S, Gallego Tejeda E, Mouren A, Bessonnard S, Mueller F, Proux C, Cohen-Tannoudji M, Navarro P. Mitotic binding of Esrrb marks key regulatory regions of the pluripotency network. Nat Cell Biol. 2016;18:1139–1148. doi: 10.1038/ncb3418. [DOI] [PubMed] [Google Scholar]
- Glotzer M, Murray AW, Kirschner MW. Cyclin is degraded by the ubiquitin pathway. Nature. 1991;349:132–138. doi: 10.1038/349132a0. [DOI] [PubMed] [Google Scholar]
- Gottesfeld JM, Forbes DJ. Mitotic repression of the transcriptional machinery. Trends in biochemical sciences. 1997;22:197–202. doi: 10.1016/s0968-0004(97)01045-1. [DOI] [PubMed] [Google Scholar]
- Halley-Stott RP, Jullien J, Pasque V, Gurdon J. Mitosis gives a brief window of opportunity for a change in gene transcription. PLoS Biol. 2014;12:e1001914. doi: 10.1371/journal.pbio.1001914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins RD, Hon GC, Lee LK, Ngo Q, Lister R, Pelizzola M, Edsall LE, Kuan S, Luu Y, Klugman S, et al. Distinct epigenomic landscapes of pluripotent and lineage-committed human cells. Cell Stem Cell. 2010;6:479–491. doi: 10.1016/j.stem.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway SL, Glotzer M, King RW, Murray AW. Anaphase is initiated by proteolysis rather than by the inactivation of maturation-promoting factor. Cell. 1993;73:1393–1402. doi: 10.1016/0092-8674(93)90364-v. [DOI] [PubMed] [Google Scholar]
- Hsiung CC, Bartman CR, Huang P, Ginart P, Stonestrom AJ, Keller CA, Face C, Jahn KS, et al. A hyperactive transcriptional state marks genome reactivation at the mitosis-G1 transition. Genes & development. 2016;30:1423–1439. doi: 10.1101/gad.280859.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi-Doi M, Zaret KS. Pioneer transcription factors in cell reprogramming. Genes & development. 2014;28:2679–2692. doi: 10.1101/gad.253443.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadauke S, Blobel GA. Mitotic bookmarking by transcription factors. Epigenetics & chromatin. 2013;6:6. doi: 10.1186/1756-8935-6-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadauke S, Udugama MI, Pawlicki JM, Achtman JC, Jain DP, Cheng Y, Hardison RC, Blobel GA. Tissue-specific mitotic bookmarking by hematopoietic transcription factor GATA1. Cell. 2012;150:725–737. doi: 10.1016/j.cell.2012.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koche RP, Smith ZD, Adli M, Gu H, Ku M, Gnirke A, Bernstein BE, Meissner A. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell Stem Cell. 2011;8:96–105. doi: 10.1016/j.stem.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooistra SM, Thummer RP, Eggen BJ. Characterization of human UTF1, a chromatin-associated protein with repressor activity expressed in pluripotent cells. Stem cell research. 2009;2:211–218. doi: 10.1016/j.scr.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Kouskouti A, Talianidis I. Histone modifications defining active genes persist after transcriptional and mitotic inactivation. The EMBO journal. 2005;24:347–357. doi: 10.1038/sj.emboj.7600516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Kao GD, Garcia BA, Shabanowitz J, Hunt DF, Qin J, Phelan C, Lazar MA. A novel histone deacetylase pathway regulates mitosis by modulating Aurora B kinase activity. Genes & development. 2006;20:2566–2579. doi: 10.1101/gad.1455006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Chen S, Wang S, Soares F, Ficher M, et al. Transcriptional landscape of the human cell cycle. PNAS. 2017 doi: 10.1073/pnas.1617636114. Early edition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang K, Woodfin AR, Slaughter BD, Unruh JR, Box AC, Rickels RA, Gao X, Haug JS, Jaspersen SL, Shilatifard A. Mitotic Transcriptional Activation: Clearance of Actively Engaged Pol II via Transcriptional Elongation Control in Mitosis. Molecular cell. 2015;60:435–445. doi: 10.1016/j.molcel.2015.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nature reviews Genetics. 2010;11:285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Balbas MA, Dey A, Rabindran SK, Ozato K, Wu C. Displacement of sequence-specific transcription factors from mitotic chromatin. Cell. 1995;83:29–38. doi: 10.1016/0092-8674(95)90231-7. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nature biotechnology. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natoli G. Maintaining cell identity through global control of genomic organization. Immunity. 2010;33:12–24. doi: 10.1016/j.immuni.2010.07.006. [DOI] [PubMed] [Google Scholar]
- Niwa H, Miyazaki J, Smith AG. Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat Genet. 2000;24:372–376. doi: 10.1038/74199. [DOI] [PubMed] [Google Scholar]
- Pauklin S, Vallier L. The cell-cycle state of stem cells determines cell fate propensity. Cell. 2013;155:135–147. doi: 10.1016/j.cell.2013.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauklin S, Madrigal P, Bertero A, Vallier L. Initiation of stem cell differentiation involves cell cycle-dependent regulation of developmental genes by Cyclin D. Genes & development. 2016;30:421–433. doi: 10.1101/gad.271452.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Tedeschi A, Schmitz J. The cohesin complex and its roles in chromosome biology. Genes & development. 2008;22:3089–3114. doi: 10.1101/gad.1724308. [DOI] [PubMed] [Google Scholar]
- Savatier P, Lapillonne H, Jirmanova L, Vitelli L, Samarut J. Analysis of the cell cycle in mouse embryonic stem cells. Methods in molecular biology. 2002;185:27–33. doi: 10.1385/1-59259-241-4:27. [DOI] [PubMed] [Google Scholar]
- Sciortino S, Gurtner A, Manni I, Fontemaggi G, Dey A, Sacchi A, Ozato K, Piaggio G. The cyclin B1 gene is actively transcribed during mitosis in HeLa cells. EMBO Rep. 2001;2:1018–1023. doi: 10.1093/embo-reports/kve223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J, Kim TW, Kim H, Kim HJ, Suh MY, Lee S, Lee HT, Kwak S, Lee SE, Lee JH, et al. Aurkb/PP1-mediated resetting of Oct4 during the cell cycle determines the identity of embryonic stem cells. eLife. 2016;5:e10877. doi: 10.7554/eLife.10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer CA, Stadtfeld M, Murphy GJ, Hochedlinger K, Kotton DN, Mostoslavsky G. Induced pluripotent stem cell generation using a single lentiviral stem cell cassette. Stem Cells. 2009;27:543–549. doi: 10.1634/stemcells.2008-1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Donahue G, Zaret KS. Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell. 2012;151:994–1004. doi: 10.1016/j.cell.2012.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, Zaret KS. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell. 2015;161:555–568. doi: 10.1016/j.cell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadtfeld M, Apostolou E, Ferrari F, Choi J, Walsh RM, Chen T, Ooi SS, Kim SY, Bestor TH, Shioda T, et al. Ascorbic acid prevents loss of Dlk1-Dio3 imprinting and facilitates generation of all-iPS cell mice from terminally differentiated B cells. Nat Genet. 2012;44:398–405. doi: 10.1038/ng.1110. S391–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabar V, Studer L. Pluripotent stem cells in regenerative medicine: challenges and recent progress. Nature reviews Genetics. 2014;15:82–92. doi: 10.1038/nrg3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- Tapia C, Kutzner H, Mentzel T, Savic S, Baumhoer D, Glatz K. Two mitosis-specific antibodies, MPM-2 and phospho-histone H3 (Ser28), allow rapid and precise determination of mitotic activity. The American journal of surgical pathology. 2006;30:83–89. doi: 10.1097/01.pas.0000183572.94140.43. [DOI] [PubMed] [Google Scholar]
- Teves SS, An L, Hansen AS, Xie L, Darzacq X, Tjian R. A dynamic mode of mitotic bookmarking by transcription factors. eLife. 2016;5 doi: 10.7554/eLife.22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valls E, Sanchez-Molina S, Martinez-Balbas MA. Role of histone modifications in marking and activating genes through mitosis. The Journal of biological chemistry. 2005;280:42592–42600. doi: 10.1074/jbc.M507407200. [DOI] [PubMed] [Google Scholar]
- van den Boom V, Kooistra SM, Boesjes M, Geverts B, Houtsmuller AB, Monzen K, Komuro I, Essers J, Drenth-Diephuis LJ, Eggen BJ. UTF1 is a chromatin-associated protein involved in ES cell differentiation. The Journal of cell biology. 2007;178:913–924. doi: 10.1083/jcb.200702058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Higgins JM. Histone modifications and mitosis: countermarks, landmarks, and bookmarks. Trends in cell biology. 2013;23:175–184. doi: 10.1016/j.tcb.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153:307–319. doi: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DW, Hassan MQ, Pratap J, Galindo M, Zaidi SK, Lee SH, Yang X, Xie R, Javed A, et al. Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature. 2007;445:442–446. doi: 10.1038/nature05473. [DOI] [PubMed] [Google Scholar]
- Young RA. Control of the embryonic stem cell state. Cell. 2011;144:940–954. doi: 10.1016/j.cell.2011.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SK, Young DW, Montecino MA, Lian JB, van Wijnen AJ, Stein JL, Stein GS. Mitotic bookmarking of genes: a novel dimension to epigenetic control. Nature reviews Genetics. 2010;11:583–589. doi: 10.1038/nrg2827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, Carroll JS. Pioneer transcription factors: establishing competence for gene expression. Genes & development. 2011;25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.