

Abstract
The use of programmable meganucleases is transforming genome editing and functional genomics. CRISPR/Cas9 was developed such that targeted genomic lesions could be introduced in vivo with unprecedented ease. In the presence of homology donors, these lesions facilitate high-efficiency precise genome editing (PGE) via homology-directed repair (HDR) pathways. However, the identity and hierarchy of the HDR (sub)pathways leading to the formation of PGE products remain elusive. Here, we established a green to blue fluorescent protein conversion system to systematically characterize oligodeoxynucleotide (ODN)-mediated PGE using Cas9 and its nickase variants in human cells. We demonstrate that, unlike double-stranded DNA (dsDNA) donors with central heterologies, ODNs generated short conversion tracts with Gaussian-like distributions. Interestingly, single-nick–induced PGE using ODN donors produced conversion tracts biased either mostly uni- or bidirectional depending on the relative strandedness of the ODNs and the nick. Moreover, the ODNs were physically incorporated into the genome only in the bidirectional, but not in the unidirectional, conversion pathway. In the presence of double-stranded genomic lesions, the unidirectional conversion pathway was preferentially utilized even though the knock-in mutation could theoretically have been converted by both pathways. Collectively, our results suggest that ODN-mediated PGE utilizes synthesis-dependent strand annealing and single-stranded DNA incorporation pathways. Both of these pathways generate short conversion tracts with Gaussian-like distributions. Although synthesis-dependent strand annealing is preferentially utilized, our work unequivocally establishes the existence of a single-stranded DNA incorporation pathway in human cells. This work extends the paradigms of HDR-mediated gene conversion and establishes guidelines for PGE in human cells.
Genome editing is the intentional alteration of the genetic information in living cells or organisms. Since Clustered Regularly Interspaced Short Palindromic Repeats/CRISPR-associated 9 (CRISPR/Cas9) (Jinek et al. 2012) was repurposed for genome editing (Cong et al. 2013; DiCarlo et al. 2013; Jinek et al. 2013), the speed of CRISPR methodology is quickly bridging the genotype and phenotype worlds and facilitating high-throughput reverse genetic studies (Wright et al. 2016). In the CRISPR age, targeted genomic lesions can be introduced with unprecedented ease, which facilitate either high-efficiency gene disruption by nonhomologous end joining (NHEJ) or, in the presence of donor DNA, precise genome editing (PGE) by homology-directed repair (HDR) (Carroll 2014). Research models of the desired genotype can be expeditiously generated for most genetically tractable organisms (Bassett et al. 2013; DiCarlo et al. 2013; Liu et al. 2014; Yang et al. 2014; Reardon 2016) and within one round of subcloning of cultured cells (Ran et al. 2013b; Byrne et al. 2014). Although HDR is the more desirable pathway for genome editing and gene therapy, its activity is much lower than NHEJ and dependent on the cell cycle status in human cells (Mao et al. 2008a,b; Lin et al. 2014; Orthwein et al. 2015). Thus, marker-free PGE still requires the laborious screening of hundreds of subclones, especially in hard-to-transfect cells or those with low HDR activity or limited tolerance of genomic lesions. Moreover, bi-allelic and multiplexed PGE is still challenging in human cells.
Complicating our understanding of PGE is the fact that HDR has multiple subpathways in eukaryotic cells (San Filippo et al. 2008). Four of these subpathways can lead to PGE without significantly sacrificing genomic stability: the double-strand break repair (DSBR, aka, Holliday junction resolution) pathway (Orr-Weaver et al. 1981; Orr-Weaver and Szostak 1983), Holliday junction dissolution (Wu and Hickson 2003; Bizard and Hickson 2014; Swuec and Costa 2014), synthesis-dependent strand annealing (SDSA) (Strathern et al. 1982; Hastings 1988; Larocque and Jasin 2010), and single-strand DNA incorporation (ssDI, also sometimes referred to as single-strand assimilation) (Radecke et al. 2006b; Storici et al. 2006; Davis and Maizels 2014) pathways (Supplemental Fig. S1). PGE is defined by the fraction of HDR events leading to the conversion of desired knock-in mutations using exogenous homology donors. Importantly, the identity and hierarchy of usage of the HDR subpathways leading to the formation of PGE products remains a knowledge barrier that hinders the improvement of PGE.
In a previous study, we demonstrated that in human somatic cells the introduction of large knock-in mutations with double-strand DNA (dsDNA) donors (including transfected plasmids or viruses with dsDNA intermediates) preferentially engages the DSBR subpathway for PGE (Kan et al. 2014). Although oligodeoxynucleotides (ODNs) probably do not serve as natural HDR donors, many laboratories have reported that PGE can be routinely obtained by exogenously transfecting ODN donors into cells (Chen et al. 2015; Richardson et al. 2016). Since ODN donors are likely unable to physically form Holliday junctions and therefore cannot engage DSBR, they must perforce utilize some form of the SDSA or ssDI pathways during PGE (Storici et al. 2006; Davis and Maizels 2014). In the absence of a targeted mega-nuclease like Cas9, the ODN donors may use spontaneously occurring genomic lesions in order to perform PGE. Indeed, it was proposed that ODNs could be incorporated into the gaps generated by nucleotide excision repair (Faruqi et al. 2000; Kuan and Glazer 2004) or lagging strand synthesis during DNA replication (Ferrara and Kmiec 2004; Wu et al. 2005; Huen et al. 2006), although the specific mechanisms are still disputed (Suzuki 2008; Jensen et al. 2011). Interestingly, both of these models involve the physical incorporation of the ODNs into the genome via an ssDI-like mechanism; a prediction that was later supported by experiments carried out by Radecke et al. (2006b). Confusingly, however, the same group of authors also demonstrated that ODNs were not physically incorporated into the genome during double-strand break (DSB)-induced PGE, suggesting the existence of an alternative mechanism(s) (Radecke et al. 2006a). Finally, in the presence of mega-nuclease–induced targeted single-strand nicks, it was proposed by Davis and Maizels that certain forms of the SDSA and the ssDI pathways could be utilized depending on the strandedness of the donor ODNs (Davis and Maizels 2011, 2014), a hypothesis that was supported by recent work from the Corn laboratory (Richardson et al. 2016). However, the exact mechanisms, conversion tracts, and hierarchy of these pathways have remained elusive.
Results
The enhanced green fluorescent protein to blue fluorescent protein conversion system
To systematically investigate the molecular mechanisms of mega-nuclease–induced PGE using ODN donors, we established the enhanced green fluorescent protein (EGFP) to blue fluorescent protein (BFP) conversion system (Fig. 1A). This system takes advantage of the sequence similarity of EGFP and BFP. By introducing T65S and Y66H mutations into the chromophore domain, EGFP can be completely converted into BFP (Katada et al. 2008). To this end, using recombinant adeno-associated virus (rAAV)-mediated PGE (Khan et al. 2011; Kan et al. 2014), we introduced the CMV-EGFP-pA expression cassette into the hypoxanthine phosphoribosyltransferase 1 (HPRT1) locus on the X Chromosome of human HCT116 cells, in both sense (S) and antisense (AS) directions with respect to the direction of the HPRT1 gene (Supplemental Fig. S2A,B). Targeted integration in either direction results in mono-allelic EGFP expression and the inactivation of the HPRT1 gene. After rAAV infection, the HPRT1-negative cells were enriched using 6-thioguanine selection, and individual subclones with bright EGFP expression were screened using designated primers (Supplemental Fig. S2A,B). The EGFP cassette was precisely knocked into the HPRT1 gene in one out of four subclones in the sense orientation and two out of four subclones in the antisense orientation (Supplemental Fig. S2C,D). One individual subclone from each orientation was designated as HPRT1-EGFP (S) and (AS) cell lines, respectively, and used for subsequent studies. Targeted genomic lesions such as nicks and DSBs were subsequently introduced into these reporter cell lines within the chromophore sequence using Cas9 or nCas9 from Streptococcus pyogenes and sgRNAs in both S and AS orientations with respect to EGFP (Fig. 1A). In the presence of ODN donors bearing the SH mutations (ACCTAC to TCTCAT), EGFP could efficiently be converted into BFP, which generated distinguishable spectra under flow cytometry (Supplemental Fig. S3B,C).
Figure 1.

The EGFP to BFP conversion system in the HPRT1-EGFP antisense cell line. (A) Schematics of the EGFP to BFP conversion system in the HPRT1-EGFP antisense cell line. An EGFP expression cassette was knocked into the HPRT1 locus of the HCT116 cell line (only the antisense orientation is diagrammed here for the sake of ease of presentation). Genomic lesions could then be introduced near the chromophore (TY residues) of the EGFP sequence using CRISPR/Cas9 cleavase, nickases, or dual nickases. HDR repair of the genomic lesions using ODN donors containing the sequence of the SH residues leads to the conversion of EGFP to BFP. The ODN donors also contained synonymous SNPs that could be coconverted with the sequence of the SH residues. Schematic elements: EGFP, green boxes; BFP, blue boxes; HPRT1, inverted yellow boxes; sequences of the chromophore residues, TY and SH; the cytomegalovirus promoter, CMV; the polyadenylation sequence, pA; Cas9 variants, red ovals with scissors; genomic lesions, lightning bolts; ODN donors, horizontal red lines; synonymous SNPs, vertical blue hash marks. (B) The efficiency of BFP conversion in the HPRT1-EGFP antisense cell line. The wild-type Cas9 and D10A variant are labeled as Cas9 and nCas9, respectively. The strandedness (S, sense; AS, antisense) of the sgRNA and ODNs (for this experiment without synonymous SNPs) are labeled with respect to the coding sequence of EGFP. Note that the Cas9 D10A variant nicks the strand complementary to that of the sgRNA. All data are shown as the mean ± SEM of three biological replicates.
ODNs stimulate efficient PGE in the presence of genomic lesions
We first compared the efficiency of ODN-mediated PGE using nCas9 with ODNs containing the SH mutation and 77 bp homology arms (BFP_S160 and BFP_AS160) (see Supplemental Fig. S10 for more details) and sgRNAs in both S and AS orientations with respect to EGFP. By transfecting the HPRT1-EGFP cell lines with the nCas9 and sgRNA expression vectors and ODN donors all in a DNA format, we could routinely induce efficient EGFP to BFP conversion as quantitated using flow cytometry (Fig. 1; Supplemental Fig. S3). In contrast, the omission of the Cas9 and/or sgRNA resulted in <0.01% conversion into blue cells (Supplemental Fig. S4). Interestingly (see the Discussion as well), the conversion frequency in the HPRT1-EGFP (AS) cell line was approximately two to four times higher than the conversion frequency in the HPRT1-EGFP (S) cell line (cf. Fig. 1 and Supplemental Fig. S3). Because the HPRT1-EGFP (AS) cell line consistently generated a higher BFP conversion efficiency, this cell line was chosen for subsequent studies. In contrast to the bias observed with HPRT1-EGFP orientation, the difference in conversion efficiency between any of the four possible sgRNA and ODN pair configurations was not as pronounced (Fig. 1B, white bars). Thus, no significant strand bias for either the nick or the ODN donor was observed, and this was also true for the HPRT1-EGFP (S) cell line (Supplemental Fig. S3).
We next examined the efficiency of ODN-mediated PGE in the presence of double-stranded genomic lesions in the HPRT1-EGFP (AS) cell line. Wild-type Cas9 cleavase generates double-stranded blunt ends (Garneau et al. 2010; Gasiunas et al. 2012; Jinek et al. 2012; Cho et al. 2013). nCas9 paired-nickases generate juxtaposed, offset nicks, and it has been proposed that chromosomes with offset nicks may dissociate into DSBs with sticky overhangs (Ran et al. 2013a; Cho et al. 2014; Shen et al. 2014). Depending on the orientations of the protospacer adjacent motif (PAM) sequences, paired-nickases are generally classified into PAM-out and PAM-in configurations, which generate 5′ and 3′ overhangs, respectively. When ODNs were used as homology donors, all of the double-stranded genomic lesions induced PGE with consistently higher efficiency (approximately twofold) than single nicks (Fig. 1B, cf. filled bars and white bars). The only exception to this observation was in the case of the dual nickases utilizing PAM-in sgRNAs and AS donor ODNs, which had a conversion frequency more comparable to that induced by single nicks. As we discuss below, this configuration is indeed a special case. In summary, these experiments demonstrated that both nicks and (even more so) double-stranded lesions can induce robust PGE in human somatic cells by utilizing donor ODNs.
Single nicks generate unidirectional conversion tracts when using complementary-strand ODNs
To map the associated conversion tracts, some of the ODNs were substituted with three-to-a-side synonymous single-nucleotide polymorphisms (SNPs) on both sides of the TY to SH mutations in either a distributive or clustered fashion (6SNPs_A and 6SNPs_B, respectively) in both S and AS orientations (Figs. 1A, 2F; see Supplemental Fig. S10 for more details). The BFP-positive cells were enriched using FACS sorting and single-cell subcloned via limiting dilution. The BFP conversion of the individual subclones was subsequently confirmed by PCR using mismatch sensitive primers (BFP_CF and BFP_ER) (Supplemental Fig. S10). The individual conversion tracts of the BFP-positive subclones were then amplified using flanking primers (BFP_EF and BFP_ER) (Supplemental Fig. S10) and sequenced using Sanger sequencing (Supplemental Tables). The SNP retention curves were compiled by plotting the frequency of SNP retention against the distance of each SNP to the center of the SH mutations. The retention frequency of SNPs on both the 6SNP_A and 6SNP_B ODNs in the same configuration were overlaid into single SNP retention curves (Figs. 2, 4; Supplemental Figs. S5, S8, S9). Kolmogorov-Smirnov tests were then performed to determine the degree of symmetry around the central dinucleotides of the ODNs (Methods).
Figure 2.
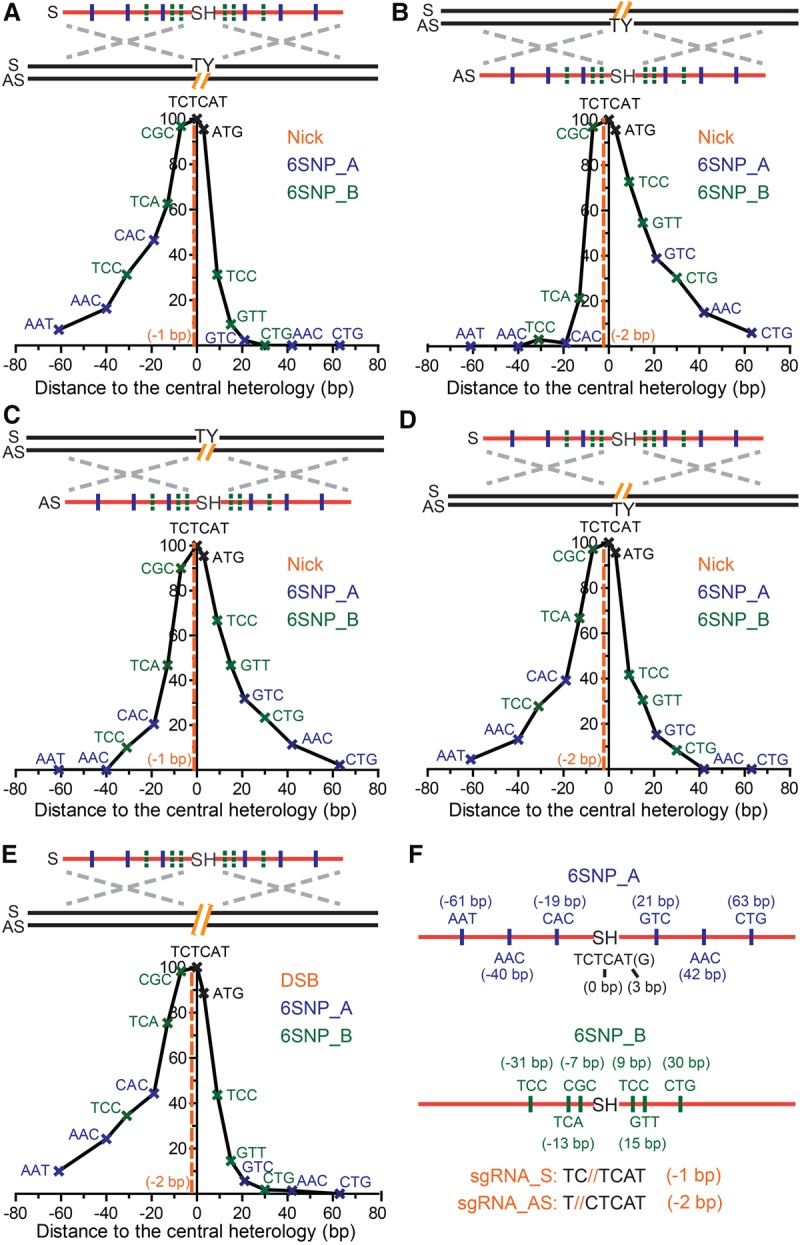
Conversion tracts of single-nick– and DSB-induced HDR using ODN donors. (A–E) The conversion tracts of single-nick–induced HDR using ODN donors complementary to (A,B) or the same strand of (C,D) the nick, and DSB-induced HDR (E). The conversion tracts were compiled by overlaying the retention frequency of each SNP in both 6SNP_A and 6SNP_B donors. The positions of the SNPs and predicted genomic lesions are labeled in reference to the center of the chromophore sequence. Schematic elements are colored as follows: genomic DNA, black; genomic lesions, orange; ODNs, red; chromophore sequences, TY and SH; SNPs on the 6SNP_A ODNs, blue; SNPs on the 6SNP_B ODNs, green; homology regions, dashed silver crosses; strandedness of DNA, S and AS. (F) Schematics of the ODN donors with synonymous SNPs. The SNPs are represented as the central nucleotide of the trinucleotides in the S orientation, and the distance between the SNPs and the center of the SH sequence (TCTCAT) is labeled. Because the last T in the SH sequence is a wobble nucleotide, it could be used as a SNP as well and is represented as the ATG at position +3. ODNs with six distributed or clustered SNPs are labeled as 6SNP_A (top) and 6SNP_B (bottom), respectively. The sequences shown at the bottom of the panel emphasize that the S nick is made at position −1 and the AS nick at position −2. The actual sequences of the ODNs can be found in Supplemental Figure S8.
In stark contrast to dsDNA donors containing a long central heterology (Kan et al. 2014), ODN-mediated PGE produced short conversion tracts with Gaussian-like distributions (Fig. 2A–E). When PGE was initiated by nCas9 and a single sgRNA, ODNs complementary to the strand with the nick produced conversion tracts with an approximately one-sided Gaussian-like distribution strongly biased in the 3′ direction relative to the nick (P = 0.001) (Fig. 2A). Impressively, the distribution of the conversion tracts was almost perfectly mirrored when the strandedness of the nick and the ODNs were simultaneously inverted (P = 0.022) (cf. Fig. 2B and Fig. 2A; Supplemental Table S2). In both cases, the SNP retention curve was composed of a mini-plateau (very high coconversion region) near the SH mutations being selected for, followed by a gradual sigmoidal decay in the 3′ direction and a steep drop-off in the 5′ direction. For example, CAC, as the first SNP on the 6SNP_A ODN 3′ to the nick, was retained in 46.5% of the conversion tracts, whereas GTC, its counterpart 5′ to the nick, was converted a mere 2.3% of the time (Fig. 2A; Supplemental Table S1). The corresponding SNPs on the 6SNP_B ODNs were converted with a similar asymmetry, although not always as dramatically as the ones on 6SNP_A. This was probably due to the fact that the SH mutations and the proximal SNPs were clustered tightly together and therefore were frequently recognized as one piece of heterology during gene conversion. The coconversion of this larger heterology perforce utilized more distal homologies and thus likely generated longer conversion tracts.
The conversion tracts generated by ODNs complementary to the strand with the nicks (Fig. 2A,B) were consistent with an SDSA model (Strathern et al. 1982; Hastings 1988; Larocque and Jasin 2010), in which gene conversion stems from unidirectional DNA synthesis using the ODN sequence as a template (Fig. 3A; Davis and Maizels 2014). After a genomic lesion such as a single-strand nick is introduced, the 3′ strand of the lesion can base-pair with the homology region on a complementary-strand ODN during the homology search. Then, the invading strand copies the downstream genetic information from the ODN donor via 5′ to 3′ DNA synthesis. Interestingly, if a small part of the selected knock-in mutations are upstream (i.e., 5′) to the genomic lesion (as 4 or 5 nt are in our system), the 3′-end of the invading strand needs to be resected to accommodate the conversion of the heterology region before DNA synthesis begins. Because we need to select for the conversion of the SH mutations and use proximal SNPs to map the conversion tracts nearby (which probably favors coconversion) in our experimental strategy, it is hard to address how often this 3′-end processing occurs in natural HDR where these constraints would not apply. Whatever the frequency, however, it nevertheless seems that the 3′-end processing rarely goes beyond 10 bp (Figs. 2A,B, 3A). Then, DNA synthesis proceeds past the knock-in mutations before the invading strand anneals back with its original partner. If DNA synthesis travels beyond the genomic lesion, a flap will be created after annealing. The flap can be cleaved by structure-specific endonucleases and the resultant gap filled by DNA synthesis and ligation as part of standard flap metabolism (Balakrishnan and Bambara 2013). In summary, part of the ODN sequence including the knock-in mutations will be converted as a result of DNA synthesis starting from up to 10 bp 5′ of the genomic lesion (Figs. 2A,B, 3A). Since the synapse formed by strand invasion cannot be stabilized by the formation of double Holliday junctions, the invading strand falls back after DNA synthesis and bridges the genomic lesion (Jasin and Rothstein 2013), generating short conversion tracts with an average of 20 bp in the 3′ direction (Figs. 2A,B, 3A).
Figure 3.

Mechanisms of single-nick–induced PGE using ODN donors. (A) Repair of complementary strand nicks via SDSA. (B) Repair of nicks on the same strand via ssDI. Schematic elements are labeled as follows: (Top) genomic DNA, black; genomic lesions and DNA ends, hatched orange lines; ODNs, red; knock-in mutations, green; resection nucleases, yellow PAC-MAN; base-pairing, purple vertical lines; DNA synthesis, red dashed arrows; processing nucleases, yellow lightning bolts; (bottom) predicted SNP retention curves, red dashed line; predicted position of genomic lesions, orange vertical dashed line; position of the knock-in mutation selected for, green vertical solid line; regions with more than 50% coconversion frequency, black double-headed arrows; strandedness of DNA, S and AS; orientations of the DNA ends, 5′ and 3′.
Single nicks generate bidirectional conversion tracts when using same strand ODNs
In contrast to ODN donors corresponding to the complementary strand, ODN donors corresponding to the same strand that was nicked produced short conversion tracts with more symmetrical two-sided Gaussian-like distributions (Fig. 2C,D; Supplemental Fig. S5). For example, CAC, the first SNP in the 5′ direction on the 6SNP_A ODN, was retained 20.5% of the time as compared to 31.8% for GTC, the corresponding first SNP in the 5′ direction (Fig. 2C; Supplemental Table S3). Again, this SNP retention curve was mirrored when the strandedness of the nick and the ODNs were simultaneously inverted (cf. Fig. 2D and Fig. 2C; Supplemental Table S4). We also noted a modest, but consistent, 5′ bias in these SNP retention curves, in which the SNPs 5′ to the nick were consistently converted with higher frequency than their downstream counterparts (Fig. 2C,D). Interestingly, this bias is opposite to the direction of common exonuclease resection, which would predict more extensive 5′ rather than 3′ resection (Symington and Gautier 2011).
The bidirectional conversion tracts generated by ODN donors that were the same as the strand with the nick were consistent with an ssDI model, in which the ODN is physically assimilated into a single-strand gap and displaces the flanking sequences on both sides (Fig. 3B; Radecke et al. 2006a; Storici et al. 2006; Davis and Maizels 2014). Although various forms of the ssDI model have been proposed (Faruqi et al. 2000; Ferrara and Kmiec 2004; Kuan and Glazer 2004; Wu et al. 2005; Huen et al. 2006), the efficiency of this pathway was so low in the absence of targeted genomic lesions that it was generally not regarded as a naturally occurring HDR pathway (also note that ssDI is different from the more familiar process of single-strand annealing [SSA]) (San Filippo et al. 2008; Jasin and Rothstein 2013). Interestingly, it has also been suggested that transcript RNA-mediated HDR occurs via a mechanism similar to ssDI, although the RNA donor has to fall off after it bridges the complementary strand genomic lesion (Keskin et al. 2014). In the presence of targeted genomic lesions, it was also not known how the lesions were processed into the single-stranded gaps required for ssDI. In our model (Fig. 3B), we propose that the initial gap is generated by a futile homology search when the 3′ end is trying to engage in the strand invasion à la the SDSA pathway (Fig. 3A). When the ODN is initially assimilated into the gap generated by a futile homology search, it will produce the subtle 5′-asymmetry/bias that was actually observed in our SNP retention curves (Figs. 2C,D, 3B). Bidirectional conversion tracts are then generated as the ODN displaces the flanking sequences via a branch migration-like activity (Murayama et al. 2008), while 5′ to 3′ exonuclease resection may also contribute to this process (Symington and Gautier 2011). Eventually, the flaps of the ODN and genomic DNA are processed by flap endonucleases (Balakrishnan and Bambara 2013), and the ODN is physically incorporated into the chromosome in both directions from the genomic lesion with a bias toward the 5′ direction from the lesion (Fig. 3B).
DSBs generate mainly unidirectional conversion tracts using ODN donors
Double-stranded genomic lesions can theoretically engage both the SDSA and ssDI subpathways in the presence of ODN donors. To elucidate the hierarchy of HDR subpathways utilized in the presence of double-stranded genomic lesions, we compiled the SNP retention curve of ODN-mediated PGE induced by wild-type Cas9 cleavase. When a DSB was introduced by Cas9, it generated a conversion tract with an approximately one-sided Gaussian-like distribution (Fig. 2E) that was virtually indistinguishable from the conversion track generated by a single-strand nick in a strand complementary to the ODN (cf. Fig. 2E and Fig. 2A; Supplemental Table S5). The approximately one-sided conversion tract suggested that, in the presence of a blunt-ended DSB directly at the position of the knock-in mutation, PGE is preferentially mediated by the SDSA pathway using the lesion complementary to the strand with the ODNs (Fig. 3A), even though the lesion can likely initiate effective ssDI as well. This result nicely explains the previous paradox that the physical incorporation of ODN donors was detected in the absence (Radecke et al. 2006b), but not presence (Radecke et al. 2006a), of targeted chromosomal DSBs; without a targeted lesion, the ODNs were likely physically incorporated via the ssDI pathways (Faruqi et al. 2000; Ferrara and Kmiec 2004; Kuan and Glazer 2004; Wu et al. 2005; Huen et al. 2006) using naturally occurring gaps, whereas with a DSB most of the PGE products are generated via the SDSA pathway as a result of DNA synthesis using ODNs only as templates (Figs. 2E, 3A).
Paired nicks generate plateaued conversion tracts using ODN donors
The use of nCas9 in conjunction with dual sgRNAs in PAM-out and PAM-in configurations generates paired nicks with putative 5′ or 3′ overhangs, respectively (Ran et al. 2013a; Cho et al. 2014; Shen et al. 2014). These overhangs can theoretically engage the ODNs via both the SDSA and ssDI pathways. In addition, since paired nicks can be effectively placed up to 60 bp away from each other, it was not known whether they are processed into double-strand gaps. To elucidate the mechanism(s) of paired-nick–induced PGE, we compiled SNP retention curves using nCas9 paired-nickases in both PAM-out and PAM-in configurations in the presence of S and AS ODN donors. We discovered that these paired-nickases generated conversion tracts with much larger plateaus (Fig. 4), compared to those of the respective single nicks (Fig. 2). The SNPs between the paired nicks were retained with an average of more than 75% frequency (Supplemental Tables S6, S7). Besides the larger plateau, the PAM-out paired-nickases generated a one-sided SNP retention curve with an approximately sigmoidal decay on the left and a much steeper drop-off on the right (Fig. 4A), whereas the PAM-in configuration with a S ODN donor generated two approximately sigmoidal curves (Fig. 4B) that were farther apart from each other compared to the SNP retention curve of the corresponding single nickase (Fig. 2D).
Figure 4.

The conversion tracts of paired-nicks–induced HDR. (A–D) The conversion tracts of paired-nicks–induced HDR using S (A,B) and AS ODNs (C,D). The conversion tracts were compiled by overlaying the retention frequency of each SNP in both 6SNP_A and 6SNP_B donors (Fig. 2F). The positions of the SNPs and predicted genomic lesions are labeled in reference to the center of the chromophore sequence. All schematic elements are colored as in the legend of Figure 2.
We propose that the 5′ overhangs generated by PAM-out paired-nickases can be processed by extensive exonuclease resection (Symington and Gautier 2011) to generate large double-stranded gaps (Fig. 5A) that ultimately yield the broad plateaued SNP retention curve (Fig. 4A). We believe that, following this resection, PGE is likely initiated by strand invasion of a now uncovered 3′ end complementary to the strand of the ODNs via the SDSA pathway (Fig. 5A). In the PGE products, DNA synthesis would have to travel a long distance to bridge the extended double-strand gaps. After annealing with the 3′ end on the other side of the lesion, the resultant flap on the AS strand is processed, and the single-strand gap on the S strand is filled in by standard gap repair. We believe that the steep drop-off on the right side of the SNP retention curve (Fig. 4A) is an SDSA SNP retention signature (Fig. 2A,B) indicative of the minimal 3′-end processing before the start of DNA synthesis and that strand invasion is initiated by the 3′ overhang on the AS strand (Fig. 5A). The broad-plateaued conversion tracts (Fig. 4A) are thus evidence that the 5′ overhangs of the PAM-out paired-nickases are processed into double-strand gaps, so that the subsequent DNA synthesis has to travel beyond these gaps to repair the genomic lesions in the PGE products (Fig. 5A).
Figure 5.

Mechanisms of paired-nick–induced PGE using the S ODNs. (A) Repair of the PAM-out double nicks via SDSA. (B) Repair of the PAM-in double nicks via ssDI. All schematic elements are colored and defined as in the legend to Figure 3.
In contrast, in the PAM-in configuration, the 3′ overhangs initially generated by paired nicking are not subject to extensive exonuclease resection (Fig. 5B; Symington and Gautier 2011). Because the 3′ overhang on the AS strand complementary to the ODNs cannot effectively convert the knock-in mutations (unless it is resected by 30 nt), the majority of the PGE products must be generated by the ssDI pathway. We propose that the presence of AS strand nicks may loosen the 3′ ends on the chromosomal S strand and mobilize it for a (in this instance, futile) homology search (Fig. 5B). This would provide a window of opportunity for the S strand ODN to anneal to the gap generated by futile homology search before further bidirectional strand displacement occurs. In total, this would produce the longer conversion tracts observed compared to those of single-nick–induced PGE via the ssDI pathway (cf. Fig. 2D and Fig. 4B).
ODNs preferentially utilize the SDSA pathway in the presence of double-stranded genomic lesions
For the paired-nickase–induced PGE using S strand ODNs, we noticed that the knock-in mutations and 3′ overhang on the AS strand were close to each other in the PAM-out configuration but far away in the PAM-in configuration (Fig. 4A,B). Even if SDSA is the preferred pathway for double-strand lesions (Fig. 2E), it cannot effectively convert the knock-in mutations using the AS strand overhang in the PAM-in configuration, unless extensive resection of the 3′ end occurs (Fig. 4B). To dissect whether the hierarchy of paired-nickases–induced PGE is determined by the PAM configuration or the relative position of the knock-in mutation and the 3′ overhang on the complementary strand, we also compiled the SNP retention curves of paired-nickases–induced PGE using AS strand ODNs (Fig. 4C,D).
PAM-out paired-nickases with AS strand ODNs produced a compound SNP retention curve with a plateau, a sigmoidal decay on the right and a linear slope on the left side (Fig. 4C; Supplemental Table S8). We inferred that the conversion tracts were predominantly generated by ssDI, because SDSA would have produced continuous conversion tracts downstream of the 3′ overhang on the S strand (and thus ACC, TCC, and CAC would have been retained with 100% frequency—which was not observed). We thus propose that the 5′ overhangs generated by PAM-out paired-nickases are first resected by exonucleases (Symington and Gautier 2011) to expose a single-strand gap for ssDI (Supplemental Fig. S6A). Since resection on both strands may occur to a random extent before an incoming ODN is assimilated to the 5′ overhang on the S strand, the left border of the single-strand gaps should be randomly distributed within a certain region before the initial ssDI occurs. Subsequently, strand displacement occurs in both directions, which creates the sigmoid decay on the right of the SNP retention curve. At the same time, the linear slope on the left is shaped by the accumulation of individual ssDI events with random left borders (Fig. 4C; Supplemental Fig. S6A).
On the other hand, PAM-in paired-nickases generated a two-sided SNP retention curve (Fig. 4D) similar to that of single-nick–induced ssDI (Fig. 2C). We propose that the bulk of the PGE products are generated via a simple SDSA process initiated by the 3′ overhang on the genomic S strand (Supplemental Fig. S6B). However, the conversion tracts were slightly wider than those from single-nick–induced SDSA (Supplemental Tables S2, S9), probably because the 3′ overhang on the S strand exceeds the first nucleotide of the SH mutations by 7 bp (Fig. 4D; Supplemental Fig. S10), which may lead to reduced gene conversion efficiency due to the requirement for relatively long 3′ end resection. As a result, some PGE products may also stem from the ssDI pathway using the nick on the AS strand, which may contribute to the higher SNP retention frequency on the left side (Fig. 4D) than is seen in an exclusively SDSA-dependent profile (Fig. 2B). Finally, steric hindrance of PAM-in paired-nickases (Ran et al. 2013a; Cho et al. 2014; Shen et al. 2014) may also result in partial nicking in a fraction of cells, and a single nick on the AS strand would have to engage the ssDI pathway. Consistent with all these notions, it is relevant to note that, of all the double-stranded lesions and donor ODN configurations analyzed in this study, the efficiency of BFP conversion in this PAM-in configuration was the lowest (Fig. 1B).
ODNs are physically incorporated into the genome in the ssDI but not in the SDSA pathway
The distribution of conversion tracts can be complicated by 3′ processing and the coconversion of proximal SNPs. Besides the directionality and composition of the conversion tracts, a definitive feature that distinguishes the SDSA and ssDI pathways is the physical incorporation of donor ODNs (Radecke et al. 2006a,b). Thus, the ODNs are physically incorporated to bridge a genomic lesion in the ssDI pathway, whereas in SDSA the genomic lesion is repaired by DNA synthesis using the ODN sequence solely as a template (Fig. 3A,B). To confirm the results of our conversion tract study, we internally labeled a T nucleotide with biotin in S strand ODNs in the center of the SH mutation (TCTCAT) (Supplemental Fig. S8) and searched for the physical incorporation of this biotinylated T in the PGE products induced by nCas9, Cas9, and nCas9 paired-nickases (Fig. 6). Shortly after transfecting HPRT1-EGFP antisense cells with Cas9 or nCas9, sgRNAs and the biotinylated ODNs, cells with bright BFP expression were isolated by fluorescence activated cell sorting (FACS). The genomic DNA from 105 BFP-positive cells was prepared, digested to completion with XhoI and XbaI restriction enzymes, and pulled down using streptavidin beads (Fig. 6A,B; Radecke et al. 2006a,b). The free and noncovalently bound DNA fragments were carefully washed away under denaturing conditions, and the beads with covalently linked fragments were used as templates in a 40-cycle PCR reaction. An internal primer and a flanking primer were used to specifically detect the ODNs incorporated into the BFP locus (BFP_CF and BFP_QR) (Supplemental Fig. S10). Using biotinylated ODNs on the S strand, the covalently linked genomic fragments were detected only with a single sgRNA on the AS strand and dual sgRNAs in the PAM-in configuration, but not with a single sgRNA on the S strand, a single sgRNA with wild-type Cas9 (Radecke et al. 2006a), nor with dual sgRNAs in the PAM-out configuration (Fig. 6C). Collectively, these results confirmed that the ODNs were physically incorporated into the chromosome only when the ssDI model was predicted by the conversion tract profiles (Figs. 2D, 4B).
Figure 6.

The biotin pull-down assay. (A,B) Schematic illustration of the biotin pull-down assay via the SDSA (A) and ssDI (B) pathways. In the case of ssDI (B), the biotinylated ODN is predicted to be incorporated into the target genomic locus, whereas in SDSA (A), it should not. The XhoI- and XbaI-digested genomic fragments covalently linked to biotin can be enriched using streptavidin beads under denaturing conditions. The primers BFP_QF and BFP_QR can specifically amplify these genomic fragments with ODN incorporation but not free ODN donors. Biotin, yellow circles; streptavidin, orange ovals; genomic DNA, black lines; ODN sequence, solid red lines; DNA synthesis, dashed red lines; chromophore sequence, TY and SH; genomic lesions, hatched orange lines; homology regions, dashed silver crosses; restriction sites, XhoI and XbaI; PCR primers, horizontal arrows. (C) Results of the biotin pull-down assay. After transfecting with biotinylated ODNs (BFP_S90_Biotin) (Supplemental Fig. S8), the BFP-positive cells were enriched using FACS sorting. The genomic DNA of the BFP-positive cells was digested with XhoI and XbaI. The fragments covalently linked to biotin were pulled down with streptavidin beads in denaturing conditions, PCR-amplified using one internal primer and one flanking primer of the ODN donors (BFP_QF and BFP_QR) (Supplemental Fig. S8), and detected on an agarose gel.
Mismatch repair activity has differential impacts on the SDSA and ssDI pathways
All of the above experiments were carried out in the human colorectal carcinoma HCT116 cell line. HCT116 cells were intentionally chosen because they are mismatch repair (MMR)-defective, which permitted an unequivocal analysis of the SNP retention profiles. To confirm and extend these observations in different cell lines, we also carried out PGE experiments in MMR-deficient DLD-1 colon carcinoma cells (Chen et al. 1983) and MMR-proficient retinal pigment epithelial (RPE) cells that had been immortalized, but not transformed, by the expression of telomerase (RPE+hTERT) (Bodnar et al. 1998). PGE in these cell lines was carried out at the phosphatidylinositol glycan anchor biosynthesis class A (PIGA) locus (Karnan et al. 2012). The inactivation of PIGA, an X-linked gene, results in the loss of glycosylphosphatidyl inositol (GPI) anchors—a phenotype that can be quantitatively assessed using reagents that bind to GPI anchors. Thus, the relative frequency of PIGA inactivation can be monitored by FACS analysis. In our case, we used a commercially available reagent, FLAER, which is an Alexa 488-conjugated aerolysin variant that binds to GPI anchors; correct targeting could be assessed by the percentage of FLAER-negative cells (Supplemental Fig. S7; Karnan et al. 2012). Since correct targeting without ODN incorporation can also lead to FLAER-negative cells, the absolute amount of PGE was then determined by next-generation sequencing. In these experiments, a single ODN containing seven SNPs was used to facilitate PIGA+ to PIGA− conversion (Supplemental Figs. S8A, S9A). When ODNs complementary to the strand with the nick were utilized, the SNP retention curves in the DLD-1 cell line were heavily biased in a one-sided manner (S9C; P = 0.019 and 0.001, respectively) (Supplemental Fig. S9B). The conversion tracts were very similar to those observed for HCT116 cells (Fig. 2B,C), although fewer data points were monitored in DLD-1. When ODNs identical to the strand with the nick were used, much more symmetric SNP retention curves were observed similar to HCT116 cells (cf. Supplemental Fig. S9D,E and Fig. 2A,C, respectively). However, the mild 5′ bias observed with HCT116 was not observed in DLD-1, probably due to the reduced number of data points analyzed and/or potential PCR biases. Collectively, however, the DLD-1 data supported (and extended) our conclusions for the mechanism of ODN-mediated PGE in MMR-deficient cells at different loci—including an independent, endogenous locus.
We have demonstrated that MMR strongly reduces PGE when HDR is carried out by the DSBR pathway (Kan et al. 2014). Consequently, PGE was performed at the PIGA locus in MMR-proficient RPE cells using the same ODN, with seven SNPs, that was used with DLD-1 cells (Supplemental Fig. S8A). First, the PGE efficiency in the RPE+hTERT cell line was robust (Supplemental Fig. S8B). This important observation, in and of itself, suggested that the cellular MMR status does not affect ODN-mediated PGE as much when the SDSA and ssDI pathways are utilized as it does when dsDNA donors and DSBR are used. Second, the SNP conversion patterns using same-sense donor ODN in the RPE+hTERT cell line were similar to those observed for HCT116 and DLD-1 cells (cf. Supplemental Fig. S8D and both Fig. 2D and Supplemental Fig. S9E). The only salient difference observed was that, in the case of PGE in RPE+hTERT cells with the same-sense donor ODNs, the overall frequency of recombination was five times lower (Supplemental Fig. S8B), a feature discussed below. However, when the complementary strand ODNs were used, we noted that the conversion tracts for RPE+hTERT cells (Supplemental Fig. S8C) were much narrower than what was observed in either HCT116 (Fig. 2A) or DLD-1 (Supplemental Fig. S9B) cells. We believe that these features may reflect the differential impact that the MMR machinery exerts on the SDSA and the ssDI pathways. The MMR system rejects heteroduplex DNA formation from dissimilar sequences in the displacement loop via its “anti-recombination” activity (Siehler et al. 2009). For SDSA, this may occur after minimal displacement synthesis, thus allowing the conversion of the central TG to GT but preventing the incorporation of distal SNPs. Thus, in this experimental set-up, the overall effect of MMR is to reduce the conversion tract length of SDSA (Supplemental Fig. S8E), but it does not affect the overall frequency of PGE nor the choice of recombination pathways. For the ssDI pathway, however, this “anti-recombination” activity directly prevents the assimilation of dissimilar ODNs to the genomic lesions and therefore reduces the overall frequency of ssDI (Supplemental Fig. S8F), consistent with our data (Supplemental Fig. S8B). In summary, our data suggest that MMR reduces the conversion tract length of SDSA as well as the efficiency of ssDI via its heteroduplex rejection activity (Supplemental Fig. S8E).
Discussion
In this study, we have systematically dissected the molecular mechanisms of ODN-mediated PGE induced by Cas9, Cas9 D10A nickase (nCas9), and paired-nickases using two lines of evidence: the directionality of the conversion tracts and the physical incorporation of the ODN donors. We demonstrate that ODN-mediated PGE utilizes the SDSA and ssDI subpathways. In contrast to DSBR, these pathways generate short conversion tracts with predominately one-sided and two-sided Gaussian-like distributions, respectively. In the presence of double-stranded genomic lesions (such as DSBs and paired nicks), the SDSA pathway is preferentially utilized.
Our results not only establish the properties of the individual SDSA and ssDI pathways but also illustrate the hierarchy of ODN-mediated PGE mediated by double-stranded genomic lesions. The SDSA and ssDI pathways have their respective effective conversion zones, as defined by the region with more than 50% conversion frequency with respect to the genomic lesion initiating the pathways (Fig. 3; also see Fig. 2). Therefore, conversion of knock-in mutations is inefficient outside these zones, and this parameter should be considered when designing gene targeting studies. Although both pathways work robustly by themselves (Fig. 1B; Supplemental Fig. S2A), it appears that they don't work equally well in the presence of DSBs when both pathways are nonetheless in their effective zone (Figs. 2E, 4; Radecke et al. 2006a). Instead, SDSA is preferentially utilized whenever the knock-in mutation is within the effective zone of the 3′ overhang complementary to the strand with the ODN donors (Figs. 2E, 4). This could be because cells tend to prevent the physical incorporation of exogenous sequences during HDR whenever possible. However, when the SDSA pathway drops out of its effective zone, ssDI is nonetheless capable of generating the majority of the PGE products (Fig. 4B,C).
As expected, double-stranded lesions generally induced PGE with a higher frequency than single-strand nicks (Fig. 1B). The only instance where a complex genomic lesion didn't induce increased PGE was in the case of PAM-in paired-nickases using ODN donors from the AS strand (Fig. 1B). As elaborated above, this was also the only instance where extensive (>7 nt) resection of the 3′ end was required for PGE to occur, and we believe that these two observations are connected, given the sharp drop-off in the SDSA conversion tracts (Fig. 2A,B). If this exception is illustrative, it may suggest that double-strand lesions, in general, loosen the local chromatin architecture more than single-strand nicks and thus permit relevant cofactors (such as resection nucleases) easier access to the 3′ end, which ultimately facilitates more vigorous homology searches.
In our nick-induced PGE experiments, no significant strand bias for either the nick or the ODN donors was observed (Fig. 1B). These observations were consistent with a previous report using zinc-finger nickases at three independent chromosomal loci (Ramirez et al. 2012) but inconsistent with a more recent one (Davis and Maizels 2014). The latter difference might be explained by the fact that the recent report employed ODN donors with a large (17-bp) heterology flanking the genomic lesion (Davis and Maizels 2014). As discussed above, a sizable heterology on both sides of the genomic lesion could strongly favor the use of ssDI as a bidirectional gene conversion pathway. In contrast, in order to be converted by the SDSA pathway, 3′ resection of at least 8–9 nt would be required and that likely occurs at a much lower frequency. Collectively, these observations suggest that strand-specific transactions such as transcription and DNA replication are unlikely to have a major influence on PGE mediated by nicks. With that said, it is important to note that, while no strand-specific biases were noted in any of our experiments, a significant two- to fourfold higher frequency of PGE was noted for the BFP reporter in the antisense orientation than in the sense orientation (Fig. 1; Supplemental Fig. S3). The reason for this bias is not completely clear, but it is unlikely to be due to conflicting transcription between the EGFP reporter and the endogenous HPRT1 locus because EGFP expression was very high in these cells. Indeed, we believe the bias was technical (and not biological) in nature since the higher EGFP (and consequently, BFP) expression may have simply allowed us to more easily isolate correctly targeted cells. Finally, we also note that a strand bias was observed when PGE was mediated by nicks and same sense ODN donors in RPE+hTERT cells (Supplemental Fig. S8B). As discussed previously, we do not believe that this is related to transcription or DNA replication issues but to the fact that these cells are MMR-proficient, which is especially inhibitory to the strand annealing step required for ssDI.
PAM-in paired-nickases are significantly less effective in producing NHEJ-related mutations than their PAM-out counterparts, which has mainly been attributed to the steric hindrance of the nCas9 proteins in the PAM-in configuration and/or the displacement loops formed by sgRNAs (Ran et al. 2013a; Cho et al. 2014; Shen et al. 2014). Our results may provide an additional or alternative explanation (Figs. 4A, 5A): PAM-out paired-nickases generate 5′ overhangs that are subject to exonuclease resection (Symington and Gautier 2011). The resection creates a double-strand gap that will normally engage the error-prone NHEJ pathway and produce deletions in between. In contrast, the 3′ overhangs produced by PAM-in paired-nickases may remain largely as separated nicks (Figs. 4B, 5B), which may be precisely repaired by standard single-strand nick repair without engaging the NHEJ pathway. Importantly, however, the PAM-in paired-nickases can induce PGE at a comparable efficiency to their PAM-out counterparts (Fig. 1B), at least when the PAM sequences are placed far enough apart from each other to avoid steric hindrance. Thus, we propose that PAM-in paired-nickases are less efficient in NHEJ-mediated gene disruption because the 3′ overhangs rarely form double-strand gaps due to the lack of 3′ to 5′ resection (Symington and Gautier 2011). Consequently, although PAM-in paired nickases are not often used for ODN-mediated PGE, they may actually be advantageous because they may induce similar levels of HDR with less NHEJ mutations compared to their PAM-out counterparts. The use of the Cas9 N863A variant further allows the efficient generation of short 3′ overhangs with PAM-out sgRNAs, minimizing the effect of steric hindrance (Nishimasu et al. 2014).
Although paired-nickases induced PGE with a higher frequency compared to a single nickase (Fig. 1B), we noticed that paired-nickases (in both PAM configurations) occasionally produced imprecise HDR products using ODN donors (Supplemental Tables S6–S9). Most of the imprecise products contained NHEJ-like mutations near the position of the distal nick with respect to the knock-in mutations. Since a single nickase can also generate NHEJ mutations (Davis and Maizels 2011, 2014), we propose that the imprecise HDR was generated by the repeated nicking of the distal nickase in the PGE products. These mutations were not found near the position of the proximal nicks because, in our system, the conversion of the knock-in mutations would destroy the binding site of the proximal nickase and prevent further nicking in the PGE products. If this hypothesis is correct, an important guideline of ODN-mediated PGE would be to introduce an additional silent mutation in the PAM sequence of the ODN donors to prevent the further binding of Cas9 or nCas9 in the PGE products. Alternatively, these nicks might be converted to frank DSBs which, through canonical end joining, might yield NHEJ-like mutations.
Our conversion tract data also provide insight into a recent paradox: although SDSA is the more efficient form of mega-nuclease–induced HDR (Li et al. 2001; Heyer et al. 2010), the majority of the PGE products are actually generated by the DSBR pathway when dsDNA donors with long central heterologies are employed (Li and Baker 2000; Li et al. 2001; Langston and Symington 2004; Kan et al. 2014). Our results and others’ demonstrate that SDSA is apparently a short-tract gene conversion pathway using chromosomal (Elliott et al. 1998; Larocque and Jasin 2010) and ODN donors (Fig. 2A,B). Also, the Gaussian-like nature of the associated conversion tracts makes it extremely inefficient in producing gene conversions longer than 100 bp (Fig. 2A,B). Thus, dsDNA donors with long central heterologies would perforce engage a long-tract gene conversion pathway. Notably, mega-nuclease–induced HDR using chromosomal donors occasionally generated unidirectional long-tract gene conversion products (Elliott et al. 1998; Larocque and Jasin 2010), which were believed to be produced by break-induced replication (BIR) (Borts and Haber 1987; Kraus et al. 2001; Llorente et al. 2008). In the case of PGE, however, BIR using exogenous homology donors leads to the formation of chromosome:donor fusions and severely compromises genomic stability (Borts and Haber 1987; Kraus et al. 2001; Llorente et al. 2008). In contrast, our previous work demonstrated that the DSBR pathway is a second (and precise) long-tract gene conversion pathway (Kan et al. 2014), which generates bidirectional conversion tracts with a linear distribution. Collectively, these results indicate that although SDSA is the more efficient HDR pathway for short-tract gene-conversion, the DSBR pathway is the predominant pathway of PGE using dsDNA donors with long central heterologies.
dsDNA donors can mediate robust PGE. We can routinely achieve about 20% marker-free PGE using Cas9-2A-GFP with associated FACS enrichment (Duda et al. 2014) and circular dsDNA donors with long central heterologies (Supplemental Figure S11). As a practical guideline, we propose that circular dsDNA should be utilized in the presence of DSBs and/or paired-nicks for converting large knock-in mutations (Fig. 7). One of the reasons that these donors work so well is that they may have a longer half-life than ODN donors, which better coincides with the kinetics of the Cas9 expressed in a DNA format. In contrast, ODN donors should be utilized for converting small modifications. Although ODN donors are optimally used for introducing SNPs, they can also be used to introduce more complex genomic lesions by producing slightly longer conversion tracts. Regardless, what is critical is that the desired knock-in mutation(s) needs to be placed within the effective conversion zones of the SDSA or ssDI pathways, or it will not be incorporated into the genome efficiently (Fig. 7). Cas9 expressed in mRNA or protein formats may be used to accommodate the shorter half-life of ODN donors and increase the efficiency of PGE.
Figure 7.

The hierarchy of mega-nuclease–induced HDR leading to PGE. PGE is defined as the fraction of HDR leading to the conversion of the desired knock-in mutations using exogenous homology donors. In the PGE products, retrospectively, the hierarchy of the HDR pathway is determined primarily by the types of homology donors and secondarily by the genomic lesions. dsDNA donors containing a sizable central heterology must be converted via the DSBR model, which generates long conversion tracts with a linear distribution. ODN donors can utilize both SDSA and ssDI pathways, depending on the strandedness of the ODNs and the relative position of the knock-in mutation to the genomic lesion. These pathways generate short conversion tracts in normal distributions. SDSA is preferentially utilized in the presence of double-stranded genomic lesions when both pathways can convert the knock-in mutation effectively.
Finally, our results demonstrated that the ODN donors are not physically incorporated into the genome in the SDSA pathway, making this pathway particularly valuable for agricultural engineering. Although plants and livestock containing CRISPR-mediated gene disruption are generally not categorized as genetically modified organisms (GMOs), PGE using exogenous HDR donors is under more stringent regulatory scrutiny (Wolt et al. 2016). An empirical distinction lies in whether the engineering process leads to the incorporation of foreign DNA into the genome (Wolt et al. 2016). We established that when an nCas9 and ODNs complementary to the strand of the nick are used in combination, the genomic modification is introduced via the endogenous DNA repair process of SDSA, whereas the ODN donors merely serve as repair templates (Figs. 2, 4, 6). These findings extend the scope of non-GMO plants and livestock to ODN-mediated PGE.
Methods
Nucleotide sequences
All sgRNA targets, ODN donors, primers, and the relevant plasmid sequences can be found in Supplemental Figure S10.
Cell culture
All cell lines utilized in this study were purchased from the American Type Culture Collection. The human HCT116 and DLD-1 cell lines were cultured in McCoy's 5A medium supplemented with 10% FBS, 1% L-glutamine, 1% penicillin/streptomycin. The human RPE+hTERT cell line was cultured in Dulbecco's modified Eagle medium supplemented with 10% FBS and 1% penicillin/streptomycin. All cells were grown with 5% CO2 at 37°C.
The HPRT1-EGFP cell lines
The rAAV EGFP knock-in vectors were constructed using an unpublished method (Y Kan, N Batada, EA Hendrickson, in prep.). Basically, the CMV-EGFP-pA cassette was amplified from EGFP-N2 (Clontech), and the homology arms flanking HPRT1 exon 3 were amplified using designated primers (Supplemental Fig. S10). The CMV-EGFP-pA cassette and homology arms were ligated into a modified version of the pAAV-MCS vector in both sense and antisense orientations with respect to HPRT1. rAAV packaging and infections were performed as described (Khan et al. 2011). The infected HCT116 cells were seeded into 10-cm tissue culture dishes and selected with 5 µg/mL 6-TG for 14 d. Individual colonies were initially scanned for EGFP expression under a fluorescence microscope and subsequently screened by PCR using the indicated primers (Supplemental Fig. S2A,B). One of the targeted clones with EGFP in either the sense or antisense direction in the HPRT1 locus was flow-sorted for high EGFP expression using a FACSAria II cell sorter (BD Biosciences) and used for subsequent studies.
The EGFP to BFP conversion system
The EGFP reporter cells were seeded at ∼50% confluency. The next day, 5 × 105 cells were transfected with a Cas9 or nCas9 expression plasmid (10 µg; #41815 and #41816, respectively, Addgene), the MLM3636 plasmid containing the designated sgRNA expression cassette (10 µg; #43860, Addgene), and the relevant ODNs (10 µg) using a Neon Transfection System (Invitrogen). For paired-nickases, cells were transfected with 7.5 µg of each sgRNA plasmid, the Cas9 expression plasmid, and ODNs. All transfections were performed using 100-µL tips under elevated conditions compared to the manufacturer's protocol: 1530V, a 10-msec pulse width, and three pulses. For the PGE efficiency experiment, cells were transfected using ODNs without flanking SNPs (Supplemental Fig. 10), and the percentage of BFP-positive cells were quantitated 2 d after transfection using a LSRFortessa cell analyzer (BD Biosciences). For the conversion tracts experiment, cells were transfected using ODNs containing flanking SNPs. Cells from five individual transfections were combined, cultured for 2 d, and sorted for BFP expression using a FACSAria II cell sorter (BD Biosciences).
Conversion tracts analysis
The BFP-positive cells were single-cell subcloned into 96-well-plates at a concentration of 3 to 10 cells per well, and 14 d later, single colonies were trypsinized, transferred into new 96-well plates, and grown to confluency. Genomic DNA was prepared using DirectPCR Lysis Reagent (Viagene). The BFP fragments containing all the potential SNPs were amplified and confirmed by PCR using primers BFP_EF X BFP_ER and BFP_CF X BFP_ER, respectively. The PCR products were purified using a QIAquick PCR purification kit. The retention of vector-borne SNPs was analyzed by Sanger sequencing.
Statistics
Kolmogorov-Smirnov tests were performed to determine the degree of symmetry around the central dinucleotides of the ODN donors for SNP retention curves induced by single-nicks. All reads from Sanger and Illumina sequencing were randomly divided into three groups. The retention frequency of all SNPs with approximately equal distance to the central dinucleotides was compiled in each group and entered into the Kolmogorov-Smirnov equations in pairs. The resultant P-value is shown on the corresponding graphs. The P-value reflects the degree of symmetry of the distributions around the y-axis, which is the central heterology of the ODN donors.
The PIGA conversion system
One half-million DLD-1 and RPE+hTERT cells were transfected with 15 µg PIGA_KO ODNs (Supplemental Figs. S6A, S8A, S9) and 15 µg Cas9 D10A-2A-mCherry with the respective S or AS sgRNA expression cassette in one backbone (adapted from PX461). Ten transfections were combined for each sgRNA, recovered for 14 d, stained with the FLAER reagent (Pinewood Scientific Services) at a concentration of 5 × 10−9 M, and FACS-sorted for PIGA negativity. Of the cells with the lowest PIGA expression, 5.6% and 6.5% were collected for S and AS sgRNA, respectively, using a nonstringent gate (Supplemental Fig. S7). These cells contain the majority of HDR and NHEJ events and some wild-type cells. Then, the genomic DNA was prepared using DirectPCR lysis (Viagen), and the target locus was amplified using flanking Nextera compatible primers (NextF & NextR) (Supplemental Fig. S10). The amplicons were gel-purified and sequenced using Nextera amplicon sequencing (paired-end 2 × 125 bp). The HDR products were characterized as the reads containing the central GT dinucleotide. The SNP retention curves were compiled by plotting the retention frequency of flanking SNPs in the HDR products versus the position of the SNPs in reference to the GT dinucleotide.
The biotin incorporation assay
The biotin incorporation assay was performed as described (Radecke et al. 2006b) with minor modifications. The EGFP reporter cells were transfected with internal biotin-labeled ODNs (BFP_S90_Biotin) and flow-sorted as described above. The genomic DNA from 1 × 105 BFP-positive cells was prepared using a Puregene Cell kit (Gentra) with a 60-min centrifugation step after isopropanol precipitation. The genomic DNA was digested to completion with XhoI and XbaI restriction enzymes (New England Biolabs). The biotinylated DNA fragments were isolated using a Dynabeads Kilobase BINDER kit (Life Technologies) according to the manufacturer's protocol, except that all reagents were supplemented with 0.1% BSA, and two additional washes using 0.1 M NaOH and 0.05 M NaCl at room temperature and one more rinse with 95°C water were performed to remove the noncovalently linked genomic fragments. The biotinylated genomic fragments were detected with a 40-cycle PCR using Phusion Hot Start II High-Fidelity DNA polymerase (Thermo Scientific). The genomic DNA preparation prior to Dynabeads purification was used as control in a 25-cycle reaction.
Data access
The sequence data generated in this study have been submitted to the NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) under accession numbers SRR5631326-SRR5631331. Sanger sequencing data have been submitted to SRA under accession number SRP108269.
Competing interest statement
E.A.H. declares that he is a member of the scientific advisory boards of Horizon Discovery, Ltd. and Intellia Therapeutics, companies that specialize in applying gene editing technology to basic research and therapeutics.
Supplementary Material
Acknowledgments
We thank Drs. Feng Zhang, Keith Joung, and George Church for sharing plasmids through Addgene. We thank Jason Motl and Nisha Shah at the University of Minnesota Flow Cytometry Core for cell sorting. This work was supported by grants from the National Institutes of Health: the National Institutes of General Medical Sciences (GM088351) and the National Cancer Institute (CA154461 and CA190492).
Author contributions: Y.K. and E.A.H. designed the study, analyzed the data, and wrote the manuscript. Y.K. performed the bulk of the experiments, and T.T. and B.R. assisted during the latter stages of the project.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.214775.116.
References
- Balakrishnan L, Bambara RA. 2013. Flap endonuclease 1. Ann Rev Biochem 82: 119–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AR, Tibbit C, Ponting CP, Liu JL. 2013. Highly efficient targeted mutagenesis of Drosophila with the CRISPR/Cas9 system. Cell Rep 4: 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bizard AH, Hickson ID. 2014. The dissolution of double Holliday junctions. Cold Spring Harb Perspect Biol 6: a016477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar AG, Ouellette M, Frolkis M, Holt SE, Chiu CP, Morin GB, Harley CB, Shay JW, Lichtsteiner S, Wright WE. 1998. Extension of life-span by introduction of telomerase into normal human cells. Science 279: 349–352. [DOI] [PubMed] [Google Scholar]
- Borts RH, Haber JE. 1987. Meiotic recombination in yeast: alteration by multiple heterozygosities. Science 237: 1459–1465. [DOI] [PubMed] [Google Scholar]
- Byrne SM, Mali P, Church GM. 2014. Genome editing in human stem cells. Methods Enzymol 546: 119–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll D. 2014. Genome engineering with targetable nucleases. Ann Rev Biochem 83: 409–439. [DOI] [PubMed] [Google Scholar]
- Chen TR, Hay RJ, Macy ML. 1983. Intercellular karyotypic similarity in near-diploid cell lines of human tumor origins. Cancer Genet Cytogenet 10: 351–362. [DOI] [PubMed] [Google Scholar]
- Chen F, Pruett-Miller SM, Davis GD. 2015. Gene editing using ssODNs with engineered endonucleases. Methods Mol Biol 1239: 251–265. [DOI] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim JM, Kim JS. 2013. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol 31: 230–232. [DOI] [PubMed] [Google Scholar]
- Cho SW, Kim S, Kim Y, Kweon J, Kim HS, Bae S, Kim JS. 2014. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res 24: 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis L, Maizels N. 2011. DNA nicks promote efficient and safe targeted gene correction. PLoS One 6: e23981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis L, Maizels N. 2014. Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc Natl Acad Sci 111: E924–E932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCarlo JE, Norville JE, Mali P, Rios X, Aach J, Church GM. 2013. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Res 41: 4336–4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda K, Lonowski LA, Kofoed-Nielsen M, Ibarra A, Delay CM, Kang Q, Yang Z, Pruett-Miller SM, Bennett EP, Wandall HH, et al. 2014. High-efficiency genome editing via 2A-coupled co-expression of fluorescent proteins and zinc finger nucleases or CRISPR/Cas9 nickase pairs. Nucleic Acids Res 42: e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott B, Richardson C, Winderbaum J, Nickoloff JA, Jasin M. 1998. Gene conversion tracts from double-strand break repair in mammalian cells. Mol Cell Biol 18: 93–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faruqi AF, Datta HJ, Carroll D, Seidman MM, Glazer PM. 2000. Triple-helix formation induces recombination in mammalian cells via a nucleotide excision repair-dependent pathway. Mol Cell Biol 20: 990–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara L, Kmiec EB. 2004. Camptothecin enhances the frequency of oligonucleotide-directed gene repair in mammalian cells by inducing DNA damage and activating homologous recombination. Nucleic Acids Res 32: 5239–5248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau JE, Dupuis ME, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, Moineau S. 2010. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468: 67–71. [DOI] [PubMed] [Google Scholar]
- Gasiunas G, Barrangou R, Horvath P, Siksnys V. 2012. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc Natl Acad Sci 109: E2579–E2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings PJ. 1988. Recombination in the eukaryotic nucleus. Bioessays 9: 61–64. [DOI] [PubMed] [Google Scholar]
- Heyer WD, Ehmsen KT, Liu J. 2010. Regulation of homologous recombination in eukaryotes. Annu Rev Genet 44: 113–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MS, Li XT, Lu LY, Watt RM, Liu DP, Huang JD. 2006. The involvement of replication in single stranded oligonucleotide-mediated gene repair. Nucleic Acids Res 34: 6183–6194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasin M, Rothstein R. 2013. Repair of strand breaks by homologous recombination. Cold Spring Harb Perspect Biol 5: a012740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen NM, Dalsgaard T, Jakobsen M, Nielsen RR, Sorensen CB, Bolund L, Jensen TG. 2011. An update on targeted gene repair in mammalian cells: methods and mechanisms. J Biomed Sci 18: 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337: 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. 2013. RNA-programmed genome editing in human cells. eLife 2: e00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kan Y, Ruis B, Lin S, Hendrickson EA. 2014. The mechanism of gene targeting in human somatic cells. PLoS Genet 10: e1004251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnan S, Konishi Y, Ota A, Takahashi M, Damdindorj L, Hosokawa Y, Konishi H. 2012. Simple monitoring of gene targeting efficiency in human somatic cell lines using the PIGA gene. PLoS One 7: e47389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katada H, Ren Y, Shigi N, Komiyama M. 2008. Site-specific gene manipulation of fluorescent proteins using artificial restriction DNA cutter. Nucleic Acids Symp Ser (Oxf) 52: 483–484. [DOI] [PubMed] [Google Scholar]
- Keskin H, Shen Y, Huang F, Patel M, Yang T, Ashley K, Mazin AV, Storici F. 2014. Transcript-RNA-templated DNA recombination and repair. Nature 515: 436–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan IF, Hirata RK, Russell DW. 2011. AAV-mediated gene targeting methods for human cells. Nat Protoc 6: 482–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus E, Leung WY, Haber JE. 2001. Break-induced replication: a review and an example in budding yeast. Proc Natl Acad Sci 98: 8255–8262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan JY, Glazer PM. 2004. Targeted gene modification using triplex-forming oligonucleotides. Methods Mol Biol 262: 173–194. [DOI] [PubMed] [Google Scholar]
- Langston LD, Symington LS. 2004. Gene targeting in yeast is initiated by two independent strand invasions. Proc Natl Acad Sci 101: 15392–15397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larocque JR, Jasin M. 2010. Mechanisms of recombination between diverged sequences in wild-type and BLM-deficient mouse and human cells. Mol Cell Biol 30: 1887–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Baker MD. 2000. Mechanisms involved in targeted gene replacement in mammalian cells. Genetics 156: 809–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Read LR, Baker MD. 2001. The mechanism of mammalian gene replacement is consistent with the formation of long regions of heteroduplex DNA associated with two crossing-over events. Mol Cell Biol 21: 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Staahl BT, Alla RK, Doudna JA. 2014. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife 3: e04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Long L, Xiong K, Yu B, Chang N, Xiong JW, Zhu Z, Liu D. 2014. Heritable/conditional genome editing in C. elegans using a CRISPR-Cas9 feeding system. Cell Res 24: 886–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorente B, Smith CE, Symington LS. 2008. Break-induced replication: what is it and what is it for? Cell Cycle 7: 859–864. [DOI] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, Gorbunova V. 2008a. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst) 7: 1765–1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Z, Bozzella M, Seluanov A, Gorbunova V. 2008b. DNA repair by nonhomologous end joining and homologous recombination during cell cycle in human cells. Cell Cycle 7: 2902–2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama Y, Kurokawa Y, Mayanagi K, Iwasaki H. 2008. Formation and branch migration of Holliday junctions mediated by eukaryotic recombinases. Nature 451: 1018–1021. [DOI] [PubMed] [Google Scholar]
- Nishimasu H, Ran FA, Hsu PD, Konermann S, Shehata SI, Dohmae N, Ishitani R, Zhang F, Nureki O. 2014. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156: 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Weaver TL, Szostak JW. 1983. Yeast recombination: the association between double-strand gap repair and crossing-over. Proc Natl Acad Sci 80: 4417–4421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Weaver TL, Szostak JW, Rothstein RJ. 1981. Yeast transformation: a model system for the study of recombination. Proc Natl Acad Sci 78: 6354–6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, Munro M, Pinder J, Salsman J, Dellaire G, et al. 2015. A mechanism for the suppression of homologous recombination in G1 cells. Nature 528: 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Radecke F, Peter I, Radecke S, Gellhaus K, Schwarz K, Cathomen T. 2006a. Targeted chromosomal gene modification in human cells by single-stranded oligodeoxynucleotides in the presence of a DNA double-strand break. Mol Ther 14: 798–808. [DOI] [PubMed] [Google Scholar]
- Radecke S, Radecke F, Peter I, Schwarz K. 2006b. Physical incorporation of a single-stranded oligodeoxynucleotide during targeted repair of a human chromosomal locus. J Gene Med 8: 217–228. [DOI] [PubMed] [Google Scholar]
- Ramirez CL, Certo MT, Mussolino C, Goodwin MJ, Cradick TJ, McCaffrey AP, Cathomen T, Scharenberg AM, Joung JK. 2012. Engineered zinc finger nickases induce homology-directed repair with reduced mutagenic effects. Nucleic Acids Res 40: 5560–5568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Lin CY, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y, et al. 2013a. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013b. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reardon S. 2016. Welcome to the CRISPR zoo. Nature 531: 160–163. [DOI] [PubMed] [Google Scholar]
- Richardson CD, Ray GJ, DeWitt MA, Curie GL, Corn JE. 2016. Enhancing homology-directed genome editing by catalytically active and inactive CRISPR-Cas9 using asymmetric donor DNA. Nat Biotechnol 34: 339–344. [DOI] [PubMed] [Google Scholar]
- San Filippo J, Sung P, Klein H. 2008. Mechanism of eukaryotic homologous recombination. Ann Rev Biochem 77: 229–257. [DOI] [PubMed] [Google Scholar]
- Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, et al. 2014. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods 11: 399–402. [DOI] [PubMed] [Google Scholar]
- Siehler SY, Schrauder M, Gerischer U, Cantor S, Marra G, Wiesmuller L. 2009. Human MutL-complexes monitor homologous recombination independently of mismatch repair. DNA Repair (Amst) 8: 242–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Snipe JR, Chan GK, Gordenin DA, Resnick MA. 2006. Conservative repair of a chromosomal double-strand break by single-strand DNA through two steps of annealing. Mol Cell Biol 26: 7645–7657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathern JN, Klar AJ, Hicks JB, Abraham JA, Ivy JM, Nasmyth KA, McGill C. 1982. Homothallic switching of yeast mating type cassettes is initiated by a double-stranded cut in the MAT locus. Cell 31: 183–192. [DOI] [PubMed] [Google Scholar]
- Suzuki T. 2008. Targeted gene modification by oligonucleotides and small DNA fragments in eukaryotes. Front Biosci 13: 737–744. [DOI] [PubMed] [Google Scholar]
- Swuec P, Costa A. 2014. Molecular mechanism of double Holliday junction dissolution. Cell Biosci 4: e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symington LS, Gautier J. 2011. Double-strand break end resection and repair pathway choice. Annu Rev Genet 45: 247–271. [DOI] [PubMed] [Google Scholar]
- Wolt JD, Wang K, Yang B. 2016. The regulatory status of genome-edited crops. Plant Biotech J 14: 510–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AV, Nunez JK, Doudna JA. 2016. Biology and applications of CRISPR systems: harnessing nature's toolbox for genome engineering. Cell 164: 29–44. [DOI] [PubMed] [Google Scholar]
- Wu L, Hickson ID. 2003. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 426: 870–874. [DOI] [PubMed] [Google Scholar]
- Wu XS, Xin L, Yin WX, Shang XY, Lu L, Watt RM, Cheah KS, Huang JD, Liu DP, Liang CC. 2005. Increased efficiency of oligonucleotide-mediated gene repair through slowing replication fork progression. Proc Natl Acad Sci 102: 2508–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Jaenisch R. 2014. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat Protoc 9: 1956–1968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.