

Abstract
Housekeeping genes of animal genomes cluster in the same chromosomal regions. It has long been suggested that this organization contributes to their steady expression across all the tissues of the organism. Here, we show that the activity of Drosophila housekeeping gene promoters depends on the expression of their neighbors. By measuring the expression of ∼85,000 reporters integrated in Kc167 cells, we identified the best predictors of expression as chromosomal contacts with the promoters and terminators of active genes. Surprisingly, the chromatin composition at the insertion site and the contacts with enhancers were less informative. These results are substantiated by the existence of genomic “paradoxical” domains, rich in euchromatic features and enhancers, but where the reporters are expressed at low level, concomitant with a deficit of interactions with promoters and terminators. This indicates that the proper function of housekeeping genes relies not on contacts with long distance enhancers but on spatial clustering. Overall, our results suggest that spatial proximity between genes increases their expression and that the linear architecture of the Drosophila genome contributes to this effect.
Eukaryotic genomes have an underlying architecture and the arrangement of genes is nonrandom (Hurst et al. 2004). The first hint of this functional organization came from the observation that the expression of a gene depends on its chromosomal location, a phenomenon known as position effects (Elgin and Reuter 2013). When X-ray mutagenesis allowed geneticists to induce chromosomal rearrangements, it was observed that the Drosophila white gene is silenced when translocated near the centromere (Muller 1930). Realizing that the genomic context can have an influence fueled the idea that the arrangement of eukaryotic genes is optimized for their expression.
This paradigm explained why heterochromatic regions such as telomeres and centromeres are generally gene-poor, but more intriguing patterns soon emerged. Chromosome staining revealed that housekeeping genes reside only in R chromosome bands in mammals (Filipski 1990). Whole genome sequencing further revealed that in animal genomes, housekeeping genes are clustered in the same chromosomal regions (Hurst et al. 2004; Vinogradov 2004). In humans, gene clusters are either tandem duplications or clusters of housekeeping genes (Lercher et al. 2002), indicating that the aggregation of housekeeping genes is one of the main features of genome organization.
More recently, large-scale mapping of chromatin proteins and histone marks revealed that housekeeping genes have their own chromatin signature in Drosophila (Filion et al. 2010; Kharchenko et al. 2010). In the five color classification by Filion et al. (2010), housekeeping genes lie in Yellow chromatin domains, which are gene-dense, typically span 3–5 genes, and map to interbands of polytene chromosomes (Zhimulev et al. 2014). In contrast, developmentally regulated genes lie in Red chromatin domains, characterized by the binding of a distinct set of proteins. It was suggested earlier that the linear clustering of housekeeping genes may facilitate the establishment of a proper configuration of chromatin (Vinogradov 2004), but so far no mechanism has been proposed.
What can bring housekeeping genes together? An obvious hypothesis is that this organization favors robust gene expression and reduces the chances of accidental silencing. At least three scenarios could support this view. In the first, Yellow chromatin may stop the spreading of nearby repressive chromatin. Linear clustering would then reduce the number of interfaces with repressive chromatin and thus stabilize expression. In the second scenario, housekeeping genes may be stimulated by specific enhancers (Zabidi et al. 2015). Clustering around the strongest enhancers would allow a large number of genes to benefit the increase in expression. Finally, in the third scenario, the transcription of a gene directly stimulates its neighbors (Feuerborn et al. 2015). Clustering would then directly increase transcription at a local scale.
These models can be tested by studying the influence of position effects on housekeeping genes. For instance, the first model implicitly assumes that genes transplanted out of Yellow chromatin should be repressed. The second and third models predict that transplanted genes should be most active in regions where they contact enhancers, and in the proximity of active genes, respectively. However, it is still an open question whether housekeeping genes are sensitive to position effects at all. Even though half of the expressed genes in any Drosophila cell type are housekeeping (Chintapalli et al. 2007), very little is known about their relationship with the genomic context.
Results
TRIP with Drosophila housekeeping promoters
Drosophila housekeeping genes form clusters of consecutive genes, which are themselves densely packed in the genome (Fig. 1A). To understand whether this configuration contributes to their expression, we used the TRIP technology (Thousands of Reporters Integrated in Parallel) (Akhtar et al. 2013) to study their sensitivity to position effects. Briefly, we randomly integrated thousands of reporter genes that are identical except for a random DNA barcode in the 3′ end of the transcription unit. These barcodes were then used to monitor the expression levels of all reporters in parallel in a pool of cells.
Figure 1.

Clusters of Drosophila housekeeping genes and experimental design. (A) Housekeeping genes (HK) are represented as purple boxes and regulated genes (REG) as gray boxes. The vertical bars on top represent the average expression of each gene across 30 conditions. Housekeeping genes form densely packed clusters interspersed by regulated genes. (B) Reporter libraries are generated by barcoding-PCR, introducing a random barcode. Upon co-electroporation with a Sleeping Beauty expression plasmid in Kc167 cells, barcoded reporters are integrated at random in the Drosophila genome. (C) Barcoded reporters are mapped by inverse PCR. Quantification of expression is performed by RT-PCR on the barcode and normalization for the copy number of each insertion by PCR with the same primers.
We constructed GFP reporter libraries tagged with random DNA barcodes (Fig. 1B; see Supplemental Methods) using an efficient Gibson ligation approach (Gibson et al. 2009). We cloned four housekeeping promoters of Drosophila upstream of GFP, inserted the barcoded reporters in the genome of Drosophila Kc167 cells by Sleeping Beauty transposition (Mátés et al. 2009), mapped them by inverse PCR, and quantified their expression by comparing barcode frequencies in the DNA and in the RNA (Fig. 1C). The promoters were chosen at random under the condition that they would drive detectable levels of GFP expression. Housekeeping promoters are usually short and self-contained (Zabidi et al. 2015) so the risk is small that the chosen 1-kb fragments lack a key element. We ruled out the presence of regulatory elements on the backbone (Supplemental Fig. S1) and ensured that barcode sequences have negligible effects on expression in more than 98% of the cases (Supplemental Fig. S2). Also, when reporters were inserted inside genes, their expression was independent of the relative orientation of the reporter, indicating that the signal does not originate from readthrough transcription (Supplemental Fig. S3A).
In total, we obtained expression data for 85,663 integrated reporters, 55,397 of which contain a promoter (Table 1), yielding a measure of position effects every 3 kb on average. Figure 2A shows that integrations have a mild bias toward introns of active genes (18% observed versus 15% expected) and away from exons (21% observed vs. 25% expected). This bias is partly accounted for by the difference in G+C content between exons and introns (48% vs. 40%) because Sleeping Beauty transposons integrate at TA dinucleotides (Mátés et al. 2009). Overall, this data set achieves unprecedented coverage and density of reporter expression (see also Supplemental Fig. S4).
Table 1.
Integration statistics and basic information about the TRIP reporters
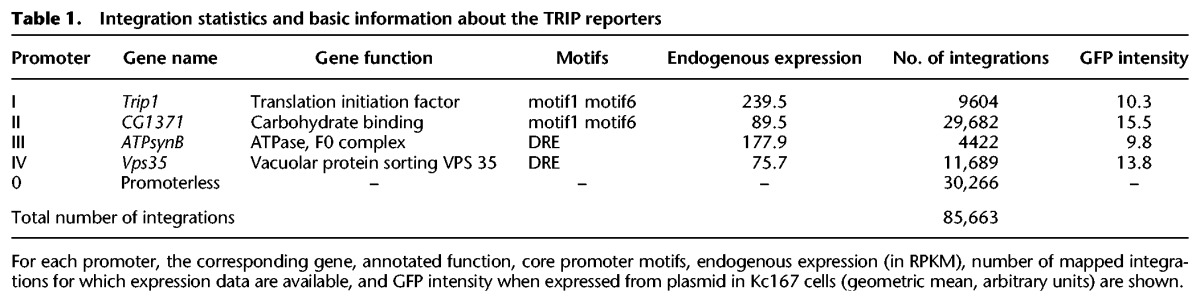
Figure 2.

TRIP reporters measure position effects. (A) The pie chart shows observed over expected percentage of insertions in promoters, exons, and introns for active and silent Drosophila genes. The observed frequency of insertion is close to the expected for each class. (B) Chromatin protein binding on the reporters is assayed simultaneously by DamID. If the protein binds the integrated reporter, Dam methylates the DNA nearby the barcode (locus 2); otherwise, it does not (locus 1). After digestion with the methylation-sensitive enzyme DpnII, only the methylated barcodes can be amplified by PCR, revealing which insertions were bound by the chromatin protein. (C) The violin plots show the DamID score for HP1 or Brahma (markers of Green and Red chromatin, respectively) when the promoter II is inserted in different regions of the Drosophila genome. HP1 is more bound when the reporters are inserted in Green chromatin and Brahma is more bound when they are inserted in Red chromatin. (D) Chromosomal structure at two loci with and without inserted reporters. The left panel shows the 4C profile with the barcode as a viewpoint; the right panel shows the corresponding slice of the Hi-C matrix (virtual 4C) without the reporter.
Once integrated, the reporters may acquire the chromatin of their surroundings or set up their own. To answer this question, we assayed the binding of key chromatin proteins on the integrated reporters simultaneously. Briefly, we used a modified DamID assay (van Steensel and Henikoff 2000), where nonmethylated barcodes are digested by the methylation-sensitive enzyme DpnII. The surviving barcodes represent integrated reporters bound by the protein of interest (Fig. 2B). We observed that the chromatin of the reporter mirrors its surrounding context (Fig. 2C; also see Supplemental Fig. S5). We also tested whether the insertion of the reporter perturbs the spatial organization of the locus. To this end, we compared Hi-C data obtained on Kc167 cells without insertion (Li et al. 2015) to 4C data performed on several integrated barcodes simultaneously (Fig. 2D). The similarity between the maps shows that the reporters hitchhike on preexisting chromosomal contacts but generally do not create their own. Our constructs thus give a readout of chromatin spreading and local chromosomal contacts, as required for reporters of position effects.
Contacts between genes drive position effects of housekeeping promoters
We first explored the global patterns of expression. As expected, reporters were less expressed in pericentric heterochromatin (Fig. 3A). In contrast, the expression of reporters integrated on chromosome arms varied widely. For each promoter, we observed coincident domains of high or low expression. We estimated that only 7% of the information was lost by pooling the promoter data sets (see Methods), indicating that the genomic context has the same influence on all the promoters of this study. This means that the Drosophila genome is divided into regions that are generally permissive or refractory to transcription. From this point, we pooled the promoter data sets. Using a Hidden Markov Model, we identified 866 domains of either high or low reporter expression (median size 48.2 and 32.6 kb, respectively) (Fig. 3B), where the mean signal differs by a factor of 5–7 (Fig. 3C). Thus, housekeeping promoters are sensitive to position effects, and the genomic context strongly influences their expression even outside pericentric heterochromatin.
Figure 3.

Magnitude of euchromatic position effects. (A) Violin plots showing the expression of reporters inserted in euchromatin versus heterochromatin. All the reporters are expressed at a lower level in heterochromatin (I-IV: promoter number, 0: no promoter control). (B) Expression profile of integrated reporters (each represented by a vertical bar). (Top) Profile of promoter II; (bottom) merged profiles of promoters I, III, and IV. Colors are assigned by a Hidden Markov Model (see Methods). Domains of either high or low expression are clearly visible. The domains coincide between profiles, showing that all the promoters have similar behaviors at the same location. (C) Violin plots showing the expression of reporters inserted in domains of high versus low expression. Labels as inA. (D) Violin plots showing the expression of the reporters inserted in different chromatin types defined as in Filion et al. (2010). The classification explains ∼15% of the variance (F test, P < 2.2 × 10−16). (E) Compaction of the chromatin fiber shown as the decay of contact frequencies. Beyond ∼5 kb, contacts decrease following a power law. Domains of high expression (green) are less compact than domains of low expression (purple). See definition of reporter expression score in Methods.
These observations prompted us to better understand how the genomic context contributes to regulate the genes. We focused on possible effects of chromatin composition as well as chromatin conformation. We found that the chromatin composition at the insertion site correlates with the expression of the reporters (Fig. 3D), and the average expression differs in each of the five chromatin types of Filion et al. (2010), reflecting the expression level of the endogenous genes. In addition, we observed that the domains of high and low expression have different three dimensional conformations (Fig. 3E). The decay of contact frequency as a function of linear distance is faster in the domains of higher expression, indicating that the chromatin fiber is more open and less compact than in the domains of low expression. This is in line with similar observations based on the expression of endogenous genes in Drosophila embryos (Sexton et al. 2012). Since both chromatin composition and chromosomal conformation could potentially explain position effects, we conducted computational analyses to determine which is the dominant mechanism.
We took a regression approach to predict the expression of the reporters from a repertoire of 112 chromatin features (van Bemmel et al. 2013), together with enhancer (Zabidi et al. 2015), promoter, and terminator contacts (Li et al. 2015). Surprisingly, the best individual predictor was the contact frequency with active terminators, closely followed by the contact frequency with active promoters (Fig. 4A). Both have a predictive power at least twice as high as any of the other factors. We observed the same for reporters inserted outside genes (Supplemental Fig. S3B). The average expression level of the endogenous genes flanking the reporters had a more than twofold lower predictive power, indicating that the three-dimensional conformation, instead of the linear proximity, contributes to position effects. For comparison purposes, we also added the predictive power of linear models based on chromatin states (Filion et al. 2010; Kharchenko et al. 2010). Even though these models have more parameters, they predict less accurately the expression of the reporters. The complete list of tested features is shown in Supplemental Table 1.
Figure 4.

Contacts with promoters and terminators of active genes best predict reporter expression. (A) Chromatin and conformational features are used individually to predict the expression level of the reporters. The feature with highest predictive power is the amount of contacts with terminators (i.e., the position of the 3′ end of each gene) of active genes, followed by contacts with promoters. Histone marks are indicated in orange, the mean expression of flanking genes in purple, and chromatin state models in pink. (B) Lasso multiple regression. By including more variables, the predictive power goes up to 0.33. Nineteen chromatin features are required to increase the predictive power from 0.25 to 0.29, and 89 are required to reach 0.32. This means that chromatin features are redundant and have multiple small effects.
In the above, contacts are inferred by proxy from Hi-C data obtained in wild-type cells (Supplemental Fig. S6). This is justified by the agreement between virtual and actual 4C (Fig. 2D), but minor variations of topology may add up to large deviations in contacts. When using the actual interaction profile of the reporters given by 4C on 73 barcodes (see Supplemental Methods), the predictive powers of the contact frequency with active promoters, terminators, and enhancers were 0.27, 0.26, and 0.18, respectively. These values are close to the estimates from Hi-C. This supports the role of contacts with active promoters and terminators in position effects.
Combining all the chromatin features with the best predictor raised the predictive power from 26% to 33%, meaning that the chromatin composition at the insertion site also influences the expression of the reporters. Beyond 20 features, the predictive power increased slowly, presumably because the predictors are redundant (Fig. 4B). Thus, the impact of the chromatin context typically results from the combination of many small and redundant effects. It is all the more striking that a single variable, the frequency of contacts with terminators, is responsible for almost 80% of the achievable predictive power. These results thus reveal that chromatin has a small but significant impact on euchromatic position effects, and they indicate that contacts with the terminators and promoters of active genes have a predominant role.
Reporters may have low expression in euchromatin
To characterize the chromatin of reporters expressed at low levels, we performed a principal component analysis (PCA) of the chromatin proteins present in the domains of low expression (purple domains in Fig. 3C). We obtained a cloud with a visible group of outliers (Fig. 5A). The chromatin features of those outliers are characteristic of active regions and contain, among others, H3K4me3 and RNA polymerase (Fig. 5A). Thus, a subset of the reporters inserted in euchromatin are expressed at low level. For this reason, we called these regions “paradoxical” chromatin domains.
Figure 5.

Paradoxical domains contain all the signatures of activity but are deficient in promoter and terminator contacts. (A) (Left) Principal component analysis of the chromatin features at insertion sites where the reporters are expressed at low levels. Highlighted: insertions with chromatin features divergent from the majority. (Right) Heat map of the chromatin features. The cloud highlighted on the PCA has the features of euchromatin. This means that some reporters expressed at low level are inserted in euchromatin (called paradoxical chromatin). (B) The violin plots show that paradoxical chromatin is rich in enhancers. The distribution of enhancer density in domains of high and low expression is shown for comparison. (C) Examples of paradoxical chromatin domains (shadowed in yellow). Reporters inserted in bun and shep are expressed at low levels (top track), yet the genes are expressed and euchromatic (bottom tracks). In contrast, the contacts with active promoters and terminators are low in bun and shep (second and third tracks). The HP1 and Polycomb (Pc) levels in bun and shep are typical of active genes. (D) Genes in paradoxical chromatin are exceptionally long (circular permutations, P < 0.001), causing a deficit of contacts with terminators. The notches and whiskers are default values from R (R Core Team 2016).
The PCA identified 304 paradoxical chromatin domains in the Drosophila genome (median size 6.5 kb, covering 3% of the genome, breakdown in chromatin colors: 73.5% Red, 18.9% Blue, 7.4% Yellow, 1.6% Black, and 0.2% Green). In total, 1319 reporters were integrated in paradoxical domains (representing 2.2%, 2.6%, 3.1%, and 3.0% of pI, pII, pIII, and pIV reporters, respectively). Of these insertions, 1200 were in genes, with a mild enrichment for the antisense orientation (652 versus 548), and 1062 were in introns. Paradoxical domains harbor expressed genes covered in active chromatin marks, and they are rich in enhancers (Fig. 5B), confirming that those regions are indeed euchromatic (also see Supplemental Fig. S7). In addition, Hi-C contact frequency decays in paradoxical domains with a power equal to −0.99, similar to domains of high reporter expression (Fig. 3E). Figure 5C shows two examples of paradoxical domains in the bun and shep genes. Both genes are expressed at high level, but the reporters inserted in their body are not. The factors that most commonly correlate with transcriptional repression, such as HP1, Polycomb, and Lamin, are not present in either bun or shep (the observed levels are typical of active genes). It is thus doubtful that the low expression of the reporters in paradoxical regions is due to a chromatin feature. It is also unlikely to be due to transcriptional interference, as reporters inserted in active genes are expressed at higher levels than reporters inserted in inactive genes (Supplemental Fig. S8) and reporters inserted in both orientations have a lower expression than average (Wilcoxon test, P < 2.2 × 10−16 each).
The one aspect that distinguishes paradoxical domains from regular euchromatin is that they contain exceptionally long genes (Fig. 5D). With a size of 98 kb, bun is more than 50 times larger than the median Drosophila gene (1.7 kb). More generally, 79% of the paradoxical domains intersect a gene longer than 10 kb. When genes are so long, insertions in their body are less likely to interact with the promoter or the terminator. Consistently, the paradoxical domains of bun and shep correspond to a depletion of contacts with promoters and terminators (Fig. 5C). These results show that an enhancer-rich and fully euchromatic environment is not sufficient to activate the reporters. This undermines the view that chromatin and enhancers play a critical role in position effects and instead gives support to the idea that they are driven by contacts with promoters and terminators.
Facilitated diffusion explains position effects
For the expression of a reporter to increase, the rate-limiting step of transcription must occur faster. Our results suggest that the contacts with active promoters and terminators supply a factor that facilitates this step. To better understand how this could happen at the molecular level, we turned to simulation modeling. Terminators contain few promoter motifs (Supplemental Table S3), so it is unlikely that they recruit transcription factors. A more reasonable hypothesis is that some of the complexes assembled during the transcription cycle can detach from the transcription unit and diffuse away. In fact, transcription termination may release mature complexes from the promoter or terminator of an active gene (Bentley 2014), which could stimulate the expression of nearby reporters.
Once unbound, active complexes follow the principles of facilitated diffusion on chromatin, i.e., they are quickly re-adsorbed because their positive charges (Brendel and Karlin 1989) are attracted to the negatively charged DNA. If another chromatin fiber is in close proximity, complexes may reattach at a different locus and “jump” over large distances on the linear genome while moving very little in physical space. These principles are well-established (Berg et al. 1981; Bénichou et al. 2011), but without the knowledge of the genome conformation, it has so far been impossible to use them for genomic analyses. Hi-C maps provide the first opportunity to model facilitated diffusion on the actual conformation of the genome, and TRIP data are the ideal readout to evaluate such models.
We assumed that rate-limiting factors are assembled at specific sites of the genome and that they diffuse from these sites until their spontaneous disassembly (Fig. 6A). Fully assembled complexes activate the transcription of the reporters they encounter, whereas individual subunits have no effect. We further assumed that the Hi-C matrix describes the probability that a complex detaching from one genomic site will then land on another genomic site. More precisely, each element Hij of the Hi-C matrix is proportional to the probability that the rate-limiting complexes land at site j after detaching from site i. Their trajectories are therefore random walks on the genome folded in the nuclear space (Avcu and Molina 2016). These modeling assumptions describe a “birth-diffusion-death” process (see Supplemental Methods), where birth and death correspond to assembly and disassembly of the complexes.
Figure 6.

The birth-diffusion-death model of position effects. (A) Sketch of the model. Rate-limiting complexes for expression are released from DNA at assembly sites and diffuse on chromatin until they dissociate. At each step of the diffusion process, the complexes “jump” to another site on the genome with a probability that is proportional to the value of the Hi-C contact matrix. (B) Models with unstable complexes better fit the expression of the reporters. The best model corresponds to release sites at both promoters and terminators, but it is only marginally better than assuming that complexes are released at terminators only. The black curve represents the predictive power of a null model where assembly sites have a uniform distribution. The half-life is measured in number of “jumps.”
We used the model to evaluate how the following hypotheses conform with our experimental data: (1) The complexes are assembled at the promoters of active genes; (2) they are assembled at the terminators of active genes; and (3) they are assembled at both the promoters and the terminators of active genes. For comparison, we also considered a null model where the complexes are assembled uniformly on the genome. We then calculated the Pearson correlation coefficient between the experimental reporter expression and the number of times a genomic site is visited by the rate-limiting complex (see Supplemental Methods).
We set the half-life of the complexes to a range of values and evaluated the models in each case. Surprisingly, the optimum was achieved for the most unstable complexes (Fig. 6B). Since the half-life of the complexes dictates the duration of the strolls after their release, this means that their diffusion is very brief and limited to a short range. This in turn means that only the reporters that are located very close to the release sites will have an increased expression.
The hypothesis that best fits the data is that the rate-limiting complexes are released at both the promoters and the terminators of active genes, with a very small margin over the hypothesis that those complexes are released only at the terminators of active genes. Because Drosophila genes are short and close to each other, it may be that contacts with active promoters always entail some contacts with the terminators. The hypothesis that position effects are driven by the release of active transcription complexes at promoters and terminators is compatible with our data, provided those complexes are very unstable. This in turn implies that the trans-activating effect of promoters and terminators act at very short range and only on the reporters that are in very close spatial proximity.
Discussion
Here, we used improved TRIP protocols and analysis tools (Zorita et al. 2015) to systematically assay the magnitude of position effects on housekeeping promoters in the Drosophila genome. We obtained a data set of ∼85,000 insertions in Kc167 cells, which, to our knowledge, is the largest of this kind to date. We discovered that housekeeping genes are subject to position effects, even away from pericentric heterochromatin. Our results also revealed that the transcriptional response is promoter-independent, indicating that the genomic context has a similar influence on different housekeeping genes. This allowed us to precisely delineate domains that are intrinsically permissive or refractory to the expression of housekeeping genes. Several types of domains were already defined in those cells (Filion et al. 2010; Kharchenko et al. 2010), but unlike those, TRIP domains, by definition, have a causal influence on transcription.
The major surprise of our results was that contacts with active promoters and terminators are the best predictors of reporter expression. The implication of terminators in position effects is in line with the observation that transcripts pile up at terminators in mammals (Kapranov et al. 2007), which suggests that they are sites of increased transcriptional activity. Unexpectedly, the chromatin at the insertion site and the contacts with enhancers were less predictive, among others because of paradoxical chromatin domains, where the reporters are expressed at low level in spite of a euchromatic environment.
Our model of birth-diffusion-death on chromatin suggests that some rate-limiting factor is released near the promoters and especially the terminators of active genes. The RNA polymerase itself is a poor candidate for this role, as it is available in large supply (Darzacq et al. 2007). Collisions between promoters and RNA polymerases happen hundreds of times per transcription cycle, so the extra molecules released at the terminator probably have a negligible effect. Instead, one of the most rate-limiting steps of transcription is the elongation checkpoint (Kwak et al. 2013), so we surmise that an elongation complex assembled at promoters and detaching at terminators may ease the elongation checkpoint on nearby promoters. It is also possible that contacts between active genes permit histone-modifying enzymes on one gene to activate another gene, as was recently suggested (Ulianov et al. 2016).
The spatial proximity of the reporters to promoters and terminators of active genes is the most significant determinant of the transcriptional activity of the reporters. Previous studies have provided clear evidence that the majority of Hi-C contacts are maintained when inhibiting transcription (Li et al. 2015). Taken together, these two observations suggest that the three-dimensional structure of the chromosomes determines the expression of the reporters and not vice versa. This conclusion is corroborated by the similarity of 3D contacts at a locus before and after integration of the reporters (Fig. 2D). Therefore, our data point to a causative role of the three-dimensional structure of the genome in determining the expression of housekeeping genes.
Spatial clustering between active genes is compatible with the view that transcription proceeds in “factories” (Rieder et al. 2012). It is possible that multiple active genes coalesce into compact structures where transcription is most efficient. However, it still remains to be determined whether the contacts activating the reporters are pairwise or involve multiple genes simultaneously.
The chromatin composition at the insertion site influences the expression of the reporters, but outside pericentric heterochromatin it consists of multiple redundant effects. This suggests that, for housekeeping genes, the chromatin context acts as a fine-tuning mechanism at a domain-wide scale. Given that Yellow chromatin is characteristic of housekeeping genes, it is surprising that it does not seem to play a more important role in their expression. The distinctive sign of Yellow chromatin is the presence of H3K36me3, which is a mark covering transcribed exons. This chromatin type is enriched on housekeeping genes because they have few introns. The 3′ end of developmentally regulated genes is typically exon-dense and covered in Yellow chromatin, even if the promoter lies in Red chromatin. It is thus possible that Yellow chromatin marks housekeeping genes without contributing to their high expression.
There is ample evidence that endogenous enhancers can activate integrated transgenes (Kvon 2015), so it is surprising that they do not show a preponderant role in this study. It is important to note that enhancer trap and STARR-seq are performed with minimal promoters (Kvon 2015; Zabidi et al. 2015), whereas the promoters used here have not been truncated. It is possible that the effect of enhancers is visible on weak promoters but that their effects are less obvious on stronger promoters. Interestingly, STARR-seq housekeeping enhancers are highly enriched in promoters of active genes. This is consistent with our findings but again raises the question of why contacts with enhancers poorly predict reporter expression. An issue may be the sensitivity of STARR-seq, since many promoters of active genes were not picked up as enhancers (Kvon 2015; Zabidi et al. 2015). Unfortunately, it is unclear if the enhancer screens were performed to saturation. It is also possible that sequences activating transcription on a plasmid have little activity in chromatin. Finally, the fact that terminators are usually not picked up by STARR-seq indicates that their activity reported here is the byproduct of transcription rather than a putative enhancer-like activity.
This study focuses on housekeeping promoters, but it would also be interesting to assay other kinds of promoters with TRIP. Developmentally regulated genes are more difficult to study because their promoters are usually larger, and the distinction between distal versus cis-regulatory elements is less clear. More generally, it would be interesting to determine whether the domains of high and low expression identified here are universal, or if some other promoters show different patterns of position effects. More TRIP maps will be required to know how much our results can be generalized to other promoters. It will also be interesting to study regulated genes and test whether they also are coregulated when they cluster in space. A key question is what makes a domain transcriptionally active or inactive. We foresee that the cocktail of transcription factors expressed in the cell will play a major role, but further experiments are required to answer this question in full.
Finally, our results suggest the following interpretation for the organization of the Drosophila genome: If frequent contacts with the terminators of active genes increase expression, housekeeping genes may benefit from being in spatial proximity to other active genes. Since genomic loci contact most frequently their closest neighbors on the chromosome, we may expect that genomes where housekeeping genes are small and lie in linear proximity to each other have a higher fitness. In contrast, developmentally regulated genes should be shielded from transcriptional interference; the same principles thus explain why they are typically long and isolated. Such an organization has the benefit of maintaining activators of transcription close to active genes, thereby reducing accidental activation of other genes. This also implies that transcription is intrinsically noisy, as active genes influence nearby genes. By creating compartments, the spatial organization of the genome may reduce this noise and make transcription more specific and fine-tuned.
Methods
Cell culture
Kc167 cells were maintained in Schneider's Drosophila medium (Gibco). Twenty-four and 48 h after electroporation (see Supplemental Methods), the expression of the Sleeping Beauty 100× transposase and of LNGFR were induced by two heat shocks at 37°C of 2 h each, with at least 4 h recovery between them. At Day 3, after electroporation, LNGFR-positive cells were selected using MACSelect LNGFR micro-beads (Miltenyi biotech), and pools of 10,000, 20,000, or 50,000 cells were plated in 25-cm2 flasks containing 5 mL of medium and grown for 2 wk, transferring to a 75-cm2 flask when the culture reached a density of 107 cells/mL. This pooling was done in replicate for each promoter-construct to account for the biological variability.
RNA-seq
RNA was extracted using TRIzol (Life Technologies). One hundred micrograms of total RNA were taken for poly(A)+ selection (Oligotex mRNA mini kit, Qiagen). Reverse transcription of the reporter RNA (ThermoScript, Life Technologies) was performed with 2 µg mRNA using primer 14 (Supplemental Table 2). PCR was performed with primers 12 and 15 (Supplemental Table 2). Using the same primers and conditions, two PCRs were done with 500 ng of DNA to amplify all the barcodes present in the cell population and normalize for barcode abundance.
Data sets for bioinformatic analyses
All analyses were performed with FlyBase genome assembly release dm3/R5, and gene model and annotations were taken as in version 57 (r5.57_FB2014_03), downloaded from ftp://flybase.org. Pericentric heterochromatin was defined according to the annotations available from the genome assembly (i.e., scaffolds with “Het” suffix). Gene expression data during Drosophila development (Brown et al. 2014) were downloaded from http://www.nature.com/nature/journal/v512/n7515/full/nature12962.html (csv file, Supplemental Data 9). The chromatin colors in Kc167 (Filion et al. 2010) were downloaded from GEO (accession GSE22069). The nine modENCODE chromatin states (Kharchenko et al. 2010) were downloaded from http://www.modencode.org/. Binding data for 112 chromatin features (van Bemmel et al. 2013) were downloaded from GEO (accession number GSE36175, file GSE36175_norm_aggregated_tiling_arrays.txt.gz; the data set consists of 107 DamID-array and five ChIP-array profiles performed in Kc167 cells). Raw Hi-C data sets (Li et al. 2015) were downloaded from GEO (accessions GSM1551442, GSM1551443, and GSM1551444). The coordinates of the STARR-seq enhancers (Zabidi et al. 2015) were downloaded from GEO (accession GSE57876) as processed data files.
Definition of housekeeping genes
Gene expression data containing 30 developmental time points and conditions were generated by the modENCODE Consortium (Brown et al. 2014). We defined a gene as housekeeping if, in every condition, its expression was higher than the 40th percentile of expression in this condition. This yielded 5161 housekeeping genes out of 15,139.
4C data processing
Sequenced reads were filtered to ensure the presence of the restriction sites for NlaIII and MluCI before mapping with GEM (Marco-Sola et al. 2012) to the Drosophila genome with options -m3 --unique-mapping. Barcodes were clustered using Starcode (Zorita et al. 2015), allowing two errors. Contaminant reads (where the barcode belongs to another promoter library) and barcodes with less than 100 reads were removed. For each barcode, the viewpoint was selected as the NlaIII fragment with the highest read count (which was always correct for mapped barcodes).
Hi-C data processing
Reads were trimmed 3′ of the first GATC before mapping to Drosophila genome release dm3/R5 with GEM (Marco-Sola et al. 2012), as for 4C. To define contact frequencies, reads were pooled in 2000-bp bins. At this resolution, over 90% of the bins had more than 1000 contacts (Supplemental Fig. S6).
Definition of domains of high and low expression
A Hidden Markov Model (HMM) with two states and Student's t emission were fitted on the pooled expression profiles of the reporters using a custom R package available as Supplemental Source Code and from https://github.com/gui11aume/HMMt (see Supplemental Methods of Filion et al. 2010). Model parameters were fitted with the Baum-Welch algorithm, and domains were called with the Viterbi algorithm as in Filion et al. (2010). The information lost by pooling the promoter data sets was estimated by bootstrapping. Four data sets of size matching Table 1 but drawn at random from the pooled data were resampled 100 times. Four HMMs were fitted as above, and the agreement between the calls of the individual HMMs and the HMM with pooled data was measured. The information lost was the difference between this average agreement and the agreement measured on the non-resampled individual promoter data sets.
Data access
The raw and processed TRIP data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE71971. To ensure reproducibility, a virtual machine containing a complete and running version of the data processing pipeline is available as a Docker image available for download from the following link: https://hub.docker.com/r/histonemark/tripeline/.
Supplementary Material
Acknowledgments
We thank Dr. Miguel Beato and Dr. Ben Lehner for their critical review of the manuscript. We also thank the Sequencing core facility of the CRG and the Genomics core facility of the NKI. Finally, we thank the anonymous reviewers for their significant contribution to the manuscript. We acknowledge the support of the CERCA Programme / Generalitat de Catalunya. This work was supported by the Spanish Ministry of Economy and Competitiveness, “Centro de Excelencia Severo Ochoa 2013–2017,” SEV-2012-0208, Plan Nacional BFU2012-37168, NWO-ALW VICI, ERC Advanced Grant 293662 (B.v.S.), ERC Synergy Grant 609989 (G.J.F.), and the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme (FP7/2007-2013) under REA grant agreement no. 608959 (R.C.).
Author contributions: M.C., A.R., and G.J.F. prepared the manuscript. G.J.F. and B.v.S. designed the experiments. M.C., A.R., and J.v.A. performed the experiments. R.C. designed the diffusion model. M.C., A.R., and G.J.F. designed and performed the bioinformatics analyses.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.211433.116.
Freely available online through the Genome Research Open Access option.
References
- Akhtar W, Waseem A, de Jong J, Pindyurin AV, Ludo P, Wouter M, de Ridder J, Anton B, Wessels LFA, van Lohuizen M, et al. 2013. Chromatin position effects assayed by thousands of reporters integrated in parallel. Cell 154: 914–927. [DOI] [PubMed] [Google Scholar]
- Avcu N, Molina N. 2016. Chromatin structure shapes the search process of transcription factors. bioRxiv 10.1101/050146. [DOI] [Google Scholar]
- Bénichou O, Chevalier C, Meyer B, Voituriez R. 2011. Facilitated diffusion of proteins on chromatin. Phys Rev Lett 106: 038102. [DOI] [PubMed] [Google Scholar]
- Bentley DL. 2014. Coupling mRNA processing with transcription in time and space. Nat Rev Genet 15: 163–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg OG, Winter RB, von Hippel PH. 1981. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry 20: 6929–6948. [DOI] [PubMed] [Google Scholar]
- Brendel V, Karlin S. 1989. Association of charge clusters with functional domains of cellular transcription factors. Proc Natl Acad Sci 86: 5698–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JB, Boley N, Eisman R, May GE, Stoiber MH, Duff MO, Booth BW, Wen J, Park S, Suzuki AM, et al. 2014. Diversity and dynamics of the Drosophila transcriptome. Nature 512: 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintapalli VR, Jing W, Dow JAT. 2007. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet 39: 715–720. [DOI] [PubMed] [Google Scholar]
- Darzacq X, Xavier D, Yaron S-T, de Turris V, Yehuda B, Shenoy SM, Phair RD, Singer RH. 2007. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol 14: 796–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgin SCR, Reuter G. 2013. Position-effect variegation, heterochromatin formation, and gene silencing in Drosophila. Cold Spring Harb Perspect Biol 5: a017780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuerborn A, Alexander F, Cook PR. 2015. Why the activity of a gene depends on its neighbors. Trends Genet 31: 483–490. [DOI] [PubMed] [Google Scholar]
- Filion GJ, van Bemmel JG, Braunschweig U, Talhout W, Kind J, Ward LD, Brugman W, de Castro IJ, Kerkhoven RM, Bussemaker HJ, et al. 2010. Systematic protein location mapping reveals five principal chromatin types in Drosophila cells. Cell 143: 212–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipski J. 1990. Evolution of DNA sequence contributions of mutational bias and selection to the origin of chromosomal compartments. In Advances in mutagenesis research (ed. Obe G), pp. 1–54. Springer, New York. [Google Scholar]
- Gibson DG, Lei Y, Ray-Yuan C, Craig Venter J, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6: 343–345. [DOI] [PubMed] [Google Scholar]
- Hurst LD, Pál C, Lercher MJ. 2004. The evolutionary dynamics of eukaryotic gene order. Nat Rev Genet 5: 299–310. [DOI] [PubMed] [Google Scholar]
- Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, Stadler PF, Hertel J, Hackermuller J, Hofacker IL, et al. 2007. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316: 1484–1488. [DOI] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Aki M, Riddle NC, Jason E, Sabo PJ, Erica L, Gorchakov AA, Tingting G, et al. 2010. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471: 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvon EZ. 2015. Using transgenic reporter assays to functionally characterize enhancers in animals. Genomics 106: 185–192. [DOI] [PubMed] [Google Scholar]
- Kwak H, Fuda NJ, Core LJ, Lis JT. 2013. Precise maps of RNA polymerase reveal how promoters direct initiation and pausing. Science 339: 950–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lercher MJ, Urrutia AO, Hurst LD. 2002. Clustering of housekeeping genes provides a unified model of gene order in the human genome. Nat Genet 31: 180–183. [DOI] [PubMed] [Google Scholar]
- Li L, Lyu X, Hou C, Takenaka N, Nguyen HQ, Ong C-T, Cubeñas-Potts C, Hu M, Lei EP, Bosco G, et al. 2015. Widespread rearrangement of 3D chromatin organization underlies polycomb-mediated stress-induced silencing. Mol Cell 58: 216–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco-Sola S, Sammeth M, Guigó R, Ribeca P. 2012. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat Methods 9: 1185–1188. [DOI] [PubMed] [Google Scholar]
- Mátés L, Chuah MKL, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A, Grzela DP, Schmitt A, Becker K, Matrai J, et al. 2009. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet 41: 753–761. [DOI] [PubMed] [Google Scholar]
- Muller HJ. 1930. Types of visible variations induced by X-rays in Drosophila. J Genet 22: 299–334. [Google Scholar]
- R Core Team. 2016. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- Rieder D, Dietmar R, Zlatko T, McNally JG. 2012. Transcription factories. Front Genet 3: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. 2012. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148: 458–472. [DOI] [PubMed] [Google Scholar]
- Ulianov SV, Khrameeva EE, Gavrilov AA, Flyamer IM, Kos P, Mikhaleva EA, Penin AA, Logacheva MD, Imakaev MV, Chertovich A, et al. 2016. Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res 26: 70–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bemmel JG, Filion GJ, Rosado A, Talhout W, de Haas M, van Welsem T, van Leeuwen F, van Steensel B. 2013. A network model of the molecular organization of chromatin in Drosophila. Mol Cell 49: 759–771. [DOI] [PubMed] [Google Scholar]
- van Steensel B, Henikoff S. 2000. Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol 18: 424–428. [DOI] [PubMed] [Google Scholar]
- Vinogradov AE. 2004. Compactness of human housekeeping genes: selection for economy or genomic design? Trends Genet 20: 248–253. [DOI] [PubMed] [Google Scholar]
- Zabidi MA, Arnold CD, Schernhuber K, Pagani M, Rath M, Frank O, Stark A. 2015. Enhancer-core-promoter specificity separates developmental and housekeeping gene regulation. Nature 518: 556–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhimulev IF, Zykova TY, Goncharov FP, Khoroshko VA, Demakova OV, Semeshin VF, Pokholkova GV, Boldyreva LV, Demidova DS, Babenko VN, et al. 2014. Genetic organization of interphase chromosome bands and interbands in Drosophila melanogaster. PLoS One 9: e101631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorita E, Cuscó P, Filion GJ. 2015. Starcode: sequence clustering based on all-pairs search. Bioinformatics 31: 1913–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.