

Abstract
Many fungal pathogens must enter plant cells for successful colonization. Barley mildew resistance locus o (Mlo) is required for host cell invasion upon attack by the ascomycete powdery mildew fungus, Blumeria graminis f.sp. hordei, and encodes the founder of a family of heptahelical integral membrane proteins unique to plants. Recessively inherited loss-of-function mutant alleles (mlo) result in effective penetration resistance to all isolates of the biotrophic parasite. We used noninvasive fluorescence-based imaging to show that fluorescently tagged MLO protein becomes redistributed in the plasma membrane (PM) and accumulates beneath fungal appressoria coincident with the initiation of pathogen entry into host cells. Polarized MLO accumulation occurs once upon attack and appears to be independent of actin cytoskeleton function. Likewise, barley ROR2 syntaxin, a genetically defined component of penetration resistance to B. graminis f.sp. hordei, and a subset of predicted PM-resident proteins become redistributed to fungal entry sites. We previously identified calmodulin, a cytoplasmic calcium sensor, as an interactor and positive regulator of MLO activity and demonstrate here by FRET microscopy an increase in MLO/calmodulin FRET around penetration sites coincident with successful host cell entry. Our data provide evidence for the formation of a pathogen-triggered PM microdomain that is reminiscent of membrane microdomains (lipid rafts) induced upon attempted entry of pathogenic bacteria in animal cells.
Keywords: FRET, lipid raft, mildew resistance locus O, pathogen entry, powdery mildew
Host cell entry defines a critical phase during pathogenesis of animal pathogenic bacteria and many fungal parasites of plants. In some cases, this stage has been shown to require particular host proteins (1, 2). In both the dicot Arabidopsis and the monocot barley, specific isoforms of the family of plasma membrane (PM)-localized mildew resistance locus O (MLO) proteins are required for entry of the respective host powdery mildew species in leaf epidermal cells (refs. 3 and 4 and C. Consonni, P.S.-L., and R.P., unpublished data). It is thought that the fungal pathogen manipulates, directly or indirectly, MLO to suppress a vesicle-associated and soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) protein-dependent resistance response at the cell periphery (2, 5, 6). Barley Ror1 and Ror2 are two genes that are required for full expression of mlo resistance and are thus assumed to be components of penetration resistance. Barley Ror2 and its Arabidopsis ortholog, PEN1, encode PM-resident syntaxins containing a SNARE domain (5). A barley synaptosome-associated protein of 25 kDa (SNAP25) homolog, HvSNAP34, is also required for penetration resistance to Blumeria graminis f.sp. hordei (Bgh) and has been shown to form a binary SNARE complex with ROR2. These findings have been interpreted as evidence for the existence of a SNARE complex-dependent resistance mechanism acting at the cell periphery against powdery mildew ingress (5, 6).
Attempted powdery mildew invasion coincides also invariably with a focal deposition of newly synthesized host cell wall material and callose at entry sites, and the timing of the deposition of these cell wall appositions appears to be delayed in Arabidopsis pen1 mutant plants (7). The concurrent induction of an oxidative microburst and the accumulation of plant phenolics at fungal entry sites might contribute to terminate fungal ingress by cell wall fortification and direct antimicrobial activities (5, 7, 8). Here, we show that the powdery mildew fungus induces at entry sites the formation of PM microdomains. This induction leads to a colocalization of both positive and negative regulatory components of penetration resistance, thereby revealing a potential strategy of the parasite to exploit a functional domain that enables avoidance from the host's immune response and allows host cell invasion.
Methods
Plant and Fungal Material. Hordeum vulgare Golden Promise (Mlo) and Ingrid mlo-3 were used for this study. Bgh (isolate K1) and Blumeria graminis f.sp. tritici were propagated on susceptible cultivars. Transgenic Arabidopsis lines expressing GFP-PEN1 were described (5). Leaves inoculated with Bgh spores were mounted in a solution of 2.5% mannitol/0.01% silwet/0.2% propidium iodide (Sigma) to stain fungal structures for visualization by means of fluorescence microscopy. Filipin (Sigma) staining of Bgh-challenged leaf sections was performed as described (9).
Plasmids and Transient Single-Cell Gene Expression. Gateway (Invitrogen) cloning technology was used to create destination vectors containing the maize ubiquitin promoter plus CFP or YFP placed either C- or N-terminally of a Gateway recombination cassette. Transient gene expression in barley epidermal cells mediated by particle bombardment was essentially performed as described (10). For delivery of multiple constructs, equimolar plasmid mixtures were coated onto the gold particles.
Fluorescence Microscopy and FRET-Acceptor Photobleaching (APB). Analysis of intracellular fluorescence was performed by confocal laser-scanning microscopy using a LSM 510 META microscopy system (Zeiss) equipped with Argon ion and He-Ne lasers. The 488-nm Argon ion laser line was used to cross-excite GFP [or yellow fluorescent protein (YFP)] with dsRed and/or propidium iodide. The emitted light was collected in the lambda spectrum mode between 494 and 644 nm. Reference spectra of GFP, YFP, dsRed, and/or propidium iodide were used to linearly unmix the relevant spectra. Unspecific autofluorescence was subtracted accordingly. Colocalization analysis and FRET-APB were carried out as described (11, 12). Calculations of FRET efficiencies were performed according to ref. 11.
For disruption of actin cytoskeleton function, cells were cobombarded with MLO-YFP (or YFP-ROR2), peroxisome-targeted dsRed, and barley HvADF3 carrying a S6A amino acid substitution to enhance activity (M.M., P.S.-L., and R.P., unpublished work). Forty-eight hours after bombardment, leaves were analyzed for peroxisome-targeted dsRed mobility to assess impairment of actin cytoskeleton function. Thereafter, leaves were inoculated with Bgh spores and analyzed at 10–12 hours postinoculation (hpi). Leaves were mounted in 0.85 M sodium chloride for 10 min to achieve plasmolysis (5).
Fluorescence Recovery After Photobleaching (FRAP). A region of interest (ROI) around focally accumulated GFP-PEN1 in Bgh-challenged leaves of transgenic Arabidopsis plants was bleached by using the 488-nm Argon laser line 25–50 times at 100% intensity. Bleaching times varied from 3 to 7 sec, depending on the size of the ROI. Before and after bleaching, images were collected in the lambda spectrum mode. Reference spectra were used to linearly unmix the fluorescence arising from GFP-PEN1 and the propidium iodide-stained spores. To minimize the effects of photobleaching due to imaging, images were collected at 1–2% laser intensity.
Fluorescence Lifetime Imaging Microscopy (FLIM). FLIM was performed by using a Coherent Mira 900-F multiphoton system in combination with a Zeiss 200M inverted microscope. Two-photon excitation (TPE) at 820 nm was used to excite CFP. TPE pulses were generated by a Ti:Sapphire laser (Coherent, Santa Clara, CA). Fluorescent light was detected by using the nondescanned single-photon counting detection. FLIM detection was performed with a Hamamatsu R3809U MCP photomultiplier (Hamamatsu City, Japan; time resolution of 50 ps). CFP emission was selected via a 480DF30 nm band-pass filter. Images were acquired by using the Becker and Hickl SPC 730 module (Berlin; ref. 13). From the intensity images, complete fluorescence lifetime decays were calculated per pixel and fitted by using a double-exponential decay model. The lifetime of one component was fixed to the value found for MLO-CFP (2.5 ns).
Results
To examine a potential pathogen-triggered subcellular compartmentalization of MLO, we translationally fused the cytoplasmic C terminus of the protein with YFP (yellow variant of GFP). In an mlo null mutant background, we examined the subcellular localization of this functional fusion protein (10) after ballistic transformation of barley leaf epidermal cells in the absence or presence of Bgh sporelings by using confocal laser-scanning microscopy (Fig. 1 A–C). In the absence of the pathogen, most of the MLO-YFP fusion protein localized at the cellular periphery (Fig. 1 A). Consistent with biosynthesis of PM-resident proteins at the ER and subsequent transport via the secretory pathway, we detected some protein also in presumptive endomembranes and at the nuclear envelope. Upon challenge with Bgh conidiospores, we observed a striking focal accumulation of the fusion protein beneath fungal appressoria at ≈10–14 hpi (Fig. 1 B and C).
Fig. 1.

Induced polarity in host cells and powdery mildew sporelings. (A–I) Pathogen-triggered cell polarity in plant cells. A barley epidermal cell expressing MLO-YFP (A–C) or YFP-ROR2 (D–F) in the absence and presence of Bgh sporelings (10–14 hpi). (G–I) Nonchallenged and Bgh-challenged Arabidopsis epidermal cells expressing GFP-PEN1. Epiphytic fungal structures (red) were stained with propidium iodide. Stages of fungal development on cross-sectioned epidermal cells are depicted schematically above the columns. Images shown are single focal planes. Close-up views of attempted fungal entry sites for MLO-YFP, YFP-ROR2, and GFP-PEN1 expressing cells are shown in C, F, and I, respectively. White arrowheads mark the positions of focally accumulated fusion proteins. s, conidiospore. (Scale bar, 20 μm.) (J–M) Filipin-mediated fluorescence in Bgh conidiospores and at pathogen entry sites. Bgh-challenged leaf sections were stained with filipin. A representative conidiospore before germination (J), overview of germinated conidia (K) and a close-up view of germinated conidiospores (L and M) are shown. In J and L, arrowheads mark opposite poles of the conidium and the septum between the spore body and AGT, respectively. Note the filipin-mediated fluorescence underneath the appressorium in the attacked host epidermal cell (M, marked by arrowhead). s, conidiospore. (Scale bar, 20 μm.) (N–U) A barley epidermal cell expressing either MLO-YFP or YFP-ROR2 together with HvADF3 S6A blocking actin cytoskeleton function. Disruption of actin cytoskeleton was monitored by coexpression of a peroxisome-targeted dsRed (p-dsRed) variant in the same cells. (N–P) Coexpression of MLO-GFP, p-dsRed, and HvADF3 S6A. (Q–S) Coexpression of YFP-ROR2, p-dsRed, and HvADF3. (T and U) Close-up views of attempted fungal entry sites in the same cells. The stage of fungal development on a cross-sectioned epidermal cell is depicted schematically above the columns. (Scale bar, 20 μm.)
Similar to fluorophore-tagged MLO fusions, we found that N-terminally YFP-tagged ROR2 (YFP-ROR2) also accumulates beneath attempted fungal penetration sites at ≈10–14 hpi (Fig. 1 D–F). Furthermore, accumulation of YFP-ROR2 occurred independent of MLO because an indistinguishable accumulation pattern was seen in an mlo null mutant background (data not shown). Next, we analyzed transgenic Arabidopsis lines expressing a functional fusion of PEN1 with GFP (GFP-PEN1), driven by the CaMV 35S promoter (in pen1-1 null mutant background) or by native 5′ upstream sequences (in Col-0 background; ref. 5). In accordance with an independent study (7), we found that in both lines PM-resident GFP-PEN1 accumulated beneath fungal appressoria (Fig. 1 G–I), indicating that the subcellular relocalization is independent of gene expression levels. In addition, this finding suggests that pathogen-triggered focal accumulation of Arabidopsis PEN1 and barley ROR2 syntaxins is a conserved feature in dicot and monocot epidermal cells. Interestingly, of the two germ tubes formed by Bgh, accumulation of MLO-YFP, YFP-ROR2, and GFP-PEN1 was seen exclusively beneath appressorial, but not below, primary germ tubes (see Discussion).
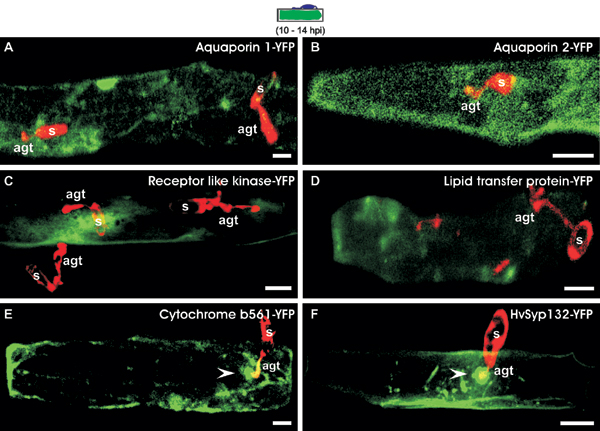
To distinguish whether focal protein accumulation upon fungal challenge is a general phenomenon of integral PM polypeptides or is restricted to a subset of these proteins, we selected six barley genes encoding predicted PM proteins known to be expressed in the leaf epidermis for further analysis (two aquaporin isoforms, a Xa21-like receptor kinase, a lipid transfer protein, cytochrome b561, and a homolog of Arabidopsis syntaxin SYP132). We fused the C terminus (except SYP132, which was fused N-terminally) of the respective genes with YFP, transiently expressed the resulting constructs in barley epidermal cells, and inoculated the leaves with Bgh conidiospores. Although each of the tested fusion proteins accumulated as expected in the periphery of leaf epidermal cells in the absence of the pathogen, we failed to detect evidence for pathogen-triggered focal accumulation upon expression of either of the two aquaporin isoforms, the Xa21-like receptor kinase, or the lipid transfer protein (Fig. 5, which is published as supporting information on the PNAS web site). Likewise, neither cytoplasmic CFP or YFP nor an anonymous PM-localized fluorophore-tagged tobacco protein (provided by K. Oparka, Scottish Crop Research Institute, Dundee, Scotland, U.K.) accumulated beneath fungal appressoria (data not shown). Unlike this finding, cytochrome b561 and the SYP132-related syntaxin were redistributed beneath attempted invasion sites. Thus, a subset of integral PM proteins becomes recruited to sites of pathogen entry, indicating the formation of a pathogen-triggered microdomain in the host PM.
The formation of PM microdomains in animals and yeast is known to be often associated with a polarization of sterol-rich domains and this can be visualized by using the fluorochrome filipin (14). We detected a striking accumulation of filipin-mediated fluorescence at opposite poles of nongerminated Bgh conidiospores (Fig. 1J). After spore germination, filipin staining became diffuse in the spore body but concentrated in appressoria, the tip of appressorial germ tubes (AGTs), suggesting that membrane sterols become polarized to the leading edge of fungal growth (Fig. 1 K–M). Filipin staining was also enriched at the septum, which is known to separate the spore body from the AGT (Fig. 1L). Collectively this pattern is essentially identical to the one described recently for Candida albicans, a human pathogenic ascomycete fungus (14). At ≈12–15 hpi, circular filipin-mediated fluorescence was also detectable beneath appressoria in epidermal cells (Fig. 1M), possibly indicating redistribution and polarization of plant sterols in the epidermal PM.
We examined whether the accumulation of proteins beneath appressoria results from lateral concentration processes of preformed PM resident proteins or from de novo-synthesized protein transported via secretory pathway(s). Because the latter is thought to require actin filament-guided vesicle trafficking, we disrupted actin cytoskeleton function in epidermal cells by overexpression of a barley actin depolymerizing factor (HvADF3), which leads to a complete arrest of actin-dependent transport processes (e.g., peroxisome movement; M.M., P.S.-L., and R.P., unpublished work). Coexpression of HvADF3, a peroxisome-targeted dsRed variant, and MLO-YFP or YFP-ROR2 showed in most inspected cells focal accumulation at attempted penetration sites, despite immotile and aggregated peroxisomes (Fig. 1 N–U). We conclude that formation of the Bgh-triggered microdomain is unlikely to involve actin filament-mediated transport processes.
Next, we asked whether pathogen-triggered polarized protein accumulation is a continuous process or is triggered once in response to the intruder. We performed FRAP experiments by using Bgh-challenged transgenic Arabidopsis plants expressing GFP-PEN1 at 12 hpi (Fig. 2 A–G). Upon bleaching of GFP-PEN1 at focal accumulation sites, there was no significant fluorescence recovery during a 6-h recovery period (Fig. 2 A–C and G). In contrast, we found a rapid recovery of GFP-PEN1 fluorescence (within 25 min) after bleaching of PM-resident GFP-PEN1 at a distance from entry sites (Fig. 2 D–F and G). These data suggest that the process of protein polarization is triggered only once after Bgh attack, whereas replenishment of GFP-PEN1 at a distance occurs rapidly and continuously. Because we could not resolve whether the accumulation of barley MLO-YFP and YFP-ROR2 occurs in the PM, in cell wall appositions, or in both, we inoculated leaves of Arabidopsis GFP-PEN1-expressing plants with Bgh conidiospores, and at 12 hpi, plasmolysed the cells to retract the protoplast from the cell wall. After plasmolysis (Fig. 2 I and J), focally accumulated GFP-PEN1 (Fig. 2H) was completely retracted beneath fungal appressoria in 95% of inspected cells, suggesting that PEN1 accumulation sites are physically separable from cell wall appositions.
Fig. 2.

Focal PEN1 accumulation is triggered once and occurs at the cytoplasmic face of biotic stress sites. (A–F) Arabidopsis leaves expressing GFP-PEN1 were inoculated with Bgh conidiospores. At 12 hpi, FRAP analysis was performed at (A–C) and away from (D–F) focal accumulation (FA) sites. White arrowheads mark the position of focally accumulated GFP-PEN1 and the yellow flash depicts the site of photobleaching. (G) Quantitative measure of fluorescence recovery over time at and away from FA sites. Note that the fluorescence recovery is incomplete (≈75%) away from FA sites because of continuous photobleaching during imaging. (H–J) Bgh-challenged Arabidopsis leaves stably expressing GFP-PEN1 were imaged at 12 hpi before (H) and after (I and J) plasmolysis. st, stomata; he, Hechtian threads. The stage of fungal development on a cross-sectioned epidermal cell is schematically depicted above the columns. (Scale bar, 20 μm.)
Pathogen-triggered focal MLO accumulation prompted us to examine the spatiotemporal interaction dynamics with calmodulin (CaM), known to be required for full MLO activity (15). An α-helical amphiphilic stretch in the cytoplasmic C-terminal tail of MLO functions as CaM-binding domain (CaMBD), and single amino acid substitutions in the CaMBD abolish CaM binding, both in vitro and in vivo. Fusion constructs of barley MLO and CaM were generated by using genes encoding YFP and CFP (cyan fluorescent protein). CFP-CaM was found to localize in the cytoplasm, the nucleus, and at the cell periphery. Upon coexpression of MLO-YFP and CFP-CaM, we detected colocalization at the cell periphery, at endomembranes, and around the nucleus, thereby revealing potential interaction sites (Fig. 3 A–C).
Fig. 3.

MLO interacts with CaM in nonchallenged barley epidermal cells. (A–C) Barley leaf epidermal cells were cobombarded with fluorescently tagged MLO (A) and CaM (B) and analyzed by confocal microscopy. The white arrowhead in A marks the position of focally accumulated MLO-YFP at an attempted fungal entry site. N, nucleus. Note that the images shown are 3D reconstructions from individual image stacks. (Scale bar, 20 μm.) (D) FRET-APB analysis of the interaction between fluorescently tagged MLO and CaM. Mean FRET efficiencies (black bars) and background FRET (white bars) ± SD from 50–100 sample sites are shown. MLO is depicted by its serpentine structure, CaM is a purple circle, and CFP and YFP fluorophores are cyan and yellow ribbon models, respectively. Red dots in the C-terminal tail of MLO symbolize the W423R and L420R/W423R amino acid substitutions in the CaMBD domain. (E–J) Pseudocolored fluorescence lifetime (FRET-FLIM) images of barley epidermal cells expressing combinations of CFP, YFP, and fluorescently tagged MLO and CaM forms. The bar at the top displays the scale of CFP donor fluorescence lifetimes in pseudocolors from blue (2.5 ns, no interaction) to red (0.0 ns, very strong interaction). (Scale bar, 20 μm.)
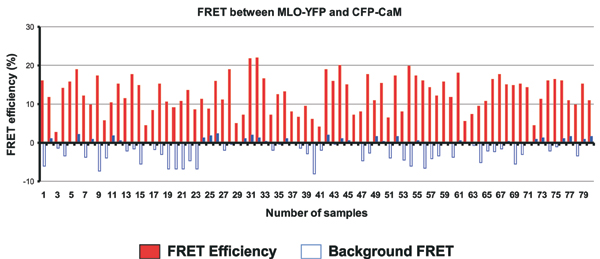
To directly study the physical interaction between MLO and CaM, the respective YFP and CFP fusion proteins were first used as donor-acceptor pairs in FRET studies in the absence of the fungus. APB and FLIM were applied to record FRET (11–13). For each pair of constructs tested, data were recorded from at least 50–100 sample sites in at least 50 transformed epidermal cells. Mean FRET efficiencies of 12.2 ± 4.6% were obtained for energy transfer between MLO-YFP as an acceptor and CFP-CaM as a donor (average ± SD, Fig. 3D; note that typical FRET-APB efficiencies for protein interaction pairs range from 10% to 30%). We also swapped acceptor and donor fluorophores and recorded an almost identical FRET efficiency of 12.3 ± 4.1% (Fig. 3D). Importantly, energy transfer between MLO-YFP and CFP-CaM was significantly reduced when mutant variants of MLO bearing either a single (W423R) or a double amino acid substitution in the CaMBD (L420R/W423R) were used for APB experiments (W423R, 2.7 ± 2.3%; L420R/W423R, 2.1 ± 2.8%). Similar low-level FRET signals were recorded in control experiments involving combinations of MLO-YFP/CFP (-0.3 ± 1.4%), YFP/CFP (2.3 ± 2.7%), or mock FRET (CFP alone; -0.4 ± 1.7%; Fig. 3D). The background FRET signal seen upon expression of CFP and YFP (2.3 ± 2.7%) is likely due to an intrinsic affinity of the two fluorochromes to form dimers at elevated concentrations (16). Collectively, the data demonstrate that the CFP-CaM fusion protein retains the ability to bind to the authentic CaMBD, and shows the requirement of an intact CaMBD for efficient FRET between MLO and CaM. The findings further suggest that, despite nonphysiological expression levels, the FRET signal reflects bona fide MLO/CaM interactions in living cells in the absence of Bgh.
We noted that FRET efficiencies varied from cell to cell and from region to region within the same cell, possibly reflecting the dynamic nature of this protein–protein interaction in living cells (Fig. 6, which is published as supporting information on the PNAS web site). To obtain spatial information about the MLO/CaM interaction in horizontal sections of a whole epidermal cell, we used time-correlated single-photon counting FLIM as an additional FRET sensor (12, 13). FRET-FLIM allows the direct study of protein–protein interactions by monitoring fluorescence lifetime of the donor (CFP) molecule. Whereas epidermal cells expressing either CFP, or CFP-CaM, or a combination of CFP and YFP, showed donor life times of ≈2.5 ns (Fig. 2 E–G), there was a marked reduction in fluorescence lifetime of the donor when CFP-CaM was coexpressed with MLO-YFP (Fig. 2 H and I). This reduction was mainly confined to the cellular periphery and the perinuclear region where the two fusion proteins colocalize. Notably, the reduction in CFP-CaM lifetime varied in different regions (from 2.5 to 1.7–2.0 ns; FRET efficiency of 20–30%), probably indicating a highly dynamic nature of the MLO/CaM interaction. Coexpression of the MLO W423R CaMBD mutant fused to YFP with CaM fused to CFP resulted in CFP lifetimes close to 2.5 ns (Fig. 2 J). Taken together, the FLIM analysis provides additional support for the existence of constitutive and dynamic interactions between MLO and CaM in the absence of the pathogen.
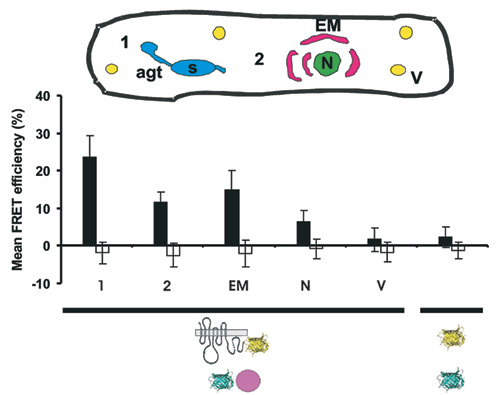
We recorded in a time-course experiment the MLO/CaM interaction in pathogen-challenged host cells. Whereas FRET-APB efficiency was essentially unaltered 4–12 hpi as compared with the noninoculated control (12.4 ± 4.1% versus 12.2 ± 4.6%), a significant increase was detected 14–24 hpi (20.8 ± 5.0%; Fig. 4A). This increase was not seen uniformly throughout the cell, showing peak FRET efficiency at the cell periphery around fungal penetration sites (23.7 ± 5.6%; Fig. 7, which is published as supporting information on the PNAS web site). Because ectopic expression of MLO permits pathogen entry in ≈90% of attacked host cells (15), the increase in FRET efficiency correlates with the switch from surface to invasive fungal growth. At later time points (24–36 hpi), FRET efficiency decreased to levels that were similar to those seen in nonchallenged cells (9.8 ± 3.2%).
Fig. 4.

Whole-cell FRET efficiency between MLO and CaM increases coincident with pathogen entry. (A) Barley epidermal cells coexpressing MLO-YFP and CFP-CaM were challenged with Bgh conidia. FRET was quantified in nonchallenged cells (first column), 4–12 (second column), 14–24 (third column), and 24–36 (fourth column) hpi by using FRET-APB. (B) Barley epidermal cells coexpressing MLO-YFP and CFP-CaM were challenged with barley (Bgh) or wheat (Bgt) powdery mildew conidiospores. FRET was quantified by using APB at 14–24 hpi. In addition to wild-type MLO, single amino acid substitution mutants MLO-10 (second column) and MLO-29 (third column) were tested. Mean FRET efficiencies (black bars) and background FRET (white bars) ± SD from 50–100 sample sites are shown. The stage of fungal development is schematically depicted above the columns. MLO-YFP and CFP-CaM are schematically indicated below the graph as shown in Fig. 3D.
To examine the correlation between pathogen entry and the increase of the MLO/CaM interaction at 14–24 hpi, we determined FRET between CaM and nonfunctional MLO mutant variants. Resistant barley mutants mlo-10 and mlo-29 encode MLO variants that either lack two amino acids in the second cytoplasmic loop (3) or possess a single amino acid replacement in the third cytoplasmic loop (17), respectively. Both mutant proteins accumulate in the PM to indistinguishable levels in comparison with wild-type MLO (18). Upon transient expression in a mlo null mutant background, the two variants exhibit drastically reduced functionality (5–15% single-cell mlo complementation; R.P., unpublished work). Coexpression of CFP-CaM and YFP fusion constructs encoding the two nonfunctional MLO variants yielded FRET efficiencies of 14.5 ± 4.4% (mlo-10) and 15.0 ± 4.4% (mlo-29) at 14–24 hpi (Fig. 4B). This finding is comparable with the level seen with wild-type MLO at the prepenetration stage (Fig. 4A). Thus, the two mutant MLO proteins, each harboring an intact CaMBD, appear to retain the ability to interact with CaM but do not exhibit the increase in FRET efficiency seen in the presence of wild-type MLO that is coincident with fungal invasion. Because the two tested nonfunctional MLO variants do not permit host cell entry of Bgh to significant levels, the amino acid substitutions in the cytoplasmic loops might either inhibit at a distance altered CaM interactions in the C-terminal tail of MLO, or the FRET efficiency enhancement requires successful fungal entry.
To test the latter hypothesis, we measured FRET between wild-type MLO-YFP and CFP-CaM upon inoculation with the wheat powdery mildew fungus, Blumeria graminis f.sp. tritici. After inoculation with this inappropriate fungus that fails to invade barley epidermal cells at most interaction sites even in the presence of wild type Mlo (19), we observed a FRET efficiency (13.2 ± 3.5%) similar to mlo-10, mlo-29, and the noninoculated control (Fig. 4B). This finding strongly suggests that the pathogen-triggered increase in MLO/CaM FRET efficiency requires successful fungal invasion into plant cells.
Discussion
We have shown that attack of the powdery mildew parasite induces in epidermal cells the formation of a PM microdomain that contains certain PM proteins and excludes others. Filipin-mediated fluorescence at these sites indicates a potential enrichment of sterols, a typical feature of lipid rafts in animal and yeast cells. Moreover, lack of FRAP in the Bgh-triggered microdomain is also reminiscent of lipid rafts in animal cells (20, 21). Although very little is known about lipid rafts in plants, biochemical evidence for the existence of lipid raft-mediated compartmentalization of PM proteins has been recently obtained by analysis of PMs derived from healthy tobacco (22). These lipid rafts were greatly enriched in glycosylceramide and a mixture of plant sterols (stigmasterol, sitosterol, 24-methylcholesterol, and cholesterol). Thus, if the microdomain beneath Bgh appressoria is an authentic lipid raft, then it is likely that retained redistribution of MLO and ROR2, despite blocked intracellular vesicle movement (Fig. 1 N–U), is driven by a clustering of PM plant sterols rather than de novo-targeted transport of vesicles to incipient fungal entry sites.
Recruitment of penetration resistance components MLO, ROR2, and PEN1 (and cytochrome b561 and the SYP132-related syntaxin) was exclusively seen beneath the tip of AGTs but not below the primary germ tube. This finding is surprising because apoplastic oxidative microbursts (revealed by hydrogen peroxide accumulation) are known to take place underneath both primary and AGTs, demonstrating that each germ tube is capable of eliciting a subcellular defense response at the cell periphery of attacked host cells (17, 23). Although the enzyme(s) generating these oxidative microbursts have not been conclusively identified in barley (24, 25), it is interesting that treatment of tobacco BY2 cells with cryptogein, a 10-kDa protein secreted by the phytoparasitic oomycete Phytophthora cryptogea, resulted in the accumulation of the NADPH oxidase NtrbohD and its presumed negative regulator, NtRac5, components of the oxidative burst machinery, in PM lipid rafts (22). One possibility is that unknown Bgh effector(s), secreted by both primary and AGTs, trigger the localization of the oxidative burst machinery underneath both fungal structures, whereas focal accumulation of MLO and ROR2 beneath Bgh AGTs might be due to effectors released specifically by appressoria. Consistent with this idea of multiple PM microdomains recruiting different sets of proteins, is the observation that lipid rafts isolated from healthy tobacco were capable of recruiting aquaporins, whereas both tested barley aquaporin isoforms, known to be expressed in the leaf epidermis, were excluded from the Bgh-triggered microdomain (ref. 22, this study, and Fig. 5).
Focal accumulation of ROR2 was found to occur in the absence of MLO. Because the latter protein suppresses ROR2 syntaxin-dependent penetration resistance (5, 26), we hypothesize that fungus-induced colocalization of both proteins beneath appressoria is part of a process leading to the establishment of a functional domain enabling avoidance from the host's immune response and allowing host cell invasion. Numerous examples in animals show that microbial agents including Escherichia coli, Mycobacterium bovis, and the simian virus 40 favor interactions with lipid rafts and their proteins to enter host cells (27). Because the establishment of a lipid raft-like functional domain at fungal entry sites has, to our knowledge, not been reported before, it should be interesting to find out whether this is a widespread phenomenon in plant and animal interactions with pathogenic fungi.
We explored the potential of FRET-APB and FRET-FLIM to study the dynamics of the interaction between MLO and CaM in epidermal cells upon Bgh attack. Both imaging methods provided evidence for a specific interaction through the CaMBD in the C-terminal cytoplasmic tail of MLO. This finding is consistent with previous data in which the significance of the protein–protein association was inferred from in vitro binding studies by using MLO CaMBD mutants and their reduced in vivo MLO activity (15). Here, we showed a marked increase in FRET efficiency upon expression of MLO-YFP and CFP-CaM at pathogen entry sites that depends on successful fungal invasion (Fig. 4), thereby providing spatiotemporal information that would be impossible to obtain using conventional biochemical or genetic methods. The retained CaM interactions seen in the presence of fully inactive MLO-10 and MLO-29 mutant forms in nonchallenged cells and their inability to confer a change of FRET efficiency upon Bgh attack, suggests that the mutations in the cytoplasmic loops might impair a novel MLO subfunction acting upstream of and enabling the dynamic interaction changes with CaM upon fungal entry into host cells. Preliminary data suggest that this subfunction might involve direct interactions with ROR2 because, unlike wild-type MLO, mutant variants containing substitutions in cytoplasmic loops two and three (including MLO-10) are impaired in their ability to associate with the membrane-resident wild-type syntaxin (R.A.B., P.S.-L., and R.P., unpublished work). Thus, we consider it realistic to unravel in future experiments the molecular mechanics of MLO-dependent fungal entry into host epidermal cells by combining the noninvasive imaging technology used in this study and the large set of subtle mutations affecting MLO, ROR2/PEN1, and CaM functions in both barley and Arabidopsis.
Supplementary Material
Acknowledgments
We thank Anja Reinstädler, Rainer Franzen, and Ulrich Martin for excellent technical assistance; Karl Oparka for providing the anonymous tobacco PM marker plasmid; and Patrick Schweizer (Institute of Plant Genetics and Crop Plant Research, Gartersleben, Germany) for providing cDNA clones of barley PM proteins. This work was supported by Deutsche Forschungsgemeinschaft Grant PA861/1-1 (to R.P. and E.S.).
Abbreviations: AGT, appressorial germ tube; APB, acceptor photobleaching; CaM, calmodulin; CaMBD, CaM-binding domain; CFP, cyan fluorescent protein; FLIM, fluorescence lifetime imaging microscopy; FRAP, fluorescence recovery after photobleaching; hpi, hours postinoculation; MLO, mildew resistance locus O; PM, plasma membrane; YFP, yellow fluorescent protein.
References
- 1.Mengaud, J., Ohayon, H., Gounon, P., Mege, R. M. & Cossart, P. (1996) Cell 84, 923-932. [DOI] [PubMed] [Google Scholar]
- 2.Panstruga, R. & Schulze-Lefert, P. (2003) Microbes Infect. 5, 429-437. [DOI] [PubMed] [Google Scholar]
- 3.Büschges, R., Hollricher, K., Panstruga, R., Simons, G., Wolter, M., Frijters, A., van Daelen, R., van der Lee, T., Diergaarde, P., Groenendijk, J., et al. (1997) Cell 88, 695-705. [DOI] [PubMed] [Google Scholar]
- 4.Devoto, A., Piffanelli, P., Nilsson, I., Wallin, E., Panstruga, R., von Heijne, G. & Schulze-Lefert, P. (1999) J. Biol. Chem. 274, 34993-35004. [DOI] [PubMed] [Google Scholar]
- 5.Collins, N. C., Thordal-Christensen, H., Lipka, V., Bau, S., Kombrink, E., Qiu, J. L., Hückelhoven, R., Stein, M., Freialdenhoven, A., Somerville, S. C. & Schulze-Lefert, P. (2003) Nature 425, 973-977. [DOI] [PubMed] [Google Scholar]
- 6.Schulze-Lefert, P. (2004) Curr. Opin. Plant Biol. 7, 377-383. [DOI] [PubMed] [Google Scholar]
- 7.Assaad, F. F., Qiu, J. L., Youngs, H., Ehrhardt, D., Zimmerli, L., Kalde, M., Wanner, G., Peck, S. C., Edwards, H., Ramonell, K., et al. (2004) Mol. Biol. Cell 15, 5118-5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von Röpenack, E., Parr, A. & Schulze-Lefert, P. (1998) J. Biol. Chem. 273, 9013-9022. [DOI] [PubMed] [Google Scholar]
- 9.Grebe, M., Xu, J., Mobius, W., Ueda, T., Nakano, A., Geuze, H. J., Rook, M. B. & Scheres, B. (2003) Curr. Biol. 13, 1378-1387. [DOI] [PubMed] [Google Scholar]
- 10.Shirasu, K., Nielsen, K., Piffanelli, P., Oliver, R. & Schulze-Lefert, P. (1999) Plant J. 17, 293-299. [Google Scholar]
- 11.Karpova, T. S., Baumann, C. T., He, L., Wu, X., Grammer, A., Lipsky, P., Hager, G. L. & McNally, J. G. (2003) J. Microsc. 209, 56-70. [DOI] [PubMed] [Google Scholar]
- 12.Bhat, R. A., Börst, J. W., Riehl, M. & Thompson, R. D. (2004) Plant Mol. Biol. 55, 239-252. [DOI] [PubMed] [Google Scholar]
- 13.Becker, W., Bergman, A., Biskup, C., Kelbaukas, L., Zimmer, T., Kloecker, N. & Bendorf, K. (2003) Proc. SPIE Int. Soc. Opt. Eng. 4963, 1-10. [Google Scholar]
- 14.Martin, S. W. & Konopka, J. B. (2004) Eukaryot. Cell 3, 675-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kim, M. C., Panstruga, R., Elliott, C., Müller, J., Devoto, A., Yoon, H. W., Park, H. C., Cho, M. J. & Schulze-Lefert, P. (2002) Nature 416, 447-451. [DOI] [PubMed] [Google Scholar]
- 16.Zacharias, D. A., Violin, J. D., Newton, A. C. & Tsien, R. Y. (2002) Science 296, 913-916. [DOI] [PubMed] [Google Scholar]
- 17.Piffanelli, P., Zhou, F., Casais, C., Orme, J., Jarosch, B., Schaffrath, U., Collins, N. C., Panstruga, R. & Schulze-Lefert, P. (2002) Plant Physiol. 129, 1076-1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Müller, J., Piffanelli, P., Devoto, A., Miklis, M., Elliott, C., Ortmann, B., Schulze-Lefert, P. & Panstruga, R. (2005) Plant Cell 17, 149-163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elliott, C., Zhou, F., Spielmeyer, W., Panstruga, R. & Schulze-Lefert, P. (2002) Mol. Plant–Microbe Interact. 15, 1069-1077. [DOI] [PubMed] [Google Scholar]
- 20.Oliferenko, S., Paiha, K., Harder, T., Gerke, V., Schwarzler, C., Schwarz, H., Beug, H., Gunthert, U. & Huber, L. A. (1999) J. Cell Biol. 146, 843-854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shvartsman, D. E., Kotler, M., Tall, R. D., Roth, M. G. & Henis, Y. I. (2003) J. Cell Biol. 163, 879-888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mongrand, S., Morel, J., Laroche, J., Claverol, S., Carde, J. P., Hartmann, M. A., Bonneu, M., Simon-Plas, F., Lessire, R. & Bessoule, J. J. (2004) J. Biol. Chem. 279, 36277-36286. [DOI] [PubMed] [Google Scholar]
- 23.Hückelhoven, R., Fodor, J., Preis, C. & Kogel, K. H. (1999) Plant Physiol. 119, 1251-1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schultheiss, H., Dechert, C., Kogel, K. H. & Hückelhoven, R. (2003) Plant J. 36, 589-601. [DOI] [PubMed] [Google Scholar]
- 25.Christensen, A. B., Thordal-Christensen, H., Zimmermann, G., Gjetting, T., Lyngkjaer, M. F., Dudler, R. & Schweizer, P. (2004) Mol. Plant–Microbe Interact. 17, 109-117. [DOI] [PubMed] [Google Scholar]
- 26.Freialdenhoven, A., Peterhänsel, C., Kurth, J., Kreuzaler, F. & Schulze-Lefert, P. (1996) Plant Cell 8, 5-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duncan, M. J., Shin, J. S. & Abraham, S. N. (2002) Cell Microbiol. 4, 783-791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}