

Abstract
Type II topoisomerases orchestrate proper DNA topology, and they are the targets of anti-cancer drugs that cause treatment-related leukemias with balanced translocations. Here, we develop a high-throughput sequencing technology to define TOP2 cleavage sites at single-base precision, and use the technology to characterize TOP2A cleavage genome-wide in the human K562 leukemia cell line. We find that TOP2A cleavage has functionally conserved local sequence preferences, occurs in cleavage cluster regions (CCRs), and is enriched in introns and lincRNA loci. TOP2A CCRs are biased toward the distal regions of gene bodies, and TOP2 poisons cause a proximal shift in their distribution. We find high TOP2A cleavage levels in genes involved in translocations in TOP2 poison–related leukemia. In addition, we find that a large proportion of genes involved in oncogenic translocations overall contain TOP2A CCRs. The TOP2A cleavage of coding and lincRNA genes is independently associated with both length and transcript abundance. Comparisons to ENCODE data reveal distinct TOP2A CCR clusters that overlap with marks of transcription, open chromatin, and enhancers. Our findings implicate TOP2A cleavage as a broad DNA damage mechanism in oncogenic translocations as well as a functional role of TOP2A cleavage in regulating transcription elongation and gene activation.
Human type II topoisomerases, of which there are α and β isoforms, are central to dynamic changes in DNA topology for replication, transcription, and chromosome organization and segregation. They relax, unknot, and untangle DNA by transiently cleaving and religating both strands of the double helix. Each subunit of the TOP2 homodimer forms a phosphodiester bond with the base 3′ to the cleavage, creating the cleavage complex, a covalent intermediate with four-base staggered DNA ends tethered by the enzyme (Pendleton et al. 2014). TOP2A increases during cell growth and is required for replication (Pendleton et al. 2014). It also is a member of the BAF complex that decatenates newly replicated sister chromatids during mitosis (Dykhuizen et al. 2013). TOP2B expression is cell cycle independent (Pendleton et al. 2014).
Roles for both isoforms have been suggested during transcription. TOP2B is necessary for transcription of developmentally regulated genes and nuclear hormone receptor target genes (Ju et al. 2006; Lyu et al. 2006). In a Burkitt lymphoma cell line, dissipation of supercoiling at transcription start sites of highly expressed genes was found to be TOP2B dependent (Kouzine et al. 2013). Both isoforms were detected at supercoiled domains during transcription in RPE1 cells (Naughton et al. 2013). TOP2A was identified in the Pol II complex (Mondal and Parvin 2001). A switch from TOP2A to TOP2B binding at promoters of long genes during neuronal differentiation (Tiwari et al. 2012; Thakurela et al. 2013) suggested promoter priming by TOP2A for murine ES cell differentiation (Thakurela et al. 2013).
Type II topoisomerases are targets of anti-cancer drugs referred to as “TOP2 poisons” that convert native TOP2 into a toxin by stabilizing cleavage complexes, which causes DNA strand breaks and induces apoptosis (Pendleton et al. 2014). TOP2 poisons also cause treatment-related leukemias with balanced translocations of the KMT2A gene and, less often, other loci (Pendleton et al. 2014). The prevailing model of the translocation mechanism involves misrepair of TOP2-mediated DNA damage (Lovett et al. 2001; Whitmarsh et al. 2003; Mistry et al. 2005; Povirk 2006; Robinson et al. 2008). Consistent with this model, TOP2 poisons increase in vitro TOP2 cleavage at or near the breakpoints (Lovett et al. 2001; Whitmarsh et al. 2003; Mistry et al. 2005; Robinson et al. 2008), and etoposide treatment induced TOP2B cleavage in a leukemia cell line 3′ in the KMT2A breakpoint cluster region (bcr) (Cowell et al. 2012). In KMT2A-rearranged (KMT2A-R) leukemia in infants, maternal-fetal exposures to dietary TOP2 interacting substances (Spector et al. 2005) and an inactivating variant in NQO1, which detoxifies the TOP2 poison p-benzoquinone (Wiemels et al. 1999; Smith et al. 2002; Lindsey et al. 2004; Guha et al. 2008), are predisposing factors.
However, studies of TOP2 have been impeded by lack of a technology to determine cleavage by these enzymes at single-base precision in cells genome-wide. A key challenge to surmount is removal of the covalently attached DNA from TOP2 at the cleavage site. Conventional ChIP cannot distinguish noncovalent TOP2 DNA binding from TOP2 covalently bound to DNA in cleavage complexes, and most TOP2 DNA binding is noncovalent (Mueller-Planitz and Herschlag 2007; Lee et al. 2012). General techniques to map DNA breaks in cells are not TOP2 specific, have low spatial resolution (Iacovoni et al. 2010; Pang et al. 2015), and may not detect TOP2 cleaved DNA ends protected by the enzyme (Crosetto et al. 2013; Baranello et al. 2014). A tiling array of select genomic regions used to analyze murine TOP2B cleavage did not distinguish double-strand breaks (DSBs) from single-strand nicks (SSNs) (Sano et al. 2008). Furthermore, in a cell line study employing TDP2 (Tyrosyl-DNA phosphodiesterase 2) to remove the DNA from TOP2, genome-wide data were not reported (Cowell et al. 2012).
Here, we develop a platform to detect the sequences at which TOP2 is actively engaged in cleavage complexes at single-base precision genome-wide, and apply this technology to characterize TOP2A cleavage in human K562 leukemia cells.
Results
Optimization of procedure to detect TOP2 cleavage complexes
Following cellular lysis, sonication, and immunocapture of TOP2A, including that in covalent cleavage complexes (Supplemental Fig. S1), we used calf intestinal alkaline phosphatase (CIP) to hydrolyze the covalent phosphodiester bond between the DNA and active site tyrosine residue of each TOP2A subunit, thus releasing the DNA ends at the 3′ sides of cleavage, i.e., +1 positions in cleavage complexes (Fig. 1A). CIP is a phosphomonoesterase commonly used to hydrolyze 5′-phosphate bonds but can act as a phosphodiesterase. The CIP used here (New England Biolabs) has phosphodiesterase activity at 3% the level of its phosphomonoesterase activity (Igunnu et al. 2011). The exposed DNA ends became ligation sites for sequencing library adapters to localize TOP2A cleavage at single-base precision (Fig. 1A).
Figure 1.

Approach, reproducibility, and assay validation. (A) TOP2 cleavage complexes detected by sequencing. After TOP2 immunocapture, CIP releases covalently attached TOP2 subunits from DNA at the +1 positions relative to the cleavage, which (+/− preamplification) become 5′ adapter-ligated ends; 5′ ends from sonication give random signals. Input control (data not shown) is sonicated lysate with random 5′ ends created by sonication. (B) Strong read count correlations in 10-kb windows between DMSO-treated biological replicates +/− preamplification (Supplemental Table S1). (C) UCSC Genome Browser images and KMT2A gene model (http://genome.ucsc.edu/) (Kent et al. 2002) showing similar read distribution in nonamplified and amplified p-benzoquinone (pBQ)–treated replicates. Black bar indicates bcr. (D) Overlap of TOP2A cleavage sites detected by sequencing with cleavage sites from the in vitro assay. Vertical line beneath the red arrow in the KMT2A bcr schematic is a translocation breakpoint hotspot from the TOP2A high-throughput sequencing assay (bottom, left) and autoradiograph inset from TOP2A in vitro cleavage assay of sense strand of same sequence (bottom, right). Colors indicate different treatments; symbols, different replicates. Arrows at peaks in sense strand (bottom, left) indicate +1 positions of cleavage sites also found in vitro (bottom, right, dashes). Connecting lines indicate sites with cleavage detected at +1 positions of both strands by sequencing (bottom, left). Coordinates, NC_000011.10 (GRCh38/hg38). (VP16) Etoposide. Bars beneath KMT2A bcr schematic, regions from both assays in Supplemental Figures S3 and S4.
We mapped TOP2A cleavage complexes after treating K562 cells with vehicle (DMSO), anti-cancer drugs (etoposide or mitoxantrone), or environmental (p-benzoquinone) or dietary (genistein) TOP2 poisons. A preamplification step was introduced after the CIP treatment before ligation of adapters to increase sensitivity in the event of limited DNA recovery. Libraries from three nonamplified and 11 preamplified samples (Supplemental Fig. S1) yielded 41 million–65 million raw reads each, with 95%–99% mapping and 68%–72% mapping uniquely to the genome (GRCh38/hg38) (Supplemental Table S1).
We found strong correlations in read densities for 10-kb windows across the genome between nonamplified and amplified biological replicates (r = 0.86, DMSO; r = 0.88, etoposide; r = 0.85; p-benzoquinone) (Fig. 1B, left; Supplemental Fig. S2). Independent nonamplified and amplified p-benzoquinone–treated replicates showed similar KMT2A bcr signal profiles (Fig. 1C). Read densities also correlated tightly between amplified replicates (r = 0.94, DMSO; r = 0.97, etoposide; r = 0.88, p-benzoquinone; r = 0.84, genistein) (Fig. 1B, right; Supplemental Fig. S2), indicating high reproducibility regardless of preamplification. Genomic bases showing greater than or equal to fourfold signal enrichment in the 5′ end of mapped reads (i.e., +1 positions of cleavage sites) with a P-value <0.05 compared with that of the input (Audic and Claverie 1997) were defined as cleavage sites.
Aggregate analyses of 650 sense-strand nucleotides in four in vitro cleavage assays (Fig. 1D; Supplemental Figs. S3, S4) identified 77 KMT2A bcr TOP2A cleavage sites also found by sequencing, including in a treatment-related leukemia translocation breakpoint hotspot (Fig. 1D; Whitmarsh et al. 2003). Because of the usage of naked DNA for the in vitro assays, chromatin determinants of cellular TOP2 cleavage, and greater sensitivity of sequencing, the sites were not entirely concordant, but the general overlap validates the phosphodiesterase activity by CIP and detection of bona fide cleavage complexes at single-base precision. Concordance with local base sequence preferences for TOP2 in vitro cleavage (Palumbo et al. 1994; Capranico and Binaschi 1998) further showed that amplification does not alter the precision in cleavage site detection (Supplemental Fig. S5).
TOP2A cleavage events are SSNs more often than DSBs
Rather than creating a concerted DSB, the two subunits of the TOP2 homodimer create coordinated nicks on each strand of DNA, forming a fleeting TOP2-bound cleaved DNA complex (Zechiedrich et al. 1989). Etoposide occupancy at both scissile bonds is required for DSBs, and most breaks formed at concentrations relevant to chemotherapy are SSNs (Bromberg et al. 2003). Consistent with reports in which the vast majority of detected TOP2 cleavage events were SSNs (Bromberg et al. 2003; Muslimovic et al. 2009), we found 1.3%–3.4% of TOP2A cleavage events were DSBs and the remainder were SSNs (Supplemental Table S1; Supplemental Fig. S2).
Genome-wide characterization of TOP2A cleavage cluster regions
The KMT2A bcr analyses suggested a clustering of reads (Fig. 1C) and TOP2A cleavage sites (Fig. 1D; Supplemental Figs. S3, S4) in compact genomic regions similar to cleavage by yeast Spo11 (Pan et al. 2011). Therefore, we adapted computational methods for identifying Spo11 hotspots (Pan et al. 2011) to uncover TOP2A cleavage cluster regions (CCRs; i.e., cleavage sites as defined by criteria above mapping within 200 bp of each other). To exclude candidates with high background signals from noncleavage sites, each CCR met further criteria of a 25-nt minimum length and a total number of reads greater than or equal to fourfold over input in the same genomic region (Pan et al. 2011).
High concordance in CCR patterns in nonamplified versus amplified or amplified versus amplified biological replicates (Supplemental Fig. S6) enabled merging of amplified replicates for CCR analyses. We identified 310,239, 202,926, 359,486, and 415,312 CCRs in merged amplified DMSO, etoposide, p-benzoquinone, and genistein data sets, respectively. The single amplified mitoxantrone-treated sample had 182,679 CCRs (Supplemental Table S1). Most CCRs (75%–90%) were 100–200 nt long (Fig. 3A, below; Supplemental Fig. S6). Comparisons to existing human single-nucleotide polymorphism (SNP) data (The 1000 Genomes Project Consortium 2015) indicated lower SNP densities in the 100 nt surrounding TOP2A CCR centers than in the surrounding 10 kb and, therefore, high genome-wide TOP2A CCR conservation across the human population (Fig. 2).
Figure 3.

Characterization and functional analysis of TOP2A CCRs. (A) CCR length distribution. Bars represent numbers of CCRs in increasing 50-bp intervals of length. Note that most CCRs are 100–200 nt long. See also Supplemental Figure S6. (B) CCR occurrences in genomic elements compared with the control (10,000 random size-matched genome segments). Note enrichment in introns and lincRNAs; note underabundance in pseudogenes, repeats, and promoters. (***) P < 2.2 × 10−16; χ2 test. (A,B) Amplified samples; same treatments merged where applicable (Supplemental Table S1). (C) Scatterplot of TOP2A CCR signal density for each chromosome sorted by chromosome length with highest density on Chr 11. VP16 treatment shown as representative. Dashed line indicates average CCR signal density for all chromosomes. See also Supplemental Figure S7. (D) GO analysis of genes overlapping with TOP2A CCRs in union set of amplified DMSO-, VP16-, mitoxantrone-, pBQ-, and genistein-treated samples. GO term categories starting at the 12 o'clock position listed clockwise; metabolic process and cellular process are most enriched.
Figure 2.
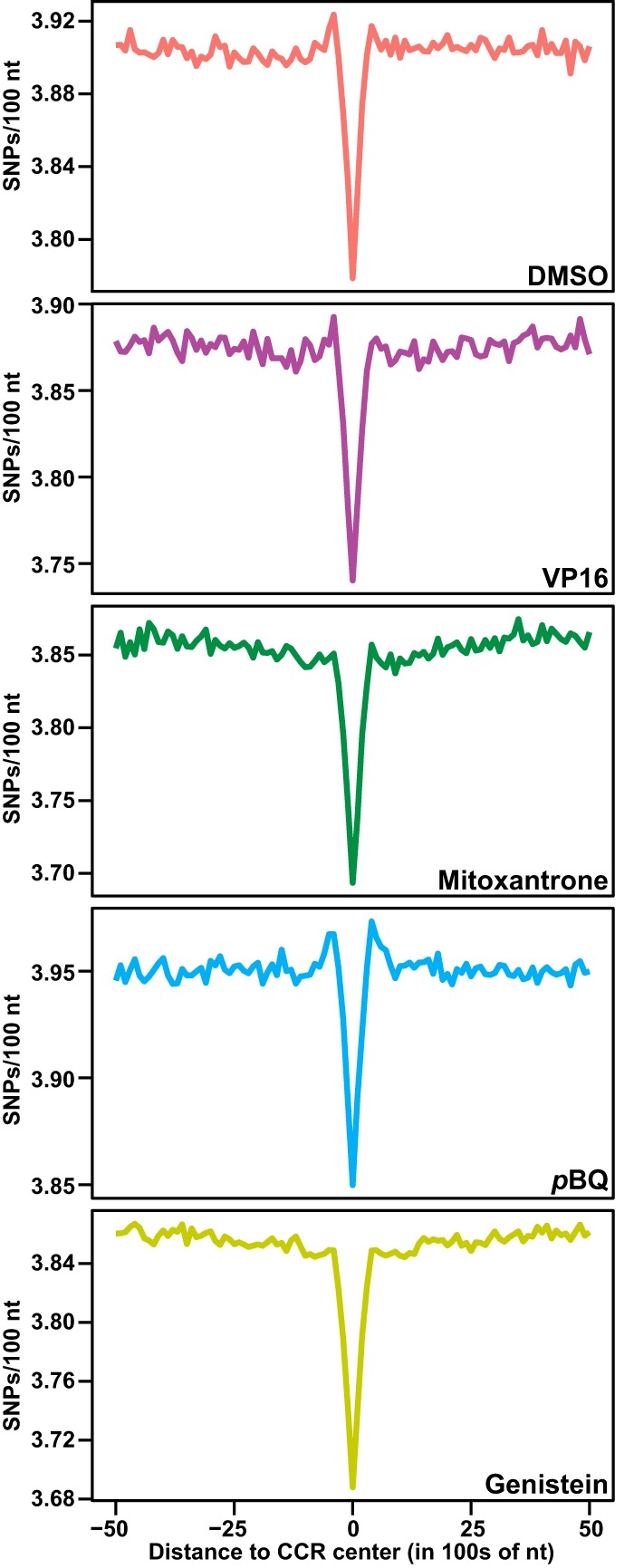
Regional TOP2A CCR sequence conservation. Lower SNP density within 100 nt at CCR centers compared with the surrounding 10 kb in 100-nt sliding windows. P < 2.2 × 10−16; Kruskal–Wallis test. Amplified samples; same treatments merged where applicable (Supplemental Table S1).
We found that TOP2A CCRs were enriched in gene introns and lincRNAs compared with genomic control regions; fewer fell in pseudogenes or repetitive elements as annotated by RepeatMasker (Fig. 3B; Smit et al. 2013–2015), suggesting a preference for transcriptionally active regions of the genome. Enrichment at introns but not promoters suggested a potential role of TOP2A cleavage in relieving torsional stress during transcription (Fig. 3B).
Certain chromosomes consistently showed more CCRs than others. Overall, Chr 11 containing KMT2A and 15 of 79 of its translocation partner genes showed the highest CCR density compared with other chromosomes (Fig. 3C; Supplemental Fig. S7).
By gene ontology (GO) analysis of proteins encoded by genes with TOP2A CCRs, the most enriched category (GO:008152) was metabolic process, which encompasses cell growth and macromolecular processes (DNA repair, replication). The second most enriched category, cellular process (GO:0009987), contained genes involved in transcriptional regulation (Fig. 3D).
Genes involved in oncogenic translocations show TOP2A CCR enrichment
Most translocation breakpoints in cancer overall (91%) (Novo et al. 2007), including KMT2A translocation breakpoints (Felix et al. 2006; Meyer et al. 2013), are in introns. The nonrandom density by chromosome and CCR intron preference led us to further study how TOP2A cleavage and translocations are related. KMT2A and its partner genes comprise the KMT2A recombinome (Meyer et al. 2013). Interestingly, we found that the proportion of KMT2A recombinome genes containing TOP2A CCRs was significantly higher than all coding genes (Fig. 4A, left). In accord with the reported precision in the reciprocal breakpoint junctions of KMT2A translocations in treatment-related leukemias that implicates DNA DSBs at/near the breakpoints (Lovett et al. 2001; Whitmarsh et al. 2003; Povirk 2006; Felix 2012), more KMT2A recombinome genes contained TOP2A DSBs (Fig. 4A, middle) than all coding genes, particularly with etoposide, the drug most commonly associated with KMT2A-R treatment–related leukemia (Felix 2012).
Figure 4.

TOP2A cleavage in genes involved in oncogenic translocations. (A, left) Larger proportions of genes containing CCRs in KMT2A recombinome compared with all coding genes. (**) P < 1 × 10−5 for DMSO, VP16, mitoxantrone; (*) P < 1 × 10−4 for pBQ, genistein; χ2 test. (A, middle) Larger proportions of genes containing TOP2A DSBs in KMT2A recombinome compared with all coding genes. (*) P < 0.05 for DMSO, mitoxantrone, pBQ, genistein; (**) P = 0.00027375 for VP16; χ2 test. (A, right) Larger proportions of cancer fusion genes (Mitelman et al. 2016) containing CCRs compared with all coding genes. (***) P < 2.2 × 10−16; χ2 test. Amplified samples; same treatments merged where applicable (Supplemental Table S1). (B–E) CCR signals (HP10M) in individual amplified samples along regions of genes involved in leukemia-associated translocations linked to TOP2 poisons (bars). Panels show sonicated input and different treatments (top in panels). Gene models from GRCh38/hg38 in the UCSC Genome Browser (bottom) (Kent et al. 2002; http://genome.ucsc.edu/) correspond to tracks shown. (B) KMT2A. Bar, 8.3-kb bcr spanning exon 7 through exon 13 positions 118,481,830–118,490,167; NC_000011.10. (C) PML. Left bar, 1.45-kb intron 3 bcr, positions 74,023,409–74,024,856; right bar, 1.06-kb intron 6 bcr, positions 74,033,415–74,034,477; NC_000015.10. (D) RARA. Bar, 16.9-kb intron 2 bcr, positions 40,332,397–40,348,315; NC_000017.11. (E) RUNX1. Left bar, 25-kb intron 6 bcr, positions 34,859,473–34,834,602; right bar, 35-kb intron 7 bcr, positions 34,834,409–34,799,463; NC_000021.9.
When CCR signal strength, i.e., the number of reads at cleavage sites per CCR expressed as hits per 10 million reads (HP10M), was examined globally, several of the highest CCR signals with p-benzoquinone treatment were in the KMT2A bcr (Supplemental Table S2). Also, p-benzoquinone and genistein, which have potential epidemiologic links to infant leukemia (Wiemels et al. 1999; Smith et al. 2002; Lindsey et al. 2004; Spector et al. 2005; Guha et al. 2008), induced TOP2A cleavage in the bcr (Supplemental Figs. S3, S4). Furthermore, most TOP2A breaks are SSNs (Supplemental Fig. S2), and it is well established that paired SSNs on opposite DNA strands and template-directed polymerization of long overhangs are required to create the KMT2A translocation breakpoint junctions with the several-hundred-base sequence duplications typical of leukemia in infants (Gillert et al. 1999; Raffini et al. 2002). These findings support a model in which TOP2A cleavage triggers KMT2A translocations in both treatment-related and infant leukemia.
The TOP2 poisons also induced stronger and distinguishable patterns of TOP2A CCRs compared with DMSO in introns of other genes besides KMT2A, e.g., PML, RARA, and RUNX1, in which TOP2-mediated DNA damage from TOP2 poison exposures has been implicated in leukemia-associated translocations (Fig. 4B–E; Roulston et al. 1998; Smith et al. 1998; Mistry et al. 2005; Ottone et al. 2009; Felix 2012). Remarkably, compared with all coding genes, known cancer fusion genes per se (i.e., genes involved in translocations) (Mitelman et al. 2016) also were more likely to contain TOP2A CCRs (Fig. 4A, right). In addition, we found high genic TOP2A CCR signal strengths (i.e., average signal strength of all CCRs within a gene in HP10M) in fusion genes linked to leukemias more broadly, as well as solid tumors (Supplemental Table S3; Mitelman et al. 2016).
CCR profiles in coding genes support role of TOP2A cleavage in transcription elongation
We extracted genome-wide transcript abundance data from two RNA-seq data sets for K562 cells (Bansal et al. 2014) to investigate whether TOP2A cleavage is associated with transcription. Whereas the distribution of coding transcript abundance is bimodal, with DMSO treatment and each TOP2 poison, we found that coding genes containing one or more CCRs were significantly enriched in the peak containing more abundant transcripts (Fig. 5A), suggesting that TOP2A cleavage may promote higher levels of transcription.
Figure 5.

Relationships between TOP2A CCRs and transcription marks in coding genes. (A) Distribution of transcript abundance density for all coding genes compared to coding genes with CCRs. Two RNA-seq data sets for untreated K562 cells (GEO accession number GSE46718) (Bansal et al. 2014) were used to plot transcript abundance. Note skew of CCR-containing genes toward peak with more abundant transcripts (colored lines) compared with bimodal distribution of transcript abundance for all coding genes (black line). P-value for DMSO and each TOP2 poison = 2.2 × 10−16; Kruskal–Wallis test. (B,C) Higher H3K36me3 (B) and POLR2A (C) signals (total mapped reads) along bodies of coding genes with (Y indicates yes; darker colors) compared with without (N indicates no; lighter colors) CCRs. Data from the ENCODE Project Consortium 2012 for H3K36me3 and POLR2A signals (Supplemental Table S4; The ENCODE Project Consortium 2012) were converted from GRCh37/hg19 to GRCh38/hg38 using liftOver (http://genome.sph.umich.edu/wiki/LiftOver) (Hinrichs et al. 2006). (Boxes) 25th to 75th percentiles; (whiskers) fifth to 95th percentiles; (horizontal lines) medians. (***) P < 2.2 × 10−16; Kruskal–Wallis test. (D) CCR distribution along gene bodies divided into 100 equally sized windows. Graphs display CCRs/window relative to total. Note distribution in middle and 3′ ends with DMSO and pBQ and proximal shifts with VP16, mitoxantrone, and genistein. (A–D) Amplified samples; same treatments merged where applicable (Supplemental Table S1).
Existing data for K562 cells (The ENCODE Project Consortium 2012) allowed us to study relationships between TOP2A CCRs in coding genes and marks of transcription. H3K36me3 is a mark of actively transcribed genes deposited by the elongating RNA Pol II–associated methyltransferase SETD2 (Li et al. 2002; Strahl et al. 2002). With DMSO treatment and each TOP2 poison, we found that TOP2A-cleaved coding genes had higher H3K36me3 and POLR2A (RNA Pol II subunit A) signals compared with those lacking CCRs (Fig. 5B,C). Because H3K36me3 and POLR2A sites are distributed across the gene body, these data suggested a role of TOP2A cleavage in transcription elongation and led us to study TOP2A CCR distribution along the gene body. In DMSO-treated and p-benzoquinone–treated cells, the TOP2A CCRs localized preferentially to the middle and 3′ end compared with the very 5′ end and increased progressively along the gene body (Fig. 5D). In contrast, etoposide, mitoxantrone, and genistein caused marked increases in TOP2A CCRs just downstream from the 5′ end and, thus, altered this distribution compared with CCRs in the absence of these poisons. Overall, these data suggest that TOP2A cleavage changes the gene body topology for transcription elongation.
TOP2A cleavage of coding genes is independently associated with gene length and transcript abundance
Next we examined interrelationships among TOP2A CCR signal strength, gene length, and transcript abundance. To address whether genic TOP2A CCR signal strength is related to gene length, we divided the genes into four length categories and defined genic CCR signal strength as the average signal strength of all CCRs within a gene to control for the tendency of longer genes to have more CCRs due to their increased sequence space. We found significant increases in CCR signal strength as a function of length category (<10, 10–49, 50–99, ≥100 kb) (Fig. 6A,B; Supplemental Fig. S8).
Figure 6.

Independent associations of CCR signal strength with coding gene length and transcript abundance. (A,B) Correlation between length and genic CCR signal strength (HP10M) in DMSO-treated (A) and VP16-treated (B) samples. Gene length from GRCh38/hg38 by categories on x-axis. (Boxes) 25th to 75th percentiles; (whiskers) fifth to 95th percentiles; (horizontal lines) median for each length interval. χ2 test P-values, top right in panels. Amplified samples; same treatments merged (Supplemental Table S1). (C) Scatter plot of protein-coding transcript abundance versus gene length based on two RNA-seq data sets for untreated K562 cells (GEO accession number GSE46718) (Bansal et al. 2014) and gene length from GRCh38/hg38. Smooth line was predicted by the gam method (Supplemental Methods). (Shading) Confidence interval around smoothed trend line. Genes with RPKM > 0.1 plotted. r-value (top right) shows slight overall negative correlation. (D) Box and whisker plots of genic CCR signal strength versus transcript abundance within indicated length categories subdivided based on </> average genic CCR signal strength. (*) P < 0.05; χ2 test. Note correlation between genic CCR signal strength and transcript abundance across all lengths. Union set of all amplified samples (Supplemental Table S1).
We then used the transcript abundance data from the two RNA-seq data sets for K562 cells (Bansal et al. 2014) to ask whether gene length and transcript abundance are related. The small negative correlation (r = −0.06) (Fig. 6C) indicates both that transcript abundance is not gene length dependent and that the overlap of TOP2A-cleaved coding genes with coding genes having higher levels of expression (Fig. 5A) is not affected by gene length. In fact, when coding genes within the length categories above were subdivided by whether genic CCR signal strength (HP10M) was more or less than average, the genes with higher than average CCR signal strength within each length category had significantly higher transcript abundance (Fig. 6D). These data demonstrate that TOP2A cleavage events are independently correlated with the length of coding genes and their level of expression. Together with the CCR enrichment in more distal regions of gene bodies (Fig. 5D), they further reinforce a role for TOP2A cleavage in transcription elongation.
TOP2A cleavage functions in lincRNA transcription
The TOP2A CCR enrichment in lincRNAs (Fig. 3B) raised questions about whether TOP2A cleavage events are important for lincRNA transcription. Like the bimodal coding transcript distribution (Fig. 5A), a probability density plot generated by extracting lincRNA transcript abundance data from the two RNA-seq data sets for K562 cells (Bansal et al. 2014) showed a bimodal distribution of lincRNA transcript abundance and, as expected, globally lower lincRNA expression compared with coding gene expression (Fig. 5A; Supplemental Fig. S9). For each treatment tested, TOP2A-cleaved lincRNA loci were more likely to have moderately abundant transcripts. Just as for coding genes, CCR signal strength increased with increasing length, and lincRNA expression and length showed a weak negative correlation (r = −0.10). Across the range of lengths, lincRNAs with a higher than average CCR signal strength also exhibited higher levels of transcription. Thus, as for coding genes, TOP2A cleavage of lincRNAs is independently associated with both length and expression levels (Supplemental Fig. S9).
TOP2A CCRs are associated with open chromatin and enhancer elements
Also, by using existing data for K562 cells (The ENCODE Project Consortium 2012), we discovered that DNase I cleavage signals in DNase I hypersensitive sites (DHSs) were stronger in DHSs that overlap with TOP2A CCRs compared with nonoverlapping DHSs (Fig. 7A). Because DHSs are marks of open chromatin (Thurman et al. 2012), this finding suggests that TOP2A cleavage aids in maintaining open chromatin. Furthermore, the stronger DNase I signals in DHSs overlapping with TOP2A CCRs in the TOP2 poison–treated samples (Fig. 7A) imply that TOP2A DNA damage with these compounds favors open chromatin.
Figure 7.

Colocalization of genome-wide TOP2A CCRs with chromatin features. (A) Higher DNase I signal density in DHSs that overlap (colored lines) with CCRs compared with DHSs that do not (black line) overlap. P < 2.2 × 10−16; Kruskal–Wallis test. (B,C) Enriched H3K4me1 (B) and H3K27ac (C) signals at CCR centers (position 0 on x-axis) compared with 1-kb upstream and downstream flanking sequences in 100-nt sliding windows. P < 0.001 for DMSO, VP16, pBQ; P < 0.01 for mitoxantrone, genistein; Kruskal–Wallis test. (A–C) Amplified samples; same treatments merged where applicable. (D) Clustering of features found within CCRs by PCA and k-means algorithms. PC1 and PC2 account for 37.84% and 14.46% of the total variance, respectively. Dots represent PCA loadings for indicated features. Colors show five clusters of features found in CCRs. Along PC1, note the separation of histone marks of gene repression (dark blue, green) from marks known to positively affect gene expression (light blue, purple, red). Along PC2, note the separation between clusters of marks of actively expressed genes (light blue, purple) from cluster of gene activating elements including enhancer marks (red). Union set of CCRs from all amplified samples. (A–D) Analyses were performed on existing data for chromatin features (The ENCODE Project Consortium 2012) after liftOver (Hinrichs et al. 2006; http://genome.sph.umich.edu/wiki/LiftOver) conversion to GRCh38/hg38. See also Supplemental Tables S1, S4.
H3K4me1 and H3K27ac are marks of active enhancers. We discovered that the signals for these marks were significantly enriched directly at the center of TOP2A CCRs, including marks of both strong (H3K4me1/H3K27ac) and weak (H3K4me1 alone) enhancers (Fig. 7B,C; Supplemental Fig. S10; Creyghton et al. 2010). These data indicate that TOP2A cleavage is also targeted to active enhancer elements.
Clustering of chromatin features overlapping with CCRs
We further characterized the chromatin features that impact CCR location by accessing data for histone marks, POLR2A sites, and DHSs (The ENCODE Project Consortium 2012) and conducting a genome-wide principal component analysis (PCA) (Huff et al. 2010). Five clusters were distinguished by two PCs (Fig. 7D). Along PC1, clusters of gene repression marks H3K9me3 and H3K27me3 (dark blue and green, respectively) are separated from clusters of marks known to positively affect expression, including DHSs and POLR2A sites (light blue) and H3K36me3, H3K9me1, H3K79me2, and H4K20me1 (purple). Along PC2, we found an intriguing separation between clusters of marks tagging actively expressed genes (light blue, purple) from a cluster of enhancer marks (e.g., H3K27ac, H3K4me1) and other gene activating elements (red). Thus, the CCRs overlapping with marks of gene bodies undergoing transcription (Fig. 5B,C) are distinct from those overlapping with marks of enhancers (Fig. 7B,C; Supplemental Fig. S10). This PCA indicates that discrete genomic environments contain CCRs associated with gene bodies or gene activating elements, and reinforces that active portions of the genome are enriched in CCRs.
Discussion
We developed a procedure to comprehensively map TOP2A cleavage sites genome-wide by high-throughput sequencing of DNA ends released from immunopurified cleavage complexes that surmounts inherent limitations in cellular approaches to study TOP2 cleavage (Sano et al. 2008; Iacovoni et al. 2010; Crosetto et al. 2013; Baranello et al. 2014; Pang et al. 2015). The procedure identifies +1 positions in cleavage complexes at single-base precision by coupling a new use of the phosphodiesterase activity in CIP preparations—to detach DNA from cleavage complexes—with the power of high-throughput sequencing.
We address the potential for low DNA yield from either low phosphodiesterase activity of CIP (O'Brien and Herschlag 2001; Igunnu et al. 2011) or restricted samples by amplifying the released DNA before library preparation without affecting reproducibility. Thus, our assay can be used even with limited DNA recovery, and the selective release of DNA directly from immunopurified cleavage complexes enhances specificity. The detection of cleavage at precise sites also found in vitro and at translocation breakpoint hotspots, as well as local base preferences for cleavage mirroring those in vitro, gave validation to our assay. Also, the 1.3%–3.4% TOP2A DNA DSBs that we found are consistent with prior studies (Zechiedrich et al. 1989; Muslimovic et al. 2009). Overall, our methodology achieved the genome-wide identification of TOP2A cleavage sites not previously accomplished.
Our procedure avoids the fixation step of conventional ChIP-seq for two reasons. First, the active site tyrosine residues of TOP2 are already covalently attached to the DNA by phosphodiester linkage. Second, fixation would preclude distinguishing noncovalent TOP2 DNA binding from the TOP2 covalently bound to DNA in cleavage complexes.
Our method differs from the procedure that employs TDP2 to release the DNA from the cleavage complex (Cowell et al. 2012). TDP2 hydrolyzes 5′ phosphodiester bonds specifically but requires TOP2 proteolytic degradation to expose the protein–DNA linkage (Gao et al. 2014; Ashour et al. 2015). The use of CIP to release the DNA is simpler because it is carried out directly on the bead-bound TOP2 without protease digestion. It is also critical for the enzyme used to hydrolyze the phosphodiester bond to have high activity at TOP2 SSNs as well as DSBs because most cellular TOP2 cleavage sites are SSNs (Zechiedrich et al. 1989; Muslimovic et al. 2009; data from the present study in Supplemental Table S1 and Supplemental Fig. S2). However, TDP2 has low activity at TOP2 SSNs compared with TOP2 DSBs (Gao et al. 2014).
Our observations expand working models of functional TOP2A–DNA interactions. Beyond confirming that TOP2 poisons confer nonrandom and different local base cleavage preferences (Capranico and Binaschi 1998), we discover that TOP2A cleavage sites cluster in CCRs that display evolutionary conservation within the human population. Also, the CCRs exhibit favoritism for introns and lincRNAs comprising transcriptionally active genome elements.
We follow the standard ChIP-seq practice of only retaining uniquely mapping reads (Kellis et al. 2014) to identify CCRs and specifically guard against falsely calling CCRs in repeats caused by multimapping. Despite the resultant reduction in reads within repeats, our analysis still identifies CCRs within repetitive elements. However, we cannot exclude that uniquely mapping reads are underrepresented in longer repeats due to the limits of read length.
Like TOP2A cleavage sites, oncogenic translocations are biased toward introns, and we discover that not only KMT2A recombinome genes but also all known cancer fusion genes are more likely to be cleaved by TOP2A, raising the intriguing possibility that TOP2A has a previously unrecognized broader role in triggering translocations. The TOP2 poisons enhance cleavage in regions of KMT2A, PML, RARA, and RUNX1 that are often translocated in leukemia, suggesting that the sequence preferences for TOP2 poison–induced cleavage set the stage for translocations. We find evidence in cells that TOP2A cleavage can generate the DSBs and SSNs required to create the breakpoint junctions found in treatment-related (Lovett et al. 2001; Whitmarsh et al. 2003) and infant (Gillert et al. 1999; Raffini et al. 2002) KMT2A-R leukemias, respectively, including that etoposide causes a higher proportion of KMT2A recombinome genes to have TOP2A DSBs relative to all genes in the genome. At a single-nucleotide scale, we discover TOP2A cleavage sites at translocation breakpoints, including at the KMT2A-R treatment–related leukemia translocation breakpoint hotspot, supporting the model in which TOP2A DNA damage is the direct precursor lesion to leukemogenic translocations.
This study also uncovers new relationships between TOP2A cleavage and coding gene and lincRNA transcription. Unlike the bimodal distribution of transcript abundance, we show skews of TOP2A-cleaved loci toward more highly expressed coding genes and a lincRNA subset with moderately abundant transcripts. We find that in coding genes, TOP2A CCRs are spatially enriched in the distal regions of gene bodies, suggesting that TOP2A cleavage is important for transcriptional elongation, and we discover that the TOP2 poisons etoposide, mitoxantrone, and genistein cause proximal shifts in this spatial distribution.
Whereas transcript abundance and length are unrelated, in coding and lincRNA loci alike we observe that TOP2A CCR signal strength is independently associated with gene length and with the abundance of transcripts produced by genes of all length categories. Additionally, we discover that genes with TOP2A CCRs have stronger signals for the marks of transcription elongation POLR2A and H3K36me3 (Li et al. 2002; Strahl et al. 2002). Others previously observed that long genes contain a higher density of supercoiled chromatin, which stalls POLR2A during transcription elongation (Joshi et al. 2012). Given that genes with CCRs display higher transcriptional activity, the greater CCR signal strength with increasing length, yet lack of correlation between length and transcript abundance, suggests that greater TOP2A cleavage is required to relax the DNA supercoils in long coding and lincRNA loci during transcription.
Thus, we conclude that TOP2A CCRs play an important role in transcription elongation of coding genes and lincRNA loci. The discoveries of TOP2A in the Pol II complex and TOP2A requirement for chromatin dependent in vitro transcription (Mondal and Parvin 2001) are in concert with this conclusion, as are the enriched TOP2 binding in highly transcribed Saccharomyces cerevisiae genes (Sperling et al. 2011) and the reduced expression of longer genes with yeast Top2 inhibition (Joshi et al. 2012). Although murine ES cells display TOP2A binding at promoters (Thakurela et al. 2013), we do not find TOP2A CCR enrichment at promoters. Because TOP2A binding does not necessarily equate to cleavage (Mueller-Planitz and Herschlag 2007; Lee et al. 2012), our study and the mouse ES cell study together suggest that TOP2A begins to track supercoil accumulation starting at the promoter but then cleaves well into the gene body during transcription elongation.
Consistent with roles of TOP2A cleavage in translocations and transcription, TOP2A CCRs are correlated with stronger DNase I sites tagging open chromatin. We also uncover another subset of TOP2A CCRs correlated with the H3K4me1/H3K27ac and H3K4me1 marks of strong and weak enhancers (Creyghton et al. 2010) that suggest a previously unsuspected function of TOP2A in long-range regulation of gene activation. Our discovery that marks of active transcription along gene bodies, marks of open chromatin, and marks of enhancers colocalize with TOP2A CCRs within separate genomic environments implicates TOP2A CCRs as chromatin remodeling orchestrators alongside multiple chromatin features.
Altogether our data indicate a new, more general DNA-damaging role of TOP2A cleavage in oncogenic translocations beyond those attributed to exposures to TOP2 poisons in leukemia (Felix 2012; Gole and Wiesmuller 2015) and a modular organization of TOP2A CCRs with factors in the epigenome and transcriptional machinery as a fundamental feature of human cells that impacts translocations and transcription. Because our methodology was developed in a transformed leukemia cell line, in its further applications, it will be important to compare genome-wide TOP2 cleavage patterns between isoforms in different primary cell types representing normal development and disease states to build on these discoveries.
Methods
Cell treatment and library preparation
Vehicle or TOP2 poison–treated K562 cells were lysed with RIPA buffer. The lysates were sonicated to fragment the DNA to 100–1500 bp and frozen overnight. TOP2A was immunocaptured with anti-TOP2A and Protein G magnetic beads, the bead-bound fraction separated, and nonbound fractions subjected to immunocapture again twice. The bead-bound fractions were combined and treated with CIP (New England Biolabs) to detach the TOP2A-bound DNA from cleavage complexes. The released DNA and input DNA were purified and used directly for library synthesis with an Illumina TruSeq ChIP library kit or preamplified with a SEQXE kit (Sigma-Aldrich) before preparing libraries for sequencing (Supplemental Fig. S1; Supplemental Methods).
Data analysis
Raw reads were aligned to the human genome version GRCh38/hg38 (released 12-2013). Reads that mapped uniquely were retained and normalized to RP10M (reads per 10 million reads mapped). Correlation coefficients of reads in 10-kb windows were determined for biological replicates. Criteria for calling TOP2A cleavage sites and CCRs, and definitions of genic TOP2A CCR signal strength, were as described in the Results. CCRs from similarly treated preamplified samples or from all preamplified samples across treatments were merged into union sets. Statistical tests used for cleavage site analyses and for comparisons of CCRs with human SNP data (The 1000 Genomes Project Consortium 2015), genomic elements, transcript abundance, and chromatin features and the method of converting data sets from the ENCODE Project Consortium 2012 for chromatin features (The ENCODE Project Consortium 2012) from GRCh37/hg19 to GRCh38/hg38 are in the Supplemental Methods.
Data access
All data from this study have been submitted to the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE79593. All data are also available at the UCSC Genome Browser (see Data Access Instructions in the Supplemental Methods) (Kent et al. 2002; http://genome.ucsc.edu/).
Competing interest statement
C.A.F. and E.F.R. hold the following unlicensed patent: Methods and Kits for Analysis of Chromosomal Rearrangements Associated with Leukemia. United States of America 6,368,791. 2002 April 09. C.A.F. holds the following unlicensed patent: Compositions and Methods for the Detection of DNA Topoisomerase II Complexes with DNA. United States of America 8,642,265 B2. 2014 February 04. C.A.F., M.L.C., and B.D.G. have submitted the following patent application: Compositions and Methods for the Detection of DNA Cleavage Complexes–Provisional Patent. United States of America patent application 61/490,975. 2012 May 27.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health (NIH) grants R01CA77683 (C.A.F.), R01CA80175 (C.A.F.), and R01GM033944 (N.O.); Children's Hospital of Philadelphia Department of Pediatrics Academic Enrichment Program (C.A.F.); and the University of Pennsylvania Center of Excellence in Environmental Toxicology Pilot Project: NIH 1P30 ES013508-06 (C.A.F.). C.A.F. is the Joshua Kahan Endowed Chair in Pediatric Leukemia Research. N.O. is the John G. Coniglio Chair in Biochemistry.
Author contributions: Conceptualization was by C.A.F. and B.D.G. Methodology was by C.A.F., B.D.G., M.L.C., K.A.U., and E.F.R. Software was provided by B.D.G. Formal analysis was done by X.Y., J.W.D., C.P.K., B.D.G., and C.A.F. Investigation was done by J.W.D., K.A.U., M.L.C., S.J.G., and C.P.K. The original draft was done by X.Y., J.W.D., B.D.G., and C.A.F. Review and editing were done by K.A.U., M.L.C., S.J.G., C.P.K., J.A.W.B., E.F.R., and N.O. Visualization was by X.Y., K.A.U., and C.A.F. Funding acquisition was by C.A.F. Resources were from J.A.W.B., N.O., and B.D.G. Data curating was by X.Y., J.W.D., B.D.G., and C.A.F. Supervision was by C.A.F. and B.D.G.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.211615.116.
References
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526: 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashour ME, Atteya R, El-Khamisy SF. 2015. Topoisomerase-mediated chromosomal break repair: an emerging player in many games. Nat Rev Cancer 15: 137–151. [DOI] [PubMed] [Google Scholar]
- Audic S, Claverie JM. 1997. The significance of digital gene expression profiles. Genome Res 7: 986–995. [DOI] [PubMed] [Google Scholar]
- Bansal H, Yihua Q, Iyer SP, Ganapathy S, Proia DA, Penalva LO, Uren PJ, Suresh U, Carew JS, Karnad AB, et al. 2014. WTAP is a novel oncogenic protein in acute myeloid leukemia. Leukemia 28: 1171–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranello L, Kouzine F, Wojtowicz D, Cui K, Przytycka TM, Zhao K, Levens D. 2014. DNA break mapping reveals topoisomerase II activity genome-wide. Int J Mol Sci 15: 13111–13122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg KD, Burgin AB, Osheroff N. 2003. A two-drug model for etoposide action against human topoisomerase IIα. J Biol Chem 278: 7406–7412. [DOI] [PubMed] [Google Scholar]
- Capranico G, Binaschi M. 1998. DNA sequence selectivity of topoisomerases and topoisomerase poisons. Biochim Biophys Acta 1400: 185–194. [DOI] [PubMed] [Google Scholar]
- Cowell IG, Sondka Z, Smith K, Lee KC, Manville CM, Sidorczuk-Lesthuruge M, Rance HA, Padget K, Jackson GH, Adachi N, et al. 2012. Model for MLL translocations in therapy-related leukemia involving topoisomerase IIβ-mediated DNA strand breaks and gene proximity. Proc Natl Acad Sci 109: 8989–8994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crosetto N, Mitra A, Silva MJ, Bienko M, Dojer N, Wang Q, Karaca E, Chiarle R, Skrzypczak M, Ginalski K, et al. 2013. Nucleotide-resolution DNA double-strand break mapping by next-generation sequencing. Nat Methods 10: 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dykhuizen EC, Hargreaves DC, Miller EL, Cui K, Korshunov A, Kool M, Pfister S, Cho YJ, Zhao K, Crabtree GR. 2013. BAF complexes facilitate decatenation of DNA by topoisomerase IIα. Nature 497: 624–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix CA. 2012. Therapy-related leukemias. In Childhood leukemias (ed. Pui C-H), pp. 723–771. Cambridge University Press, Cambridge, UK. [Google Scholar]
- Felix CA, Kolaris CP, Osheroff N. 2006. Topoisomerase II and the etiology of chromosomal translocations. DNA Repair (Amst) 5: 1093–1108. [DOI] [PubMed] [Google Scholar]
- Gao R, Schellenberg MJ, Huang SY, Abdelmalak M, Marchand C, Nitiss KC, Nitiss JL, Williams RS, Pommier Y. 2014. Proteolytic degradation of topoisomerase II (Top2) enables the processing of Top2.DNA and Top2.RNA covalent complexes by tyrosyl-DNA-phosphodiesterase 2 (TDP2). J Biol Chem 289: 17960–17969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillert E, Leis T, Repp R, Reichel M, Hosch A, Breitenlohner I, Angermuller S, Borkhardt A, Harbott J, Lampert F, et al. 1999. A DNA damage repair mechanism is involved in the origin of chromosomal translocations t(4;11) in primary leukemic cells. Oncogene 18: 4663–4671. [DOI] [PubMed] [Google Scholar]
- Gole B, Wiesmuller L. 2015. Leukemogenic rearrangements at the mixed lineage leukemia gene (MLL)-multiple rather than a single mechanism. Front Cell Dev Biol 3: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha N, Chang JS, Chokkalingam AP, Wiemels JL, Smith MT, Buffler PA. 2008. NQO1 polymorphisms and de novo childhood leukemia: a HuGE review and meta-analysis. Am J Epidemiol 168: 1221–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinrichs AS, Karolchik D, Baertsch R, Barber GP, Bejerano G, Clawson H, Diekhans M, Furey TS, Harte RA, Hsu F, et al. 2006. The UCSC Genome Browser Database: update 2006. Nucleic Acids Res 34: D590–D598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huff JT, Plocik AM, Guthrie C, Yamamoto KR. 2010. Reciprocal intronic and exonic histone modification regions in humans. Nat Struct Mol Biol 17: 1495–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovoni JS, Caron P, Lassadi I, Nicolas E, Massip L, Trouche D, Legube G. 2010. High-resolution profiling of gammaH2AX around DNA double strand breaks in the mammalian genome. EMBO J 29: 1446–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igunnu A, Osalaye DS, Olorunsogo OO, Malomo SO, Olorunniji FJ. 2011. Distinct metal ion requirements for the phosphomonoesterase and phosphodiesterase activities of calf intestinal alkaline phosphatase. Open Biochem J 5: 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joshi RS, Pina B, Roca J. 2012. Topoisomerase II is required for the production of long Pol II gene transcripts in yeast. Nucleic Acids Res 40: 7907–7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, Rosenfeld MG. 2006. A topoisomerase IIβ-mediated dsDNA break required for regulated transcription. Science 312: 1798–1802. [DOI] [PubMed] [Google Scholar]
- Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, et al. 2014. Defining functional DNA elements in the human genome. Proc Natl Acad Sci 111: 6131–6138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12: 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzine F, Gupta A, Baranello L, Wojtowicz D, Ben-Aissa K, Liu J, Przytycka TM, Levens D. 2013. Transcription-dependent dynamic supercoiling is a short-range genomic force. Nat Struct Mol Biol 20: 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Jung SR, Heo K, Byl JA, Deweese JE, Osheroff N, Hohng S. 2012. DNA cleavage and opening reactions of human topoisomerase IIα are regulated via Mg2+-mediated dynamic bending of gate-DNA. Proc Natl Acad Sci 109: 2925–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Moazed D, Gygi SP. 2002. Association of the histone methyltransferase Set2 with RNA polymerase II plays a role in transcription elongation. J Biol Chem 277: 49383–49388. [DOI] [PubMed] [Google Scholar]
- Lindsey RH Jr, Bromberg KD, Felix CA, Osheroff N. 2004. 1,4-Benzoquinone is a topoisomerase II poison. Biochemistry 43: 7563–7574. [DOI] [PubMed] [Google Scholar]
- Lovett BD, Lo Nigro L, Rappaport EF, Blair IA, Osheroff N, Zheng N, Megonigal MD, Williams WR, Nowell PC, Felix CA. 2001. Near-precise interchromosomal recombination and functional DNA topoisomerase II cleavage sites at MLL and AF-4 genomic breakpoints in treatment-related acute lymphoblastic leukemia with t(4;11) translocation. Proc Natl Acad Sci 98: 9802–9807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu YL, Lin CP, Azarova AM, Cai L, Wang JC, Liu LF. 2006. Role of topoisomerase IIβ in the expression of developmentally regulated genes. Mol Cell Biol 26: 7929–7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer C, Hofmann J, Burmeister T, Groger D, Park TS, Emerenciano M, Pombo de Oliveira M, Renneville A, Villarese P, Macintyre E, et al. 2013. The MLL recombinome of acute leukemias in 2013. Leukemia 27: 2165–2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry AR, Felix CA, Whitmarsh RJ, Mason A, Reiter A, Cassinat B, Parry A, Walz C, Wiemels JL, Segal MR, et al. 2005. DNA topoisomerase II in therapy-related acute promyelocytic leukemia. N Engl J Med 352: 1529–1538. [DOI] [PubMed] [Google Scholar]
- Mitelman F, Johansson B, Mertens F. 2016. Mitelman database of chromosome aberrations and gene fusions in cancer. https://cgap.nci.nih.gov/Chromosomes/Mitelman.
- Mondal N, Parvin JD. 2001. DNA topoisomerase IIα is required for RNA polymerase II transcription on chromatin templates. Nature 413: 435–438. [DOI] [PubMed] [Google Scholar]
- Mueller-Planitz F, Herschlag D. 2007. DNA topoisomerase II selects DNA cleavage sites based on reactivity rather than binding affinity. Nucleic Acids Res 35: 3764–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muslimovic A, Nystrom S, Gao Y, Hammarsten O. 2009. Numerical analysis of etoposide induced DNA breaks. PLoS One 4: e5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naughton C, Avlonitis N, Corless S, Prendergast JG, Mati IK, Eijk PP, Cockroft SL, Bradley M, Ylstra B, Gilbert N. 2013. Transcription forms and remodels supercoiling domains unfolding large-scale chromatin structures. Nat Struct Mol Biol 20: 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novo FJ, de Mendibil IO, Vizmanos JL. 2007. TICdb: a collection of gene-mapped translocation breakpoints in cancer. BMC Genomics 8: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien PJ, Herschlag D. 2001. Functional interrelationships in the alkaline phosphatase superfamily: phosphodiesterase activity of Escherichia coli alkaline phosphatase. Biochemistry 40: 5691–5699. [DOI] [PubMed] [Google Scholar]
- Ottone T, Hasan SK, Montefusco E, Curzi P, Mays AN, Chessa L, Ferrari A, Conte E, Noguera NI, Lavorgna S, et al. 2009. Identification of a potential “hotspot” DNA region in the RUNX1 gene targeted by mitoxantrone in therapy-related acute myeloid leukemia with t(16;21) translocation. Genes Chromosomes Cancer 48: 213–221. [DOI] [PubMed] [Google Scholar]
- Palumbo M, Mabilia M, Pozzan A, Capranico G, Tinelli S, Zunino F. 1994. Conformational properties of topoisomerase II inhibitors and sequence specificity of DNA cleavage. J Mol Recognit 7: 227–231. [DOI] [PubMed] [Google Scholar]
- Pan J, Sasaki M, Kniewel R, Murakami H, Blitzblau HG, Tischfield SE, Zhu X, Neale MJ, Jasin M, Socci ND, et al. 2011. A hierarchical combination of factors shapes the genome-wide topography of yeast meiotic recombination initiation. Cell 144: 719–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang B, de Jong J, Qiao X, Wessels LF, Neefjes J. 2015. Chemical profiling of the genome with anti-cancer drugs defines target specificities. Nat Chem Biol 11: 472–480. [DOI] [PubMed] [Google Scholar]
- Pendleton M, Lindsey RH Jr, Felix CA, Grimwade D, Osheroff N. 2014. Topoisomerase II and leukemia. Ann N Y Acad Sci 1310: 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Povirk LF. 2006. Biochemical mechanisms of chromosomal translocations resulting from DNA double-strand breaks. DNA Repair (Amst) 5: 1199–1212. [DOI] [PubMed] [Google Scholar]
- Raffini LJ, Slater DJ, Rappaport EF, Lo Nigro L, Cheung NK, Biegel JA, Nowell PC, Lange BJ, Felix CA. 2002. Panhandle and reverse-panhandle PCR enable cloning of der(11) and der(other) genomic breakpoint junctions of MLL translocations and identify complex translocation of MLL, AF-4, and CDK6. Proc Natl Acad Sci 99: 4568–4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson BW, Cheung NK, Kolaris CP, Jhanwar SC, Choi JK, Osheroff N, Felix CA. 2008. Prospective tracing of MLL-FRYL clone with low MEIS1 expression from emergence during neuroblastoma treatment to diagnosis of myelodysplastic syndrome. Blood 111: 3802–3812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulston D, Espinosa R III, Nucifora G, Larson RA, Le Beau MM, Rowley JD. 1998. CBFA2(AML1) translocations with novel partner chromosomes in myeloid leukemias: association with prior therapy. Blood 92: 2879–2885. [PubMed] [Google Scholar]
- Sano K, Miyaji-Yamaguchi M, Tsutsui KM, Tsutsui K. 2008. Topoisomerase IIβ activates a subset of neuronal genes that are repressed in AT-rich genomic environment. PLoS One 3: e4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley R, Green P. 2013–2015. RepeatMasker Open-4.0 2013–2015. http://www.repeatmasker.org/.
- Smith MT, Zhang L, Wang Y, Hayes RB, Li G, Wiemels J, Dosemeci M, Titenko-Holland N, Xi L, Kolachana P, et al. 1998. Increased translocations and aneusomy in chromosomes 8 and 21 among workers exposed to benzene. Cancer Res 58: 2176–2181. [PubMed] [Google Scholar]
- Smith MT, Wang Y, Skibola CF, Slater DJ, Lo Nigro L, Nowell PC, Lange BJ, Felix CA. 2002. Low NAD(P)H:quinone oxidoreductase activity is associated with increased risk of leukemia with MLL translocations in infants and children. Blood 100: 4590–4593. [DOI] [PubMed] [Google Scholar]
- Spector LG, Xie Y, Robison LL, Heerema NA, Hilden JM, Lange B, Felix CA, Davies SM, Slavin J, Potter JD, et al. 2005. Maternal diet and infant leukemia: the DNA topoisomerase II inhibitor hypothesis: a report from the children's oncology group. Cancer Epidemiol Biomarkers Prev 14: 651–655. [DOI] [PubMed] [Google Scholar]
- Sperling AS, Jeong KS, Kitada T, Grunstein M. 2011. Topoisomerase II binds nucleosome-free DNA and acts redundantly with topoisomerase I to enhance recruitment of RNA Pol II in budding yeast. Proc Natl Acad Sci 108: 12693–12698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strahl BD, Grant PA, Briggs SD, Sun ZW, Bone JR, Caldwell JA, Mollah S, Cook RG, Shabanowitz J, Hunt DF, et al. 2002. Set2 is a nucleosomal histone H3-selective methyltransferase that mediates transcriptional repression. Mol Cell Biol 22: 1298–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakurela S, Garding A, Jung J, Schubeler D, Burger L, Tiwari VK. 2013. Gene regulation and priming by topoisomerase IIα in embryonic stem cells. Nat Commun 4: 2478. [DOI] [PubMed] [Google Scholar]
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, et al. 2012. The accessible chromatin landscape of the human genome. Nature 489: 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari VK, Burger L, Nikoletopoulou V, Deogracias R, Thakurela S, Wirbelauer C, Kaut J, Terranova R, Hoerner L, Mielke C, et al. 2012. Target genes of Topoisomerase IIβ regulate neuronal survival and are defined by their chromatin state. Proc Natl Acad Sci 109: E934–E943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitmarsh RJ, Saginario C, Zhuo Y, Hilgenfeld E, Rappaport EF, Megonigal MD, Carroll M, Liu M, Osheroff N, Cheung NK, et al. 2003. Reciprocal DNA topoisomerase II cleavage events at 5′-TATTA-3′ sequences in MLL and AF-9 create homologous single-stranded overhangs that anneal to form der(11) and der(9) genomic breakpoint junctions in treatment-related AML without further processing. Oncogene 22: 8448–8459. [DOI] [PubMed] [Google Scholar]
- Wiemels JL, Pagnamenta A, Taylor GM, Eden OB, Alexander FE, Greaves MF. 1999. A lack of a functional NAD(P)H:quinone oxidoreductase allele is selectively associated with pediatric leukemias that have MLL fusions. United Kingdom Childhood Cancer Study Investigators. Cancer Res 59: 4095–4099. [PubMed] [Google Scholar]
- Zechiedrich EL, Christiansen K, Andersen AH, Westergaard O, Osheroff N. 1989. Double-stranded DNA cleavage/religation reaction of eukaryotic topoisomerase II: evidence for a nicked DNA intermediate. Biochemistry 28: 6229–6236. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.