

Abstract
Drosophila melanogaster is a widely used genetic model organism in developmental biology. While this model organism has been intensively studied at the RNA level, a comprehensive proteomic study covering the complete life cycle is still missing. Here, we apply label-free quantitative proteomics to explore proteome remodeling across Drosophila’s life cycle, resulting in 7952 proteins, and provide a high temporal-resolved embryogenesis proteome of 5458 proteins. Our proteome data enabled us to monitor isoform-specific expression of 34 genes during development, to identify the pseudogene Cyp9f3Ψ as a protein-coding gene, and to obtain evidence of 268 small proteins. Moreover, the comparison with available transcriptomic data uncovered examples of poor correlation between mRNA and protein, underscoring the importance of proteomics to study developmental progression. Data integration of our embryogenesis proteome with tissue-specific data revealed spatial and temporal information for further functional studies of yet uncharacterized proteins. Overall, our high resolution proteomes provide a powerful resource and can be explored in detail in our interactive web interface.
Drosophila melanogaster is among the best-described model organisms for development and aging. During its life cycle, it progresses through well-defined stages including embryo, larva, pupa, and adult, undergoing a complete phenotypic metamorphosis (Lawrence 1992). These transitions are based on tightly regulated gene expression at the transcriptional, epigenetic, and translational level. Currently, most developmental gene expression studies in Drosophila rely on in situ hybridization of RNA (Lécuyer et al. 2007; Tomancak et al. 2007), transcriptome analysis using large-scale microarray/RNA-seq data sets (Chintapalli et al. 2007; Kalinka et al. 2010; Graveley et al. 2011; Brown et al. 2014), or a combination of both (Jambor et al. 2015). However, mRNAs are further translated into proteins, which perform the actual cellular functions. It has been shown in multiple species such as Saccharomyces cerevisiae (Griffin et al. 2002), Trypanosoma brucei (Butter et al. 2013), Caenorhabditis elegans (Grün et al. 2014), and human (Schwanhäusser et al. 2011), as well as in Drosophila melanogaster (Bonaldi et al. 2008), that transcript levels are only a moderate predictor for protein expression as they do not account for post-transcriptional processes such as translational regulation or protein stability (Vogel and Marcotte 2012; Liu et al. 2016). Recently, this has also been addressed with a developmental perspective in Caenorhabditis elegans (Grün et al. 2014), Xenopus laevis (Peshkin et al. 2015), and Trypanosoma brucei (Dejung et al. 2016), but not yet in Drosophila.
The number of fly proteins with available antibodies increased in the last decade from around 450 (Adams et al. 2000) to 1586 (listed in FlyBase version 6.01) but still covers only a small fraction of expressed genes. To accelerate protein studies in Drosophila, several tagging strategies were devised. Around 100 genes have been fused with GFP using piggyBac transposition (Morin et al. 2001), and 400 GFP-tagged fly lines have been established using MiMICs (Minos Mediated Integration Cassettes) to permit systematic protein investigations (Nagarkar-Jaiswal et al. 2015). In an alternative approach, BAC TransgeneOmics allowed the creation of 880 lines and the systematic study of 207 GFP-tagged fly proteins (Sarov et al. 2016). In principle, all protein-coding genes can be investigated, but this requires the establishment of a line for each protein. Additionally, a putative caveat of tagging strategies is altered protein behavior like mislocalization, changes in protein stability, or a dominant negative regulatory effect (Margolin 2012).
The fruit fly is one of the model species investigated by modENCODE (The modENCODE Consortium et al. 2010), and thus several large data sets are available for mapping histone modifications (Kharchenko et al. 2011), global RNA levels during development (Graveley et al. 2011), and tissue-specific splicing (Brown et al. 2014). In contrast, proteomic studies in Drosophila have been restricted to certain developmental stages. For example, changes in the proteome during aging from eclosure to 60-d-old flies (Sowell et al. 2007), the adult itself (Sury et al. 2010; Xing et al. 2014), larva and pupa (Chang et al. 2013), the embryo (Fabre et al. 2016), and the oocyte-to-embryo transition (Kronja et al. 2014) have been investigated. However, these studies have relatively low proteome coverage (around 2000 proteins), do not cover the complete developmental process, and are not directly comparable because of technical differences.
Applying label-free quantitative proteomics (Cox et al. 2014), we here measured protein expression throughout the Drosophila life cycle with a coverage of 7952 proteins to provide insight into proteome remodeling. With embryogenesis being a focus in Drosophila developmental studies, we amended the life cycle proteome with an embryogenesis proteome of 5458 proteins with high temporal resolution. Finally, data integration with tissue-specific (Lécuyer et al. 2007) and developmental transcriptomic studies (Graveley et al. 2011) allows investigation of the importance of spatial and translational regulation.
Results
Proteomics screen of the life cycle
We collected whole-animal samples at 15 representative time points during the Drosophila life cycle (Fig. 1A). The embryonic time points were chosen according to major stages of embryonic development: prior to zygotic gene activation (0–2 h, E02), gastrulation (4–6 h, E06), organogenesis (10–12 h, E10), and the late stages of embryogenesis (18–20 h, E20). For larva, the three different instar larvae (L1, L2, and early L3) and a late stage (L3 crawling larva) were examined. Pupae were collected daily starting with the white pupa, and, for adults, the virgin males and females (up to 4 h after eclosure) as well as 1-wk-old animals of each sex were chosen. All samples were collected as biological quadruplicates and processed by mechanical disruption with a universal protein extraction protocol. For each replicate, a 5-h mass spectrometry (MS) run was used, resulting in 340 h of measurement (68 MS runs). We searched the resulting eight million MS/MS spectra against a Saccharomyces cerevisiae and Drosophila melanogaster database using the MaxQuant software suite (Cox and Mann 2008). Overall, we identified 9627 protein groups (a protein group contains proteins indistinguishable by the peptides that were identified) with 144,067 unique peptide sequences at a FDR < 0.01. This number includes 1078 yeast and 8549 Drosophila protein groups (Supplemental Fig. S1A). The identification of yeast proteins is nearly exclusively restricted to the larval stages, where it is a food source (Supplemental Fig. S1B). The number of 8549 identified fly proteins is comparable to a previous in-depth measurement of multiple sources of Drosophila material reaching 9124 proteins (Brunner et al. 2007). After filtering for robust detection in at least two replicates of any time point, we performed our subsequent analysis on a set of 7952 protein groups (Fig. 1B; Supplemental Table S1).
Figure 1.
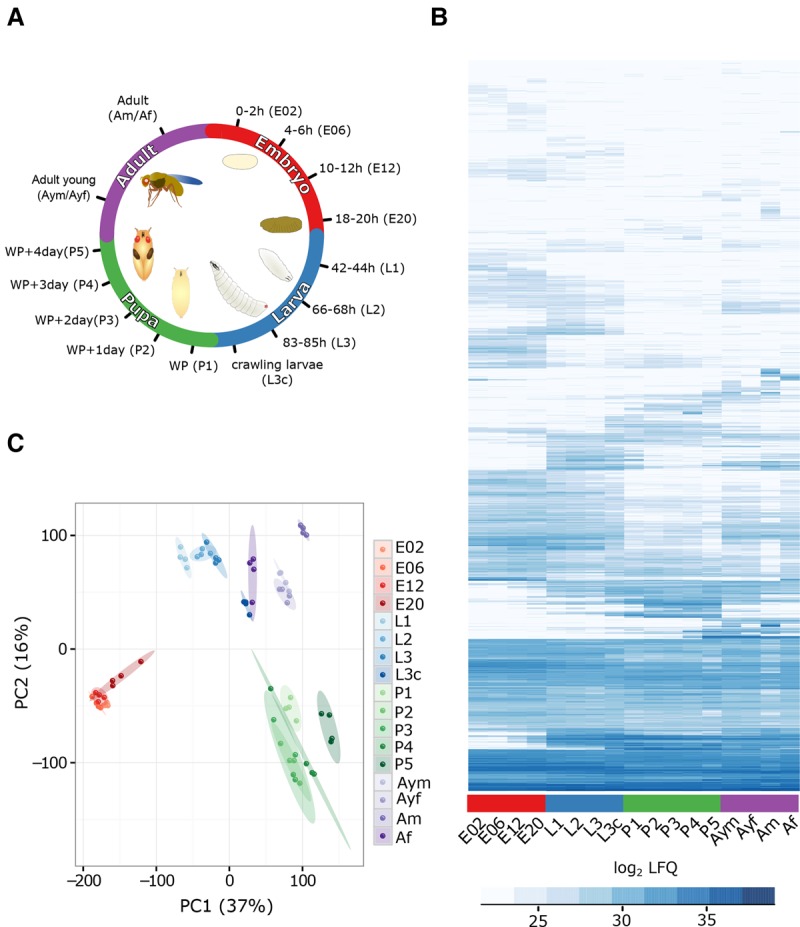
Drosophila developmental life cycle proteome. (A) Scheme depicting the collected time points throughout the four major metamorphic stages of Drosophila (embryo [red], larva [blue], pupa [green], and adult [violet]). (WP) White pupa, (L3c) crawling third instar larva. (B) Heat map of log2 LFQ values of the 7952 protein groups quantified during fly development. (C) Visualization of the first two principal components separating samples according to their developmental stage. The biological replicates are indicated in the same color, with elliptic areas representing the standard error of the two depicted components.
Developmental processes are tightly regulated and thus highly reproducible in each organism. Nevertheless, to visualize biological variability of this process, we performed correlation and principal component analysis (PCA). To increase quantitation reliability, all label-free quantitation (LFQ) values were solely based on unique peptide intensities for each protein group. Despite the fact that our replicates are originating from different egg-laying events, are being processed independently, and are measured several days apart on the mass spectrometer, we find a very high correlation within the time points (R = 0.84–0.98) (Supplemental Fig. S1C) and clear formation of clusters in PCA (Fig. 1C). These findings demonstrate a very high reproducibility of our experimental conditions from the biological system to the mass spectrometry measurement.
Core proteome and protein expression dynamics
To identify a core proteome, i.e., proteins detected at all stages of development, we grouped the proteins according to their presence in the four major stages of the life cycle (Fig. 2A). We found 4627 protein groups, more than half of our proteome, to be detectable in all stages. To obtain an overview of the functionality of these continuously expressed proteins, we performed gene ontology (GO) annotation enrichment analysis and reduced the GO term complexity to uncover major descriptors (Fig. 2B). As expected, our core proteome is enriched for metabolic and cellular processes describing the basic activities of any cellular system, exemplified by covering all known proteins for such essential processes as tRNA aminoacetylation, endosome transport via a multivesicular body sorting pathway, cell junction maintenance, nuclear pore organization, and ribosome assembly (Fig. 2B; Supplemental Fig. S2A; Supplemental Table S2). We also analyzed developmental expression dynamics for all proteins with an averaged abundance above the detection limit, log2 LFQ intensity > 25 (Fig. 2C; Supplemental Table S3). We additionally applied a Gini coefficient filter of 0.1, which divided our proteome into 1386 stably expressed proteins throughout life cycle and 1978 differentially expressed proteins. Consistent with a previous developmental study in Xenopus, we see that the dynamicity decreases with protein abundance (Fig. 2C; Peshkin et al. 2015). We show examples of highly dynamic and stably expressed proteins (Fig. 2D; Supplemental Fig. S2D). The stable proteins include the widely accepted loading controls: tubulins, actins, heat-shock proteins, Gapdh1, Gapdh2, and Vinculin.
Figure 2.

Characteristics of the developmental proteome. (A) Overlap of quantified protein groups between developmental stages results in a core proteome of 4627 proteins. (B) Clusters of enriched GO terms obtained from the core proteome are plotted in a coordinate system defined by the first two dimensions of a multidimensional scaling according to their similarity scores. The color of the circle represents the GO cluster with a representative term highlighted. The diameter of the circle is proportional to the size of the GO category. (C) The density plot relates protein abundance with a dynamicity score during developmental protein expression (log10 transformed Gini index). In the lower-right quadrant, highly stable proteins are represented, while the upper-right quadrant contains proteins with changing expression levels during development. (D) Expression profiles for two highly dynamic (upper panel) and two stably expressed (lower panel) proteins highlighted in red in the dynamicity plot.
Developmental expression profiles of highly abundant proteins
We first characterized the 100 most abundant proteins per stage, comprising around 10% of the total protein mass (Supplemental Table S1). Among proteins with the highest LFQ values, we find ribosomal proteins, being especially prevalent in the top 100 list during embryogenesis, a phase of rapid cell proliferation. The fly uses different storage proteins at specific developmental stages: yolk proteins (Yp1, Yp2, and Yp3) in embryogenesis and Lsp proteins, whose protein abundance rises drastically in L3. Among these highly abundant proteins, there are several preliminary annotated genes that are not further characterized. CG1850, representing the most abundant protein in the pupal stage, shares a small stretch of similarity to the cuticular protein Cpr72Eb (BLAST E-value: 0.019) (Supplemental Fig. S2B). Interestingly, some other highly expressed computed genes (CG) also show similar protein expression patterns to well-studied cuticular proteins like Cpr72Ea (CG1850 and CG13023), Cpr64Aa and Cpr64Ac (CG34461 and CG42323), and Cpr66D (CG16886 and CG30101). While thus far we have looked at the most highly expressed 100 proteins, our proteome can be interrogated to reveal the temporal expression pattern of any quantified protein.
Proteome remodeling throughout the life cycle
Our proteome covers a dynamic range of more than six orders of magnitude, showing expression changes of individual proteins of more than 100,000-fold (Supplemental Fig. S2C). We interrogated our data set for stage-specific proteins by applying ANOVA (FDR < 0.01) on the log2 LFQ values (Fig. 3A). The majority of these 1535 differentially regulated protein groups are found in adult flies (556), followed by embryos (473), pupae (317), and larvae (189). To connect the proteome differences to stage-specific biological functions, we performed GO enrichment analysis on clustered protein expression profiles (Fig. 3A; Supplemental Tables S4, S5). The most enriched GO terms during embryogenesis include mitotic cell cycle regulation and nuclear division represented by cyclins (CycE, CycA, CycB) and developmental kinases, such as Loki (Lok), Greatwall (Gwl), and Grapes (Grp). By this clustering, we were able to separate an early and late embryogenesis phase (Fig. 3B). The early phase (0–6 h) is characterized by high expression of proteins involved in cytoskeleton organization (Dgt4, AlphaTub67C, and GammaTub37C), microtubule binding proteins (Mars and Wee Augmin [Wac]), as well as the classical examples Bicaudal C (BicC) and Cup, important in translational regulation of the oskar mRNA. In contrast, proteins involved in tissue morphogenesis, such as Bazooka (Baz), Fat (Ft), Ribbon (Rib), and Tramtrack (Ttk), are up-regulated in later phases (12–20 h). Stage-specific proteins in larvae and pupae include expected structural constituents of the chitin-based cuticle: Lcp, Tweedle (Twd), and cuticular proteins. Intriguingly, several proteins that are highly up-regulated only at a single pupal stage, like CG13376, CG13082, and CG42449, are poorly characterized (Fig. 3B; Supplemental Fig. S3A). In the adult, odorant-binding proteins (Obp83b and Obp57a), proteins involved in light perception and phototransduction (Arr1 and Arr2), and the retinal degeneration protein A (RdgA) show strong expression, consistent with the adult fly having a fully developed light sensory system. Also, proteins involved in muscle contraction, like flightin (Fln) and Eaat1, increase their expression 100-fold in adult stages (Fig. 3B).
Figure 3.

Stage-specific proteins and ecdysone-induced developmental regulation. (A) Heat map showing 1535 protein groups found to be differentially (ANOVA, FDR < 0.01) regulated during the life cycle. These protein groups were clustered into up to 12 stage-specific profiles. Average profiles of the individual clusters for each developmental stage are shown. (B) Heat map showing log2 LFQ abundance of proteins with stage-specific expression profiles discussed in the text. (C) Schematic representation of ecdysone pulses during fly development (upper panel) and heat map of log2 LFQ expression levels of selected proteins of 20-hydroxyecdysone regulated genes (lower panel). (D) For the Eig71E and Sgs gene family, RNA expression profiles (dotted line) differ from protein levels (solid line) during the pupal phase, demonstrating prolonged protein stability. (E) Three examples showing single protein expression burst, but more broadly detectable RNA indicating more tightly controlled protein expression.
Overall, our data are in agreement with previously published studies and connects protein expression with well-described morphological changes during Drosophila development. Therefore, our screen defines the developmental stage to study molecular or phenotypic effects of yet uncharacterized proteins. All protein profiles can be interrogated using the interactive web interface (http://www.butterlab.org/flydev).
Developmentally regulated functions: ecdysone-induced proteins and cuticle formation
The regulation of molting by endogenous 20-hydroxyecdysone (20E) is a prototype example of hormonal gene regulation pathways in insects (Yamanaka et al. 2013). Previous microarray studies focused on 20E-induced gene regulation of mRNA transcripts between the L3 larval stage and 12 h after puparium formation (Beckstead et al. 2005; Gonsalves et al. 2011). However, for the ecdysone-induced gene family 71E (Eig71E), we find intriguing differences between the expression profiles of mRNA and protein in pupae. Messenger RNA expression is detectable in three different waves: Eig71Ee spikes at L3c, another group represented by Eig71Ed at P1, and a later group represented by Eig71Ek at P2 (Fig. 3D; Supplemental Fig. S3B; Graveley et al. 2011). While the mRNA is detectable only in early pupal stages, the corresponding Eig71E proteins show prolonged high expression levels until P5 (Fig. 3C,D). Likewise, second puff genes display a similar transcriptome versus proteome pattern. A 1000-fold up-regulation of glue proteins (Sgs5, Sgs7, and Sgs8) at late L3 concordant with the detection of their mRNA in a narrow window of ∼24 h between crawling L3 and P1 (Beckstead et al. 2005) is followed by the presence of the protein in all pupal stages (Fig. 3D; Supplemental Fig. S3C). Our data show that for selected puff proteins, protein stability is the major determinant of their expression patterns during development. In contrast, in a high number of cases, we detect the protein at a single time point, while the RNA is detectable at multiple time points (Fig. 3E). In the aforementioned cases, protein levels cannot be directly predicted by transcriptomics, which demonstrates the necessity of proteome data for studying fly development.
Comparison of sex-specific protein patterns in adult flies
Sex-specific proteins are of high interest and have already been investigated by several proteomics studies (Dorus et al. 2006; Takemori and Yamamoto 2009; Sury et al. 2010; Wasbrough et al. 2010). To benchmark our label-free quantitative approach, we compared our adult time point to the published SILAC data set (Sury et al. 2010) and found a high overlap of sex-specific proteins (R = 0.84) (Supplemental Fig. S4A), showing that our developmental proteome recapitulates previous studies that are more specialized. To identify sex-specific proteins, we defined a fourfold expression difference with a P-value > 0.01 between male and female flies (1 wk old) and found 308 male- and 374 female-specific proteins (Fig. 4A; Supplemental Table S6). The 308 male proteins include Tektin-A and Tektin-C as sperm-specific flagellar proteins (Amos 2008), several less characterized genes known to be expressed in fly testes and seminal vesicles (Dorus et al. 2006; Takemori and Yamamoto 2009), and some proteins functioning in male development, like Lectin-46Ca, Lectin-46Cb, and Lectin-30A. For some proteins, like Hsp60B, Hsp60C, and the male fertility factor Kl-5, as well as Aquarius (Aqus) and Antares (Antr), an essential role in sperm development or sperm storage has already been demonstrated. The list of 374 female-specific proteins include vitelline membrane (Vm32E) and chorion proteins (Cp15, Cp18 and Cp36), which are important for eggshell assembly, the vitellogenins (Yp1, Yp2, and Yp3), and the fatty acid desaturase Fad2.
Figure 4.

Sex-specific proteome and maternally loaded proteins. (A) Volcano plot comparing protein expression levels between 1-wk-old male and female flies. Candidates discussed in the text are highlighted (filled black circles). Dashed lines indicate a fourfold expression difference with P < 0.01. (B) Volcano plot comparing protein expression levels between young male and female flies (<4 h old after eclosure) shows very few female-specific proteins. Candidates discussed in the text are highlighted (filled black circles). Dashed lines indicate a fourfold expression difference with P < 0.01. (C) Developmental expression profile of the female-specific protein CG31862 shows detection of mRNA (dotted line) in late pupal stage, while the protein (solid line) is also found in female flies. (D) Integration of mRNA levels with embryo-specific proteins allows identifying maternally loaded proteins. The mRNA levels of the adult female flies compared to embryos (x-axis) and males (y-axis) distinguishes cases in which either both the mRNA and protein (x = 0, y > 2), or only the protein (darker shaded area) is maternally loaded. (E) Relative embryonic hatching rate (four biological replicates) of CG17018 knockdown embryos compared to wild type. (F) Image of representative wild type and the CG17018 knockdown embryo with fused dorsal appendages. (G) Cuticle preparation of embryos revealed absence of denticle belts patterning in the CG17018 knockdown line.
Additionally, our developmental proteome allows the investigation of young flies, which were collected as virgins within 4 h after eclosure (Supplemental Table S7). The majority of proteins are equally expressed in both sexes (Supplemental Fig. S4B). While we detect only 21 female-specific proteins in young flies, there are 155 proteins with higher expression in its male counterpart (Fig. 4B; Supplemental Fig. S4C,D). In agreement with this observation, a previous transcriptomic study showed up-regulation of genes in female flies after mating, suggestively triggered by sperm and seminal fluid proteins (McGraw et al. 2008). In contrast, the majority of male-specific proteins are already present in young male flies prior to mating (Fig. 4B; Supplemental Fig. S4D). Interestingly, the only two proteins with more than 30-fold up-regulation in virgin females compared to males are not characterized: CG31862 and CG12288. Noteworthily, CG31862 is found in P5 and shows a continuously high protein level, while its RNA expression is restricted to the late pupal phase (Fig. 4C).
Maternally loaded proteins
While there is ample knowledge about maternally loaded RNA in Drosophila embryos (Tadros and Lipshitz 2005), no systematic analysis for maternally loaded proteins has been conducted yet. We interrogated our data for proteins enriched during embryogenesis whose RNA levels were higher in adult females compared to adult males. Among this subset of likely maternally loaded material should be candidates that have a functional importance during early development. In most cases, protein and mRNA are present in 2-h-old embryos, suggesting that both are maternally loaded (Fig. 4D; Supplemental Table S8). These include well-known examples such as Oskar (Osk), String (Stg), Piwi, Aubergine (Aub), Extra sexcombs (Esc), Dorsal (Dl), Mothers against dpp (Mad), and Swallow (Swa) (Chao et al. 1991; Edgar and Datar 1996; Luschnig et al. 2004; Simmons et al. 2010; Mani et al. 2014). However, also yet undescribed candidates like CG11674, CG5568, CG17018, CG15047, Zpg, GammaTub37C, and Tosca found in this set represent interesting candidates with a putative role in oogenesis and early embryogenesis. In order to investigate potential germline-specific functions of these candidates, we performed RNAi-mediated knockdown using the driver nanos-GAL4 and specific transgenic lines expressing double-stranded RNA from inverted repeats (shRNAs). Germline-specific expression of two independent shRNAs targeting CG17018 RNA revealed drastic effects on the embryonic hatching rate. While the number of laid eggs was unaffected (Supplemental Fig. S4E), hatching was reduced by almost 80% (Fig. 4E). In addition, ∼30% of unhatched eggs displayed defective dorsal appendages that are fused (Fig. 4F). Cuticle preparations showed that CG17018 knockdown embryos miss the denticle belts, revealing an absence of patterning at early stages (Fig. 4G). Also of note, CG17018 knockdown ovaries were indistinguishable from wild-type ones, as we could not detect any obvious morphological or differentiation defects (Supplemental Fig. S4F). Taken together, our findings imply a critical role of CG17018 during early embryogenesis.
Furthermore, our proteomic data set allows a comprehensive classification of maternally loaded proteins when the RNA is not present. The most prominent proteins include the major egg yolk vitellogenins (Yp1, Yp2, and Yp3), Dec-1, Cp36, and Cp7Fb as part of the chorion, the oxidoreductase family member CG12398 for which a role in vitelline membrane formation has been previously suggested (Fakhouri et al. 2006), the serine protease Nudel (Ndl), the sensor protein Obp19c, and the female-specific protein Fit, as well as two uncharacterized candidates, CG14309 and CG14834 (Fig. 4D; Supplemental Table S8).
Small proteins in the developmental proteome
Recently, there has been an increased interest in small proteins and translated small ORFs (smORFs) with up to 100 amino acids (aa) (Ramamurthi and Storz 2014), as their protein-coding potential is difficult to assess bioinformatically (Ladoukakis et al. 2011). These small proteins localize to specific subcellular compartments and perform cellular functions as any other protein (Magny et al. 2013). Our data set detects 268 small proteins (Fig. 5A), of which 84% have two or more unique peptides and temporal expression information (Supplemental Fig. S5A; Supplemental Table S1). This number is similar to a previous investigation using ribosome profiling (Aspden et al. 2014), demonstrating that mass spectrometry-based proteomics is on par with next generation sequencing approaches to detect translation of small proteins.
Figure 5.

Small proteins and peptides from noncoding regions of the genome. (A) Protein length distribution of identified (green, not enough quantitation values) and quantified (orange) protein groups of the life cycle proteome. Most proteins have quantitation values (>90%), and this fraction only marginally depends on protein length. The red line demarcates the fraction of 268 small proteins (<100 aa). (B) Representative MS/MS spectrum with annotated b- and y-ions of the peptide INILKSVNK(2+) from the putative noncoding gene CR43476. (C) Sequence comparison of Cyp9f2 and the “pseudogene” product Cyp9f3Ψ with amino acid substitution between both proteins marked in orange. Coverage of peptides for either protein is shown (yellow, more intense regions have overlapping peptides).
Peptides originating from noncoding regions of the genome
Peptides originating from putative noncoding regions have been reported in diverse organisms. Therefore, we re-analyzed our data including ncRNA sequences from FlyBase, which we in silico translated for open reading frames of at least 20 aa. Overall, we identified 29 putative proteins that unambiguously map to nontranslated transcripts at a FDR < 0.01 (Supplemental Table S9). Due to short open reading frames of these small proteins, we usually detect a single peptide per transcript. However, only two of these ncRNA-derived peptides showed a good MS2 fragmentation pattern and were independently identified with more than 10 different MS/MS spectra in several replicates and time points. One of these, FBtr0340701, has also been found in a control experiment using human cell lysate (data not shown), classifying it as a false positive identification originating from a contaminant. The only remaining peptide with strong evidence of identification matches to CR43476 (Fig. 5B).
Other genes classified as nonexpressed are pseudogenes. These genes have mutations in their promoter regions or other functional elements that make their expression unlikely (Harrison et al. 2003). We checked for protein evidence of the 2902 reported pseudogenes (FlyBase 6.01) and found nine protein groups in our data set to include peptides unambiguously mapping to pseudogenes. Whereas most of these proteins are represented by a single peptide (Supplemental Table S10), the most prominent hit, FBtr0082602, encoding Cyp9f3Ψ, is supported by 23 peptides including five unique sequences. The measured peptides match to the N-terminal and C-terminal regions, demonstrating that the complete pseudogene is most likely translated (Fig. 5C). Furthermore, Cyp9f3Ψ and Cyp9f2 present distinct expression patterns, further indicating that, despite their close genomic vicinity, they are differently regulated during development (Supplemental Fig. S5B).
Despite an extremely low expression of peptides originating from ncRNA transcripts, only very few detected peptides map to noncoding regions of the genome, illustrating a low false discovery rate in our screen and a carefully curated gene annotation of the Drosophila melanogaster genome (Matthews et al. 2015).
Highly temporal-resolved embryogenesis proteome
Because they are intensely studied, we were particularly interested in proteome changes during embryogenesis. To investigate the process in a high time-resolved and systematic manner, we collected whole embryos at narrow intervals: every hour after egg laying for up to 6 h and then every 2 h until 20 h (Fig. 6A). These 14 time points were also measured in four independent biological replicates to account for technical, biological and environmental variation. To control for our collections, we staged embryos of selected time points by morphology and Engrailed antibody staining (Supplemental Fig. S6A; Campos-Ortega and Hartenstein 1997). Protein expression levels were determined using label-free quantitation based on unique peptides provided by MaxLFQ (Cox et al. 2014). We detected 6487 expressed protein groups, of which 5458 were quantified in at least two replicates of any time point (Supplemental Table S11). PCA revealed that embryo stages correlate well with our collected time points (R = 0.93), showing a developmental progression through embryogenesis (Fig. 6A; Supplemental Fig. S6C). Noteworthily, all four independent biological replicates show very high reproducibility (R = 0.92–0.96) (Supplemental Fig. S6B,C). We also validated the expression profiles of seven proteins by immunostaining with antibodies against endogenous proteins (Fig. 6C; Supplemental Fig. S6D).
Figure 6.

The embryogenesis proteome time course. (A) Scheme indicating the collected time points. PCA shows high reproducibility of replicates, and the first component shows high correlation with developmental progression (R = 0.93). (B) Heat map of log2 LFQ expression values for 1644 developmentally regulated protein groups in embryogenesis. (C) Western blots of seven selected proteins validate their temporal expression profile from the proteomics screen. (D) Dot plot connecting the selected enriched GO terms with developmental progression. The circle size indicates the odds ratio of each GO term category. (E) The regulated protein groups were assigned automatically to 70 clusters based on expression profiles, of which four representative clusters with an up-regulation at 2–3 h (cluster 41), 5 h (cluster 10), 10 h (cluster 60), and 20 h (cluster 57) are shown. (F) Profiles of tissue-specific protein expression created by integrating RNA fluorescence in situ hybridization data. Muscle and central nervous system (CNS) clusters were chosen as examples. (G) Ubiquitous (tubulin-GAL4) and mesodermal (24B- and mef2-GAL4) but not neuronal (elav-GAL4) knockdown of CG1674 results in reduced locomotion activity (Dunnett's test; [***] P-value < 0.001).
Expression profiles during embryogenesis
We analyzed the time course data using a multivariate empirical Bayes approach and identified 1644 protein groups with differential expression during embryogenesis (Fig. 6B). To obtain a functional overview on the embryogenesis process, we performed GO enrichment analysis on this set of differentially expressed proteins. Based on this analysis, we observed enrichment of terms related to very early embryogenesis cellular processes (0–1 h), such as zygotic determination of anterior/posterior axis and syncytial blastoderm mitotic cell cycle (Fig. 6D). Additionally, proteins involved in ribosome biogenesis up-regulate at 2 h to initiate active translation concomitant with zygotic gene activation (ZGA) starting at 2 h. We also noted high enrichment of proteins involved in cell cycle and cytoskeleton organization during early phases of embryogenesis (2–3 h). While proteins involved in nervous system development are highly present at 3 h, muscle structure development proteins are more prominent later in embryogenesis, at 14 h.
As an alternative approach to analyze the data, we automatically clustered the differentially expressed proteins with similar temporal profiles, resulting in 70 distinct clusters, and performed GO enrichment analysis on these clusters (Fig. 6E; Supplemental Tables S12, S13). As a result, the known embryonic developmental program can be followed by temporal alignment of individual clusters (Fig. 6E), possibly hinting at putative functions of not yet characterized proteins.
Integrating the developmental proteome and spatial expression
To integrate spatial information, we fused our proteome profiles with tissue-specific RNA expression data from fluorescence in situ hybridizations (Lécuyer et al. 2007). We chose muscle development to highlight the value provided by the merged data. In the muscle-specific clusters (Fig. 6F; Supplemental Tables S14, S15), we noted up-regulation of proteins involved in muscle development such as Mlc2, Mp20, and Mlp60A (Sandmann et al. 2006) at 14 h. Later in embryogenesis (20 h), we found high expression of Eaat1 and EcR, which control muscle contraction at larval stages. Furthermore, this data integration allowed us to identify similarly expressed, not yet characterized proteins (CG1674, CG6040, and CG15022), shown to localize in muscle tissue, suggesting a role in muscle development. In order to test this hypothesis, we performed RNAi-mediated knockdown of two candidates. Remarkably, mesodermal knockdown of either CG1674 or CG6040 severely affects locomotion behavior of adult flies (Fig. 6G; Supplemental Fig. S6E). Likewise, the complete CG6040 loss-of-function produces viable flies that display similar climbing defects, confirming the specificity of the RNAi phenotype. Importantly, neuronal knockdown of both genes did not impair locomotion performance, supporting their muscle-specific functions. We next performed in situ hybridization on embryos and observed a strong enrichment of CG1674 mRNA in muscle tissue, more specifically in somatic and pharyngeal muscles, whereas CG6040 exhibits moderate ubiquitous expression (Supplemental Fig. S6F). Altogether, our findings strongly suggest a muscular function for CG1674 and CG6040. However, further investigation will be required to understand their specific role in muscle development.
Alternatively, other tissue data can be inspected for biological insights. The analysis of the central nervous system (CNS) revealed an up-regulation of proteins involved in neural development (Roughest [Rst], Smooth [Sm], and Erect wing [Ewg]) at 8–12 h and in synapse organization and axon ensheathment (Ank2 and Wrapper) at 14 h (Fig. 6F; Supplemental Tables S14, S15). Likewise, all 21 tissue clusters can be examined in our web interface.
Comparing transcriptome and proteome to study translational delay
We compared our embryogenesis proteome with the transcriptome generated as part of the modENCODE Project (Graveley et al. 2011). In agreement with the transcriptome analysis, we found that the general protein complexity is increased during embryogenesis (Fig. 7A; Supplemental Fig. S7A). Similar to a previous study in yeast (Fournier et al. 2010), we found only a moderate correlation (maximum R = 0.5) between transcriptome and proteome and noted that the best correlation is nonsynchronous, showing a 4- to 5-h proteome delay (Fig. 7B).
Figure 7.

Temporal transcriptome/proteome dynamics and isoform quantitation. (A) Plot showing the number (bars) of detected transcripts (orange) and proteins (green) at each time point. The solid line depicts the cumulative sum of unique transcripts (orange) and proteins (green). The dashed line represents the median across all time points. (B) Heat map displaying the Pearson correlation between transcript and protein expression levels. Matching time points between the two data sets are indicated by orange boxes. (C) Median scaled quantification plotted after clustering of the first PCA component of RNA (orange) and protein (green) expression into six different categories. Shaded regions display the standard error of the fitted line. (D) Expression profiles with isoform-specific information of three proteins: Lola, Mod(mdg4), and Rtnl1. Isoforms are colored according to the legend. (E) Validation of Lola-RAA/Lola-RI isoform quantitation by immunoblotting against Lola at four selected time points. Protein lysate of lola-RAA/lola-RI mutant embryos at 20 h were used to identify the isoform-specific band (arrow) corresponding to Lola-RAA/Lola-RI. Beta-tubulin was used as a loading control. (F) RNA levels were determined by in situ hybridization at the selected time points with a specific probe for lola-RAA/lola-RI.
By multidimensional scaling followed by clustering, we subgrouped the RNA/protein expression profiles into six clusters (Fig. 7C; Supplemental Table S16). In the majority of cases, the mRNA is more abundant at early time points, while the protein expression peaks at later stages. Except for cluster 1, the remaining clusters illustrate different behavior of RNA and protein during embryogenesis. We observed a temporal proteome delay in clusters 5 and 6: while the RNA expression peaks around 7 h, proteins steadily up-regulate later in embryogenesis, putatively due to translational control mechanisms (Fig. 7C).
Quantification of protein isoforms during embryogenesis
As distinct protein isoforms may show differential developmental regulation, we mined our proteomic data for protein isoforms. We found 34 genes with various quantified isoforms, some of them showing differential expression, such as lola, mod(mdg)4, and Rtnl1 (Fig. 7D; Supplemental Fig. S7B). We further validated our isoform quantitation by immunoblotting following the expression of Lola- RAA/RI (also known as Lola-K) (Giniger et al. 1994). While Lola- RAA/RI is highly expressed at 20 h (Fig. 7E), its mRNA shows an expression peak at 14 h shown by in situ hybridization (Fig. 7F). This underscores again the importance of a developmental proteome as an addition to transcriptomic studies.
Discussion
We generated high-quality proteome data sets for embryogenesis and the full life cycle of Drosophila melanogaster that close the gap for systematic developmental investigation of protein expression. Both proteomes cover nearly 8000 and 5500 protein groups during the life cycle and embryogenesis, respectively, accounting for at least one-third of annotated Drosophila genes. However, while these two data sets are larger than previous ones, they are not complete. Especially low abundant proteins or proteins that are highly expressed in a restricted number of small tissues will likely not be present in our proteomes. Thus, a not-quantified protein can either be absent in this stage or expressed below our limit of detection (LOD) enforced by the mass spectrometry measurement. Nevertheless, these large-scale data sets allow us to assess the developmental expression of proteins and protein isoforms, report maternally provided proteins, validate small proteins (≤100 aa), identify Cyp9f3Ψ as an expressed protein-coding gene, and describe peptides originating from noncoding regions.
We scored significant developmentally regulated protein groups: 1535 for the whole life cycle and 1644 for embryogenesis. Nearly half of them are not characterized in depth, suggesting a large area of developmental gene regulation still to be discovered.
We used our data to follow the well-characterized regulation by the hormone ecdysone at a protein level. This revealed intriguing differences to previously reported transcriptome analysis (Beckstead et al. 2005; Gonsalves et al. 2011). For several ecdysone-induced genes, the protein abundance relies on protein stability rather than the presence of RNA transcripts. Overall, transcript abundance and protein levels correlate only modestly. The same observation holds true even when considering the temporal delay between transcript and protein expression. The temporal difference in RNA and protein expression needs to be taken into account when studying phenotypic differences of protein-coding genes using mRNA as a proxy.
As previous transcriptomic studies reported maternally loaded RNAs, our proteomic data enable systematic identification of maternally provided proteins. Here, we catalog not yet reported maternally loaded proteins such as CG14309, CG14834, and CG12398, whose functions in early development need further investigation. For instance, the knockdown of the maternally loaded protein CG17018 results in a severe defect in embryo development.
To gain further insight, we complemented our data sets with other available published data. For example, to de-convolute tissue-specific expression information, we merged our embryogenesis proteome with RNA in situ hybridization data (Lécuyer et al. 2007). This allowed us to pinpoint individual proteins showing tissue-specific developmental regulation, as exemplified with the impaired muscular phenotypes of CG1674 and CG1640 knockdown lines. Additionally, this analysis can be extended to other tissues to uncover currently unknown proteins that might play an essential role in the development of a specific tissue. This underscores the power to combine available high-quality Drosophila data sets to achieve a more holistic model for developmental gene regulation.
Methods
Collection of embryos, larvae, pupae, and adult flies
Population cages of wild-type Oregon R flies containing only fertilized females were maintained at 25°C. For the whole life cycle comparative analysis, embryos were collected from cages on agar apple juice plates in 2-h laying time windows and processed immediately (0–2 h) or aged at 25°C for the required time (4–6, 10–12, and 18–20 h). Early larval collections were performed from embryo plates, whereas crawling larvae and pupae stages were collected directly from flasks at indicated time points. Virgin young flies within 4 h after eclosure were collected separately for males and females, as well as 1-wk-old flies (adult flies). For the time course analysis, embryos were collected on apple juice agar plates in 30-min laying time windows, processed immediately (0 h time point) or aged at 25°C for the required time. All samples were mechanically lysed prior to mass spectrometry sample preparation (see Supplemental Material for detailed descriptions).
Mass spectrometry measurement and label-free analysis
Peptides were separated by nanoflow liquid chromatography on an EASY-nLC 1000 system (Thermo) coupled to a Q Exactive Plus mass spectrometer (Thermo). Separation was achieved by a 25-cm capillary (New Objective) packed in-house with ReproSil-Pur C18-AQ 1.9-µm resin (Dr. Maisch). Peptides were separated chromatographically by a 280-min gradient from 2% to 40% acetonitrile in 0.5% formic acid with a flow rate of 200 nL/min. Spray voltage was set between 2.4 and 2.6 kV. The instrument was operated in the data-dependent mode (DDA) performing a top15 MS/MS per MS full scan. Isotope patterns with unassigned and charge state 1 were excluded. MS scans were conducted with 70,000 and MS/MS scans with 17,500 resolution. The raw measurement files were analyzed with MaxQuant 1.5.2.8 standard settings except LFQ (Cox et al. 2014) and match between run options were activated, as well as quantitation was performed on unique peptides only. The raw data were searched against the translated Ensembl transcript databases (release 79) of D. melanogaster (30,362 translated entries) and the S. cerevisiae protein database (6692 entries). Known contaminants, protein groups only identified by site, and reverse hits of the MaxQuant results were removed. In the life cycle data set, the imputation was performed in two distinct ways for proteins with a measured intensity (raw) missing an LFQ intensity or proteins with no intensity value. In the first case, values were imputed from a normal distribution with a mean value shifted by −0.6 from the mean value of all measured LFQ intensities and half of the standard deviation. In contrast, proteins with no intensity value were replaced with the smallest measured value in the set. For the embryo time course, missing values were drawn from a distribution calculated with the logspline R package (https://cran.r-project.org/package=logspline). For cases where three or more replicates were measured, the mean of the measured replicates was used as the mean parameter of the distribution. Otherwise, the average of the two neighboring time points was used. In cases of no measured values in neighboring time points or for proteins measured only in one replicate with no surrounding values, a fixed value of 22.5 close to the LOD was used.
Bioinformatics analysis
Significant changes during the life cycle were calculated by analysis of variance (ANOVA), flagging as stage-specific proteins those with FDR < 0.01 (Benjamini–Hochberg procedure) and present in either one unique stage or differing in one stage compared to the rest (log2 LFQ FC > 4 in all stages, allowing only one not fulfilling the condition). The effect of the differences was assessed calculating Cohen's effect size and the Tukey HSD post-hoc test. The Gini ratio was used to measure the stability of protein abundance throughout the time course. Automatic clustering of genes and samples was performed using Affinity Propagation (Frey and Dueck 2007) on the significant proteins, taking negative squared Euclidean distances as a measure of similarity. The goodness of the clusters was assessed from the Silhouette information according to the given clustering. Gene set enrichment analysis (GSEA) was done in R (R Core Team 2017), followed by a strategy of scoring similar (redundant) terms calculating the information content (IC) between two terms. Results were presented as a treemap or a scatterplot of terms clustered based on the first two components of a PCA of the IC similarity scores. For the embryo development, significant changes of protein abundance along the time course were assessed. FPKM levels for FlyBase 5.12 Transcripts from short poly(A)+ RNA-seq (Graveley et al. 2011) and localization data from http://fly-fish.ccbr.utoronto.ca were integrated with our proteome data.
Data access
The mass spectrometry raw data from this study have been submitted to the ProteomeXchange (http://www.proteomexchange.org) under the data set identifiers PXD005691 (life cycle) and PXD005713 (embryogenesis).
Supplementary Material
Acknowledgments
We thank Junaid Akhtar for training, as well as Marion Scheibe, Anja Freiwald, and Christian Berger for critical reading of the manuscript. Antibodies were obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH or kindly provided by Christian Berger. We acknowledge support of the Zentrum für Datenverarbeitung (ZDV) at the University of Mainz in hosting the web application. The study was partly funded by the Rhineland Palatinate Forschungsschwerpunkt GeneRED (Gene Regulation in Evolution and Development). D.K. is supported by the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.213694.116.
References
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. 2000. The genome sequence of Drosophila melanogaster. Science 287: 2185–2195. [DOI] [PubMed] [Google Scholar]
- Amos LA. 2008. The tektin family of microtubule-stabilizing proteins. Genome Biol 9: 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aspden JL, Eyre-Walker YC, Phillips RJ, Amin U, Mumtaz MAS, Brocard M, Couso J-P. 2014. Extensive translation of small Open Reading Frames revealed by Poly-Ribo-Seq. eLife 3: e03528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckstead RB, Lam G, Thummel CS. 2005. The genomic response to 20-hydroxyecdysone at the onset of Drosophila metamorphosis. Genome Biol 6: R99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaldi T, Straub T, Cox J, Kumar C, Becker PB, Mann M. 2008. Combined use of RNAi and quantitative proteomics to study gene function in Drosophila. Mol Cell 31: 762–772. [DOI] [PubMed] [Google Scholar]
- Brown JB, Boley N, Eisman R, May GE, Stoiber MH, Duff MO, Booth BW, Wen J, Park S, Suzuki AM, et al. 2014. Diversity and dynamics of the Drosophila transcriptome. Nature 512: 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunner E, Ahrens CH, Mohanty S, Baetschmann H, Loevenich S, Potthast F, Deutsch EW, Panse C, de Lichtenberg U, Rinner O, et al. 2007. A high-quality catalog of the Drosophila melanogaster proteome. Nat Biotechnol 25: 576–583. [DOI] [PubMed] [Google Scholar]
- Butter F, Bucerius F, Michel M, Cicova Z, Mann M, Janzen CJ. 2013. Comparative proteomics of two life cycle stages of stable isotope-labeled Trypanosoma brucei reveals novel components of the parasite's host adaptation machinery. Mol Cell Proteomics 12: 172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos-Ortega JA, Hartenstein V. 1997. The embryonic development of Drosophila melanogaster, 2nd ed Springer, Berlin. [Google Scholar]
- Chang YC, Tang HW, Liang SY, Pu TH, Meng TC, Khoo KH, Chen GC. 2013. Evaluation of Drosophila metabolic labeling strategies for in vivo quantitative proteomic analyses with applications to early pupa formation and amino acid starvation. J Proteome Res 3: 2138–2150. [DOI] [PubMed] [Google Scholar]
- Chao YC, Donahue KM, Pokrywka NJ, Stephenson EC. 1991. Sequence of swallow, a gene required for the localization of bicoid message in Drosophila eggs. Dev Genet 12: 333–341. [DOI] [PubMed] [Google Scholar]
- Chintapalli VR, Wang J, Dow JAT. 2007. Using FlyAtlas to identify better Drosophila melanogaster models of human disease. Nat Genet 39: 715–720. [DOI] [PubMed] [Google Scholar]
- Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 26: 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. 2014. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol Cell Proteomics 13: 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejung M, Subota I, Bucerius F, Dindar G, Freiwald A, Engstler M, Boshart M, Butter F, Janzen CJ. 2016. Quantitative proteomics uncovers novel factors involved in developmental differentiation of Trypanosoma brucei. PLoS Pathog 12: e1005439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorus S, Busby SA, Gerike U, Shabanowitz J, Hunt DF, Karr TL. 2006. Genomic and functional evolution of the Drosophila melanogaster sperm proteome. Nat Genet 38: 1440–1445. [DOI] [PubMed] [Google Scholar]
- Edgar BA, Datar SA. 1996. Zygotic degradation of two maternal Cdc25 mRNAs terminates Drosophila’s early cell cycle program. Genes Dev 10: 1966–1977. [DOI] [PubMed] [Google Scholar]
- Fabre B, Korona D, Groen A, Vowinckel J, Gatto L, Deery MJ, Ralser M, Russell S, Lilley KS. 2016. Analysis of Drosophila melanogaster proteome dynamics during embryonic development by a combination of label-free proteomics approaches. Proteomics 16: 2068–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhouri M, Elalayli M, Sherling D, Hall JD, Miller E, Sun X, Wells L, LeMosy EK. 2006. Minor proteins and enzymes of the Drosophila eggshell matrix. Dev Biol 293: 127–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier ML, Paulson A, Pavelka N, Mosley AL, Gaudenz K, Bradford WD, Glynn E, Li H, Sardiu ME, Fleharty B, et al. 2010. Delayed correlation of mRNA and protein expression in rapamycin-treated cells and a role for Ggc1 in cellular sensitivity to rapamycin. Mol Cell Proteomics 9: 271–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey BJ, Dueck D. 2007. Clustering by passing messages between data points. Science 315: 972–976. [DOI] [PubMed] [Google Scholar]
- Giniger E, Tietje K, Jan LY, Jan YN. 1994. lola encodes a putative transcription factor required for axon growth and guidance in Drosophila. Development 120: 1385–1398. [DOI] [PubMed] [Google Scholar]
- Gonsalves SE, Neal SJ, Kehoe AS, Westwood JT. 2011. Genome-wide examination of the transcriptional response to ecdysteroids 20-hydroxyecdysone and ponasterone A in Drosophila melanogaster. BMC Genomics 12: 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR, Brooks AN, Carlson JW, Duff MO, Landolin JM, Yang L, Artieri CG, van Baren MJ, Boley N, Booth BW, et al. 2011. The developmental transcriptome of Drosophila melanogaster. Nature 471: 473–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin TJ, Gygi SP, Ideker T, Rist B, Eng J, Hood L, Aebersold R. 2002. Complementary profiling of gene expression at the transcriptome and proteome levels in Saccharomyces cerevisiae. Mol Cell Proteomics 1: 323–333. [DOI] [PubMed] [Google Scholar]
- Grün D, Kirchner M, Thierfelder N, Stoeckius M, Selbach M, Rajewsky N. 2014. Conservation of mRNA and protein expression during development of C. elegans. Cell Rep 6: 565–577. [DOI] [PubMed] [Google Scholar]
- Harrison PM, Milburn D, Zhang Z, Bertone P, Gerstein M. 2003. Identification of pseudogenes in the Drosophila melanogaster genome. Nucleic Acids Res 31: 1033–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jambor H, Surendranath V, Kalinka AT, Mejstrik P, Saalfeld S, Tomancak P. 2015. Systematic imaging reveals features and changing localization of mRNAs in Drosophila development. eLife 4: e05003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinka AT, Varga KM, Gerrard DT, Preibisch S, Corcoran DL, Jarrells J, Ohler U, Bergman CM, Tomancak P. 2010. Gene expression divergence recapitulates the developmental hourglass model. Nature 468: 811–814. [DOI] [PubMed] [Google Scholar]
- Kharchenko PV, Alekseyenko AA, Schwartz YB, Minoda A, Riddle NC, Ernst J, Sabo PJ, Larschan E, Gorchakov AA, Gu T, et al. 2011. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 471: 480–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronja I, Whitfield ZJ, Yuan B, Dzeyk K, Kirkpatrick J, Krijgsveld J, Orr-Weaver TL. 2014. Quantitative proteomics reveals the dynamics of protein changes during Drosophila oocyte maturation and the oocyte-to-embryo transition. Proc Natl Acad Sci 111: 16023–16028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladoukakis E, Pereira V, Magny EG, Eyre-Walker A, Couso JP. 2011. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biol 12: R118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence PA. 1992. The making of a fly. The genetics of animal design. Blackwell Scientific, Oxford. [Google Scholar]
- Lécuyer E, Yoshida H, Parthasarathy N, Alm C, Babak T, Cerovina T, Hughes TR, Tomancak P, Krause HM. 2007. Global analysis of mRNA localization reveals a prominent role in organizing cellular architecture and function. Cell 131: 174–187. [DOI] [PubMed] [Google Scholar]
- Liu Y, Beyer A, Aebersold R. 2016. On the dependency of cellular protein levels on mRNA abundance. Cell 165: 535–550. [DOI] [PubMed] [Google Scholar]
- Luschnig S, Moussian B, Krauss J, Desjeux I, Perkovic J, Nüsslein-Volhard C. 2004. An F1 genetic screen for maternal-effect mutations affecting embryonic pattern formation in Drosophila melanogaster. Genetics 167: 325–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magny EG, Pueyo JI, Pearl FMG, Cespedes MA, Niven JE, Bishop SA, Couso JP. 2013. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science 341: 1116–1120. [DOI] [PubMed] [Google Scholar]
- Mani SR, Megosh H, Lin H. 2014. PIWI proteins are essential for early Drosophila embryogenesis. Dev Biol 385: 340–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin W. 2012. The price of tags in protein localization studies. J Bacteriol 194: 6369–6371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews BB, Dos Santos G, Crosby MA, Emmert DB, St Pierre SE, Gramates LS, Zhou P, Schroeder AJ, Falls K, Strelets V, et al. 2015. Gene model annotations for Drosophila melanogaster: impact of high-throughput data. G3 (Bethesda) 5: 1721–1736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGraw LA, Clark AG, Wolfner MF. 2008. Post-mating gene expression profiles of female Drosophila melanogaster in response to time and to four male accessory gland proteins. Genetics 179: 1395–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The modENCODE Consortium, Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, Lin MF, et al. 2010. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science 330: 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin X, Daneman R, Zavortink M, Chia W. 2001. A protein trap strategy to detect GFP-tagged proteins expressed from their endogenous loci in Drosophila. Proc Natl Acad Sci 98: 15050–15055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagarkar-Jaiswal S, Lee P-T, Campbell ME, Chen K, Anguiano-Zarate S, Gutierrez MC, Busby T, Lin W-W, He Y, Schulze KL, et al. 2015. A library of MiMICs allows tagging of genes and reversible, spatial and temporal knockdown of proteins in Drosophila. eLife 4: e05338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peshkin L, Wühr M, Pearl E, Haas W, Freeman RM, Gerhart JC, Klein AM, Horb M, Gygi SP, Kirschner MW. 2015. On the relationship of protein and mRNA dynamics in vertebrate embryonic development. Dev Cell 35: 383–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.R-project.org/. [Google Scholar]
- Ramamurthi KS, Storz G. 2014. The small protein floodgates are opening; now the functional analysis begins. BMC Biol 12: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandmann T, Jensen LJ, Jakobsen JS, Karzynski MM, Eichenlaub MP, Bork P, Furlong EEM. 2006. A temporal map of transcription factor activity: mef2 directly regulates target genes at all stages of muscle development. Dev Cell 10: 797–807. [DOI] [PubMed] [Google Scholar]
- Sarov M, Barz C, Jambor H, Hein MY, Schmied C, Suchold D, Stender B, Janosch S, Kj VV, Krishnan RT, et al. 2016. A genome-wide resource for the analysis of protein localisation in Drosophila. eLife 5: e12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. 2011. Global quantification of mammalian gene expression control. Nature 473: 337–342. [DOI] [PubMed] [Google Scholar]
- Simmons MJ, Thorp MW, Buschette JT, Peterson K, Cross EW, Bjorklund EL. 2010. Maternal impairment of transposon regulation in Drosophila melanogaster by mutations in the genes aubergine, piwi and Suppressor of variegation 205. Genet Res (Camb) 92: 261–272. [DOI] [PubMed] [Google Scholar]
- Sowell RA, Hersberger KE, Kaufman TC, Clemmer DE. 2007. Examining the proteome of Drosophila across organism lifespan. J Proteome Res 6: 3637–3647. [DOI] [PubMed] [Google Scholar]
- Sury MD, Chen J-X, Selbach M. 2010. The SILAC fly allows for accurate protein quantification in vivo. Mol Cell Proteomics 9: 2173–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadros W, Lipshitz HD. 2005. Setting the stage for development: mRNA translation and stability during oocyte maturation and egg activation in Drosophila. Dev Dyn 232: 593–608. [DOI] [PubMed] [Google Scholar]
- Takemori N, Yamamoto M-T. 2009. Proteome mapping of the Drosophila melanogaster male reproductive system. Proteomics 9: 2484–2493. [DOI] [PubMed] [Google Scholar]
- Tomancak P, Berman BP, Beaton A, Weiszmann R, Kwan E, Hartenstein V, Celniker SE, Rubin GM. 2007. Global analysis of patterns of gene expression during Drosophila embryogenesis. Genome Biol 8: R145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel C, Marcotte EM. 2012. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat Rev Genet 13: 227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasbrough ER, Dorus S, Hester S, Howard-Murkin J, Lilley K, Wilkin E, Polpitiya A, Petritis K, Karr TL. 2010. The Drosophila melanogaster sperm proteome-II (DmSP-II). J Proteomics 73: 2171–2185. [DOI] [PubMed] [Google Scholar]
- Xing X, Zhang C, Li N, Zhai L, Zhu Y, Yang X, Xu P. 2014. Qualitative and quantitative analysis of the adult Drosophila melanogaster proteome. Proteomics 14: 286–290. [DOI] [PubMed] [Google Scholar]
- Yamanaka N, Rewitz KF, O'Connor MB. 2013. Ecdysone control of developmental transitions: lessons from Drosophila research. Annu Rev Entomol 58: 497–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.