

Abstract
Inducible nitric oxide synthase (iNOS) generates nitric oxide (NO) in myeloid cells that acts as a defense mechanism to suppress invading microorganisms or neoplastic cells. In tumor-bearing mice, elevated iNOS expression is a hallmark of myeloid-derived suppressor cells (MDSC). MDSCs use NO to nitrate both the T cell receptor and STAT1, thus inhibiting T cell activation and the anti-tumor immune response. The molecular mechanisms underlying iNOS expression and regulation in tumor-induced MDSCs are unknown. We report here that deficiency in IRF8 results in diminished iNOS expression in both mature CD11b+Gr1− and immature CD11b+Gr1+ myeloid cells in vivo. Strikingly, although IRF8 was silenced in tumor-induced MDSC, iNOS expression was significantly elevated in tumor-induced MDSC, suggesting that the expression of iNOS is regulated by an IRF8-independent mechanism under pathological conditions. Furthermore, tumor-induced MDSC exhibited diminished STAT1 and NF-κB Rel protein levels, the essential inducers of iNOS in myeloid cells. Instead, tumor-induced MDSC showed increased SETD1B expression as compared to their cellular equivalents in tumor-free mice. Chromatin immunoprecipitation revealed that H3K4me3, the target of SETD1B, was enriched at the nos2 promoter in tumor-induced MDSC, and inhibition or silencing of SETD1B diminished iNOS expression in tumor-induced MDSC. Our results show how tumor cells use the SETD1B-H3K4me3 epigenetic axis to bypass a normal role for IRF8 expression in activating iNOS expression in MDSC, when they are generated under pathological conditions.
Keywords: iNOS, SETD1B, IRF8, MDSCs, Myeloid cells, H3K4me3
Introduction
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous population of immature myeloid cells (IMCs) that include progenitors and precursors of dendritic cells, macrophages, and granulocytes of various differentiation stages (1). Under physiological conditions, IMCs undergo a steady state myelopoiesis and differentiate into mature dendritic cells, macrophages, and granulocytes (2, 3). Various pathological conditions, including cancer, can perturb myelopoiesis and interrupt IMC differentiation, resulting in accumulation of MDSCs (2, 4–9). In human cancer patients and mouse tumor models, massive accumulation of MDSCs is a hallmark of tumor progression (10–16). MDSCs are therefore key targets in cancer immunotherapy (1, 17–21).
In mice, MDSCs were originally defined as CD11b+Gr1+ myeloid cells to reflect their myeloid origin, immune-suppressive function, and systemic expansion in a cancer host (22). Recent discoveries have expanded the definition of CD11b+Gr1+ MDSCs into polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs) based on their phenotypic and morphological features (4). MDSCs use several mechanisms to suppress T cell activation and function (23, 24). Although the specific roles of these pathways in the inhibitory activity of MDSC subpopulations remain unclear, both PMN-MDSCs and M-MDSCs inhibit T cell activation and function through nitric oxide-related pathways. PMN-MDSCs produce peroxynitrite to nitrate the T cell receptor to render T cells unresponsive to antigen stimulation (25). M-MDSCs express high level of inducible nitric oxide synthase (iNOS) to mediate nitration of STAT1 to block IFNγ signaling pathway, a key component of the host T cell cancer immune surveillance system (26). Therefore, iNOS is a key mediator of M-MDSCs suppressive function.
Although the expression regulation of iNOS has been extensively studied in various types of cells, including myeloid cells in vitro (27–30), the molecular mechanism underlying iNOS expression regulation in tumor-induced MDSCs is essentially unknown. We report here that the histone methyltransferase SETD1B regulates trimethylation of histone H3 lysine 4 (H3K4Me3) at the nos2 promoter to activate iNOS expression in tumor-induced MDSCs.
Materials and Methods
Tumor cells, mouse models, and human specimen collection
The mouse mammary carcinoma cell line, 4T1 (BALB/c mouse origin), was obtained from American Type Culture Collection (ATCC) (Manassas, VA) in 2004 and was stored in liquid nitrogen in aliquots. ATCC has characterized this cell line by morphology, immunology, DNA fingerprint, and cytogenetics. The AT3 cell line was derived from C57BL/6 mice and was kindly provided by Dr. Scott Abrams (Roswell Park Cancer Institute, NY) and was characterized as previously described (31). All cell lines in the laboratory are tested approximately every two months for mycoplasma. 4T1 and AT3 cells used in this study are mycoplasma-negative. Cells were used within 30 passages after thawing an aliquot of cells from liquid nitrogen. 4T1 cells were injected subcutaneously into the mammary glands of BALB/c mice (1×104 cells/mouse) to establish the orthotopic breast tumors. AT3 cells were injected subcutaneously into the mammary glands of C57BL/6 mice (2×105 cells/mouse) to establish the orthotopic breast tumors. IRF8 KO mice were kindly provided by Dr. Keiko Ozato (National Institutes of Health, MD) and maintained at the Augusta University animal facility. All mouse studies are performed according to protocols approved by Augusta University Institutional Animal Care and Use Committee. Peripheral blood specimens were collected from consented healthy donors at the Shepeard Community Blood Center and from de-identified colon cancer patients at the Georgia Cancer Center Cancer Clinic. All studies of human specimens were performed according to protocols approved by Augusta University Institutional Human Research Protection Committee.
Treatment of tumor-bearing mice with chaetocin
Tumor-bearing mice were treated daily with an i.p. injection of either solvent (10% Cremophor, 5% ethanol, and 85% PBS) or chaetocin (Sigma-Aldrich, St Louis, MO) starting at day 9 and day 21, respectively, at a dose of 0.5 mg/kg body weight for 3 days, followed by treatment at a dose of 0.25 mg/kg body weight for 4 more days.
Purification of tumor-induced MDSCs
Spleens cells were mixed with CD11b MicroBeads and loaded to LS columns (Miltenyi Biotech). MDSCs were eluted according to the manufacturer’s instructions. The purified cells were stained with either IgG or CD11b- and Gr1-specific mAbs (BioLegend, San Diego, CA) and analyzed by flow cytometry.
Flow cytometry analysis
Spleen, lymph nodes, thymus, and bone marrow (BM) were collected from mice. Cells were stained with fluorescent dye-conjugated antibodies that are specific for mouse CD11b-, Gr1-, Ly6G-, and Ly6C- (BioLegend). Stained cells were analyzed by flow cytometry.
Cell sorting
Spleens, BM, and tumor cells were collected from WT and IRF8 KO C57BL/6 mice. Tumor tissues were digested with collagenase solution (collagenase 1 mg/ml, hyaluronidase 0.1 mg/ml, and DNase I 30 U/ml). The buffy coat was prepared from human blood and red cells were lysed with red cell lysis buffer. Mouse cells were stained with CD11b- and Gr1-specific mAbs (BioLegend). Human cells were stained with HLA-DR-, CD11b-, and CD33-specific mAbs (BioLegend). Stained cells were sorted using a BD FACSAria II SORP or a Beckman Coulter MoFlo XDP cell sorter to isolate myeloid cell subsets.
In vitro T cell activation and co-culture with MDSCs
BM cells were collected from WT tumor-free mice and seeded at a density of 6×106 cells in a 10-cm dish. 4T1 condition media was diluted with fresh culture medium at a 1:2 ratio and added to the BM cell culture. CD3+ T cells were purified from spleen cells using the MojoSort mouse CD3+ T cell isolation kit (BioLegend) according to the manufacturer’s instructions. For T cell activation, a 96-well culture plate was coated with anti-mouse CD3 and anti-mouse CD28 mAbs at 37°C for two hours. The purified T cells were labeled with 0.25 μM CFSE (Life Technologies) and then seeded in the coated plate at a density of 1.5×105 cells/well in RPMI medium plus 10% FBS. Tumor culture supernatant-induced MDSCs were then added to the culture at a 1:2 ratio. Cells were then analyzed 3 days later by flow cytometry for T cell CFSE intensity.
Gene Expression Analysis
Cells were homogenized in Trizol (Life Technologies) to isolate total RNA. cDNA was synthesized from total RNA and used to analyze gene expression levels using gene-specific primers (Table S1) by semi-quantitative PCR or quantitative PCR (qPCR) in the StepOne Plus Real-Time PCR System (Applied Biosystems). β-actin was used as an internal control in each of the qPCR reactions. The expression level of each gene was normalized to the internal β-actin in each reaction.
Western blotting analysis
Western blotting analysis was performed as previously described (32). Anti-STAT1 and anti-pSTAT1 antibodies were obtained from BD Biosciences (San Diego, CA). Anti-p100/52, anti-p65, anti-p50, anti-RelB, and anti-cRel antibodies were obtained from Santa Cruz Biotech (Dallas, TX). Anti-H3K4me3, anti-H3, and anti-p-p65 antibodies were obtained from Cell Signaling (Danvers, MA). Anti-β-actin was obtained from Sigma-Aldrich (St Louis, MO).
Inhibition of SETD1B enzymatic activity by chaetocin in vitro
Chaetocin was tested in 10-dose IC50 mode with 3-fold serial dilution starting at 10 μM at Reaction Biology (Malvern, PA). Reactions were carried out with recombinant histone H3.3 (5 μM), recombinant SETD1B protein (5 μM), and [3H]S-adenosyl-L-methionine (1 μM) in reaction buffer (50 mM Tris-HCI, pH 8.5, 50 mM NaCl, 5 mM MgCI2, 1 mM DTT, 1 mM PMSF, and 1% DMSO). Reaction mixtures were incubated for 60 min at 30°C and then spotted onto a Whatman cellulose filter paper and counted in a scintillation counter.
Gene silencing
BM cells from WT, tumor-free mice were cultured in the presence of 4T1 conditioned media for 6 days and transiently transfected with either scramble or two different mouse SETD1B-specific siRNAs using Lipofectamine 2000 (Life Technologies) for 2 days. Cells were collected and analyzed by qPCR for SETD1B and iNOS mRNA levels. The scramble and SETD1B-specific siRNA sequences are listed in Table S1.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) was performed using anti-H3K4me3 antibody and protein A agarose beads (Millipore, Temecula, CA) according to the manufacturer’s instructions as previously described (33). The immunoprecipitated genomic DNA was amplified by semi-quantitative PCR using four pairs of PCR primers (Table S1) covering the region from −3000 to +1000 relative to the nos2 transcription initiation site at the nos2 promoter region. The PCR band intensities were quantified using NIH Image J. The ratio of band intensities of the H3K4me3-specific antibody-immunoprecipitated DNA over input DNA was used to represent the H3K4me3 levels at the specific promoter region. The ChIP experiment was repeated and analyzed by qPCR using the nos2 promoter DNA-specific primers as above.
Statistical analysis
Statistical analysis was performed by two-sided student’s t test using GraphPad Prism program (GraphPad Software, Inc., CA). A p<0.05 was taken as statistically significant.
Results
IRF8 is an essential transcriptional activator of both mature and immature myeloid cells under physiological conditions
Spleen, lymph nodes (LN), thymus, and bone marrow (BM) cells were collected from tumor-free WT and IRF8 KO mice and analyzed for CD11b+ and Gr1+ cells. Flow cytometry analysis validated that CD11b+Gr1+ cells increased dramatically in the spleen in IRF8 KO mice as compared to WT mice (Fig. 1A) (34). To determine the function of IRF8 in the regulation of iNOS expression in both mature and immature myeloid cells under physiological conditions, we sorted CD11b+Gr1− and CD11b+Gr1+ myeloid cells from the spleens of both WT and IRF8 KO mice. Few CD11b−Gr1+ cells were present in IRF8 KO mice and were not sorted (Fig. 1B). Analysis of CD11b+Gr1− and CD11b+Gr1+ cells by qPCR revealed that iNOS expression is diminished in both CD11b+Gr1− and CD11b+Gr1+ myeloid cells in IRF8 KO mice as compared to WT mice (Fig. 1B). CD11b+Gr1− and CD11b+Gr1+ myeloid cells were also isolated from BM cells of WT and IRF8 KO mice and analyzed for iNOS expression. iNOS expression level was lower to undetectable in both WT and IRF8 KO CD11b+Gr1− and CD11b+Gr1+ BM cells (Fig. 1C). These observations indicate that IRF8 is an essential transcriptional activator of iNOS in both mature and immature myeloid cells in vivo.
Figure 1. IRF8 is an essential transcriptional activator of iNOS in myeloid cells.
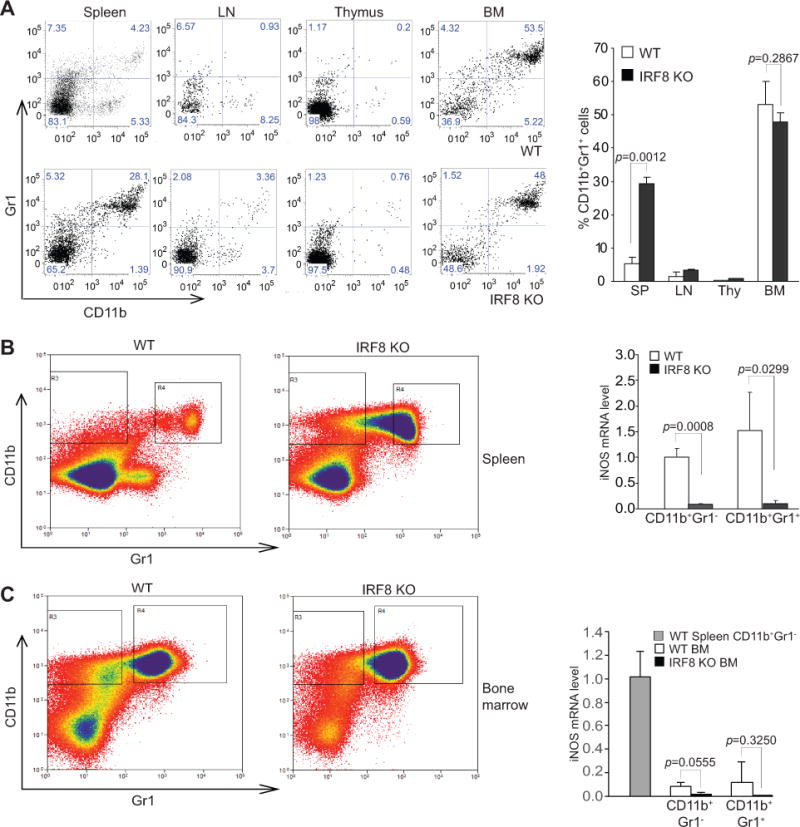
A. Spleen, LN, thymus and BM cells from tumor-free WT and IRF8 KO C57BL/6 mice were stained with CD11b- and Gr1-specific mAbs and analyzed by flow cytometry. Shown are representative images of CD11b+Gr1+ cells from the indicated tissues. The CD11b+Gr1+ cells in the indicated tissues from WT (n=3) and IRF8 KO (n=3) as shown at the left panel were quantified and presented in the right panel. Column: mean; Bar: SD. B. Spleen cells of WT and IRF8 KO C57BL/6 mice were stained with CD11b- and Gr1-specific mAbs and sorted for subsets of CD11b+Gr1− and CD11b+Gr1+ cells. Left panel shows gating of the sorted subsets of cells. The sorted CD11b+Gr1− and CD11b+Gr1+ cells from WT (n=3) and IRF8 KO (n=3) mice were analyzed by qPCR for iNOS mRNA level and presented at the right. Column: mean; Bar: SD. C. BM cells of WT and IRF8 KO mice were stained with CD11b- and Gr1-specific mAbs and sorted for subsets of CD11b+Gr1− and CD11b+Gr1+ cells. Left panel shows gating of the sorted subsets of cells. The sorted BM CD11b+Gr1− and CD11b+Gr1+ cells from WT (n=3) and IRF8 KO (n=3) mice were analyzed by qPCR for iNOS mRNA level and presented at the right. CD11b+Gr1− cells sorted from WT spleen were used as an iNOS positive control. Column: mean; Bar: SD
Tumor induced MDSCs exhibit silenced IRF8 but elevated iNOS expression
To induce MDSCs, 4T1 tumor cells were injected into the mammary glands of BALB/c mice to generate orthotopic breast cancer (35). Analysis of lymphoid organs of tumor-free and tumor-bearing mice validated that CD11b+Gr1+ MDSCs massively accumulate in spleen, BM, and blood of tumor-bearing mice (31, 36). IRF8 expression is significantly lower in MDSCs from tumor-bearing mice as compared to their equivalent in tumor-free mice (Fig. 2A). However, iNOS expression is significantly higher in MDSCs from tumor-bearing mice than their equivalent in tumor-free mice (Fig. 2B). These observations indicate that IRF8 expression is diminished whereas iNOS expression is elevated in MDSCs from tumor-bearing mice. Therefore, iNOS expression is regulated by an IRF8-independent mechanism under pathological conditions.
Figure 2. IRF8 is silenced but iNOS is upregulated in MDSCs of tumor-bearing mice.
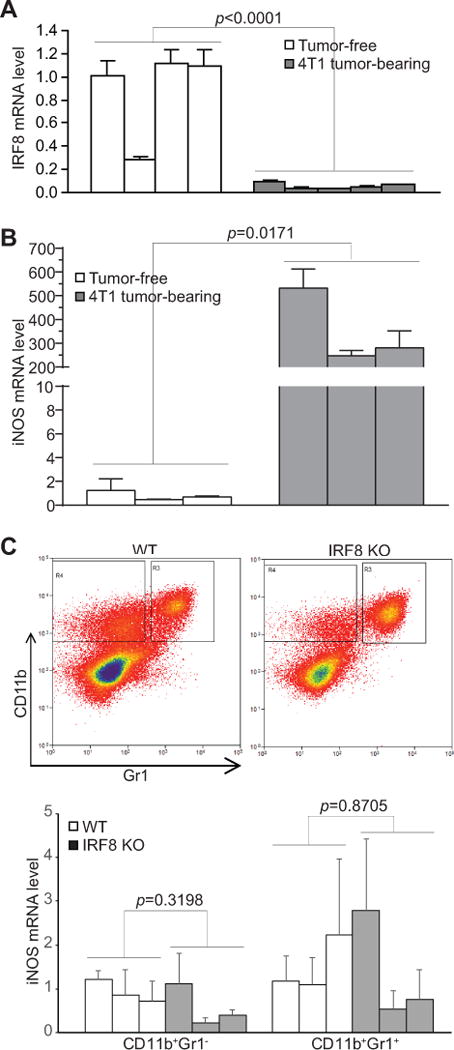
A. CD11b+Gr1+ cells were purified from spleens of tumor-free (n=4) and 4T1 tumor-bearing (n=5) BALB/c mice and analyzed by qPCR for IRF8 mRNA level. B. CD11b+Gr1+ cells were purified from spleens of tumor-free (n=3) and 4T1 tumor-bearing (n=3) BALB/c mice and analyzed by qPCR for iNOS mRNA level. C. Tumors were excised from AT3 tumor-bearing WT (n=3) and IRF8 KO (n=3) C57BL/6 mice and a single cell suspension was prepared. Tumor mixtures were then sorted for infiltrating CD11b+Gr1− and CD11b+Gr1+ cells. Top panel shows gating of sorted cells. Bottom panel shows analysis of the sorted cells by qPCR for iNOS mRNA level.
To compare iNOS expression level in tumor-bearing WT and IRF8 KO mice, AT3 cells were injected into WT and IRF8 KO mice. Tumor-infiltrating CD11b+Gr1− and CD11b+Gr1+ cells were isolated and analyzed for iNOS expression level. No significant differences were observed in iNOS expression in both subsets of tumor-infiltrating myeloid cells between tumor-bearing WT and IRF8 KO mice (Fig. 2C). These observations further indicate that IRF8 plays no significant role in the regulation of iNOS expression in tumor-induced MDSCs in tumor-bearing mice in vivo.
iNOS expression and the IFNγ and NF-κB signaling pathways
In myeloid cells, iNOS expression is up-regulated by IFNγ and NF-κB under physiological conditions (27–29). To determine whether the elevated iNOS expression in MDSCs is regulated by the IFNγ and NF-κB, MDSCs from spleens of tumor-bearing mice and their equivalent in tumor-free mice were analyzed by Western blotting. STAT1, the key mediator of the IFNγ signaling pathway, is actually down-regulated in MDSCs of tumor-bearing mice as compared to their equivalent from tumor-free mice. No activated STAT1 (pSTAT1) protein was detected in MDSCs from tumor-bearing and their equivalent in tumor-free mice (Fig. 3A). NF-κB has five Rel subunits. The canonical NF-κB complex can be combinations of any of the 4 following Rel subunits: p65, p50, RelB, and cRel. Western blotting analysis showed that the protein levels of all four subunits are lower in MDSCs from tumor-bearing mice (Fig. 3A). Similarly, the p52 subunit of the alternative NF-κB is also undetectable in MDSCs from tumor-bearing mice (Fig. 3A).
Figure 3. The IFNγ and NF-κB signaling pathways and MDSCs.
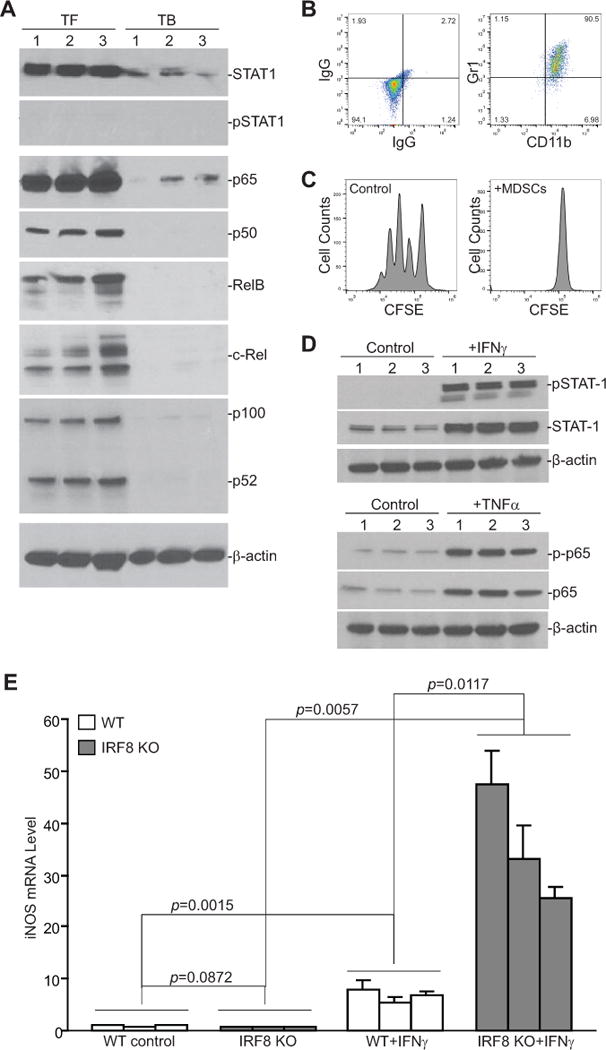
A. MDSCs were purified from the spleens of 4T1 tumor-bearing (TB, n=3) and their equivalent in tumor-free (TF, n=3) mice and analyzed by Western blotting for STAT1, pSTAT1, p65, p50, RelB, c-Rel, and p100/52. β-actin was used as a normalization control. B. 4T1 conditioned media was collected from 4T1 tumor cell culture flasks. BM cells from three WT mice were cultured in the presence of 4T1 conditioned media for 6 days. Cells were stained with IgG- or CD11b- and Gr1-specific mAb and analyzed by flow cytometry. Shown is the phenotype of the 4T1 conditioned media-induced MDSCs. C. CD3+ T cells were purified from the spleen of a tumor-free WT mouse and labeled with CFSE. The labeled T cells were then cultured in the absence or presence of 4T1 conditioned media-induced MDSCs at a 2:1 ratio for 3 days and analyzed for CFSE intensity by flow cytometry. Shown are representative data of proliferation of T cells from one of three replicates. D. BM cells from three WT mice were cultured in the presence of 4T1 conditioned media for 6 days. The resultant MDSCs were then either untreated (control) or treated with IFNγ (100 U/ml) or TNFα (100 U/ml) for approximately 20 hours, respectively. Cells were then analyzed by Western blotting for the indicated proteins. β-actin was used as a normalization control. E. BM cells from WT (n=3) and IRF8 KO (n=3) mice were cultured in the presence of 4T1 conditioned media for 6 days and then treated with IFNγ (100 U/ml) for approximately 20 hours. Cells were then analyzed by qPCR for iNOS mRNA level.
Next, BM cells were cultured in the presence of 4T1 conditioned media to induce MDSC differentiation. 4T1 conditioned media effectively induced CD11b+Gr1+ MDSC differentiation (Fig. 3B). These MDSCs exhibit potent inhibitory activity against T cell activation and proliferation (Fig. 3C), indicating that these MDSCs phenotypically and functionally resemble tumor-induced MDSCs. These BM-derived MDSCs were then analyzed for IFNγ and NF-κB signaling components. Western blotting analysis detected STAT1 and p65 proteins in these MDSCs. IFNγ treatment induced STAT1 phosphorylation and TNFα induced phosphorylation of p65 in these BM-derived MDSCs (Fig. 3D). There is no significant difference in iNOS expression level in BM-derived MDSCs between WT and IRF8 KO mice. IFNγ treatment significantly increased iNOS expression in these BM-derived MDSCs from both WT and IRF8 KO mice, however, BM-derived MDSCs from IRF8 KO mice exhibited significantly higher IFNγ-induced iNOS expression than the MDSCs from WT mice (Fig. 3E). These observations suggest that IRF8 functions as a repressor in IFNγ induction of iNOS expression in BM-derived MDSCs ex vivo.
SETD1B is up-regulated and H3K4me3 is increased in tumor-induced MDSCs
We then hypothesized that iNOS might be regulated by an epigenetic mechanism in MDSCs of tumor-bearing mice. H3K4me3 is often associated with active chromatin and gene activation (37). We then analyzed H3K4me3 levels in MDSCs from the spleen of tumor-bearing mice and their equivalent in tumor-free mice. Western blotting analysis revealed indeed that H3K4me3 is higher in MDSCs from tumor-bearing mice as compared to their equivalent in tumor-free mice (Fig. 4A). H3K4me3 is catalyzed by five histone methyltransferases (38, 39). Analysis of these five histone methyltransferases showed that the expression level of SETD1B is up-regulated in MDSCs from tumor-bearing mice as compared to their equivalent in tumor-free mice (Fig. 4B & C).
Figure 4. SETD1B and H3K4me3 levels are elevated in tumor-induced MDSCs.
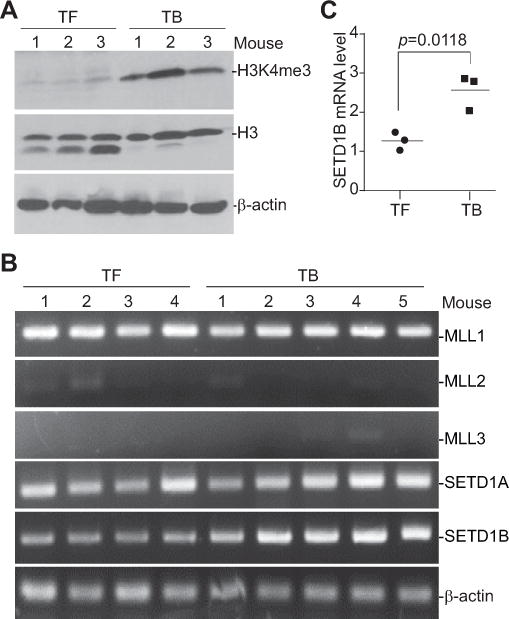
A. 4T1 tumor cells were injected s.c. into the mammary glands of BALB/c mice. Spleens were collected from tumor-free (TF, n=3) and 4T1 tumor-bearing (TB, n=3) mice. CD11b+Gr1+ cells were purified from spleen cells and analyzed for H3K4me3 level by Western blotting. Histone 3 (H3) and β-actin proteins were used as normalization controls. B. CD11b+Gr1+ cells were purified from the spleens of TF (n=4) and TB (n=5) mice and analyzed by RT-PCR with gene-specific primers as indicated. β-actin was used as a normalization control. C. CD11b+Gr1+ cells were purified from spleens of TF (n=3) and TB (n=3) mice and analyzed by qPCR for SETD1B expression level.
SETD1B increases H3K4me3 level to up-regulate iNOS expression in tumor-induced MDSCs
We then made use of a SETD1B inhibitor chaetocin. Chaetocin inhibits SETD1B at IC50 of 0.238 μM (Fig. 5A). Chaetocin treatment of tumor-bearing mice significantly suppresses tumor growth when the tumor sizes are approximately 98–139 mm3 at the time of the start of the treatment (Fig. 5B, left panel). Because MDSC accumulation is associated with tumor size (40), to minimize the effects of tumor size on MDSC accumulation and SETD1B expression, tumors were allowed to grow to approximately 1004–1058 mm3. Chaetocin treatment did not significantly decrease the tumor size of the tumor-bearing mice with this extensive tumor burden (Fig. 5B, right panel). Chaetocin treatment did not change the levels of general MDSCs (Fig. S1A), the PMN-MDSCs, or the M-MDSCs (Fig. S1B). MDSCs were then purified from spleens of control and chaetocin-treated mice. The purity of MDSCs is about 90% (Fig. 5C), and similar between the control and chaetocin-treated mice (Fig. 5D). Western blotting analysis showed that chaetocin treatment decreases H3K4me3 levels in MDSCs in tumor-bearing mice (Fig. 5E). qPCR analysis revealed that chaetocin treatment also diminished iNOS expression in MDSCs in tumor-bearing mice (Fig. 5F). Therefore, iNOS up-regulation is at least in part regulated by SETD1B catalyzed H3K4me3 in MDSCs in tumor-bearing mice.
Figure 5. Inhibition of SETD1B diminishes H3K4me3 level and iNOS expression in MDSCs in tumor-bearing mice.
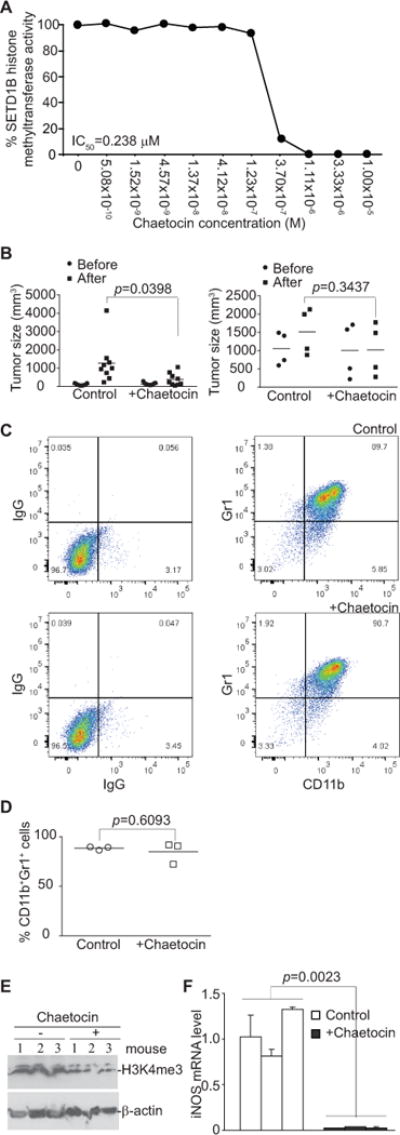
A. Chaetocin was tested in a 10-dose IC50 mode with 3-fold serial dilutions using [3H]S-adenosyl-methionine as a substrate with recombinant human HMTases as indicated. The enzyme activity was then analyzed and plotted against chaetocin concentrations. IC50 was calculated using the GraphPad Prism program. B. 4T1 tumor cells were injected s.c. into the mammary glands of BALB/c mice. Tumor-bearing mice were treated i.p. 9 (left panel, small tumor group) and 21 (right panel, large tumor group) days after tumor cell injection with solvent control (9 day tumor-bearing mice: n=9. 21 day tumor-bearing mice: n=4) or chaetocin (9 day tumor-bearing mice: n=9. 21 day tumor-bearing mice: n=4) daily for 7 days. Shown is the quantification of tumor sizes before and after chaetocin treatment. C. Spleens were collected from control and chaetocin-treated tumor-bearing mice from the large tumor group. CD11b+Gr1+ cells were isolated from the spleens using CD11b Microbeads and LS columns. Purity of the isolated cells was determined by staining cells with IgG or CD11b- and Gr1-specific mAbs and flow cytometry analysis. Shown are representative images of IgG isotype control and CD11b-/Gr1-specific mAb staining of the purified cells from control mice (top panel) and chaetocin-treated mice (bottom panel). D. Comparison of MDSC purity from control and chaetocin-treated mice as shown in C. E. The purified MDSCs from control and chaetocin-treated mice as shown in C were analyzed by Western blotting for total cellular H3K4me3 levels. β-actin was used as normalization control. F. MDSCs were purified from spleens of control (n=3) and chaetocin-treated (n=3) tumor-bearing mice with large tumors and analyzed for iNOS mRNA level by qPCR.
SETD1B regulates iNOS expression in tumor-induced MDSCs
A complementary approach was then used to validate the above finding that SETD1B regulates iNOS expression in tumor-induced MDSCs. BM cells were cultured in the presence of 4T1 conditioned media to induce MDSC differentiation. SETD1B expression was then silenced by two SETD1B-specific siRNAs (Fig. 6A). Silencing SETD1B diminished iNOS expression in MDSCs (Fig. 6B). Taken together, these observations indicate that SETD1B regulates iNOS expression in tumor-induced MDSCs.
Figure 6. SETD1B regulates iNOS expression in tumor-induced MDSCs.
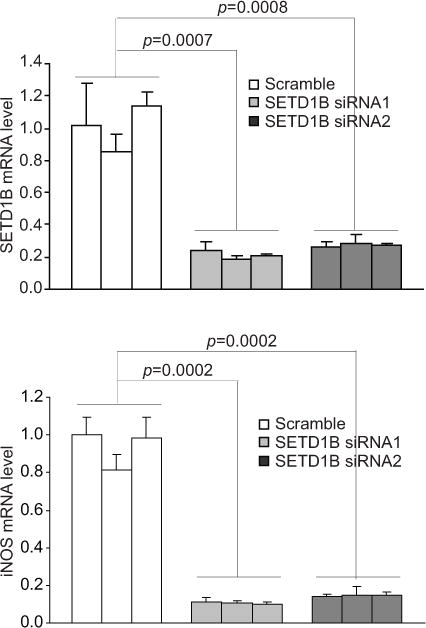
BM cells from three mice were cultured in the presence of 4T1 conditioned media for 6 days. The BM-derived MDSCs were transiently transfected with scramble siRNA and SETD1B-specific siRNAs for 48h and analyzed for SETD1B (top panel) and iNOS (bottom panel) mRNA level by qPCR. Each column represents data of 4T1 conditioned media-induced MDSCs from one mouse. Column: Mean. Bar: SD.
To determine whether SETD1B regulates iNOS expression in human MDSCs, we isolated HLA-DR−CD11b+CD33+ myeloid cells from peripheral blood specimens of healthy human donors and colon cancer patients (Fig. S2A). The level of HLA-DR−CD11b+CD33+ MDSCs varies greatly, ranging from 18.2% to 49.7% among the five colon cancer patients. The equivalent population of cells in normal donors ranges from 1.32% to 2.67%, which is lower than all five colon cancer patients (Fig. S2B). SETD1B level also varies greatly in MDSCs among colon cancer patients, but its expression level is higher in three of the five cancer patients as compared to the three normal donors (Fig. S2C). There is no correlation between the percentage of MDSCs and SETD1B expression level (Fig. S2B & C). iNOS is undetectable in the MDSCs under the conditions used here.
SETD1B increases H3K4me3 levels at the nos2 promoter region in tumor-induced MDSCs
The above observation that inhibition of SETD1B enzyme activity decreases H3K4me3 levels and iNOS expression level in tumor-induced MDSCs in vivo suggests that SETD1B might directly regulate H3K4me3 at the nos2 promoter to activate nos2 transcription. To test this hypothesis, we performed ChIP analysis of H3K4me3 levels at the nos2 promoter region. Four pairs of PCR primers were designed to cover the region of the nos2 promoter from −3000 to +1000 relative to nos2 transcription initiation site (Fig. 7A). Purified MDSCs from the spleens of the control and chaetocin-treated mice were used to prepare chromatin fragments and H3K4me3-specific antibody was used to immunoprecipitate the chromatin fragments. PCR analysis with the immunoprecipitated genomic DNA with the four primer pairs revealed that H3K4me3 is enriched at the nos2 promoter region upstream of the transcription start site (Fig. 7B & C). Chaetocin treatment significantly decreased H3K4me3 level in the region immediately upstream of nos2 transcription initiation site (Fig. 7B & C). Taken together, our data determine that SETD1B expression is up-regulated in MDSCs and SETD1B regulates H3K4me3 at the nos2 promoter region to activate nos2 transcription in MDSCs in tumor-bearing mice.
Figure 7. Inhibition of SETD1B significantly decreases H3K4me3 level at the nos2 promoter region in tumor-induced MDSCs in vivo.
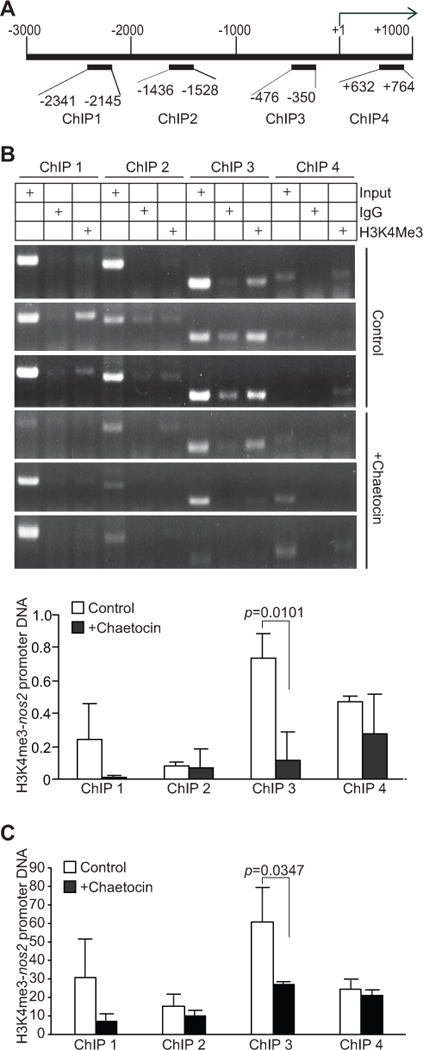
A. Structure of the nos2 promoter region. The number above the bar indicates nucleotide locations relative to the nos2 transcription initiation site. The ChIP PCR primer regions are also indicated. B. CD11b+Gr1+ cells from control (n=3) and chaetocin-treated (n=3) 4T1 tumor-bearing mice were analyzed by ChIP using IgG control antibody and H3K4me3-specific antibody, respectively, followed by PCR analysis with nos2 promoter specific PCR primers as shown in A. Input DNA was used as an normalization control. The intensities of H3K4me3 ChIP, IgG ChIP, and input PCR bands as shown in B were quantified using Image J. The IgG background was subtracted from the H3K4me3 band intensities, which was then normalized to the respective input band intensities and presented at the bottom panel. C. In a separate experiment, CD11b+Gr1+ cells from control (n=3) and chaetocin-treated (n=3) tumor-bearing mice were analyzed by ChIP anti-H3K4me3-specific antibody. The immunoprecipitated DNA was then analyzed by qPCR in triplicates. The IgG background was subtracted from H3K4me3-DNA level. The input of each ChIP primer set was arbitrarily set at 1 and the H3K4me3 was normalized to input DNA level. Column: average of 3 mice. Bar: SD.
Discussion
iNOS expression regulation varies depending on cell types and species (30, 41). In myeloid cells, particularly in macrophages, TNFα and LPS/TLR ligands-activated NF-κB is a major regulator of iNOS expression (27, 28, 42, 43). NF-κB, once activated by LPS or TNFα, can activate iNOS expression (28, 44). The p65 and p50 homodimers of the canonical NF-κB directly binds to the nos2 promoter region to activate nos2 transcription in myeloid cells (28). In addition to NF-κB, inflammatory cytokines such as IFN-γ have also been shown to regulate iNOS expression (27, 45). It has been shown that IFNγ regulates iNOS expression in an IRF8-dependent mechanism. Overexpression of IRF8 dramatically increased IFNγ-induced iNOS activation in macrophages and this activation was abolished in IRF8-deficient macrophages. Furthermore, transduction of IRF8-deficient macrophages with IRF8-expressing retrovirus rescued IFNγ-induced iNOS gene expression, whereas transduction of wild type and IRF8-deficient macrophages with IRF8-expressing retrovirus in the absence of IFNγ activation did not induce iNOS expression (27). These observations indicate that: 1) IRF8 mediates IFNγ induction of iNOS expression in macrophages in vitro; and 2) constitutive IRF8 does not activate iNOS expression in macrophages in vitro.
In this study, we isolated both mature and immature primary myeloid cells from WT and IRF8 KO mice (34) and observed that IRF8 deficiency results in diminished iNOS expression in both mature and immature primary myeloid cells. It is unlikely that these myeloid cells are exposed to IFNγ since both WT and IRF8 KO mice are healthy mice without any treatment. Therefore, our observations suggest that constitutively expressed IRF8 functions as an iNOS transcription activator in myeloid cells in vivo. IRF8 is both constitutively expressed and IFNγ-inducible in myeloid cells (27, 45). The observations that IRF8 does not regulate iNOS expression in the absence of IFNγ in macrophages in vitro (27) but regulates iNOS expression in myeloid cells in vivo suggest that IRF8 functions differently in in vitro cultured myeloid cells than in primary myeloid cells in vivo.
IRF8 is known to be silenced in tumor-induced MDSCs from both human patients and tumor-bearing mice (31, 36). However, iNOS expression is elevated in MDSCs from tumor-bearing mice (26, 46). Thus, IRF8 expression is inversely correlated with iNOS expression in MDSCs of tumor-bearing mice, which is in contrast to IRF8 function in iNOS expression in myeloid cells in tumor-free mice under physiological conditions. On the other hand, although no pSTAT1 and p-p65 proteins are detected in tumor-induced MDSCs in vivo, tumor condition medium-induced and BM-derived MDSCs ex vivo respond to IFNγ and TNFα to activate STAT1 and NF-κB p65, suggesting that MDSCs are responsive to the IFNγ and NF-κB signaling pathways, but the IFNγ and NF-κB signaling pathways are not activated in MDSCs in the tumor-bearing mice in vivo. In contrast to what was observed in myeloid cells from tumor-free WT and IRF8 KO mice, tumor conditioned medium-induced MDSCs from WT and IRF8 KO mice exhibit no significant difference in iNOS expression. Furthermore, IFNγ induced significantly higher iNOS expression in BM-derived MDSCs from IRF8 KO mice than from WT mice, which is in contrast to what was reported in the in vitro cultured macrophages (27). Because IFNγ can also induce IRF8 expression in WT myeloid cells (27, 45), these observations suggest that IRF8 function as an iNOS repressor in tumor-induced MDSCs which might use IRF8 silencing as a mechanism to increase iNOS expression under pathological conditions. IRF8 is an essential transcription factor for myeloid and T cell lineage differentiation and maturation and can function as either a transcription activator or repressor depending on which co-factors it binds to under physiological conditions (34, 43, 47–50). The contrasting functions of IRF8 in iNOS expression regulation and IFNγ induction of iNOS expression in myeloid cells and tumor-induced MDSCs might be controlled by different components of the IRF8 protein complexes in these cells, which require further studies.
We observed here that the expression level of SETD1B, a histone methyltransferase that catalyzes H3K4me3, is up-regulated in tumor-induced MDSCs. Consistent with elevated SETD1B expression levels, H3K4me3 mark is enriched at the nos2 promoter region in tumor-induced MDSCs. Furthermore, inhibition of SETD1B decreased H3K4me3 level at the nos2 promoter region and diminished iNOS expression in MDSCs in tumor-bearing mice. Our data thus determine that the SETD1B-H3K4me3epigenetic axis regulates iNOS expression in MDSCs in tumor-bearing mice, which represent a novel molecular mechanism underlying iNOS expression regulation in tumor-induced MDSCs. However, how SETD1B is up-regulated in MDSCs in tumor-bearing mice also requires further study. Nevertheless, our data suggest that tumor-induced MDSCs might use the up-regulation of the SETD1B-H3K4me3 pathway to activate iNOS expression and execute its immune suppressive function. Thus, targeting SETD1B expression in MDSCs might represent an effective approach to inhibit MDSC function in immune suppression and thus to improve the efficacy of cancer immunotherapy.
Supplementary Material
Acknowledgments
We thank Dr. Jeanene Pihkala at the Medical College of Georgia Flow Cytometry Core Facility and Dr. Ningchun Xu at Georgia Cancer Center Flow Cytometry Core Facility for assistance in cell sorting. We would also like to thank Dr. Wei Xiao for his assistance in flow cytometry. In addition, we thank Jennifer Parks and Susan Dewes for their assistance in obtaining blood specimen from consented healthy donors at the Shepeard Community Blood Center.
Financial support: NIH CA133085 and NIH CA182518 (to K. Liu), VA Merit Review Award I01BX001962 (to K. Liu.), and NIAID AI120487 (to A.V. Paschall).
Footnotes
Conflict of interest: The authors declare no potential conflicts of interest
References
- 1.Stiff A, Trikha P, Wesolowski R, Kendra K, Hsu V, Uppati S, et al. Myeloid-derived suppressor cells express Bruton’s tyrosine kinase and can be depleted in tumor bearing hosts by ibrutinib treatment. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-15-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12:253–68. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paschall AV, Zhang R, Qi CF, Bardhan K, Peng L, Lu G, et al. IFN Regulatory Factor 8 Represses GM-CSF Expression in T Cells To Affect Myeloid Cell Lineage Differentiation. J Immunol. 2015;194:2369–79. doi: 10.4049/jimmunol.1402412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bronte V, Brandau S, Chen SH, Colombo MP, Frey AB, Greten TF, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. doi: 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beury DW, Carter KA, Nelson C, Sinha P, Hanson E, Nyandjo M, et al. Myeloid-Derived Suppressor Cell Survival and Function Are Regulated by the Transcription Factor Nrf2. J Immunol. 2016;196:3470–8. doi: 10.4049/jimmunol.1501785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elpek KG, Cremasco V, Shen H, Harvey CJ, Wucherpfennig KW, Goldstein DR, et al. The tumor microenvironment shapes lineage, transcriptional, and functional diversity of infiltrating myeloid cells. Cancer Immunol Res. 2014;2:655–67. doi: 10.1158/2326-6066.CIR-13-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Umansky V, Utikal J, Gebhardt C. Predictive immune markers in advanced melanoma patients treated with ipilimumab. Oncoimmunology. 2016;5:e1158901. doi: 10.1080/2162402X.2016.1158901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Abrams SI, Netherby CS, Twum DY, Messmer MN. Relevance of Interferon Regulatory Factor-8 Expression in Myeloid-Tumor Interactions. J Interferon Cytokine Res. 2016;36:442–53. doi: 10.1089/jir.2015.0174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu C, Redd PS, Lee JR, Savage N, Liu K. The expression profiles and regulation of PD-L1 in tumor-induced myeloid-derived suppressor cells. Oncoimmunology. 2016;5:e1247135. doi: 10.1080/2162402X.2016.1247135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Condamine T, Kumar V, Ramachandran IR, Youn JI, Celis E, Finnberg N, et al. ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis. J Clin Invest. 2014;124:2626–39. doi: 10.1172/JCI74056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Youn JI, Kumar V, Collazo M, Nefedova Y, Condamine T, Cheng P, et al. Epigenetic silencing of retinoblastoma gene regulates pathologic differentiation of myeloid cells in cancer. Nat Immunol. 2013;14:211–20. doi: 10.1038/ni.2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shvedova AA, Kisin ER, Yanamala N, Tkach AV, Gutkin DW, Star A, et al. MDSC and TGFbeta Are Required for Facilitation of Tumor Growth in the Lungs of Mice Exposed to Carbon Nanotubes. Cancer Res. 2015;75:1615–23. doi: 10.1158/0008-5472.CAN-14-2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Limagne E, Euvrard R, Thibaudin M, Rebe C, Derangere V, Chevriaux A, et al. Accumulation of MDSC and Th17 cells in patients with metastatic colorectal cancer predict the efficacy of a FOLFOX-bevacizumab drug treatment regimen. Cancer Res. 2016 doi: 10.1158/0008-5472.CAN-15-3164. [DOI] [PubMed] [Google Scholar]
- 14.de Goeje PL, Bezemer K, Heuvers ME, Dingemans AC, Groen HJ, Smit EF, et al. Immunoglobulin-like transcript 3 is expressed by myeloid-derived suppressor cells and correlates with survival in patients with non-small cell lung cancer. Oncoimmunology. 2015;4:e1014242. doi: 10.1080/2162402X.2015.1014242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang C, Wang S, Liu Y, Yang C. Epigenetics in myeloid derived suppressor cells: a sheathed sword towards cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vila-Leahey A, Oldford SA, Marignani PA, Wang J, Haidl ID, Marshall JS. Ranitidine modifies myeloid cell populations and inhibits breast tumor development and spread in mice. Oncoimmunology. 2016;5:e1151591. doi: 10.1080/2162402X.2016.1151591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dufait I, Van Valckenborgh E, Menu E, Escors D, De Ridder M, Breckpot K. Signal transducer and activator of transcription 3 in myeloid-derived suppressor cells: an opportunity for cancer therapy. Oncotarget. 2016 doi: 10.18632/oncotarget.8311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Srivastava MK, Dubinett S, Sharma S. Targeting MDSCs enhance therapeutic vaccination responses against lung cancer. Oncoimmunology. 2012;1:1650–1. doi: 10.4161/onci.21970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morello S, Miele L. Targeting the adenosine A2b receptor in the tumor microenvironment overcomes local immunosuppression by myeloid-derived suppressor cells. Oncoimmunology. 2014;3:e27989. doi: 10.4161/onci.27989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Haas N, de Koning C, Spilgies L, de Vries IJ, Hato SV. Improving cancer immunotherapy by targeting the STATe of MDSCs. Oncoimmunology. 2016;5:e1196312. doi: 10.1080/2162402X.2016.1196312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu F, Li X, Lu C, Bai A, Bielawski J, Bielawska A, et al. Ceramide activates lysosomal cathepsin B and cathepsin D to attenuate autophagy and induces ER stress to suppress myeloid-derived suppressor cells. Oncotarget. 2016 doi: 10.18632/oncotarget.13438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, et al. The terminology issue for myeloid-derived suppressor cells. Cancer Res. 2007;67:425. doi: 10.1158/0008-5472.CAN-06-3037. author reply 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, et al. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 2015;12:244–57. doi: 10.1016/j.celrep.2015.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Movahedi K, Guilliams M, Van den Bossche J, Van den Bergh R, Gysemans C, Beschin A, et al. Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood. 2008;111:4233–44. doi: 10.1182/blood-2007-07-099226. [DOI] [PubMed] [Google Scholar]
- 25.Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, et al. Altered recognition of antigen is a mechanism of CD8+ T cell tolerance in cancer. Nat Med. 2007;13:828–35. doi: 10.1038/nm1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mundy-Bosse BL, Lesinski GB, Jaime-Ramirez AC, Benninger K, Khan M, Kuppusamy P, et al. Myeloid-derived suppressor cell inhibition of the IFN response in tumor-bearing mice. Cancer Res. 2011;71:5101–10. doi: 10.1158/0008-5472.CAN-10-2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiong H, Zhu C, Li H, Chen F, Mayer L, Ozato K, et al. Complex formation of the interferon (IFN) consensus sequence-binding protein with IRF-1 is essential for murine macrophage IFN-gamma-induced iNOS gene expression. J Biol Chem. 2003;278:2271–7. doi: 10.1074/jbc.M209583200. [DOI] [PubMed] [Google Scholar]
- 28.Simon PS, Sharman SK, Lu C, Yang D, Paschall AV, Tulachan SS, et al. The NF-kappaB p65 and p50 homodimer cooperate with IRF8 to activate iNOS transcription. BMC Cancer. 2015;15:770. doi: 10.1186/s12885-015-1808-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schmidt N, Pautz A, Art J, Rauschkolb P, Jung M, Erkel G, et al. Transcriptional and post-transcriptional regulation of iNOS expression in human chondrocytes. Biochem Pharmacol. 2010;79:722–32. doi: 10.1016/j.bcp.2009.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Douguet L, Bod L, Lengagne R, Labarthe L, Kato M, Avril MF, et al. Nitric oxide synthase 2 is involved in the pro-tumorigenic potential of gammadelta17 T cells in melanoma. Oncoimmunology. 2016;5:e1208878. doi: 10.1080/2162402X.2016.1208878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, et al. Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest. 2013;123:4464–78. doi: 10.1172/JCI68189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paschall AV, Yang D, Lu C, Choi JH, Li X, Liu F, et al. H3K9 Trimethylation Silences Fas Expression To Confer Colon Carcinoma Immune Escape and 5-Fluorouracil Chemoresistance. J Immunol. 2015 doi: 10.4049/jimmunol.1402243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bardhan K, Paschall AV, Yang D, Chen MR, Simon PS, Bhutia YD, et al. IFNgamma induces DNA methylation-silenced GPR109A expression via STAT1/300 and H3K18 acetylation in colon cancer. Cancer Immunol Res. 2015 doi: 10.1158/2326-6066.CIR-14-0164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Holtschke T, Lohler J, Kanno Y, Fehr T, Giese N, Rosenbauer F, et al. Immunodeficiency and chronic myelogenous leukemia-like syndrome in mice with a targeted mutation of the ICSBP gene. Cell. 1996;87:307–17. doi: 10.1016/s0092-8674(00)81348-3. [DOI] [PubMed] [Google Scholar]
- 35.Paschall AV, Liu K. An Orthotopic Mouse Model of Spontaneous Breast Cancer Metastasis. J Vis Exp. 2016 doi: 10.3791/54040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu X, Bardhan K, Paschall AV, Yang D, Waller JL, Park MA, et al. Deregulation of apoptotic factors Bcl-xL and Bax confers apoptotic resistance to myeloid-derived suppressor cells and contributes to their persistence in cancer. J Biol Chem. 2013;288:19103–15. doi: 10.1074/jbc.M112.434530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, et al. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell. 2013;152:1021–36. doi: 10.1016/j.cell.2013.01.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sandstrom RS, Foret MR, Grow DA, Haugen E, Rhodes CT, Cardona AE, et al. Epigenetic regulation by chromatin activation mark H3K4me3 in primate progenitor cells within adult neurogenic niche. Sci Rep. 2014;4:5371. doi: 10.1038/srep05371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rao RC, Dou Y. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 2015;15:334–46. doi: 10.1038/nrc3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Danna EA, Sinha P, Gilbert M, Clements VK, Pulaski BA, Ostrand-Rosenberg S. Surgical removal of primary tumor reverses tumor-induced immunosuppression despite the presence of metastatic disease. Cancer Res. 2004;64:2205–11. doi: 10.1158/0008-5472.can-03-2646. [DOI] [PubMed] [Google Scholar]
- 41.MacMicking J, Xie QW, Nathan C. Nitric oxide and macrophage function. Annu Rev Immunol. 1997;15:323–50. doi: 10.1146/annurev.immunol.15.1.323. [DOI] [PubMed] [Google Scholar]
- 42.Lu G, Zhang R, Geng S, Peng L, Jayaraman P, Chen C, et al. Myeloid cell-derived inducible nitric oxide synthase suppresses M1 macrophage polarization. Nat Commun. 2015;6:6676. doi: 10.1038/ncomms7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ouyang X, Zhang R, Yang J, Li Q, Qin L, Zhu C, et al. Transcription factor IRF8 directs a silencing programme for TH17 cell differentiation. Nat Commun. 2011;2:314. doi: 10.1038/ncomms1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hayes JB, Sircy LM, Heusinkveld LE, Ding W, Leander RN, McClelland EE, et al. Modulation of Macrophage Inflammatory Nuclear Factor kappaB (NF-kappaB) Signaling by Intracellular Cryptococcus neoformans. J Biol Chem. 2016;291:15614–27. doi: 10.1074/jbc.M116.738187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao J, Kong HJ, Li H, Huang B, Yang M, Zhu C, et al. IRF-8/interferon (IFN) consensus sequence-binding protein is involved in Toll-like receptor (TLR) signaling and contributes to the cross-talk between TLR and IFN-gamma signaling pathways. J Biol Chem. 2006;281:10073–80. doi: 10.1074/jbc.M507788200. [DOI] [PubMed] [Google Scholar]
- 46.Ito H, Ando T, Seishima M. Inhibition of iNOS activity enhances the anti-tumor effects of alpha-galactosylceramide in established murine cancer model. Oncotarget. 2015;6:41863–74. doi: 10.18632/oncotarget.6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurotaki D, Yamamoto M, Nishiyama A, Uno K, Ban T, Ichino M, et al. IRF8 inhibits C/EBPalpha activity to restrain mononuclear phagocyte progenitors from differentiating into neutrophils. Nat Commun. 2014;5:4978. doi: 10.1038/ncomms5978. [DOI] [PubMed] [Google Scholar]
- 48.Kurotaki D, Tamura T. Transcriptional and Epigenetic Regulation of Innate Immune Cell Development by the Transcription Factor, Interferon Regulatory Factor-8. J Interferon Cytokine Res. 2016;36:433–41. doi: 10.1089/jir.2015.0138. [DOI] [PubMed] [Google Scholar]
- 49.Tamura T, Nagamura-Inoue T, Shmeltzer Z, Kuwata T, Ozato K. ICSBP directs bipotential myeloid progenitor cells to differentiate into mature macrophages. Immunity. 2000;13:155–65. doi: 10.1016/s1074-7613(00)00016-9. [DOI] [PubMed] [Google Scholar]
- 50.Sun L, St Leger AJ, Yu CR, He C, Mahdi RM, Chan CC, et al. Interferon Regulator Factor 8 (IRF8) Limits Ocular Pathology during HSV-1 Infection by Restraining the Activation and Expansion of CD8+ T Cells. PLoS One. 2016;11:e0155420. doi: 10.1371/journal.pone.0155420. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.