

Abstract
Recent advances in cellular profiling have demonstrated substantial heterogeneity in the behaviour of cells once deemed ‘identical’, challenging fundamental notions of cell ‘type’ and ‘state’. Not surprisingly, these findings have elicited substantial interest in deeply characterizing the diversity, interrelationships and plasticity among cellular phenotypes. To explore these questions, experimental platforms are needed that can extensively and controllably profile many individual cells. Here, microfluidic structures—whether valve-, droplet- or nanowell-based—have an important role because they can facilitate easy capture and processing of single cells and their components, reducing labour and costs relative to conventional plate-based methods while also improving consistency. In this article, we review the current state-of-the-art methodologies with respect to microfluidics for mammalian single-cell ‘omics’ and discuss challenges and future opportunities.
The phenotypic identity of a cell is informed by many factors, including the abundance, distribution and dynamics of its internal components and the spatiotemporal pattern of signals it receives from its environment. Scientists have long attempted to classify cells into distinct types based on their defining characteristics. At first this classification relied on macroscopic observables (such as anatomical location, gross morphology, origin or distinct behaviours) but has gradually become driven by more nuanced molecular characteristics (such as what proteins or mRNAs the cells express). However, recent advances in the processing and profiling of cellular components have uncovered previously unappreciated heterogeneities in seemingly ‘uniform’ cell populations and complex tissues1–8. In many instances, these findings have altered existing cellular classification schemes (introducing new categories, redefining their breadth, uncovering more informative features or suggesting previously unappreciated interrelationships); in other instances, they have challenged some of our atomistic operating assumptions and long-held rubrics9,10.
Accurate cellular classification is complicated by the considerable difficulties associated with characterizing the properties of single cells. Indeed, the resolving power of any individual measurement is limited by technical problems associated with handling and profiling the minute inputs obtained from just one cell, as well as the stochasticity inherent in biological processes11 (FIG. 1). Small processing losses (technical noise) that are inconsequential at the population level can be disastrous when attempting to accurately score single cells (FIG. 1a). Similarly, differences in the timing of individual cellular events, driven by the biological, physical and temporal properties that control their generation (intrinsic noise12), can average cleanly at the ensemble level but render any single measurement an unreliable marker of the identity of a specific cell (FIG. 1b). Moreover, given the broad range of factors that can potentially affect cellular phenotype (and hence a cell’s classification), several variables can be required for accurate description.
Figure 1. Technical and biological noise in single-cell measurements.
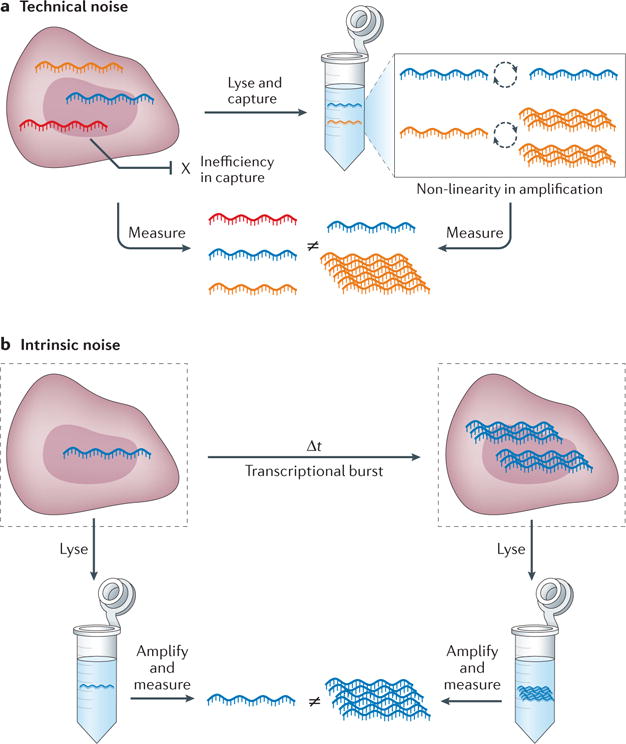
a | Technical errors in cellular processing (‘technical noise’), such as failure to reverse transcribe an mRNA transcript or over-amplification during the ensuing PCR, can dramatically affect the utility of the measured value of any single gene in a single-cell experiment. b | Similarly, the physical, spatial and temporal processes governing biological phenomena (‘intrinsic noise’), such as the ‘burstiness’ of mRNA transcription11, can limit the information content in any single instantaneous end-point measurement.
One strategy for overcoming the noise that is inherent in single-cell measurements is to increase the number of cells profiled. Although any given cellular measurement is subject to systematic (technical noise) and random (intrinsic noise) artefacts, improved throughput, coupled with a fundamental understanding of the limitations of the specific assay in use, can empower studies of the distribution of a variable across a population. Microfluidic devices, tailored to approximately the size of individual cells, can help to achieve this, enhancing experimental scale by miniaturizing, parallelizing and integrating methodological steps. This substantially reduces labour and reagent costs, simplifies workflows and improves consistency.
A second approach is to increase the number of variables that are measured from a single cell so that a more coherent picture can be achieved. The expression of any single gene may be an unreliable indicator, but the collective expression of a set of genes that co-vary across cells is more buffered from noise and thus may more effectively reveal the type, state or properties of a cell3,6,13,14. Over the past few years, several new technologies have been developed that exploit this principle, driven, in part, by the reduced cost and improved accessibility of next-generation sequencing (NGS), a currently preferred method for investigating several variables at once. Microfluidic devices can also considerably improve the preparation of single-cell analytes for NGS-based readouts.
In this Review, we describe the most common microfluidic methods and their operational principles, and assess their relative strengths and weaknesses. We examine how each has been used to address questions of cost, quality, throughput and multiplexing across different single-cell ‘omics’ — including genomics, epigenomics, transcriptomics and proteomics — with a focus on sequencing-enabled approaches. Last, we discuss future opportunities for the field in terms of efficiency, scale and integration that may help to realize a deeper understanding of cellular phenotypes.
Single-cell microfluidic approaches
In recent years, scientists have adapted micromanipulation schemes and microfluidic devices to address concerns of efficiency, cost and labour in single-cell preparation and analysis. The fundamental elements of these devices are typically valves, droplets or nanolitre-scale wells (nanowells). Each of these can be used to establish boundaries between single cells, capture their specific products, retain their components upon lysis or perform manipulations. Importantly, given their small size, these features can be used to process many single cells in a compact physical space, reducing reagent requirements (and thus costs) and increasing analyte concentrations (and thus assay efficiency when limited kinetically or by background; for example, in protein detection by proximity extension assay (PEA)15,16 or reverse enzyme-linked immunosorbent assay (ELISA)17,18). Here, we begin by introducing each of the three major microfluidic confinement strategies, and discuss their advantages and drawbacks.
Valves
The first major method couples microfluidic channels with pressure-controlled valves19,20. Valve-based systems typically rely on a soft elastomeric membrane that can be deflected with pressure to block flow through a microchannel (FIG. 2a). In this way, valves can be used to seal a channel, confine a cell, and more21. The major advantage of valve-based methods is the degree of control that they afford. Several valves can be combined to actuate complex series of operations in space and time, such as adding, removing or mixing reagents20. Moreover, they can be coupled with other devices to enable on-chip processing or detection22–24. Integrated fluidic circuits (IFCs), which contain multiple channels and valves, can process many individual cells with limited manual input3,25. However, valve-based systems rely on dedicated support structures, such as pressure controllers, for operation; similarly, they have structural constraints that limit the number of channels that can be fitted into a given area. Moreover, these systems are complex to design and fabricate, require larger volumes than both droplet- and nanowell-based technologies, and are subject to biofouling, which limits repeated use. Hence, valve-based microfluidics are preferred for complex, integrated workflows where control is vital26,27.
Figure 2. Overview of the major microfluidic device types: valves, droplets and nanowells.
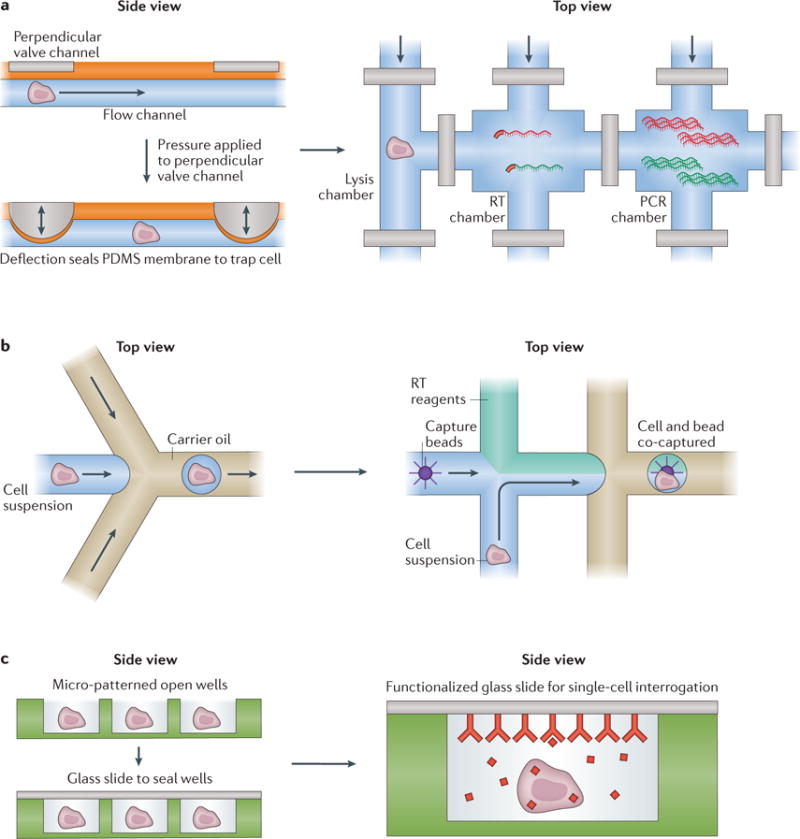
Basic microfluidic structures for processing cells (left) and example implementations (right). a | Valve-based microfluidics operate by aligning two perpendicular channels, one for cell (or cellular component) and solution flow and another for control, separated by a thin membrane. To isolate a chamber, pressure is applied to the control channel, deflecting a membrane into the flow channel to block it, and hence trap its contents. These channels can be arrayed, multiplexed and coupled to external computer-controlled pressure regulators to reliably execute complex workflows such as whole-transcriptome amplification (see, for example, REF. 3). b | In many implementations, droplet-based strategies rely on flowing single cells (or cellular components) in aqueous medium into a co-flowed oil phase. Due to pressure-driven effects, small aqueous droplets that can contain cells (or their components) are formed through encapsulation with oil, isolating each individual cell or cellular component from its peers. During or after this initial encapsulation, additional reagents, such as barcoded oligo(dT) mRNA capture beads (see, for example, REF. 31), can be incorporated or removed to enable more complex operations. c | Nanowells confine cells by gravity, and can subsequently be sealed with a membrane or glass slide to isolate single cells and their components. These cells or components can then be picked out of the wells for further processing (see, for example, REF. 5) or characterized in-well using, for example, a functionalized seal. PDMS, polydimethylsiloxane; RT, reverse transcription.
Droplets
The second major technique for confining single cells and their components is based on using microfluidic devices to encapsulate surfactant-stabilized aqueous droplets in an inert carrier oil28,29 (FIG. 2b). In this scheme, droplets envelop cells or their components at sufficiently low concentrations that most droplets contain, at most, one element (with loading occupancy following a Poisson distribution). The use of carrier oil limits device biofouling and, once formed, droplets can be moved, merged, split, added to, subtracted from, stored, thermocycled, and more28,29. Profiling can be performed in droplet (for example, measuring fluorescence)30 or, after the cells or their components have been processed, the droplets can be broken to profile en masse31,32. A major advantage of droplet-based approaches is their scale and speed. Droplets can compartmentalize up to thousands of cells or cellular components per second. However, specific control over any one droplet is difficult to achieve. Thus, droplet-based devices are ideal for situations where high throughput is of the essence.
Nanowells
A third major approach uses arrays of nanowells (FIG. 2c). Nanowells are roofless, miniature containers fabricated from polydimethylsiloxane (PDMS), glass or other, related materials. In a similar way to droplet-based devices, cells or cellular components can be loaded at low densities to achieve, at most, a single element per well, but here, loading can be achieved using gravity alone. Subsequently, the wells can be sealed by the addition of a roof — for example, a glass slide — thus isolating each cell from its neighbours. If desired, this cap can be selectively permeable or functionalized to help profile cellular analytes, such as secreted cytokines or antibodies33. A major advantage of nanowells is their operational simplicity and sample efficiency. The system requires few peripherals, and small starting numbers of cells can be used. Additionally, the fixed spatial locations of each well can be used to link several discrete measurements. Nevertheless, nanowells have lower overall throughput than droplets, and support less precise environmental manipulations than valve-based systems. Ultimately, these features make nanowell-based devices a first choice for situations in which simplicity is essential.
Preparing and detecting analytes from single cells
Historically, single-cell molecular analysis strategies began by probing biological components directly on or in cells, relying on the presence of the plasma membrane to define cellular boundaries. In most cases, cellular factors were detected optically, with spectral overlap limiting the number of variables that could be measured at once. Although several methods, such as fluorescence microscopy and flow cytometry1,34, are available for discerning the cellular properties (such as the level of a specific protein) of intact cells, even in this setting, microdevices provided early value by confining cellular products so that they could be detected20 and aiding in the processing of low-input samples5. Illustrative applications range from nanowell- and droplet-based methods for measuring the amount of antibody produced by individual B cells33 to characterizing surface marker expression in rare populations of cells using nanowells35.
However, working with intact cells limits the accessibility of their molecular components. Thus, parallel profiling efforts have focused on processing and studying intracellular variables from single-cell lysates. Whereas early work relied on isolating one cell from another in distinct wells of tube strips or multiwell plates, cellular separation and processing was soon ported to micro-devices, which could confine cellular lysates in smaller volumes, thus increasing scale, reducing cost and, for certain assays, improving sensitivity by increasing their signal-to-noise ratios15,17.
At first, microdevices were used to perform common, low-multiplex analyses on cellular lysates in a more economical, scalable, sensitive or quantitative format. For example, as described below, the abundance of one or many DNAs can be probed in parallel with a fraction of the reagent and in much less time than is required for conventional multiwell-plate-based assays36,37; ELISAs can be made ultra-sensitive by concentrating and boosting the capture of secreted proteins or antigens33,38,39; low levels of enzymatic turnover can be detected more easily by confining signal40; and RNA molecules can be quantified with digital precision41,42.
Consider, for the moment, nucleic acid detection (or the detection of another species, such as a protein, that has been mapped into nucleic acid space using, for example, oligonucleotide-tagged antibodies15,16) when working with cellular lysates. Here, to detect one or a few components, quantitative real-time PCR (qPCR) is the method of choice. At the single-cell level, qPCR measures the number of PCR cycles required to detect a primer-amplified sequence of DNA or complementary DNA (cDNA) in real time, calculating the relative abundance of a primer-targeted sequence in a single cell (or amplified single-cell material) using fluorescence. This assay has been ported to valve-, droplet- and nanowell-based platforms to measure the distribution of a transcript or the abundance of a mutation across single cells27,30,43. In instances in which greater quantitative accuracy is desired, digital PCR (dPCR) has been used instead. In dPCR44, individual molecules from a single cell (or fraction thereof) are loaded at low density into many compartments (valves, droplets or nanowells). After PCR, the proportion of chambers in which fluorescence is detected (and hence, the fraction that contains the molecule of interest) is counted, and the initial concentration is calculated based on the fact that, at low density, chamber loading follows Poisson statistics.
Although qPCR and dPCR have been used in many single-cell applications, as mentioned above, methods that are focused on quantifying a few variables in populations of single cells only enable characterization of the distributions of those variables. Indeed, the measured value of any given molecular component is, by itself, only so informative (FIG. 1), and it is often the presence of and interplay between several factors that defines cell identity. Thus, tracking many variables may be necessary for accurate phenotypic characterization. Additionally, making multiple measurements simultaneously can afford further confidence by buffering the noise associated with any one measurement and enabling imputation-based correction strategies13.
Multiplexed qPCR or dPCR is one potential solution. A valve-based system can be used to assay multiple samples combinatorially with multiple primer pairs by first splitting each sample into multiple aliquots and then mixing each aliquot with a different primer set in distinct spatial locations ( spatial encoding)24. However, given spatial considerations and design constraints, there are upper limits on multiplexing. Multiplexing capabilities can be improved by strategies that encode probes with combinations of coloured fluorophores (multi-colour combinatorial labelling; spectral encoding), but ultimately there remains an upper limit (in the hundreds of targets) for PCR-based readouts45. Additionally, qPCR- and dPCR-based detection requires the use of preselected probes, making them ideal for quantitative, targeted measurements — due to their accuracy, reliability and cost-effectiveness — but not genome-wide assays or target discovery.
Over the past few years, advances in NGS have inspired new approaches for extracting more information per cell by coupling spatial and spectral barcoding (as used in qPCR and dPCR) with temporal encoding strategies. Sequencing-based methods can directly analyse (read) the series of bases that comprise each of many millions of DNA fragments derived from one or more samples or their products. Furthermore, by identifying the genes or constructs to which these sequences correspond and using computational methods to determine their relative abundance, the levels of many targets can be quantified simultaneously46. Additionally, mapping different molecular variables to stable DNA-based reporters — such as protein levels to DNA abundance using oligonucleotide-labelled antibodies15,16,47 — can enable simultaneous profiling of different cellular ‘-omes’, such as the genome and transcriptome or the transcriptome and proteome, leading to a more holistic understanding of cellular identity and the interrelationship between the factors that define it15,48–50.
Crucially, unlike with ensemble samples, sequencing is a viable approach for profiling each single cell, even at scale, because the total information content of a single cell is substantially less than that of a population. Thus, although any sequencer is ultimately limited in bandwidth, it can nevertheless be sufficient to simultaneously profile large numbers of single cells. To give a more intuitive feel for this point, consider the following: if examining the full transcriptome of a single cell, assuming there are ~0.5 million transcripts51 and a 10% capture efficiency, there should only be, on average, 50,000 unique 3′ cDNA fragments per cell (instead of 500 million for a population of 10,000 cells). Beyond ~250,000 sequencing reads, the detection of novel transcripts (cDNAs) is unlikely (assuming perfect alignment, ~95% will have been detected with P < 1 × 10−5), suggesting that the optimal cell loading for a sequencing run of 500 million reads would be at least ~2,000 cells for many applications. Moreover, if a researcher’s goal was simply to analyse a single locus or pair of loci (for example, to determine the sequences of T cell receptor-α (TCRα) and TCRβ each T cell was expressing), only around 1,000 gene-specific reads may be needed to establish consensus, and thus ~50,000 cells should be run simultaneously to most efficiently utilize the same 500 million reads52. In general, given the need for accurate quantification and the limited utility of any one cell or measurement (owing to the conflating technical noise associated with single-cell processing and the limited biological information content contained in any single measurement), reads must be wisely allocated to balance the information collected on any cell (read depth) with the number of cells profiled4 (TABLE 1), as sequencing deeper will often not afford greater biological resolution.
Table 1.
Single-cell ‘omic’ profiling: the current state of the art and future opportunities
| Type | Information content (millions of bases) | Reads per cell (millions) | Cells per 500 million reads | Method | Experimental throughput (cells per run) | Throughput saturated? | Refs |
|---|---|---|---|---|---|---|---|
| DNA | |||||||
| Whole genome | 3,000 (REFS 148,149) | 15–20 | 25–33 | Valves | 125 | Yes | 23 |
| Valves (C1 IFC) | 96 | Yes | 22 | ||||
| Droplets | 1,000 | Yes | 59 | ||||
| Whole exome | 30 (REFS 148,149) | 1–5 | 100–500 | Droplets | 1,000 | Yes | 61 |
| Droplets | 96 | No | 150 | ||||
| Wells | 10 | No | 5 | ||||
| Epigenome | |||||||
| Methylome | 10–30 (REF. 151) | 0.05–0.2 | 2,500–10,000 | Valves (C1 IFC) | 96 | No | 78 |
| Open chromatin | 2–10 (REFS 152,153) | 0.05–0.2 | 2,500–10,000 | Valves (C1 IFC) | 96 | No | 79 |
| Chromatin immunoprecipitation | 2–10 (REFS 152,153) | 0.05–0.2 | 2,500–10,000 | Droplets | 100 | No | 80 |
| RNA | |||||||
| Full length | 10 (REF. 51) | 1–4 | 125–500 | Valves (C1 IFC) | 96 | No | 3 |
| 3′ End counting (digital gene expression) | 10 | 0.025–0.25 | 2,000–20,000 | Droplets (Drop-Seq) | 5,000 | Yes | 31 |
| Droplets (InDrop) | 1,000 | No | 32 | ||||
| Wells | 2,500 | No | 99 | ||||
| Wells (Seq-Well) | 1,000 | No | 96 |
Listed are the various single-cell ‘omics’ that have been profiled with sequencing-based readouts using microfluidic devices, with corresponding estimates of their total information content, number of reads needed per single cell, and the number of cells that can hypothetically be profiled with 500 million reads (approximately one run of an Illumina NextSeq 500 or 550 sequencing machine). Also presented are examples in which valve-, droplet- and well-based microfluidic devices have been used to profile each of these omics, as well as the throughput of the method and whether the method is saturated. IFC, integrated fluidic circuit.
Microfluidic devices for the omics
Genome
The complexity of the human genome means that tens of millions of reads are needed to examine the whole-genome amplification (WGA) products derived from a single cell. Although exome sequencing strategies can reduce this burden, the need for depth limits throughput to tens of cells per sequencing run (TABLE 1). As a result, valve-based approaches — which were the first micro-fluidic approach to be developed and have a throughput that can easily saturate sequencing capacity — continue to dominate this application.
Valves
Much of the pioneering work in microfluidic-device-assisted single-cell genomic profiling relied on valve-based implementations. In these first iterations, custom IFC architectures were designed to isolate and subsequently manipulate single cells (FIG. 3a). For example, Wang and co-workers23 developed a chip for performing WGA that they used to profile 91 single sperm cells from one individual to characterize heterogeneity in his gamete genome and create a personal recombination map. Similar architectures were also used to perform direct deterministic phasing (DDP)20 of the haplotype of diploid cells by isolating individual chromosomes rather than cells. By performing DDP on single metaphase cells from lymphoblastoid cell lines derived from a father–mother–daughter trio in the CEPH European (CEU) 1463 family that had been previously genotyped by the International HapMap Project, the authors were able to directly phase the ~1 million single-nucleotide poly morphisms (SNPs) normally measured by genotyping arrays and thereby observe recombination events in the trio.
Figure 3. Selected examples of microfluidic devices used to measure single-cell genomes and epigenomes.
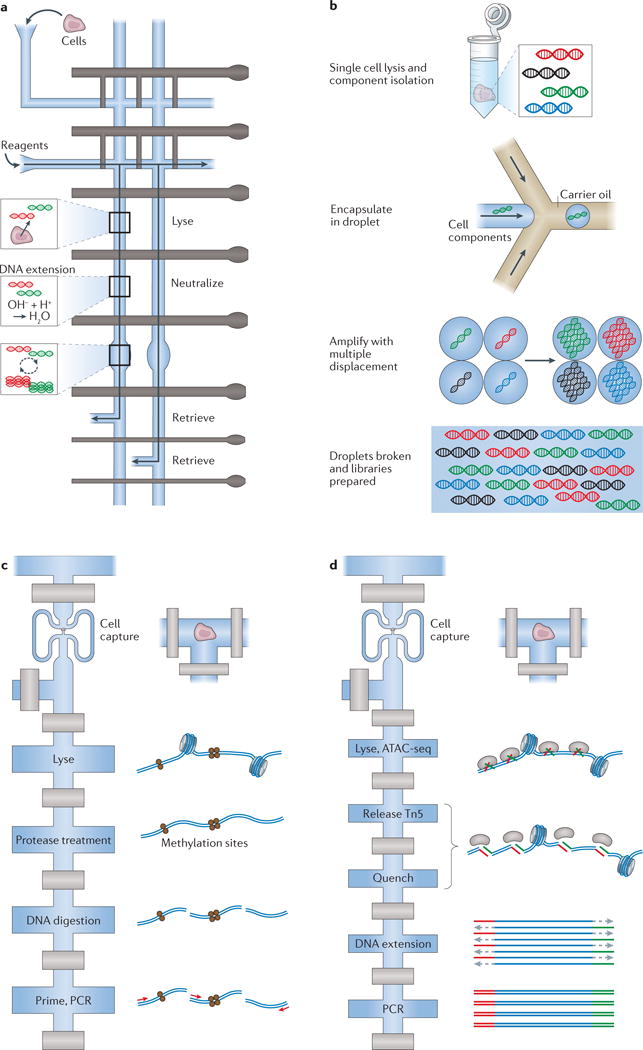
a | A diagram (top view) of the valve-based microfluidic device used by Wang et al.23 for amplification of genomic material from single cells. In the implementation, individual cells were captured and lysed, genomic DNA was then stabilized and amplified and, finally, cellular products were collected independently to ensure single-cell resolution of information. b | Emulsion multiple-displacement amplification (eMDA), as performed by Fu et al.61 (top view). Here, genomic material from single-cell lysates was encapsulated in droplets, within which MDA was performed, enabling more uniform coverage of the chromosomal profile (relative to MDA performed en masse) of the individual cell after the droplets have been broken. c | A modified diagram representing the valve-based microfluidic device implemented by Cheow et al.78 to profile the DNA methylome and transcriptome (only the methylome is shown here) of single cells (top view). d | Single-cell assay for transposase-accessible chromatin using sequencing (ATAC-seq) by Buenrostro et al.79, which uses a Tn5 transposase to identify regions of open chromatin and was performed using a commercial C1 valve-based integrated fluidic circuit from Fluidigm (top view). Part a is adapted with permission from REF. 23, Elsevier. Part c is adapted with permission from REF. 78, Macmillan Publishers. Part d is adapted with permission from REF. 79, Macmillan Publishers.
Following these initial successes, commercial valve-based solutions began to emerge for profiling single-cell genetic information. In 2014, Fluidigm released the C1 AutoPrep System, which controls an IFC that is capable of performing automated single-cell capture and processing on up to 96 single cells in parallel. Gawad and colleagues22 leveraged this platform with Phi29 polymerase-based WGA and targeted library construction to examine 1,479 single tumour cells from six people with acute lymphoblastic leukaemia (ALL) and computationally realize a deeper understanding of the sequence of genetic events that inform childhood leukaemo genesis. Other valve-based studies have collectively demonstrated the utility of this approach for performing whole or targeted genome studies in single cells53–56. Although current methods are achieving ever more efficient cover age and uniformity, further improvements can still be made to enable truly genome-wide profiling, reduce amplification errors and allow more-confident calling of copy-number variants (CNVs) and single-nucleotide variants (SNVs)26,57,58.
Droplets
Strategies for transferring single-cell genomic profiling to a droplet-based platform were introduced by the Mathies laboratory, which leveraged a microdroplet generator to flow aqueous-based PCR reagents, cells and primer-laden beads through carrier oil, achieving, with 1% efficiency, encapsulation of one cell and one bead within a droplet59. Using this approach, Kumaresan and colleagues59 generated amplified DNA libraries for single-cell Sanger sequencing of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and DNA gyrase, subunit B (gyrB) in human and Escherichia coli cells, respectively.
Since then, newer strategies have gone beyond performing amplification within droplets by enabling droplet-based pipelines for detection. In 2012, Zhu and colleagues60 devised a method to perform PCR in agarose droplets, with primers conjugated to hybridization-activated fluorescent dyes. Droplets containing cells with successful amplification of specific targets were subsequently analysed by modified fluorescence-activated cell sorting (FACS).
In parallel, droplets have been used to improve single-cell WGA quality. In this work, Fu and colleagues61 were able to generate high-quality multiple-displacement amplification (MDA) libraries through WGA of single cells using in-droplet MDA (emulsion MDA (eMDA); (FIG. 3b)). By separating the genomic fragments from a single cell into multiple droplets and then performing MDA in each one, rather than confining the entire genome of a single cell in one droplet for downstream amplification, the authors were able to saturate amplification while preventing any specific sequence from dominating the process — thus improving the overall uniformity of amplification. Additionally, Leung and colleagues62 in the Hansen laboratory presented a new technique that integrates valve- and droplet-based techniques to perform MDA in the absence of rapid flow. They controllably manipulated cells onto a substrate surface, then deposited reagent directly into the cell droplets to achieve in-drop MDA. Leung et al.62 used this approach to investigate CNVs and SNPs in single-patient-derived high-grade ovarian cancer cells. For droplets, future work will be needed to improve the overall efficiency or throughput of cell or cellular component capture, respectively.
Nanowells
Nanowells have also been used for single-cell genomic profiling. For example, Lohr et al.5 isolated single circulating tumour cells from pre-enriched peripheral blood mononuclear cells by depositing them in nanowells and using a micropipette to pick out cells that expressed epithelial cell adhesion molecule (EpCAM) on their surface. Subsequent WGA and whole-exome sequencing enabled the study of mutational patterns and lineage assignation of the different circulating tumour cells. Gole and colleagues63 performed cell lysis and WGA in a nanowell array before using a micropipette to extract amplicons from individual wells. By fluorescently monitoring DNA amplification with SYBR Green I and selecting wells in which fluorescence arose from single, uncontaminated cells, they analysed CNVs in singe nuclei from human neurons isolated from post-mortem brain samples. Importantly, although nanowells, like droplets, generally have higher throughput than valve-based approaches, in each instance only a small set of cells has been selected for downstream profiling; despite this, nanowells have helped to identify cells of interest for plate-based processing or successful on-chip reactions, respectively. As sequencer bandwidth increases, future work will be needed to automate and scale current manual-selection strategies.
Epigenome
Heterogeneous genetic profiles have been used to identify lineal relationships64,65 and distinct clones in seemingly homogeneous populations66,67. At times, genetic differences alone may be sufficient to effectively categorize cells (for example, drug-responsive versus unresponsive cancer cell clones68,69 or which T cells will be antigen specific70); in other instances (for example, distinguishing which isogenetic dendritic cells will respond in a particular way to pathogen exposure3), it may be the accessibility or modifications of the genome that are relevant71,72. Accordingly, there has also been substantial interest in understanding how genomic structure informs cellular phenotype, as well as how it is established and maintained. At the single-cell level, differences in gross DNA folding73, DNA methylation74–78, chromatin accessibility79 and histone modifications80 have all been measured, but only the latter three have been implemented using microfluidic devices81.
In each implementation, unlike with DNA, only a limited number of reads has been required to examine each single cell (<200,000 (REFS 75,79,80)), given biological and technical considerations. As a result, each method is amenable to additional scaling (TABLE 1), which is likely to inspire efforts to move single-cell epigenome characterization from valve-based microfluidic devices to droplet- and nanowell-based ones.
Valves
Lorthongpanich et al.75 used the Fluidigm Biomark system to investigate DNA methylation patterns in single cells from chimeric murine models and discovered epigenetic shifts during early embryonic development. Cheow et al.78 ported a variant of this technique to the Fluidigm C1 AutoPrep IFC22 to investigate the relationship between transcriptome and DNA methylome profiles in single fibroblast cells. Through analysis of these data, they discovered a distinct methylation structure that was associated with pluripotency and was present in a subpopulation of reprogramming fibroblasts (FIG. 3c). Buenrostro et al.79 also leveraged the C1 AutoPrep IFC to perform a single-cell assay for transposase-accessible chromatin using sequencing (ATAC-seq), a recently developed method that relies on Tn5 transposase to identify accessible chromatin regions (FIG. 3d). By profiling eight different cell lines to an average depth of ~75,000 reads, the authors linked cis- and trans-effectors to variability in accessibility profiles in individual epigenomes. For both methods, future work will be needed to improve DNA fragment recovery and increase throughput to overcome the effects of zero inflation, as well as to reduce mitochondrial DNA contamination.
Droplets
Rotem et al.80 used a droplet-based micro-fluidic strategy to execute single-cell chromatin profiling. They encapsulated single cells in droplets, wherein they lysed, and digested chromatin with micrococcal nuclease (MNase). By fusing a second droplet containing barcoded oligonucleotides and performing a ligation reaction, they achieved in-droplet barcoding, uniquely indexing each cell’s fragments. Afterwards, they broke the droplets, pooled the barcoded cellular analytes (with carrier chromatin) and carried out chromatin immunoprecipitation followed by sequencing (ChIP-seq) for histone H3 lysine 4 dimethylation (H3K4me2) and H3K4me3 in the bulk sample. Last, they took advantage of the barcode region of the sequencing reads to deconvolute the cell of origin for each individual read to restore single-cell resolution. By applying this approach to mouse embryonic stem cells (expanded in serum), the authors identified three subpopulations with distinct epigenetic landscapes. Here, improvements in efficiency are needed given the sparsity of the data in this measurement78.
Transcriptome
Arguably the most common molecular variable used to examine cellular phenotype at the genomic scale is mRNA. The presence of polyadenylated tails on mRNAs allows universal priming using a single oligo(dT)-based scheme while avoiding excess ribosomal RNA contamination. This enables transcriptome-wide mRNA profiling and the identification of distinct cell types, states and circuits through patterns in gene-expression covariation1,2,4,13,31,82–86.
In most instances, single-cell transcriptional profiling is performed by first isolating and lysing individual cells of interest, reverse transcribing their mRNA into cDNA and amplifying that material. Afterwards, amplified cDNA, obtained either by specific target amplification (STA) or by whole-transcriptome amplification (WTA), can be profiled using established methods, such as qPCR, dPCR or sequencing. One of the first demonstrations of single-cell transcriptomics using microfluidics was performed by Warren and colleagues44: following FACS-based sorting of single haematopoietic stem cells and fetal liver kinase 2 (FLK2; also known as FLT3)+ and FLK2− common myeloid progenitor cells into strip-tubes for reverse transcription PCR (RT-PCR), they used a custom valve-based IFC to array each single-cell cDNA sample and perform on-chip quantification of Pu.1 (also known as Spi1) and Gapdh levels by dPCR.
qPCR and dPCR work has continued to find niche applications, but sequencing now dominates. Importantly, foundational work31,32,87–89 has helped to demonstrate that tens to hundreds of thousands of sequencing reads are sufficient for gene expression analyses in end-tagging and full-length protocols, respectively. Thus, whereas once the field was dominated by protocols involving the sorting of tens to hundreds of single cells into multiwell plates using FACS1,2,4,90, scientists have now, in part, begun to embrace valve-based microfluidics for profiling hundreds of cells, and droplet- and nanowell-based approaches for studying thousands of cells (TABLE 1). For all approaches, further work will be needed to realize methods for studying non-polyadenylated RNAs, such as small RNAs84,86,91.
Valves
As mentioned above, valve-based methods initially focused on processing and profiling single-cell transcriptional information. For example, White and co-workers43 developed a valve-based IFC for single-cell reverse transcription qPCR (RT-qPCR) that enabled up to two transcripts to be measured on-chip for up to 50 single cells from each of six independent sample loading lanes. Here, the number of genes that could be simultaneously assayed was limited owing to the decision to detect on-chip, although off-chip detection using other microfluidic chips24 or sequencing4 is possible. The C1 AutoPrep IFC, meanwhile, has transcriptome-compatible protocols that facilitate an order of magnitude improvement in throughput over plate-based methods with an order of magnitude reduction in cost. Leveraging the scale afforded by this platform, researchers have been able to, for example, uncover rare immune cell states3, survey neuronal diversity85 and assess cellular hierarchy within lung epithelia92.
However, further scaling of this and related systems, which will be necessary to study increasingly complex cellular ensembles, has been hindered by the reliance on separate microfluidic channels to deliver processing reagents to each single cell in parallel. Moreover, for single-cell RNA sequencing (scRNA-Seq), multiwell-plate-based methods are still superior with respect to transcript capture93. For instances in which emphasis is placed on efficiency, defined as the percentage of input mRNAs recovered, plate-based methods are preferred. Still, in many instances, more cells, even if captured with lower fidelity, are preferred because, collectively, they minimize the impact of the technical and biological noise associated with any single cell or measurement on global analyses. Among the microfluidic strategies, valve-based systems still provide the greatest molecular efficiency and are useful for applications in which the highest-quality transcriptomes are needed at moderate scale94; newer methods that enable on-chip pooling should help to decrease the throughput gap between valve-based and droplet- or well-based approaches.
Droplets
To address the fundamental shortcomings of scale presented by valve-based microfluidic systems and plate-based methods, several platforms have emerged that leverage the power of early cellular barcoding to achieve high-throughput single-cell transcriptomics. By tagging the mRNAs from each cell with a unique barcode during reverse transcription, these methods simplify library preparation by enabling ensemble processing with single-cell resolution through barcode-based computational deconvolution. To perform early barcoding, microfluidic devices have been used to capture single cells in droplets with uniquely barcoded mRNA capture beads31,32. Illustratively, in one version, Macosko et al.31 presented a method called Drop-Seq that uses barcoded acrylic mRNA capture beads to achieve bead-bound cDNA replicas of the individual cells with which they are co-encapsulated; this allows user control over the number of cells sequenced and archiving for subsequent requerying. Using a mixture of human and mouse cell lines (species mixing experiments), the authors demonstrated the feasibility of Drop-Seq, obtaining >95% cell-of-origin specificity, >7,000 recovered genes per cell and >12% RNA capture. By collecting 44,808 single-cell profiles from the mouse retina over 4 days of experiments, they uncovered, with high reproducibility, ~39 subtypes and their molecular markers, and subsequently validated select observations in situ. However, to avoid cell or bead doublets, Drop-Seq requires that both cells and beads be loaded at low densities into the co-flow device used to confine cells, lyse them and capture cellular mRNAs by hybridization, before breaking the emulsion. As a result, only a small fraction of the cells encapsulated are effectively used.
A second technique developed by Klein et al.32 called InDrop overcomes this inefficiency through the use of hydrogel capture particles that can be more efficiently loaded, filling about 90% of the drops (FIG. 4a). As a result, most encapsulated cells are efficiently used. In InDrop, barcodes are released after cell lysis, and reverse transcription occurs in-droplet before the breaking of the emulsion. Species mixing validations showed 96% cell-of-origin specificity, 5,000–15,000 recovered genes per cell and >7% RNA capture. Klein and co-workers32 then used this method to probe population structure and cellular heterogeneity in 11,149 mouse embryonic stem cells during their differentiation in response to leukaemia inhibitory factor (LIF) withdrawal. Nevertheless, the InDrop workflow necessitates that all captured cells be processed in a single reaction, eliminating the ability to tune sequencing library complexity and complicating the re-sampling of selected cells. Additionally, both Drop-Seq and InDrop can only accommodate one sample at a time, and process and lyse cells in series, rather than in parallel, leading to time-dependent biases that can obfuscate underlying biology. Some of these limitations have been overcome by a more user-controllable device introduced by 1-Cell, which is commercializing the InDrop method, and 10× Genomics, which has brought a hybrid of the Drop-Seq and InDrop methods to market95. Future work will be needed to improve cell and transcript capture efficiency and the uniformity of the final library so that each cell gets similar coverage.
Figure 4. Selected examples of microfluidic devices used to measure single-cell transcriptomes and proteomes.
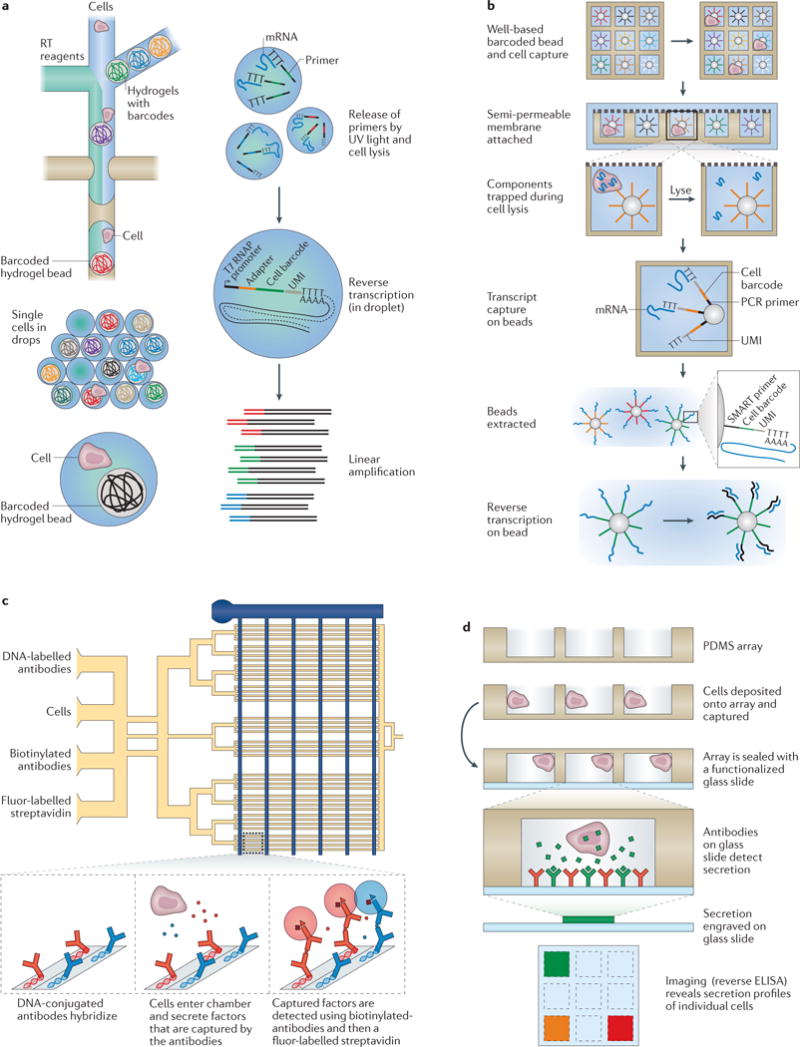
a | InDrop, developed by Klein et al.32, is a droplet-based single-cell transcriptomics method that works by co-confining single cells with hydrogel beads containing uniquely barcoded primers (top view). On UV-light-mediated release of those primers, reverse transcription (RT) and barcoding is performed in droplet, and the resulting complementary DNA (cDNA) is collected after the droplets have been broken for subsequent processing. b | Seq-Well, a massively parallel single-cell RNA sequencing (scRNA-Seq) method from Gierahn et al.96, combines early bead-based barcoding with nanowells to generate thousands of single-cell libraries (the top row shows a top view; below this are side views). Single cells are gravity-loaded onto an array that has been preloaded with uniquely barcoded oligo(dT) capture beads, sealed with a membrane (which permits buffer exchange but not mRNA escape) and lysed; mRNAs are then captured by oligo(dT) primers bound to the surface of the barcoded beads, and the beads are removed for off-chip RT, amplification, library preparation and sequencing. c | The single-cell barcode chip (SCBC) developed by Ma et al.102 uses a valve-based strategy to isolate single cells and interrogate their secreted factors (top view). In each enclosed chamber, individual cells are exposed to antibodies that are specific for various extracellular protein targets. Following incubation of cells in the chip, streptavidin-bound fluorophores and biotinylated secondary antibodies are flowed into the channels, enabling a sandwich enzyme-linked immunosorbent assay (ELISA)-based fluorescent readout of protein abundance. d | Nanowells were coupled with capture-antibody-coated glass coverslips by Love et al.33 to measure the abundance of several secreted factors from single cells, such as active hybridomas in a reverse ELISA format (side view). PDMS, polydimethylsiloxane; T7 RNAP, T7 RNA polymerase; UMI, unique molecular identifier. Part a is adapted with permission from REF. 32, Elsevier. Part b is adapted with permission from REF. 96, Macmillan Publishers. Part c is adapted with permission from REF. 102, Macmillan Publishers. Part d is adapted with permission from REF. 33, Macmillan Publishers.
Nanowells
Arrays of nanowells can also be used to mate single cells and barcoded beads. Wells rely on gravity-assisted cell and bead loading, reducing the need for peripherals. By matching bead and well size, more efficient bead loading can be achieved, which minimizes the frequency of cells without beads and improves sampling efficiency. In a method dubbed CytoSeq, Fan et al.14 demonstrated a system for co-confining cells and beads in unsealed nanowell arrays, enabling targeted transcriptional profiling from approximately 5,000 human peripheral blood mononuclear cells. However, the use of an open well design considerably limited capture efficiency and increased cross-contamination between individual libraries96.
DeKosky et al.97 used a sealed well-based device to isolate and capture mRNA from single B cells with pools of magnetic poly(dT) beads. After bead removal, the heavy and light chain sequences on each bead were linked using emulsion PCR and sequenced to determine the B cell receptor (BCR) pairs found across individual cells. To enable unique determination of the full transcriptomes of single cells, Bose et al.98 used a single, uniquely barcoded bead per well, as in CytoSeq, Drop-Seq and InDrop14,31,32,76. Unlike in CytoSeq, here the authors integrated their nanowell array into a microfluidic circuit to enable oil-based well sealing, allowing them to profile hundreds of cells from a cancer cell line with reduced cross-contamination. A more recent improvement by Yuan and Sims99 further increased cell and transcript capture efficiency. Still, in both implementations, operation required integrated temperature and pressure controllers, and the use of oil-based sealing limited the exchange of reagents. To circumvent the need for oil-based sealing, Gierahn et al.96 (FIG. 4b) demonstrated improved transcript capture and portability using a simple-to-implement, semi-permeable-membrane-based nanowell sealing approach (Seq-Well) that envisions global application of scRNA-Seq to precious samples. In each instance, further improvements can be made in transcript capture. Similarly, in instances in which multiple measurements have been made on the same cell (for example, immunophenotyping and transcriptional profiling in Seq-Well96), experimental strategies are needed to link these mutually informative metrics.
Proteome
Although a single affinity reagent can be used to profile all cellular mRNAs, any given transcript may have limited utility for characterizing the phenotype of a cell, given noise4,15. By comparison, proteins, for which consistent and universal probes (typically antibodies) are unavailable, show, on average, higher expression levels than their cognate mRNAs100, greater stability56 and buffering from transcriptional noise57. These considerations, coupled with a potentially more direct role in cellular behaviour27 and the feasibility of non-destructive detection (for secreted and cell-surface-bound factors), have often made protein expression the de facto cellular characteristic. Accordingly, a substantial body of work has already established how microfluidic devices can be used to measure protein abundance and activity.
Valves
Building on the DNA-encoded antibody library (DEAL) approach originally developed in the Heath laboratory for profiling the protein contents of solutions101, Ma et al.102 implemented a single-cell barcode chip (SCBC) to examine functional heterogeneity in the expression of 12 different proteins among immune cells (FIG. 4c). Using microfluidic channels, the authors patterned strips of capture antibodies (spatial encoding) on a glass slide, which they then capped with orthogonal microfluidic channels that could be partitioned using active valves. By flowing cells into the channels, isolating them by activating valves, capturing secreted protein with the DEAL antibodies, releasing the cells, staining the arrays in a sandwich ELISA format and, finally, imaging, the authors were able to examine inflammatory cytokine secretion by lipopolysaccharide (LPS)-stimulated macrophages and effector molecule secretion by tumour-antigen-specific cytotoxic T lymphocytes. This strategy was recently extended by Lu et al.103, who used spatial and spectral encoding to co-detect 42 immune effector proteins secreted from single differentiated macrophages stimulated with LPS. Wang et al.104 used a similar approach with higher numbers of cells loaded to examine the distance dependence of cell–cell interactions in glioma cell lines. Shi et al.38 also applied a similar design with the addition of a channel for delivering lysis buffer to examine the activity of, and interactions among, 11 proteins in the phosphoinositide 3-kinase (PI3K) signal transduction pathway in three isogenic glioblastoma cell lines.
Huang et al.105 developed a valve-based microfluidic device for manipulating, lysing, labelling, separating and quantifying the protein contents of a single cell using single-molecule fluorescence counting. Their design relies on a three-state valve to restrain a cell while adding cell lysis buffer. After lysing single SF9 insect cells, Huang et al.105 added fluorescently labelled antibodies targeted to β2 adrenergic receptors, removed unbound antibodies using electrophoresis and used cylindrical optics to count β2 adrenergic receptors on the basis of fluorescence. They performed a similar analysis to examine the phycobiliprotein (phycocyanin (PC) and allophycocyanin (APC)) contents of individual cyanobacterial cells (Synechococcus sp. PCC 7942) by measuring the natural fluorescence of PC and APC instead of labelled antibodies.
In another approach, Blazek and colleagues106 used proximity ligation assays (PLAs)107 to measure the abundance of a single protein in a few cells. Unlike DEAL, PLA relies on the binding of two oligonucleotide-tagged antibodies in proximity to generate, with a helper bridge oligonucleotide, a DNA reporter with low background noise108. This reporter is stable, amplifiable and quantitatively detectable by dPCR, qPCR or sequencing. The complexity of PLAs has been reduced by the introduction of the PEA109, which relies on pairs of monoclonal or polyclonal antibodies functionalized with either of two complementary DNA oligonucleotides that can hybridize and be extended with a DNA polymerase to generate a protein-indexed DNA molecule when colocalized by binding to their target protein. Genshaft et al.15 demonstrated that PEA could be coupled with STA transcriptome profiling to measure the abundance of 27 proteins and 89 RNAs from more than 200 stimulated single cells in a single series of reactions. Although single DNA-tagged antibodies have been used to measure protein abundance (for example, by light-mediated cellular barcoding50,110), they have yet to be deployed in an IFC. Next steps include improving cellular throughput and expanding the number of antibodies that can be simultaneously quantitatively distinguished at the single-cell level.
Droplets
Albayrak et al.111 recently used PLA coupled with reverse transcription–digital droplet PCR (RT-ddPCR) in reverse-emulsion droplets to quantify the levels of a single protein and mRNA in a few hundred individual cells. Here, by fractionating the contents of a single cell into multiple small droplets and digitally detecting amplification, the authors obtained high sensitivity, overcoming issues with amplification efficiency that affect qPCR measurements, and examined correlations between RNA and protein expression for CD147. Future improvements will be needed to scale the number of cells and targets in this approach, given the reliance on multiple droplets per cell and digital detection, respectively.
Nanowells
In the context of nanowells, Hughes and co-workers112 used an array of 6,720 wells (with a diameter of 20 μm) to perform single-cell western blotting. By constructing the array out of photoactive polyacrylamide, they photo-captured proteins in a gel matrix after open well lysis of single cells (resulting in protein losses of ~40% within compartments) and performed short-separation-distance PAGE. Subsequent rounds of probing with primary and secondary antibodies followed by stripping enabled them to examine the abundance of nine different targets per cell. Importantly, the use of electrophoresis to separate proteins by size before staining reduced the impact of false-positive off-target background binding in this method.
Love et al.33 capped arrays of nanowells containing single hybridoma cells with antibody- or antigen-functionalized glass slides to identify single cells secreting antibodies with specificity for a particular antigen (FIG. 4d); in their approach, the slide was removed from the nanowell array for imaging, and the positions of the positive secretion profiles on the slide reported which nanowells contained live cells with intended antibody specificities for subsequent cell recovery and clonal expansion. By coating the capping slide with cells that could be targeted by a pseudotyped HIV virus, Ozkumur et al.113 were able to test not only the antigen specificity of antibodies secreted by single cells in each well but also their ability to neutralize infection. Han and colleagues114 extended this approach to monitor the kinetic expression of four cytokines over 17 hours from thousands of activated T cells, demonstrating that cytokine release kinetics can predict T cell effector phenotype. Similar work by Liao et al.115 in human neurons and astrocytes derived from induced pluripotent stem cells isolated from healthy donors and people with familial Alzheimer disease enabled quantification of secreted amyloid-β and soluble amyloid precursor protein-α, demonstrating unexpected secretion patterns across single cells. In each instance, further improvements in detection efficiency, target number and throughput would extend the utility of these approaches.
Extensions
Much of the focus of this Review has been on mammalian cells. However, single-cell omics methods are by no means limited to mammalian cells, and there are several important reasons to explore other cell types. Bacteria can be probed using single-cell methods116, which may be necessary if the bacterial isolate is difficult to culture and extremely diverse117. Basic approaches for profiling bacterial DNA are similar to those used in mammalian cells. Nevertheless, given that the average bacterial genome is around a thousand times smaller (approximately a few megabases) than the human genome (approximately a few gigabases), many more bacteria (thousands) can be sequenced on a single sequencing run, necessitating high-throughput preparation strategies. With other measurements, differences between bacteria and mammalian cells similarly need to be considered. For example, most bacterial mRNA is not polyadenylated118, which means that random priming strategies are needed during reverse transcription; thus, ribosomal RNA and its elimination, which is normally achieved in mammalian cells through oligo(dT)-based priming, becomes a major problem. In addition, a key challenge with bacteria is cell lysis, which can be extremely difficult for several types of bacteria and can limit protocol applicability119–121.
Viral load has been quantified using microfluidic devices122, but it should also be feasible to probe even very small biological entities such as single virions. This should make metagenomics studies of viruses more reliable because the sequence information obtained will be directly associated with each individual virus, and will not rely solely on bioinformatic assumptions. It is also possible to co-confine a cell with either a pathogen or another cell in a microfluidic device, making it practicable to investigate interactions between and within species at the single-cell level, such as cooperativity in killing of target cells by natural killer cells123 (FIG. 5a).
Figure 5. Future directions and extensions for microfluidic devices.
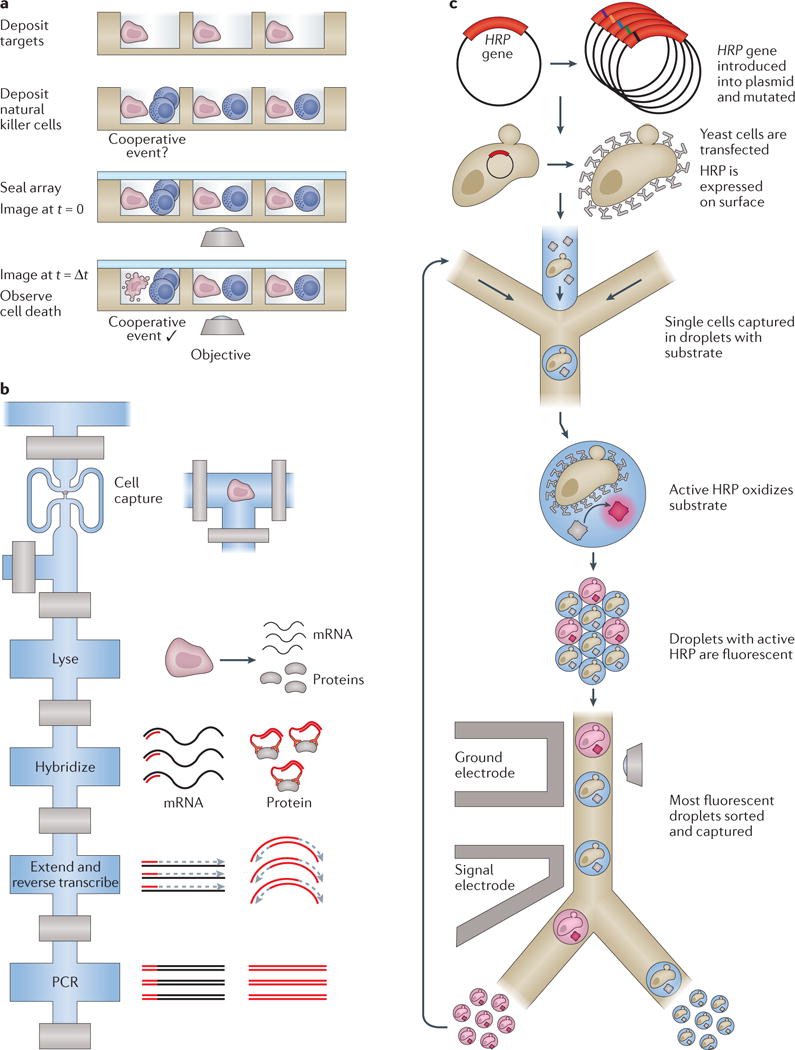
a | Multiple cells can be deposited onto well-based profiling platforms, enabling the examination of multicellular and cooperative events, such as the killing of target cells by natural killer cells by Yamanaka et al.123 (side view). b | The Fluidigm C1 valve-based integrated fluidic circuit (IFC; top view) was used to simultaneously detect several transcripts and proteins from the same single cell in a single series of reactions15. During lysis, targeted primers and complementary DNA-tagged antibodies were introduced, and reverse transcription and DNA extension reactions were carried out on both species to yield uniquely detectable, PCR-amplifiable DNAs for both cellular analytes. c | The directed evolution of enzymes in single cells can be probed with an activity assay in droplets136. Here, horseradish peroxidase (HRP) surface proteins from mutated vectors were interrogated for substrate turnover with single-cell resolution (top view). Following capture of the most active cells, the interrogation was repeated to study their evolution over time. Part a is adapted from REF. 123 with permission of The Royal Society of Chemistry. Part b is adapted with permission from REF. 15, Macmillan Publishers Limited. Part c is adapted with permission from REF. 136 (Agresti, J. J. et al. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc. Natl Acad. Sci. USA 107, 4004–4009; 2010).
In addition to enabling studies of host–pathogen and host–host interactions, microfluidic devices are now beginning to empower multi-omic studies that examine the interrelationships between distinct classes of cellular components. Illustratively, recent efforts have begun to characterize how RNA expression patterns correlate with and are driven by the levels and activity of various protein species10,15,124–126 (FIG. 5b). Whereas early studies featured fluorescent- or isotope-labelling strategies for proteomic readout, including FACS and mass cytometry6, a more recent approach leverages PEA-based protein detection, which can be coupled with STA using the valve-based design of the C1 AutoPrep IFC15. Similar methodologies, focused on simultaneously profiling mutually informative classes of variables, such as DNA and RNA or RNA and the epigenome78, seem to be just around the corner (BOX 1), and may soon guide our understanding of cellular function. Importantly, many other informative properties of cells have been measured with microfluidic devices — for example, morphology127, proliferation dynamics128, motility129, invasiveness35, interactions123, calcium dynamics130 and familial relationships131 — all of which may help to determine cellular identity in the future.
Box 1. Considerations for porting single-cell ‘omic’ assays into microdevices.
Before attempting to develop a new microfluidic device or port an established assay to an existing one, it is important to consider the relative utility of miniaturization and integration in the context of the problem at hand. Indeed, each cellular analyte poses unique challenges with respect to detection and readout (TABLE 1); similarly, each assay has nuances that inform whether and how it should be adapted.
First, microfluidic devices can improve assay implementations by increasing their scale. In some instances, such as constructing cellular atlases with single-cell RNA sequencing (scRNA-Seq)1,142, a researcher may need to profile thousands of cells to build as comprehensive a catalogue as sequencer bandwidth allows. In such cases, considerations of scale may surpass others, such as capture efficiency, especially given the limited resolving power of any single cell in light of technical and intrinsic noise sources4,88,143. Alternatively, for whole-genome or whole-exome sequencing, a sequencing run may only support full coverage of a few cells61.
In addition to increasing scale, microdevices can benefit biological assays in other ways, such as by increasing control and reducing background noise. For example, in the context of whole-genome sequencing, valve-based microdevices can be used to isolate individual chromosomes from a single cell, enabling direct analysis of haplotypes, which would be difficult using a multiwell-plate-based approach20. Similarly, for assays that have a constant background noise per unit volume due to non-specific hybridization, such as the proximity extension assay (PEA)15,16, miniaturization can reduce the noise floor by increasing the relative frequency of positive detection events to spurious ones. Thus, even when scale is not the driving factor, microfluidic devices may have utility.
Ultimately, when deciding when and how to deploy microfluidic devices, researchers must carefully weigh the pertinent costs and benefits. At present, valve-, droplet- and well-based devices each offer unique advantages, making them preferentially suited for different biological tasks. Valve-based devices enable exquisite control over cells and their components, and are often most appropriate when careful manipulation of analytes is imperative, such as in PEAs and direct deterministic phasing15,16,20. Droplet-based methods do not engender the same degree of precision but facilitate dramatic scaling, making them most appropriate when attempting to profile thousands of cells31,32,95,142. Well-based platforms, meanwhile, offer simplicity at an intermediate scale with defined spatial locations that ease the coupling of multiple discrete readouts from the same cell96.
Collectively, these and related considerations will influence how existing assays are ported and the choice of device for future efforts. Although it is hard to fully envision what the future holds, we predict that future applications will include new single-cell measurements (such as chromatin three-dimensional organization through Hi-C chromosome conformation capture (probably using valves))73,144, biophysical parameters such as cell mass132,133,135,145 (which requires tracking but not isolation), lineage determination131 (using hydrodynamic traps), integrated multi-‘omic’ profiling (probably most easily implemented using valve-based15 or well-based96 devices) and studies of how microenvironmental considerations (soluble factors and other cells)146,147 affect cellular behaviour.
Emerging applications
The precision of microfluidic devices holds promise for simultaneously profiling multiple analytes across thousands of single cells. New strategies for improved multi-omics are probably on the horizon, as are approaches for collecting additional relevant cellular metadata, such as mass132–135. Additionally, high-throughput methods, such as droplets and nanowells, provide a powerful means of probing and selecting single cells from large ensembles. For example, using droplets, it is possible to directly identify antibody-producing B cells without the need to first immortalize them through the production of hybridomas33. This enables a much faster and deeper search of the antibodies produced by an immunized animal. Similarly, these methods make it possible to screen enzymatic activity and search large libraries of enzymes, either produced in a cell-free manner or expressed in cells, to perform directed evolution for iteratively exploring a much larger library size136 (FIG. 5c). Relatedly, microfluidic devices that couple perturbations and omic readouts in many single cells have begun to fundamentally transform our studies of cellular phenotype and function by allowing us to uncover how different cellular components affect cellular behaviours82,137–139. Future work, both experimental and computational, will be required to integrate these profiling and screening approaches to fully leverage the comprehensive characterizations of cellular phenotype they afford.
Conclusion
In this Review, we have discussed how microfluidic devices have helped to transform single-cell omics from a specialized and underused approach to one that is now increasingly mainstream. Whether they use valves, droplets or wells, microfluidic tools can substantially increase the throughput and efficiency of several single-cell processing and profiling methods, while reducing financial and human costs. Still, before diving in, it is important to first weigh the costs and benefits of porting any single-cell assay into a microfluidic device (BOX 1). As these advances continue to mature, there are likely to be substantial improvements in the efficiency of processing both cells and cellular analytes. An important, linked question will be how best to balance the number of cells profiled and the depth to which each is sampled, given a fixed number of reads per sequencing run. Further progress will result in overcoming the technical obstacles associated with preparing different classes of single-cell omics. This will lead to a deeper understanding of how the identity of a cell is influenced by its components and their interactions, and the cellular microenvironment, as well as questions of how to balance the number of reads afforded to each different analyte. In parallel, improved computational algorithms, enhanced statistical frameworks and advanced visualizations will be needed to truly maximize the insights that single-cell omic profiling strategies promise to afford and guide the development of future devices6,13,140,141. Overall, the coming decade should be an extremely exciting time for single-cell biology and the interdisciplinary science that it enables.
Acknowledgments
The authors thank J. C. Love, A. S Genshaft, K. E. Kolb, C. G. K. Ziegler and S. L. Carroll for helpful comments and suggestions. This work was supported by the Searle Scholars Program (A.K.S.), the Beckman Young Investigator Program (A.K.S.), a US National Institutes of Health (NIH) New Innovator Award DP2OD020839 (A.K.S.), NIH grants U24 AI118672 (A.K.S.), P50HG006193 (A.K.S.), P01GM096971 (D.A.W.), P01HL120839 (D.A.W.) and R01EB014703 (D.A.W.), US National Science Foundation (NSF) Materials Research Science and Engineering Center grant DMR-1420570 (D.A.W.) and NSF grant DMR-1310266 (D.A.W.).
Glossary
- Spatial encoding
Stratifying analytes (cells or multiple cellular products) by physical confinement on a microfluidic chip.
- Spectral encoding
Stratifying analytes (multiple cellular products) using different colours of fluorescence.
- Temporal encoding
Stratifying analytes (cells or multiple cellular products) by measuring them sequentially.
- Exome sequencing
Selective amplification and sequencing of the protein-coding regions of the genome using exon-specific priming.
- Direct deterministic phasing (DDP)
The chromosomes of individual cells are partitioned, isolated and amplified using multiple displacement amplification. The products are then flushed and analysed by molecular haplotyping
- Transposase
An enzyme that catalyses the movement of a transposable DNA element into another DNA sequence (for example, a genome) by a cut and paste mechanism. A hyperactive variant of the Tn5 transposase is now commonly used to insert adaptor sequences for next-generation sequencing library preparation. If performed on native chromosomes, the transposase can only bind to exposed DNA, revealing accessible DNA regions
- Zero inflation
Owing to inefficiencies in detection, the distribution of counts for several detected genes can be artificially inflated by the abundance of ‘zeros’ (detection failures) during normalization
- Micrococcal nuclease (MNase)
An endo-exo nuclease derived from Staphylococcus aureus. When applied to single-stranded and double-stranded DNA, MNase will digest all accessible DNA, enabling the study of which DNA regions are occluded with chromatin
- Mass cytometry
Cells are tagged with antibodies that have been labelled with rare earth metal isotopes, nebulized and passed through a quadrapole mass spectrometer for detection. The abundance of a rare earth metal can thus be used as a proxy for the level of the protein in the single cell. Importantly, whereas the number of proteins that can be detected at once with fluorescent labels is limited owing to overlap between emission spectra, mass cytometry can detect tens of proteins in parallel because individual isotopes of rare earth metals have well-defined, non-overlapping masses
- Directed evolution
A technique in which a series of genetic variants is introduced into a cell population, which are then screened with a fitness assay to isolate a desired phenotype. The genetic variants responsible for the greatest fitness are identified by sequencing the surviving cell population for the target gene
Footnotes
Competing interests statement
The authors declare competing interests: see Web version for details.
References
- 1.Tirosh I, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patel AP, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shalek AK, et al. Single-cell RNA-seq reveals dynamic paracrine control of cellular variation. Nature. 2014;510:363–369. doi: 10.1038/nature13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shalek AK, et al. Single-cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature. 2013;498:236–240. doi: 10.1038/nature12172. A demonstration of how covariation in gene expression in scRNA-Seq data can be used to identify cellular circuits and molecular drivers of behaviour in seemingly identical immune cells. It also nicely demonstrates the use of microfluidic devices for targeted single-cell mRNA measurements (STA). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lohr JG, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol. 2014;32:479–484. doi: 10.1038/nbt.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bendall SC, et al. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen AA, et al. Dynamic proteomics of individual cancer cells in response to a drug. Science. 2008;322:1511–1516. doi: 10.1126/science.1160165. [DOI] [PubMed] [Google Scholar]
- 8.Feinerman O, et al. Single-cell quantification of IL-2 response by effector and regulatory T cells reveals critical plasticity in immune response. Mol Syst Biol. 2010;6:437. doi: 10.1038/msb.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wurtzel O, et al. A generic and cell-type-specific wound response precedes regeneration in planarians. Dev Cell. 2015;35:632–645. doi: 10.1016/j.devcel.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bjorklund AK, et al. The heterogeneity of human CD127+ innate lymphoid cells revealed by single-cell RNA sequencing. Nat Immunol. 2016;17:451–460. doi: 10.1038/ni.3368. [DOI] [PubMed] [Google Scholar]
- 11.Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods. 2008;5:877–879. doi: 10.1038/nmeth.1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. A fundamental work that describes how to think about the two non-technical sources (intrinsic and extrinsic) of noise that influence single-cell measurements and their interpretation. [DOI] [PubMed] [Google Scholar]
- 13.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33:495–502. doi: 10.1038/nbt.3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fan HC, Fu GK, Fodor SP. Expression profiling. Combinatorial labeling of single cells for gene expression cytometry. Science. 2015;347:1258367. doi: 10.1126/science.1258367. [DOI] [PubMed] [Google Scholar]
- 15.Genshaft AS, et al. Multiplexed, targeted profiling of single-cell proteomes and transcriptomes in a single reaction. Genome Biol. 2016;17:188. doi: 10.1186/s13059-016-1045-6. These authors used commercial valve-based microfluidics to perform multiplexed single-cell transcriptome and protein profiling from the same cell, an important demonstration of simultaneous single-cell multi-omic profiling. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Darmanis S, et al. Simultaneous multiplexed measurement of RNA and proteins in single cells. Cell Rep. 2016;14:380–389. doi: 10.1016/j.celrep.2015.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramirez L, Herschkowitz JI, Wang J. Stand-sit microchip for high-throughput, multiplexed analysis of single cancer cells. Sci Rep. 2016;6:32505. doi: 10.1038/srep32505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varadarajan N, et al. Rapid, efficient functional characterization and recovery of HIV-specific human CD8+ T cells using microengraving. Proc Natl Acad Sci USA. 2012;109:3885–3890. doi: 10.1073/pnas.1111205109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Whitesides GM. The origins and the future of microfluidics. Nature. 2006;442:368–373. doi: 10.1038/nature05058. [DOI] [PubMed] [Google Scholar]
- 20.Fan HC, Wang J, Potanina A, Quake SR. Whole-genome molecular haplotyping of single cells. Nat Biotechnol. 2011;29:51–57. doi: 10.1038/nbt.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hong JW, Quake SR. Integrated nanoliter systems. Nat Biotechnol. 2003;21:1179–1183. doi: 10.1038/nbt871. A review on the mechanics, operation and utility of valve-based microfluidic platforms, with a detailed discussion of applications in many fields, including biology. [DOI] [PubMed] [Google Scholar]
- 22.Gawad C, Koh W, Quake SR. Dissecting the clonal origins of childhood acute lymphoblastic leukemia by single-cell genomics. Proc Natl Acad Sci USA. 2014;111:17947–17952. doi: 10.1073/pnas.1420822111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Fan HC, Behr B, Quake SR. Genome-wide single-cell analysis of recombination activity and de novo mutation rates in human sperm. Cell. 2012;150:402–412. doi: 10.1016/j.cell.2012.06.030. An early demonstration of single-cell genome sequencing applied to microfluidic devices, in which de novo mutations in human sperm were identified and quantified with a custom valve-based device. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dalerba P, et al. Single-cell dissection of transcriptional heterogeneity in human colon tumors. Nat Biotechnol. 2011;29:1120–1127. doi: 10.1038/nbt.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gaublomme JT, et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell. 2015;163:1400–1412. doi: 10.1016/j.cell.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17:175–188. doi: 10.1038/nrg.2015.16. A review of the current state of the art in single-cell genomics, including key insights into the limitations of handling genetic material from a single cell and important work in the field. [DOI] [PubMed] [Google Scholar]
- 27.Chattopadhyay PK, Gierahn TM, Roederer M, Love JC. Single-cell technologies for monitoring immune systems. Nat Immunol. 2014;15:128–135. doi: 10.1038/ni.2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zeng Y, Novak R, Shuga J, Smith MT, Mathies RA. High-performance single cell genetic analysis using microfluidic emulsion generator arrays. Anal Chem. 2010;82:3183–3190. doi: 10.1021/ac902683t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo MT, Rotem A, Heyman JA, Weitz DA. Droplet microfluidics for high-throughput biological assays. Lab Chip. 2012;12:2146–2155. doi: 10.1039/c2lc21147e. [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Jenkins G, Zou Y, Zhu Z, Yang CJ. Massively parallel single-molecule and single-cell emulsion reverse transcription polymerase chain reaction using agarose droplet microfluidics. Anal Chem. 2012;84:3599–3606. doi: 10.1021/ac2033084. [DOI] [PubMed] [Google Scholar]
- 31.Macosko EZ, et al. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015;161:1202–1214. doi: 10.1016/j.cell.2015.05.002. A high-throughput scRNA-Seq platform that uses droplet-based capture and early bead-based barcoding to profile thousands of single cells in a cost-efficient manner. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klein AM, et al. Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell. 2015;161:1187–1201. doi: 10.1016/j.cell.2015.04.044. A high-throughput scRNA-Seq platform, distinguished from the platform in reference 31 through its use of hydrogel beads that enable more efficient bead loading and linear amplification. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Love JC, Ronan JL, Grotenbreg GM, van der Veen AG, Ploegh HL. A microengraving method for rapid selection of single cells producing antigen-specific antibodies. Nat Biotechnol. 2006;24:703–707. doi: 10.1038/nbt1210. Pioneering work that applies nanowell arrays to enable high-throughput screening of hybridomas for specific antibodies. [DOI] [PubMed] [Google Scholar]
- 34.Buettner F, et al. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat Biotechnol. 2015;33:155–160. doi: 10.1038/nbt.3102. [DOI] [PubMed] [Google Scholar]
- 35.Yao X, et al. Functional analysis of single cells identifies a rare subset of circulating tumor cells with malignant traits. Integr Biol (Camb) 2014;6:388–398. doi: 10.1039/c3ib40264a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Citri A, Pang ZP, Sudhof TC, Wernig M, Malenka RC. Comprehensive qPCR profiling of gene expression in single neuronal cells. Nat Protoc. 2011;7:118–127. doi: 10.1038/nprot.2011.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Diehn M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi Q, et al. Single-cell proteomic chip for profiling intracellular signaling pathways in single tumor cells. Proc Natl Acad Sci USA. 2012;109:419–424. doi: 10.1073/pnas.1110865109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dura B, et al. Profiling lymphocyte interactions at the single-cell level by microfluidic cell pairing. Nat Commun. 2015;6:5940. doi: 10.1038/ncomms6940. [DOI] [PubMed] [Google Scholar]
- 40.Cai L, Friedman N, Xie XS. Stochastic protein expression in individual cells at the single molecule level. Nature. 2006;440:358–362. doi: 10.1038/nature04599. An early microfluidic assay that confined secreted cellular products to effectively probe the abundance of β-galactosidase in E. coli with single-molecule resolution and sensitivity. [DOI] [PubMed] [Google Scholar]
- 41.Hindson CM, et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat Methods. 2013;10:1003–1005. doi: 10.1038/nmeth.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hindson BJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.White AK, et al. High-throughput microfluidic single-cell RT-qPCR. Proc Natl Acad Sci USA. 2011;108:13999–14004. doi: 10.1073/pnas.1019446108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warren L, Bryder D, Weissman IL, Quake SR. Transcription factor profiling in individual hematopoietic progenitors by digital RT-PCR. Proc Natl Acad Sci USA. 2006;103:17807–17812. doi: 10.1073/pnas.0608512103. An early demonstration of single-cell transcriptomics performed in microfluidic devices, with RT–qPCR used to perform in-device quantification of transcripts. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang Q, et al. Multicolor combinatorial probe coding for real-time PCR. PLoS ONE. 2011;6:e16033. doi: 10.1371/journal.pone.0016033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kazane SA, et al. Site-specific DNA-antibody conjugates for specific and sensitive immuno-PCR. Proc Natl Acad Sci USA. 2012;109:3731–3736. doi: 10.1073/pnas.1120682109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ackermann AM, Wang Z, Schug J, Naji A, Kaestner KH. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Mol Metab. 2016;5:233–244. doi: 10.1016/j.molmet.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Macaulay IC, et al. G&T-seq: parallel sequencing of single-cell genomes and transcriptomes. Nat Methods. 2015;12:519–522. doi: 10.1038/nmeth.3370. [DOI] [PubMed] [Google Scholar]
- 50.Agasti SS, Liong M, Peterson VM, Lee H, Weissleder R. Photocleavable DNA barcode-antibody conjugates allow sensitive and multiplexed protein analysis in single cells. J Am Chem Soc. 2012;134:18499–18502. doi: 10.1021/ja307689w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Marinov GK, et al. From single-cell to cell-pool transcriptomes: stochasticity in gene expression and RNA splicing. Genome Res. 2014;24:496–510. doi: 10.1101/gr.161034.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Redmond D, Poran A, Elemento O. Single-cell TCRseq: paired recovery of entire T-cell alpha and beta chain transcripts in T-cell receptors from single-cell RNAseq. Genome Med. 2016;8:80. doi: 10.1186/s13073-016-0335-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu Z, Lu S, Huang Y. Microfluidic whole genome amplification device for single cell sequencing. Anal Chem. 2014;86:9386–9390. doi: 10.1021/ac5032176. [DOI] [PubMed] [Google Scholar]
- 54.van den Bos H, et al. Single-cell whole genome sequencing reveals no evidence for common aneuploidy in normal and Alzheimer’s disease neurons. Genome Biol. 2016;17:116. doi: 10.1186/s13059-016-0976-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang Y, Swennenhuis JF, Rho HS, Le Gac S, Terstappen LW. Parallel single cancer cell whole genome amplification using button-valve assisted mixing in nanoliter chambers. PLoS ONE. 2014;9:e107958. doi: 10.1371/journal.pone.0107958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szulwach KE, et al. Single-cell genetic analysis using automated microfluidics to resolve somatic mosaicism. PLoS ONE. 2015;10:e0135007. doi: 10.1371/journal.pone.0135007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ning L, et al. Quantitative assessment of single-cell whole genome amplification methods for detecting copy number variation using hippocampal neurons. Sci Rep. 2015;5:11415. doi: 10.1038/srep11415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ni X, et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc Natl Acad Sci USA. 2013;110:21083–21088. doi: 10.1073/pnas.1320659110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kumaresan P, Yang CJ, Cronier SA, Blazej RG, Mathies RA. High-throughput single copy DNA amplification and cell analysis in engineered nanoliter droplets. Anal Chem. 2008;80:3522–3529. doi: 10.1021/ac800327d. [DOI] [PubMed] [Google Scholar]
- 60.Zhu Z, et al. Highly sensitive and quantitative detection of rare pathogens through agarose droplet microfluidic emulsion PCR at the single-cell level. Lab Chip. 2012;12:3907–3913. doi: 10.1039/c2lc40461c. [DOI] [PubMed] [Google Scholar]
- 61.Fu Y, et al. Uniform and accurate single-cell sequencing based on emulsion whole-genome amplification. Proc Natl Acad Sci USA. 2015;112:11923–11928. doi: 10.1073/pnas.1513988112. Single-cell genome profiling with droplet-based capture that demonstrates the utility of isolating components from individual cells to perform amplification in droplet, improving coverage relative to amplification en masse. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leung K, et al. Robust high-performance nanoliter-volume single-cell multiple displacement amplification on planar substrates. Proc Natl Acad Sci USA. 2016;113:8484–8489. doi: 10.1073/pnas.1520964113. High-throughput single-cell MDA performed in nanolitre volumes with commercial liquid dispensers, highlighting robust coverage of single-cell genomes in ovarian cancer cell lines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gole J, et al. Massively parallel polymerase cloning and genome sequencing of single cells using nanoliter microwells. Nat Biotechnol. 2013;31:1126–1132. doi: 10.1038/nbt.2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mikkelsen TS, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jordan IK, Makarova KS, Spouge JL, Wolf YI, Koonin EV. Lineage-specific gene expansions in bacterial and archaeal genomes. Genome Res. 2001;11:555–565. doi: 10.1101/gr.166001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Qi Q, et al. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci USA. 2014;111:13139–13144. doi: 10.1073/pnas.1409155111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paguirigan AL, et al. Single-cell genotyping demonstrates complex clonal diversity in acute myeloid leukemia. Sci Transl Med. 2015;7:281re2. doi: 10.1126/scitranslmed.aaa0763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gao R, et al. Punctuated copy number evolution and clonal stasis in triple-negative breast cancer. Nat Genet. 2016;48:1119–1130. doi: 10.1038/ng.3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–160. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ranasinghe S, et al. Antiviral CD8+ T cells restricted by human leukocyte antigen class II exist during natural HIV infection and exhibit clonal expansion. Immunity. 2016;45:917–930. doi: 10.1016/j.immuni.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Corces MR, et al. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet. 2016;48:1193–1203. doi: 10.1038/ng.3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nagano T, et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502:59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Guo H, et al. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res. 2013;23:2126–2135. doi: 10.1101/gr.161679.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lorthongpanich C, et al. Single-cell DNA-methylation analysis reveals epigenetic chimerism in preimplantation embryos. Science. 2013;341:1110–1112. doi: 10.1126/science.1240617. [DOI] [PubMed] [Google Scholar]
- 76.Farlik M, et al. Single-cell DNA methylome sequencing and bioinformatic inference of epigenomic cell-state dynamics. Cell Rep. 2015;10:1386–1397. doi: 10.1016/j.celrep.2015.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Smallwood SA, et al. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat Methods. 2014;11:817–820. doi: 10.1038/nmeth.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Cheow LF, et al. Single-cell multimodal profiling reveals cellular epigenetic heterogeneity. Nat Methods. 2016;13:833–836. doi: 10.1038/nmeth.3961. [DOI] [PubMed] [Google Scholar]
- 79.Buenrostro JD, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523:486–490. doi: 10.1038/nature14590. These authors used a commercial valve-based microfluidic platform to perform single-cell ATAC-seq, a transposase-based assay that generates NGS libraries of open chromatin regions with single-base-pair resolution. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rotem A, et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol. 2015;33:1165–1172. doi: 10.1038/nbt.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aguilar CA, Craighead HG. Micro- and nanoscale devices for the investigation of epigenetics and chromatin dynamics. Nat Nanotechnol. 2013;8:709–718. doi: 10.1038/nnano.2013.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dixit A, et al. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell. 2016;167:1853–1866.e17. doi: 10.1016/j.cell.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Trapnell C, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–386. doi: 10.1038/nbt.2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yu M, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012;487:510–513. doi: 10.1038/nature11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zeisel A, et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science. 2015;347:1138–1142. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- 86.Grun D, et al. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 2015;525:251–255. doi: 10.1038/nature14966. [DOI] [PubMed] [Google Scholar]
- 87.Pollen AA, et al. Low-coverage single-cell mRNA sequencing reveals cellular heterogeneity and activated signaling pathways in developing cerebral cortex. Nat Biotechnol. 2014;32:1053–1058. doi: 10.1038/nbt.2967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Heimberg G, Bhatnagar R, El-Samad H, Thomson M. Low Dimensionality in gene expression data enables the accurate extraction of transcriptional programs from shallow sequencing. Cell Syst. 2016;2:239–250. doi: 10.1016/j.cels.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Bacher R, Kendziorski C. Design and computational analysis of single-cell RNA-sequencing experiments. Genome Biol. 2016;17:63. doi: 10.1186/s13059-016-0927-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kowalczyk MS, et al. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015;25:1860–1872. doi: 10.1101/gr.192237.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Faridani OR, et al. Single-cell sequencing of the small-RNA transcriptome. Nat Biotechnol. 2016;34:1264–1266. doi: 10.1038/nbt.3701. [DOI] [PubMed] [Google Scholar]
- 92.Treutlein B, et al. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014;509:371–375. doi: 10.1038/nature13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ziegenhain C, et al. Comparative analysis of single-cell RNA-sequencing methods. Mol Cell. 2017;65:631–643. doi: 10.1016/j.molcel.2017.01.023. [DOI] [PubMed] [Google Scholar]
- 94.Stubbington MJ, et al. T cell fate and clonality inference from single-cell transcriptomes. Nat Methods. 2016;13:329–332. doi: 10.1038/nmeth.3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zheng GX, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8:14049. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gierahn TM, et al. Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat Methods. 2017 doi: 10.1038/nmeth.4179. http://dx.doi.org/10.1038/nmeth.4179. [DOI] [PMC free article] [PubMed]
- 97.DeKosky BJ, et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat Biotechnol. 2013;31:166–169. doi: 10.1038/nbt.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bose S, et al. Scalable microfluidics for single-cell RNA printing and sequencing. Genome Biol. 2015;16:120. doi: 10.1186/s13059-015-0684-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yuan J, Sims PA. An automated microwell platform for large-scale single cell RNA-Seq. Sci Rep. 2016;6:33883. doi: 10.1038/srep33883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003;4:117. doi: 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fan R, et al. Integrated barcode chips for rapid, multiplexed analysis of proteins in microliter quantities of blood. Nat Biotechnol. 2008;26:1373–1378. doi: 10.1038/nbt.1507. A single-cell barcoded chip that used antibodies labelled with nucleotides to enable detection of a panel of secreted proteins from individual cells isolated from whole blood. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ma C, et al. A clinical microchip for evaluation of single immune cells reveals high functional heterogeneity in phenotypically similar T cells. Nat Med. 2011;17:738–743. doi: 10.1038/nm.2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lu Y, et al. Highly multiplexed profiling of single-cell effector functions reveals deep functional heterogeneity in response to pathogenic ligands. Proc Natl Acad Sci USA. 2015;112:E607–E615. doi: 10.1073/pnas.1416756112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang J, et al. Quantitating cell–cell interaction functions with applications to glioblastoma multiforme cancer cells. Nano Lett. 2012;12:6101–6106. doi: 10.1021/nl302748q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Huang B, et al. Counting low-copy number proteins in a single cell. Science. 2007;315:81–84. doi: 10.1126/science.1133992. [DOI] [PubMed] [Google Scholar]
- 106.Blazek M, et al. Proximity ligation assay for high-content profiling of cell signaling pathways on a microfluidic chip. Mol Cell Proteomics. 2013;12:3898–3907. doi: 10.1074/mcp.M113.032821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fredriksson S, et al. Protein detection using proximity-dependent DNA ligation assays. Nat Biotechnol. 2002;20:473–477. doi: 10.1038/nbt0502-473. [DOI] [PubMed] [Google Scholar]
- 108.Landegren U, et al. Opportunities for sensitive plasma proteome analysis. Anal Chem. 2012;84:1824–1830. doi: 10.1021/ac2032222. [DOI] [PubMed] [Google Scholar]
- 109.Assarsson E, et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE. 2014;9:e95192. doi: 10.1371/journal.pone.0095192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ullal AV, et al. Cancer cell profiling by barcoding allows multiplexed protein analysis in fine-needle aspirates. Sci Transl Med. 6:219ra9–2014. doi: 10.1126/scitranslmed.3007361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Albayrak C, et al. Digital quantification of proteins and mRNA in single mammalian cells. Mol Cell. 2016;61:914–924. doi: 10.1016/j.molcel.2016.02.030. [DOI] [PubMed] [Google Scholar]
- 112.Hughes AJ, et al. Single-cell western blotting. Nat Methods. 2014;11:749–755. doi: 10.1038/nmeth.2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ozkumur AY, Goods BA, Love JC. Development of a high-throughput functional screen using nanowell-assisted cell patterning. Small. 2015;11:4643–4650. doi: 10.1002/smll.201500674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Han Q, et al. Polyfunctional responses by human T cells result from sequential release of cytokines. Proc Natl Acad Sci USA. 2012;109:1607–1612. doi: 10.1073/pnas.1117194109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Liao MC, et al. Single-cell detection of secreted Aβ and sAPPa from human IPSC-derived neurons and astrocytes. J Neurosci. 2016;36:1730–1746. doi: 10.1523/JNEUROSCI.2735-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Spencer SJ, et al. Massively parallel sequencing of single cells by epicPCR links functional genes with phylogenetic markers. ISME J. 2016;10:427–436. doi: 10.1038/ismej.2015.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brito IL, et al. Mobile genes in the human microbiome are structured from global to individual scales. Nature. 2016;535:435–439. doi: 10.1038/nature18927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Steege DA. Emerging features of mRNA decay in bacteria. RNA. 2000;6:1079–1090. doi: 10.1017/s1355838200001023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Clingenpeel S, Clum A, Schwientek P, Rinke C, Woyke T. Reconstructing each cell’s genome within complex microbial communities – dream or reality? Front Microbiol. 2015;5:771. doi: 10.3389/fmicb.2014.00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Blainey PC. The future is now: single-cell genomics of bacteria and archaea. FEMS Microbiol Rev. 2013;37:407–427. doi: 10.1111/1574-6976.12015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.McLean JS, Lasken RS. Single cell genomics of bacterial pathogens: outlook for infectious disease research. Genome Med. 2014;6:108. doi: 10.1186/s13073-014-0108-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Sun B, et al. Mechanistic evaluation of the pros and cons of digital RT-LAMP for HIV-1 viral load quantification on a microfluidic device and improved efficiency via a two-step digital protocol. Anal Chem. 2013;85:1540–1546. doi: 10.1021/ac3037206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yamanaka YJ, et al. Single-cell analysis of the dynamics and functional outcomes of interactions between human natural killer cells and target cells. Integr Biol (Camb) 2012;4:1175–1184. doi: 10.1039/c2ib20167d. [DOI] [PubMed] [Google Scholar]
- 124.Taniguchi Y, et al. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Satija R, Shalek AK. Heterogeneity in immune responses: from populations to single cells. Trends Immunol. 2014;35:219–229. doi: 10.1016/j.it.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Jovanovic M, et al. Immunogenetics. Dynamic profiling of the protein life cycle in response to pathogens. Science. 2015;347:1259038. doi: 10.1126/science.1259038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Frisk TW, Khorshidi MA, Guldevall K, Vanherberghen B, Onfelt B. A silicon-glass microwell platform for high-resolution imaging and high-content screening with single cell resolution. Biomed Microdevices. 2011;13:683–693. doi: 10.1007/s10544-011-9538-2. [DOI] [PubMed] [Google Scholar]
- 128.Zaretsky I, et al. Monitoring the dynamics of primary T cell activation and differentiation using long term live cell imaging in microwell arrays. Lab Chip. 2012;12:5007–5015. doi: 10.1039/c2lc40808b. [DOI] [PubMed] [Google Scholar]
- 129.Khorshidi MA, et al. Analysis of transient migration behavior of natural killer cells imaged in situ and in vitro. Integr Biol (Camb) 2011;3:770–778. doi: 10.1039/c1ib00007a. [DOI] [PubMed] [Google Scholar]
- 130.Wheeler AR, et al. Microfluidic device for single-cell analysis. Anal Chem. 2003;75:3581–3586. doi: 10.1021/ac0340758. [DOI] [PubMed] [Google Scholar]
- 131.Kimmerling RJ, et al. A microfluidic platform enabling single-cell RNA-seq of multigenerational lineages. Nat Commun. 2016;7:10220. doi: 10.1038/ncomms10220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cermak N, et al. High-throughput measurement of single-cell growth rates using serial microfluidic mass sensor arrays. Nat Biotechnol. 2016;34:1052–1059. doi: 10.1038/nbt.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Stevens MM, et al. Drug sensitivity of single cancer cells is predicted by changes in mass accumulation rate. Nat Biotechnol. 2016;34:1161–1167. doi: 10.1038/nbt.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Godin M, et al. Using buoyant mass to measure the growth of single cells. Nat Methods. 2010;7:387–390. doi: 10.1038/nmeth.1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Burg TP, et al. Weighing of biomolecules, single cells and single nanoparticles in fluid. Nature. 2007;446:1066–1069. doi: 10.1038/nature05741. [DOI] [PubMed] [Google Scholar]
- 136.Agresti JJ, et al. Ultrahigh-throughput screening in drop-based microfluidics for directed evolution. Proc Natl Acad Sci USA. 2010;107:4004–4009. doi: 10.1073/pnas.0910781107. Droplet-based screening of single cells undergoing directed evolution, with most active cells selected based on their fluorescence on exposure to substrates that initiate turnover of proteins on their surface. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Adamson B, et al. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell. 2016;167:1867–1882.e21. doi: 10.1016/j.cell.2016.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bodenmiller B, et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat Biotechnol. 2012;30:858–867. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Jaitin DA, et al. Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-Seq. Cell. 2016;167:1883–1896e15. doi: 10.1016/j.cell.2016.11.039. [DOI] [PubMed] [Google Scholar]
- 140.Bendall SC, et al. Single-cell trajectory detection uncovers progression and regulatory coordination in human B cell development. Cell. 2014;157:714–725. doi: 10.1016/j.cell.2014.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wagner A, Regev A, Yosef N. Revealing the vectors of cellular identity with single-cell genomics. Nat Biotechnol. 2016;34:1145–1160. doi: 10.1038/nbt.3711. This review highlights computational frameworks for inferring cellular states from single-cell genomic profiling data, with a detailed survey of current methods and the computational challenges that accompany improved experimental throughput. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Baron M, et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 2016;3:346–360.e4. doi: 10.1016/j.cels.2016.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kim JK, Kolodziejczyk AA, Ilicic T, Teichmann SA, Marioni JC. Characterizing noise structure in single-cell RNA-seq distinguishes genuine from technical stochastic allelic expression. Nat Commun. 2015;6:8687. doi: 10.1038/ncomms9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nagano T, et al. Single-cell Hi-C for genome-wide detection of chromatin interactions that occur simultaneously in a single cell. Nat Protoc. 2015;10:1986–2003. doi: 10.1038/nprot.2015.127. [DOI] [PubMed] [Google Scholar]
- 145.Burg TP, Manalis SR. Suspended microchannel resonators for biomolecular detection. Appl Phys Lett. 2003;83:2698–2700. [Google Scholar]
- 146.Zhao W, et al. Cell-surface sensors for real-time probing of cellular environments. Nat Nanotechnol. 2011;6:524–531. doi: 10.1038/nnano.2011.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Todhunter ME, et al. Programmed synthesis of three-dimensional tissues. Nat Methods. 2015;12:975–981. doi: 10.1038/nmeth.3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470:187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 149.Lander ES, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 150.Leung K, et al. A programmable droplet-based microfluidic device applied to multiparameter analysis of single microbes and microbial communities. Proc Natl Acad Sci USA. 2012;109:7665–7670. doi: 10.1073/pnas.1106752109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lovkvist C, Dodd IB, Sneppen K, Haerter JO. DNA methylation in human epigenomes depends on local topology of CpG sites. Nucleic Acids Res. 2016;44:5123–5132. doi: 10.1093/nar/gkw124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Thurman RE, et al. The accessible chromatin landscape of the human genome. Nature. 2012;489:75–82. doi: 10.1038/nature11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Rodgers-Melnick E, Vera DL, Bass HW, Buckler ES. Open chromatin reveals the functional maize genome. Proc Natl Acad Sci USA. 2016;113:E3177–E3184. doi: 10.1073/pnas.1525244113. [DOI] [PMC free article] [PubMed] [Google Scholar]