

Here, Lv et al. report that the CBL family E3 ubiquitin ligases down-regulate JAK2 stability and signaling via the adaptor protein LNK/SH2B3. Their results reveal a novel signaling axis that regulates JAK2 in normal and malignant HSPCs and suggest new therapeutic strategies for treating CBLmut myeloid malignancies.
Keywords: hematopoietic stem cells (HSCs), Janus kinase 2 (JAK2), ubiquitin, signaling, chronic myelomonocytic leukemia (CMML), juvenile myelomonocytic leukemia (JMML), acute myeloid leukemia (AML)
Abstract
Janus kinase 2 (JAK2) is a central kinase in hematopoietic stem/progenitor cells (HSPCs), and its uncontrolled activation is a prominent oncogenic driver of hematopoietic neoplasms. However, molecular mechanisms underlying the regulation of JAK2 have remained elusive. Here we report that the Casitas B-cell lymphoma (CBL) family E3 ubiquitin ligases down-regulate JAK2 stability and signaling via the adaptor protein LNK/SH2B3. We demonstrated that depletion of CBL/CBL-B or LNK abrogated JAK2 ubiquitination, extended JAK2 half-life, and enhanced JAK2 signaling and cell growth in human cell lines as well as primary murine HSPCs. Built on these findings, we showed that JAK inhibitor (JAKi) significantly reduced aberrant HSPCs and mitigated leukemia development in a mouse model of aggressive myeloid leukemia driven by loss of Cbl and Cbl-b. Importantly, primary human CBL mutated (CBLmut) leukemias exhibited increased JAK2 protein levels and signaling and were hypersensitive to JAKi. Loss-of-function mutations in CBL E3 ubiquitin ligases are found in a wide range of myeloid malignancies, which are diseases without effective treatment options. Hence, our studies reveal a novel signaling axis that regulates JAK2 in normal and malignant HSPCs and suggest new therapeutic strategies for treating CBLmut myeloid malignancies.
Janus kinase 2 (JAK2) is the key tyrosine kinase in signaling from an array of hematopoietic receptors, including thrombopoietin (TPO), erythropoietin (EPO), interleukin-3 (IL-3), and granulocyte–macrophage colony-stimulating factor (GM-CSF) receptors. Once activated by cytokines, JAK2 phosphorylates their cognate receptors and triggers a cascade of signaling events involving STATs, PI-3K/AKT, and MAPK (Kaushansky 2003). JAK2-deficient cells fail to respond to multiple cytokines, and Jak2-null mice die of anemia embryonically, demonstrating the essential role of JAK2 in cytokine receptor signaling.
While JAK2 plays an essential role in hematopoietic development (Parganas et al. 1998), uncontrolled JAK2 signaling results in hematopoietic malignancies. Myeloproliferative neoplasms (MPNs) are clonal hematopoietic stem cell (HSC) diseases, characterized by unrestrained proliferation, leading to overproduction of one or more myeloid lineages. Activating mutations in JAK2 (V617F) are found at high frequencies in subtypes of MPNs (Levine et al. 2007). The JAK2V617F mutation is a canonical driver mutation in human MPNs, and loss of JAK2 abrogates MPN in engineered mice. However, current Food and Drug Administration (FDA)-approved JAK inhibitors (JAKis) only moderately reduce the JAK2VF allele burden, and the majority of patients does not achieve molecular remission (Mesa et al. 2014), highlighting the need for a better understanding of the regulation of JAK2 to enhance the efficacy of JAK2 inhibitors. Despite a number of E3 ubiquitin (Ub) ligases for JAK2 having been suggested, none have been shown to impact JAK2 protein levels or HSC numbers in vivo. Hence, molecular mechanisms underlying the regulation of JAK2 stability and signaling remain poorly established.
Work from our laboratory and others identified LNK (also called SH2B3) as a direct and critical negative regulator of TPO receptor MPL and its associated JAK2 in hematopoietic stem/progenitor cells (HSPCs) (Tong and Lodish 2004; Buza-Vidas et al. 2006; Seita et al. 2007; Bersenev et al. 2008). Lnk−/− mice exhibit elevated white blood cell (WBC) and platelet counts and progenitor cell expansion in the bone marrow (BM) (Velazquez et al. 2002). Remarkably, Lnk deficiency leads to a >10-fold increase in HSC numbers (Ema et al. 2005; Bersenev et al. 2008). Aged Lnk−/− mice show mitigated HSC aging and spontaneously develop a chronic myelogenous leukemia (CML)-like MPN (Bersenev et al. 2010, 2012). Importantly, LNK loss-of-function mutations have been identified in human MPN and acute myeloid leukemia (AML) patients with aberrant STAT signaling (Oh et al. 2010); thus, studying LNK regulatory functions in normal and malignant HSPCs will shed significant insights into JAK2 signaling. In this study, we found that LNK recruits Casitas B-cell lymphoma (CBL) family E3 Ub ligases to regulate JAK2 ubiquitination, stability, and signaling.
CBL proteins are a highly conserved family of RING finger (RF) E3 Ub ligases that regulate the signaling of multiple tyrosine kinases. CBL (also known as C-CBL) and the closely related CBL-B are expressed in hematopoietic cells. They possess a tyrosine–kinase-binding (TKB) domain, a linker region (L), and a RF. The RF domain binds to E2 Ub-conjugating enzymes and catalyzes the transfer of Ub from the E2 to the substrate. Both the L region and the RF domain are required for E3 activity. The foremost function of the TKB domain is to determine CBL's substrate specificity, which includes receptor tyrosine kinases (RTKs), such as EGFR, PDGFR, c-KIT, and FLT3, and non-RTKs, such as ZAP70 and SYK (Thien and Langdon 2005; Mohapatra et al. 2013). Ubiquitination of phosphorylated tyrosine kinases marks them for endocytic traffic and subsequent degradation in lysosomes or for proteasomal degradation.
Notably, CBL deletions and loss-of-function mutations have been found in diverse myeloid malignancies, including myelodysplastic syndrome (MDS) (Bejar et al. 2011), MPN, AML, and particularly MDS/MPN overlap syndrome, a distinct diagnostic category within myeloid malignancies with features of both MDS and MPN (Caligiuri et al. 2007; Makishima et al. 2009; Sanada et al. 2009). MDS/MPN subtypes include atypical CML (aCML), juvenile myelomonocytic leukemia (JMML), and chronic myelomonocytic leukemia (CMML), in which CBL mutations are most frequent (∼20%) (Loh et al. 2009; Makishima et al. 2009; Muramatsu et al. 2012; Tiu and Sekeres 2014; Merlevede et al. 2016). The prognosis of CMML is poor, with a high propensity for AML progression and no effective treatment options. Most CBL missense mutations are located in the L or RF domains, attesting to the importance of the E3 ligase activity of CBL in restricting kinase signaling and neoplasms (Sanada et al. 2009).
Cbl−/− mice phenocopy Lnk−/− mice, exhibiting elevated platelet counts as well as an expanded HSPC pool with superior transplantability (Rathinam et al. 2008). Cbl−/− HSPCs are hyperresponsive to a variety of cytokines, including SCF, TPO, IL3, and FLT3 ligand (Rathinam et al. 2008; Sanada et al. 2009). Unlike Cbl or Cbl-b single-knockout mice, conditional double-knockout mice develop an aggressive MPN that closely resembles CMML/JMML, indicating the redundant but essential roles of CBL and CBL-B in MPNs (Naramura et al. 2010; An et al. 2016). It has been shown that CBL E3-dead mutants, when overexpressed, lead to enhanced and prolonged activation of STAT5; however, the protein levels of JAK2 remain unchanged in Cbl−/− or E3-inactive cells (Rathinam et al. 2008; Sanada et al. 2009). Thus, whether CBL regulates JAK2 directly or indirectly through the receptor remains poorly understood. Recently, LNK/SH2B3 was also found mutated in ∼10% of JMML patients (Stieglitz et al. 2015), attesting to the importance and relevance of our finding of this novel CBL/LNK/JAK2 signaling axis.
Here we investigated the molecular mechanism by which the adaptor protein LNK attenuates JAK2 signaling. We show that JAK2 is promptly ubiquitinated and degraded upon cytokine stimulation, and these processes are regulated by CBL and CBL-B via LNK. Using a novel inducible double-knockout mouse of Cbl;Cbl-b, we further show the therapeutic potential of JAKi in treating an aggressive CMML-like disease. Finally, our mechanistic finding was translated into primary human CBLmut AMLs. Taken together, our studies mechanistically shed light on new therapeutic strategies in treating CBLmut myeloid malignancies.
Results
CBL family E3 ligases interact with LNK
LNK/SH2B3 has emerged as a critical regulator of self-renewal as well as a suppressor of MPNs (Bersenev et al. 2008, 2010). We reported previously that LNK constrains TPO-induced JAK2 signaling in HSPCs (Bersenev et al. 2008); however, the molecular mechanisms have remained enigmatic. LNK does not possess enzymatic activity; rather, it contains several protein–protein interaction domains. Therefore, we focused our efforts on deciphering LNK signaling networks. We established JAK2V617F+ human erythroid leukemia (HEL) cells that stably express Flag-HA-tagged human LNK and subsequently performed large-scale protein purification using tandem affinity purification followed by mass spectrometry (Supplemental Fig. S1A). LNK-interacting proteins that we reported previously with mouse LNK in murine 32D cells were also pulled down in HEL cells (Supplemental Table S1). In fact, the peptide numbers for JAK2 were higher in HEL cells than in 32D cells (Jiang et al. 2012). We speculate that this is due to higher phosphorylated and total JAK2 levels in HEL cells (JAK2V617F+) than in 32D cells (JAK2WT). Importantly, CBL and CBL-B were among the most abundant LNK partners besides JAK2 (Supplemental Fig. S1B; Supplemental Table S1). We confirmed the LNK–CBL interaction by immunoprecipitation and Western blotting (Supplemental Fig. S1C).
Combined CBL/CBL-b loss increases JAK2 levels and promotes cell growth and cytokine signaling
We next assessed whether CBL and CBL-B influence JAK2 protein levels and JAK-dependent cell growth. To this end, we used the human cytokine-dependent hematopoietic progenitor cell line TF-1. TF-1 cells are maintained in GM-CSF, in which JAK2 is essential for GM-CSF receptor (GM-CSFR) signaling and growth. We engineered stable TF-1 cells expressing hMPL, rendering them TPO-dependent. We next generated TF-1/MPL cells with stable shRNA-mediated knockdown of CBL and CBL-b individually or in combination. Dual knockdown of CBL and CBL-b led to a small but consistent increase in JAK2 levels compared with single knockdown (Supplemental Fig. S2A). Dual knockdown also moderately increased cytokine-dependent cell growth (Supplemental Fig. S2B). We noted that residual CBL-b protein remained after knockdown; we thus deleted CBL-b using CRISPR/Cas9 and combined it with CBL knockdown (Dko + d). We found that dual depletion of CBL and CBL-b resulted in a marked increase in JAK2 protein levels (Fig. 1A) and enhanced growth responsiveness to TPO compared with single depletion (Fig. 1B). Moreover, the double depletion conferred a slight resistance to the JAK1/2 inhibitor ruxolitinib, although the cells remained sensitive to JAK inhibition (Fig. 1D; Supplemental Fig. S2C).
Figure 1.

CBL depletion or inactivation increases JAK2 level and promotes cell growth. (A) TF-1/hMPL cells with single or double depletion of CBL and CBL-b were analyzed for JAK2 levels by Western blot. (Luc) Luciferase; (gRNA) guide RNA. (B–D) Cells were cultured in different concentrations of TPO (B), GM-CSF (C), or ruxolitinib in the presence of TPO (D). Cell numbers after 3 d of culture were determined by 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) absorbance. (D) The left panel shows the absolute MTT values, while the right panel shows the relative MTT values normalized to 0 nM ruxolitinib in each cell line. (E) TF-1/MPL cells stably expressing either control vector, CBL wild type, or the E3-dead mutant C381A were examined for JAK2 levels by Western blot. (exp) Exposure. (F–H) MTT assay of the indicated TF-1 cell lines cultured in different concentrations of TPO (F), GM-CSF (G), or ruxolitinib in the presence of TPO (H). (H) The left panel shows the absolute MTT values, while the right panel shows the relative MTT values normalized to 0 nM ruxolitinib in each cell line. Error bars denote mean ± SD. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001.
CBL mutations are frequently found in CMML and JMML, and one defining biologic characteristic of JMML/CMML is hypersensitivity to GM-CSF (Kotecha et al. 2008; Niemeyer et al. 2010; Padron et al. 2013). Indeed, CBL Dko + d cells showed markedly enhanced growth in response to low doses of GM-CSF (Fig. 1C), indicating that CBL and CBL-B play redundant roles in JAK2 regulation. Moreover, the dosage effects of CBL family proteins that we observed explain the lack of a phenotype in JAK2 perturbation in single-knockout mice.
CBL E3 ligase activity is critical for JAK2 regulation and cytokine-dependent cell growth
Since CBL proteins contain multiple functional domains, we asked whether the E3 ligase activity mediates its function in JAK2 regulation and cytokine-mediated growth. We established TF-1/MPL cells stably expressing CBL wild type or E3 Ub ligase-dead mutant C381A (Xiong et al. 2011). Wild-type CBL expression slightly reduced JAK2 protein level and suppressed TPO- or GM-CSF-mediated cell growth, while the C381A mutant markedly elevated JAK2 levels and enhanced cell growth in response to both TPO and GM-CSF (Fig. 1E–G). Of note, both cell lines remained sensitive to growth inhibition by ruxolitinib (Fig. 1H). These data suggest that the E3 ligase activity of CBL proteins is important for regulating cytokine-mediated cell growth through modulating JAK2 protein levels.
CBL proteins regulate TPO-stimulated JAK2 ubiquitination and degradation
To investigate the mechanism by which CBL E3 Ub ligases regulate JAK2 level and MPL/JAK2-mediated cell growth, we examined the effects of CBL depletion on the JAK2 and MPL half-lives. We found that, under starved conditions, the half-life of JAK2 was extended. Upon TPO stimulation, JAK2 was promptly degraded, as determined by treatment with cycloheximide (CHX), a protein synthesis inhibitor (Fig. 2A,B). Of note, the MPL half-life is independent of TPO stimulation (Fig. 2A,B). In line with the role of CBL E3 ligases on JAK2 protein level (Fig. 1A), dual depletion of CBL and CBL-b as well as overexpression of the E3-dead CBL mutant C381A markedly increased JAK2 half-life (Fig. 2C,D; Supplemental Fig. S2D). The C381A mutant also exhibited a longer half-life than wild-type CBL, consistent with the notion that CBL regulates its own ubiquitination and degradation (Fig. 2D). In contrast, CBL depletion or inactivation failed to affect MPL half-life (Fig. 2C,D), although it increased basal MPL surface levels and impaired TPO-induced down-regulation of the cell surface MPL (Supplemental Fig. S3), consistent with a previous report (Saur et al. 2010). Thus, our data suggest that CBL proteins regulate activation-dependent JAK2 degradation through a mechanism distinct from that of MPL receptor.
Figure 2.

CBL E3 ligases mediate TPO-stimulated JAK2 ubiquitination, degradation and signaling. (A) TF-1/MPL cells were starved and stimulated with or without TPO in the presence of CHX for indicated times. JAK2 and MPL protein levels were determined by Western blot. (B) Quantification of JAK2 and MPL half-lives with or without TPO as shown in A. Error bars denote mean ± SEM. (**) P < 0.01; (***) P < 0.001. (C) JAK2 and MPL half-lives were analyzed in TF-1/hMPL cells with control or double depletion of CBL/CBL-b (Dko + d). (D) JAK2, MPL, and CBL half-lives were analyzed in TF-1/hMPL cells stably expressing empty vector (EV), CBL wild type, or the C381A mutant. (E) TF-1/MPL cells with shLuc or CBL/CBL-b (Dko + d) were treated with or without TPO for 10 min. Lysates were incubated with Flag-K63Ub-TUBE (tandem-repeated Ub-binding entity) and then precipitated with anti-Flag beads. The precipitates were subjected to Western blot with the indicated antibodies. “C” indicates control immunoprecipitation with Flag beads. (F) TF-1/MPL cells stably expressing either EV alone or wild-type or kinase-inactive mutant (Y1007/1008F [YF]) myc-JAK2 were stimulated with or without TPO for 10 min. Cell lysates were pulled down with Flag-K63Ub-TUBE as described in E. Precipitates (top) and total cell lysates (bottom) were subjected to Western blot with the indicated antibodies. The asterisk indicates a possible pTYK2 band that cross-reacts with pJAK2 antibodies. (Endo) Endogenous. (G) TF-1/MPL cells were pretreated with DMSO or 1 µM dasatinib for 1 h and then stimulated with or without TPO for 10 min. Cell lysates were precipitated with anti-pTyr antibody 4G10 followed by anti-CBL blot. Total cell lysates were subjected to Western blot with the indicated antibodies. (H) JAK2 or CBL half-lives were examined in TF-1/MPL cells pretreated with DMSO or 1 µM dasatinib followed by CHX assay. (I,J) TF-1/MPL cells with shLuc or CBL/CBL-b (Dko + d) were stimulated with a graded concentration of TPO for 10 min (I) or with 100 ng/mL TPO for the indicated times (J). Cell lysates were subjected to Western blot with the indicated antibodies.
JAK2 has been found to be ubiquitinated using pan-Ub antibodies (Ungureanu et al. 2002). However, the types of Ub chains on endogenous JAK2 and their potential function have not been determined. To address this issue, we first cotransfected JAK2 along with wild-type Ub or Ub mutants that specifically abolish Lys48-linked (K48R) or Lys63-linked (K63R) Ub into 293T cells. We found that JAK2 ubiquitination was significantly reduced in cells expressing mutant Ubs (Supplemental Fig. S4A), suggesting that JAK2 is both K48- and K63-ubiquitinated in a heterologous overexpression system. To examine endogenous JAK2 poly-Ub status, we used Ub sensors (Sims et al. 2012; van Wijk et al. 2012), which are tandem-repeated Ub-binding entities (TUBEs) with a preferential affinity for K63Ub over K48Ub (LifeSensors, Inc.) (Hjerpe et al. 2009). With this approach, we failed to detect JAK2 K48 ubiquitination using K48-specific TUBEs (Supplemental Fig. S4B). In contrast, we found that endogenous JAK2 was ubiquitinated upon TPO stimulation with pull-downs using TUBE1 that recognizes pan-Ub but showed a 10 times preference for K63-Ub over K48-Ub (Supplemental Fig. S4C). The new-generation K63-specific TUBE exhibits 1000-fold to 10,000-fold selectivity for K63-Ub over other types of poly-Ub chains (Sims and Cohen 2009). Using this K63-TUBE, we showed for the first time that endogenous JAK2 was K63-ubiquitinated promptly upon TPO stimulation (10 min) even in the absence of the proteasome inhibitor MG132 (Fig. 2E, middle panel). Furthermore, JAK2 Ub is associated with phosphorylation and activation (pY1007/1008-JAK2) (Fig. 2E, top panel). Importantly, CBL/CBL-b dual depletion abrogated JAK2 ubiquitination (Fig. 2E).
To further confirm JAK2 activation-dependent K63-Ub, we stably expressed myc-tagged wild-type JAK2 and the kinase-inactive mutant Y1007/1008F in TF-1/MPL cells and then performed K63-TUBE pull-down assay followed by Western blot analysis using anti-myc antibodies. The results revealed that wild-type but not kinase-inactive JAK2 is K63-ubiquitinated (Fig. 2F). Since CBL family proteins are activated by Src family kinases such as LYN (Javadi et al. 2013), we investigated whether the Src kinase inhibitor dasatinib regulates JAK2 stability. Dasatinib efficiently inhibited LYN phosphorylation (Fig. 2G), which resulted in a reduction in CBL phosphorylation (Fig. 2G). Furthermore, inhibition of LYN kinase led to a reduction in CBL E3 function, as evidenced by increased half-lives for both JAK2 and CBL (Fig. 2H). Taken together, our data revealed a critical role for CBL proteins in regulating the activation/phosphorylation-dependent ubiquitination and degradation of JAK2.
CBL depletion or inactivation enhances JAK2 signaling
Given the central role of JAK2 in cytokine signaling, we further investigated JAK2 signaling amplitude and kinetics in response to cytokines. We first stimulated cells with graded concentrations of TPO and found that CBL/CBL-b-depleted cells were more sensitive to low concentrations of TPO (Fig. 2I). Next, cells were stimulated with TPO for different time periods. Consistently, dual depletion of CBL and CBL-b enhanced and prolonged JAK2 activation and downstream signaling (Fig. 2J; Supplemental Fig. S2E).
To examine the specificity of the knockdown and how CBL E3 ligase activity affects JAK2 signaling, we introduced shRNA-resistant CBL wild type or the E3-dead C381A mutant in CBL knockdown cells. As expected, CBL knockdown resulted in a prolonged activation of JAK2 signaling and enhanced TPO sensitivity in cell growth (Supplemental Fig. S5). Re-expression of wild-type CBL normalized cytokine-dependent JAK2 signaling and cell growth; in contrast, the E3-dead C381A mutant exacerbated the effects of CBL knockdown (Supplemental Fig. S5). Thus, these data suggest that CBL E3 ligase activity regulates cytokine-dependent JAK2 signaling.
LNK depletion increases JAK2 half-life and TPO sensitivity and promotes cell growth
Since LNK appears to serve as an adaptor between CBL/CBL-b and JAK2, the effects of LNK depletion are expected to be similar to those of CBL depletion. We thus generated LNK-null TF-1/MPL cells using CRISPR/Cas9. Out of three guide RNAs (gRNAs) to LNK, we identified gRNA2 (LNK knockout hereafter) to be the most efficient in excising LNK, as confirmed by DNA sequencing (data not shown) and loss of LNK protein expression by Western blot (Fig. 3A). LNK knockout cells showed hypersensitivity to TPO in promoting cell growth (Fig. 3B). Furthermore, LNK depletion resulted in prolonged JAK2 but not MPL half-life (Fig. 3C) and correspondingly abolished JAK2 ubiquitination (Fig. 3D).
Figure 3.
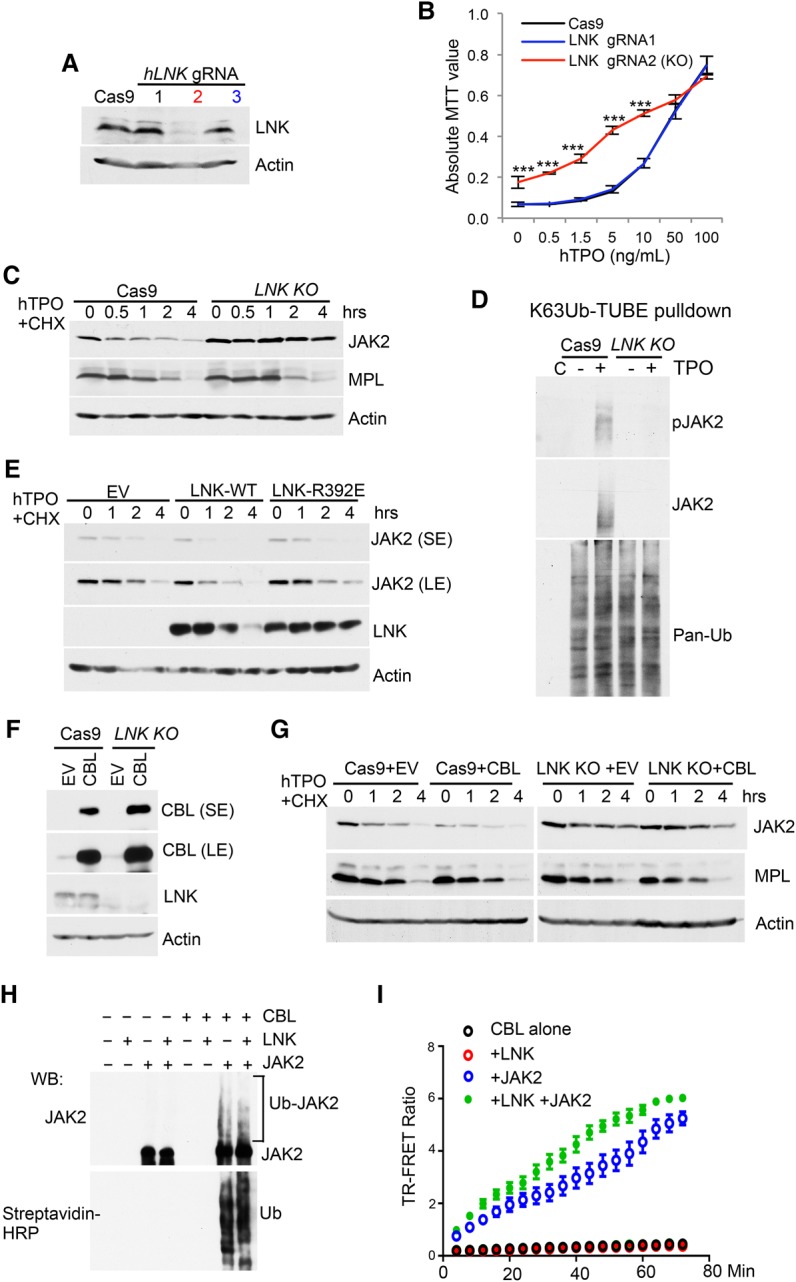
CBL E3 ligases regulate JAK2 stability through the adaptor protein LNK. (A) TF-1/MPL cells expressing either Cas9 alone or Cas9 plus three different gRNAs to LNK were analyzed for depletion efficiency using Western blot. (B) The graph shows the MTT growth assay of the indicated TF-1/hMPL cell lines cultured in TPO. Note that the black and blue lines overlap. (***) P < 0.001, two-tailed Student's t-test. (C) JAK2 and MPL half-lives were determined in TF-1/hMPL cells expressing Cas9 alone or LNK knockout by CHX assay. (D) Cas9 or LNK knockout TF-1/MPL cells were treated with or without TPO for 10 min. Lysates were pulled down with Flag-K63Ub-TUBE and anti-Flag beads. The precipitates were subjected to Western blot with the indicated antibodies. “C” indicates control immunoprecipitation with Flag beads. (E) JAK2 half-lives were determined in TF-1/MPL cells overexpressing EV, LNK wild type, or LNK-R392E by CHX assay. (F) Cas9 control or LNK knockout TF-1/MPL cells were stably infected with retroviruses expressing either EV or CBL wild type. LNK depletion efficiency and CBL expression were analyzed by Western blot. (G) JAK2 and MPL half-lives were determined in the indicated cell lines from F by CHX chase assays. (H) In vitro ubiquitination of JAK2 by CBL was assayed with recombinant JAK2, E1, E2, Ub-biotin, and the CBL TKB + RING domain. The reactions were subjected to Western blot analysis with anti-JAK2 antibodies and streptavidin-HRP. (I) The activity and kinetics of CBL E3 Ub ligase in ubiquitinating JAK2 were assayed using a homogeneous time-resolved fluorescence resonance energy transfer (TR-FRET) assay.
We demonstrated previously that LNK binds specifically to phosphorylated JAK2 through its SH2 domain, whereby disruption of the SH2 domain by a R392E point mutation dissociates JAK2 from LNK (Bersenev et al. 2008; Jiang et al. 2012). We reasoned that if LNK were the bridge between JAK2 and CBL, disruption of the LNK–JAK2 interaction would dampen the effect of LNK on JAK2 degradation. To test this hypothesis, we compared the half-life of JAK2 in LNK wild-type or R392E-expressing TF-1/MPL cells. Consistent with our hypothesis, overexpression of wild-type LNK accelerated JAK2 degradation, while LNK R392E failed to affect JAK2 half-life (Fig. 3E). In a complementary approach, we examined whether loss of LNK could mitigate CBL's effects on JAK2 stability. We stably expressed wild-type CBL in either control or LNK knockout cells (Fig. 3F). Consistently, CBL overexpression accelerated JAK2 degradation; however, in LNK-deficient cells, JAK2 stability was sustained even in the presence of CBL overexpression (Fig. 3G).
To address whether JAK2 is a direct CBL substrate and whether LNK facilitates this post-translation modification, we performed in vitro ubiquitination reconstitution assays. The recombinant JAK2 was robustly ubiquitinated by CBL in vitro (Fig. 3H), indicating that JAK2 is a direct CBL substrate. Furthermore, we performed time-resolved fluorescence resonance energy transfer (TR-FRET) assay to investigate JAK2 ubiquitination kinetics. The data revealed that JAK2 was promptly ubiquitinated by CBL and that LNK further promoted JAK2 ubiquitination (Fig. 3I). Collectively, these data reveal an adaptor role for LNK in bridging CBL and JAK2 interaction, therefore mediating JAK2 ubiquitination and degradation.
Cbl−/−;Cbl-b−/− HSPCs show increased JAK2 protein levels, signaling, and reconstituting ability
To further examine the physiological function of CBL proteins and their regulation of JAK2, we turned to transgenic mouse models with loss of function of Cbl and Cbl-b. Germline double-knockout mice for Cbl and Cbl-b are embryonic-lethal. Previously engineered Cbl-b−/−;Cblf/f;MMTV-cre mice (germline Cbl-b−/− and conditional Cbl) develop an aggressive CMML-like MPN due to an unexpected Cre activation in a fraction of HSCs (Naramura et al. 2010). We recently generated conditional double-knockout alleles for both Cbl and Cbl-b (Cblf/f;Cbl-bf/f) (Goetz et al. 2016), which show normal survival in the uninduced state. We found that pan-hematopoietic Vav-cre-mediated deletion causes early lethality (day 10), preventing us from studying adult hematopoiesis (An et al. 2016). Therefore, we generated Cblf/f;Cbl-bf/f;CreERT2 mice that allow efficient and stringently controlled deletion of both Cbl and Cbl-b genes only upon tamoxifen (TAM) treatment.
We sorted LSK (Lineage−Kit+Sca1+) HSPCs and LKS− (Lineage−Kit+Sca1−) myeloid progenitor cells and cultured them in 4-hydroxytamoxifen (4-OHT)-containing medium for 2 d to allow Cre-recombinase activation (Fig. 4A). To examine knockout efficiency, we screened individual colonies for Cbl and Cbl-b excision after plating LSK cells onto semisolid methylcellulose culture. Induced Cre expression led to a near 100% deletion of Cbl and 70% deletion of Cbl-b (Supplemental Fig. S6A). In both LSK and LKS− cells, ex vivo deletion of Cbl and Cbl-b resulted in elevated JAK2 protein levels and signaling, including activated STAT5, AKT, and ERK (Fig. 4B), which is consistent with what was observed in TF-1/MPL cells.
Figure 4.

Cbl−/−;Cbl-b−/− HSPCs show increased JAK2 protein level, signaling, and reconstituting ability and give rise to CMML-like myeloid malignancies. (A) Experimental scheme of in vitro excision of Cbl/Cbl-b followed by Western blot or BM transplantation (BMT). Sorted LSK or LKS− cells from Cblf/f;Cbl-bf/f (f/f) or Cblf/f;Cbl-bf/f;CreERT2 (f/f;ERT) mice were cultured in 4-OHT-containing medium for 2 d. Resultant cells from 2000 LSKs after culture were injected into each recipient along with competitors. (B) Two days after culture in 4-OHT, LSK or LKS− cells were analyzed for JAK2 level and signaling by Western blot. (C) Donor chimerisms in the peripheral blood of Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 LSK transplanted mice were measured at 4, 8, 12, and 16 wk. (D) Complete blood count (CBC) analysis of peripheral blood of Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 LSK transplanted mice at 12 wk after transplantation. (E,F) The left panels show the donor percentages in the BM (E) or spleens (F) of Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 LSK transplanted mice 16 wk after transplantation. The right panels show lineage distribution within the donor fraction in the BM (E) or spleens (F). (G) The numbers of donor-derived long-term HSCs (LT-HSCs) in the BM of Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 LSK transplanted mice 16 wk after transplantation are shown. (H) The numbers of donor-derived LT-HSC, short-term HSC (ST-HSC), and various multipotent progenitor (MPP) populations in the spleens of Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 LSK transplanted mice 16 wk after transplantation are shown. (I) Colony-forming unit (CFU)-GM (left) or CFU-E (right) progenitors in the spleens of Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 LSK transplanted mice are shown. (J) BM cells from Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 LSK transplanted mice were injected into secondary recipients. Donor chimerisms in the peripheral blood of recipient mice were measured at 4, 8, 12, and 16 wk. Each symbol represents an individual mouse. Horizontal lines indicate mean frequencies. Error bars indicate SE. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (ns) not significant.
Next, we studied the in vivo function of LSK cells from Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 mice upon Cbl;Cbl-b excision ex vivo. LSK cells were cultured in 4-OHT for 2 d and then injected with competitor BM cells into lethally irradiated mice. Reconstitution and lineage distribution of donor cells in the peripheral blood of recipients were analyzed every 4 wk by flow cytometry. In comparison with control LSK cells, Cbl−/−;Cbl-b−/− LSK cells displayed superior transplantability, and the differences became more pronounced with time (Fig. 4C). Furthermore, there was a markedly biased differentiation toward myeloid progeny, as demonstrated by increased Gr1+/Mac1+ percentage and decreased B220+ percentage or CD4+/8+ percentage (Supplemental Fig. S6B,C). Complete blood count (CBC) analysis also showed a pronounced increase in WBC numbers in Cbl−/−;Cbl-b−/− LSK transplanted mice, with marked elevation in myeloid cells and platelets but not lymphocytes (Fig. 4D). At 16 wk, we analyzed the lineage distributions of donor-derived cells in the recipients’ BM and spleens. BM cells from Cbl−/−;Cbl-b−/− LSK transplanted mice exhibited a significant increase in total donor and myeloid percentages but a decrease in lymphoid percentage (Fig. 4E, right panel). A similar lineage bias was observed in the spleen (Fig. 4F, right panel). Taken together, these data strongly suggest that ex vivo excision of Cbl and Cbl-b in primary HSPCs increased JAK2 levels and signaling and gave rise to enhanced HSC reconstitution and myeloid expansion.
Cbl;Cbl-b dual deletion increases HSCs and myeloid-biased MPPs (multipotent progenitors), leading to a myeloid expansion
To study the role of CBL proteins in HSPCs, we analyzed the frequency and number of primitive hematopoietic compartments in the BM and spleens of control or Cbl−/−;Cbl-b−/− LSK transplanted mice using established cell surface markers (Supplemental Fig. S6D; Pietras et al. 2015). BM cells from Cbl−/−;Cbl-b−/− LSK transplanted mice had a significant increase in the number of long-term HSCs (LT-HSCs; defined as Flk2−CD150+CD48− LSK) (Fig. 4G) but not in other HSPC subpopulations (data not shown). Strikingly, in the spleen, we observed a marked (>100-fold) increase in primitive and myeloid/erythroid-biased HSC and MPP compartments, including LT-HSC, short-term HSC (ST-HSC; defined as Flk2− CD150−CD48− LSK), MPP2 (Flk2−CD150+CD48+ LSK, megakaryocyte/erythroid-biased MPP), and MPP3 (Flk2− CD150−CD48+ LSK, myeloid-biased MPP) but not MPP4 (Flk2+CD150− LSK, lymphoid-biased MPP) (Fig. 4H). The increase in MPP2 and MPP3 is consistent with an increase in myeloid cells and platelets in the peripheral blood (Fig. 4C,D). To examine committed progenitor cells, we plated total BM or spleen cells onto semisolid methylcellulose medium to quantify colony-forming-unit (CFU) progenitors. There was no significant difference in either CFU-GM or CFU-E progenitors from the BM (data not shown). In marked contrast, spleens from Cbl−/−;Cbl-b−/− LSK transplanted mice produced >10-fold more CFU-GM colonies and CFU-E colonies than those from control mice (Fig. 4I).
To further examine the impact of loss of Cbl/Cbl-b on the functionality and self-renewal of HSCs, we performed secondary transplantations. Cells from Cbl−/−;Cbl-b−/− LSK transplanted mice exhibited a greater donor contribution in the peripheral blood in secondary recipients (Fig. 4J). In addition, we also observed a myeloid-biased expansion and a decrease in lymphoid repopulation as determined by flow cytometry and CBC analysis (Supplemental Fig. 7S). Together, these data suggest that dual deletion of Cbl and Cbl-b in HSPCs results in an expansion of LT-HSCs, myeloid-biased MPPs, and committed myeloid progenitors, leading to an overall expansion of mature myeloid cells, thus resembling a CMML-like disease.
In vivo excision of Cbl and Cbl-b gives rise to an aggressive CMML-like disease
To further elucidate the role of CBL proteins in myeloid development and myeloid malignancies, we performed studies in mice with Cbl/Cbl-b excision in vivo. Total BM cells from Cblf/f;Cbl-bf/f or Cblf/f;Cbl-bf/f;CreERT2 mice were transplanted into lethally irradiated recipients followed by intraperitoneal injection of TAM 4 wk after transplant (Fig. 5A). The deletion efficiency was high, as evaluated by colony PCR (Supplemental Fig. S8A). Twelve days after TAM treatment, we observed a myeloid expansion in the peripheral blood of Cblf/f;Cbl-bf/f;CreERT2 mice (Supplemental Fig. S8B,C). By 45 d after TAM delivery, we observed a dramatic increase in donor-derived myeloid cells in peripheral blood of Cblf/f;Cbl-bf/f;CreERT2 mice, as determined by flow cytometry (Fig. 5B). Consistent with these results, CBC analysis showed that in vivo excision of Cbl/Cbl-b in BM hematopoietic cells resulted in a marked increase in WBCs, including myeloid cells and platelets, while the numbers of lymphocytes remained unchanged in comparison with the control mice (Fig. 5C). Cbl;Cbl-b conditional double-knockout mice also exhibited anemia, a feature of the MDS/MPN overlap syndrome in which CBL mutations are mostly identified. Lineage distribution of donor cells in the BM and spleen also displayed a pronounced increase in myeloid cells and decrease in lymphoid cells, consistent with that in the peripheral blood (Fig. 5D,E). The mice succumbed to a CMML-like disease with a median survival of 60 d (Fig. 5H). Splenomegaly (Fig. 5F) and myeloid infiltration in the liver, lungs, and skin were observed in Cbl;Cbl-b conditional double knockouts but not the controls (Supplemental Fig. S8D; data not shown). The BM, spleen, and liver showed marked increased maturing myelomonocytic cells and atypical megakaryocytes that are consistent with a CMML-like MPN (Supplemental Fig. S8E). Furthermore, colony assays showed that Cbl;Cbl-b conditional double-knockout spleens had ∼40-fold expansion of CFU-GM and ∼15-fold expansion of CFU-E progenitors (Fig. 5G). Taken together, our data indicate that in vivo excision of Cbl and Cbl-b in hematopoietic cells acts as a driver for an aggressive CMML-like disease, analogous to CBL mutated MDS/MPN.
Figure 5.

JAKi blunts CMML development in Cbl;Cbl-b−/− transplanted mice. (A) Experimental scheme of in vivo excision of Cbl and Cbl-b in transplanted mice. Two million total BM cells from Cblf/f;Cbl-bf/f (f/f) or Cblf/f;Cbl-bf/f;CreERT2 (f/f;ERT) mice were injected into lethally irradiated recipient mice. Four weeks after transplantation, mice were treated with TAM. (B) Donor percentages in different blood lineages in Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 transplanted mice were analyzed by flow cytometry 45 d after TAM administration. (C) CBC analysis of Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 transplanted mice 45 d after TAM. (D,E) Donor percentages in different lineages in the BM (D) or spleens (E) from Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 transplanted mice 20–65 d after TAM injection. (F) The left panel shows representative images of spleens from Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 transplanted mice after TAM injection. The right graph shows spleen weights. (G) CFU-G/M (left) or CFU-E (right) progenitors in the spleens of Cblf/f;Cbl-bf/f and Cblf/f;Cbl-bf/f;CreERT2 transplanted mice after TAM injection are shown. n = 9. (H) A Kaplan-Meier survival curve of Cblf/f;Cbl-bf/f (n = 20) and Cblf/f;Cbl-bf/f;CreERT2 (n = 19) transplanted mice after TAM injection is shown. P-value was calculated by log-rank t-test. (I) BM cells from Cblf/f;Cbl-bf/f;CreERT2 transplanted mice were injected into cohorts of recipients. Secondarily transplanted mice were subjected to either vehicle, ruxolitinib, or quizartinib treatment. CBC analyses 6 wk after drug treatment are shown. (J) The numbers of various donor-derived HSPC populations in the spleens of Cblf/f;Cbl-bf/f;CreERT2 transplanted mice with different drug treatments are shown. (K) Cell numbers of myeloid and lymphoid lineages in the spleens of drug-treated mice were quantified by cell count and flow cytometry. Each symbol represents an individual mouse. Horizontal lines indicate mean frequencies. Error bars indicate SE. (*) P < 0.05; (**) P < 0.01; (***) P < 0.001; (ns) not significant.
JAK inhibition abrogates Cbl−/−;Cbl-b−/− CMML and inhibits the expansion of myeloid MPPs
The development of a CMML-like disease that closely resembles human CBL mutated myeloid malignancies in Cbl;Cbl-b conditional double-knockout mice suggests their potential for preclinical testing. Of note, inhibitors to two known CBL targets in hematopoietic cells, KIT and FLT3, have been tested in the RF knock-in CblC379A/− mice that develop a similar disease but with a longer latency (Rathinam et al. 2010). While the FLT3 inhibitor (FLT3i) quizartinib temporarily alleviated CMML development in this model, the c-Kit/Src inhibitor dasatinib failed to impact the disease in CblC379A/− mice (Taylor et al. 2012; Duyvestyn et al. 2014). Based on our new findings, we set out to test whether JAK2 inhibition could abrogate CMML development in Cbl;Cbl-b conditional double-knockout mice. We isolated BM cells from Cbl;Cbl-b conditional double-knockout mice that developed CMML and transplanted them into cohorts of secondary recipients. Mice were then divided randomly into three groups, and vehicle, ruxolitinib, or quizartinib was delivered to each group through oral gavage once daily for 6 wk. Ruxolitinib significantly reduced neutrophil counts in Cbl;Cbl-b conditional double-knockout transplanted mice while sparing lymphoid cells or red blood cells (RBCs). Different from the effect of ruxolitinib, quizartinib significantly reduced the lymphocyte and RBC counts in addition to reducing the neutrophil counts (Fig. 5I), suggesting a less specific myelosuppressive effect. Our data raise an interesting possibility that JAKis might be superior to FLT3is in blunting Cbl;Cbl-b-null CMML development.
Since JAK2 signaling is crucial for HSPC development, we evaluated the effects of these drugs on various stem and progenitor compartments. Strikingly, we found that ruxolitinib decreased the numbers of myeloid-biased MPP3 and MegE-biased MPP2 while alleviating the suppression of lymphoid-biased MPP4 in the spleen (Fig. 5J). In contrast, quizartinib failed to significantly affect HSPCs (Fig. 5J). In agreement with the effects on HSPCs, ruxolitinib markedly reduced mature myeloid cells but had negligible effects on B cells or T cells in the spleen (Fig. 5K). In contrast, quizartinib failed to significantly impact mature myeloid cells; instead, it decreased B-cell and T-cell numbers (Fig. 5K). Morphological analysis of BM and spleens from ruxolotinib-treated mice showed decreased myelomonocytes and increased erythrocytes that are consistent with reduced disease burden (Supplemental Fig. S9). Hence, our data indicate that JAKi is effective in treating Cbl;Cbl-b-deficient CMML in mice, potently suppressing myeloid-biased MPPs.
Primary human CBLmut AMLs show elevated JAK2 levels and signaling and are sensitive to JAK inhibition
CBL mutations are found in a variety of myeloid malignancies, particularly in MDS/MPNs (Niemeyer et al. 2010). Our new findings prompted us to investigate JAK2 regulation in human CBLmut myeloid neoplasms. We found that healthy BM mononuclear cells (MNCs) showed very little signaling response to GM-CSF in comparison with CBLmut leukemia cells (secondary AML after MDS/MPN with CBL loss-of-function mutations) (Supplemental Fig. S10; Supplemental Table S2). Thus, we compared primary CBLmut AMLs with CBLWT myelomonocytic AMLs (Supplemental Table S2). Our data demonstrated that freshly thawed CBLmut AML cells exhibited elevated JAK2 levels and an extended JAK2 half-life in comparison with CBLWT AMLs (Fig. 6A,B). Moreover, CBLmut AMLs showed enhanced sensitivity and more sustained JAK2/STAT5 signaling in response to GM-CSF compared with CBLWT AMLs (Fig. 6C,D). To examine the effects of kinase inhibition on AML growth, we subjected primary AML cells to treatment with increasing concentrations of JAKi or FLT3i. Notably, CBLmut AMLs were more sensitive to JAKi (ruxolitinib) but not to FLT3i (quizartinib) compared with CBLWT AMLs in growth assays (Fig. 6E,F). Hence, our data suggest that JAK2 is an important mediator of CBL functions in myeloid malignancies that can be exploited for therapeutic purposes.
Figure 6.

CBLmut AMLs show enhanced STAT5 activation, elevated JAK2 levels, and hypersensitivity to JAKi compared with CBLWT AMLs. (A) Equal numbers of peripheral blood-derived MNCs and BM-derived MNCs from CBLWT and CBLmut AMLs were subjected to Western blot for JAK2 levels. (B) JAK2 half-lives in CBLWT and CBLmut AML cells were examined by Western blot upon GM-CSF and CHX treatment. (C,D) CBLWT and CBLmut AML cells were stimulated with GM-CSF for the indicated times (C) or with a graded dose of GM-CSF for 10 min (F). Cell lysates were subjected to Western blot analysis with the indicated antibodies. (E,F) AML cells were plated in cytokines and a graded dose of ruxolitinib (E) or quizartinib (F). Forty-eight hours later, live-cell numbers were quantified by MTT. CBLmut samples are labeled as “Cbl” in red lines, while CBLWT samples are indicated in blues lines.
Discussion
JAK2 plays an essential role in normal and malignant HSPCs. JAK2 protein level and activity are tightly regulated to ensure its critical function in normal hematopoiesis and prevent malignant transformation. Thus far, several E3 Ub ligases for JAK2 have been proposed. SOCS proteins, especially SOCS1 and SOCS3, have been reported to be STAT target genes and promote JAK2 proteasomal degradation in a negative feedback loop where the E3 ligase VHL interacts with SOCS1 to mediate proteasome-dependent JAK2 degradation (Russell et al. 2011). In addition, Notch-induced Asb2 is reported to bridge JAK2 and Cullin1, 5-base E3 ligases, therefore mediating JAK2 degradation in proteasomes (Nie et al. 2011). However, all of these experiments have relied on overexpression studies, and we found that JAK2 stability was not affected by Cullin inhibitors (data not shown). Structural studies demonstrated that SOCS3 simultaneously binds to JAK2 and the cytokine receptor to which it is engaged, revealing how specificity is generated in SOCS action (Babon et al. 2012; Kershaw et al. 2013). In addition, genetic studies showed that SOCS proteins affect only a limited number of cytokine receptors; e.g., SOCS1 regulates IFNγ and EPOR signaling (Alexander et al. 1999), while SOCS3 regulates IL-6 and G-CSF signaling (Croker et al. 2003, 2004). None of these proteins, when lost in vivo, increases JAK2 protein levels in HSPCs, expands HSC pools, or enhances multiple lineage hematopoiesis. Thus, the E3 ligases regulating JAK2 stability and activity in HSPCs have remained elusive.
In this study, we identified a novel signaling axis indicating that CBL family E3 Ub ligases regulate JAK2 stability and signaling amplitude/duration through the adaptor protein LNK. One important distinction between SOCS and CBL is that SOCS proteins are transcriptionally induced by JAK/STAT, while LNK/CBL actions are membrane-proximal signaling events for immediate and rapid down-modulation of JAK signaling. Surprisingly, we found that JAK2 degradation is independent of that of MPL despite its reliance on the receptor for activation. Depletion of CBL/CBL-B increases the cell surface levels of MPL under basal conditions and impairs its down-regulation from the cell surface upon ligand-induced activation, consistent with a previous report (Saur et al. 2010). However, we showed that CBL proteins do not affect MPL degradation kinetics but regulate JAK2 stability. Our results imply that different pools of MPL may exist on the cell surface, associated with either inactive or active JAK2. The inactive JAK2 proteins are not ubiquitinated and consequently are stable, while the phosphorylated/active JAK2 proteins interact with CBL proteins via LNK for their prompt ubiquitination and degradation. Activated JAK2 may be sorted through routes for degradation that are distinct from MPL.
We showed for the first time that endogenous JAK2 is K63-ubiquitinated in a cytokine-inducible manner that correlates with JAK2 phosphorylation, activation, and prompt degradation as a negative feedback mechanism. We found that, in the heterologous overexpression system, JAK2 is both K48- and K63-ubiquitinated. However, we were able to detect only K63 ubiquitination of endogenous JAK2 in hematopoietic cells. Nonetheless, JAK2 may be subjected to modifications with multiple types of poly-Ub chains and also regulated by multiple E3 ligases. Thus, different types of poly-Ub chains could coexist but be conjugated with different kinetics and subjected to chain editing. CBL proteins are known to promote K63-Ub of RTKs for their trafficking and lysosomal degradation as well as K48-Ub-mediated proteasomal degradation (Thien and Langdon 2005; Mohapatra et al. 2013). Detailed and comprehensive biochemical studies to identify CBL protein-dependent-specific Ub chain linkages on JAK2, the signals that trigger JAK2 degradation, and its degradation routes warrant future studies.
It has been shown that CBL, but not RF mutants, causes decreased JAK2 (and LYN) protein expression and increased proteasomal degradation (Nagao et al. 2011; Javadi et al. 2013). However, studies of mechanism have not been reported previously. This study reveals a novel CBL–LNK–JAK2 signaling complex that regulates JAK2 ubiquitination, stability, and activity. Using in vitro ubiquitination assays, we demonstrated that JAK2 is a direct substrate of CBL. In the in vitro settings where high concentrations and proximity can drive ubiquitination of the substrates, LNK is dispensable for the ubiquitination of JAK2 by CBL. Although not required, LNK further promotes JAK2 ubiquitination kinetics in vitro. We believe that, in cells, LNK plays an important role in bridging the JAK2–CBL interaction. We showed previously that LNK binds to phosphorylated JAK2 upon TPO stimulation (Bersenev et al. 2008), which is consistent with the activation/phosphorylation-dependent JAK2 ubiquitination and degradation mediated by CBL. Previous studies have suggested that Src family kinases, such as LYN, are activators of CBL (tyrosine phosphorylation) and substrates of CBL for ubiquitination (Javadi et al. 2013). We found that Src inhibition reduces CBL phosphorylation as well as CBL E3 function, as evidenced by increased half-lives for both JAK2 and CBL, indicating LYN as an activator of CBL. CBL regulates its own ubiquitination and degradation in addition to its substrates, which include positive signaling proteins such as JAK2, KIT, and FLT3 as well as negative regulators such as LYN (Javadi et al. 2013) and LNK (this work). Taken together, previous work and our data suggest a complex self-inhibitory signaling circuit that curtails cytokine receptor/kinase signaling as well as CBL activity.
We found that dual depletion of CBL and CBL-b results in more pronounced effects on JAK2 stability and JAK2-mediated cell growth than individual depletion, providing a plausible explanation for the moderate phenotypes observed in single-knockout cells and mice. Importantly, in vitro excision of Cbl and Cbl-b in primary murine HSPCs increased JAK2 levels and signaling. Cbl/Cbl-b-null HSCs phenocopy Lnk−/− HSCs, exhibiting superior transplantability and enhanced HSC self-renewal. In vivo excision of Cbl and Cbl-b in mice transplanted with total BM cells led to a lethal CMML-like disease, similar to our previous results (Naramura et al. 2010; An et al. 2016). It is noted that in vivo excision of Cbl/Cbl-b gave rise to a more severe disease than that of in vitro excision, although both resulted in a myeloid-biased expansion of HSPCs as well as mature myeloid cells. The more severe disease may be due to the higher numbers and the types of HSPCs/progenitors that are subjected to Cbl/Cbl-b deletion. Higher numbers of HSPCs in conjunction with committed myeloid progenitors in the transplanted whole BM versus those in purified HSPCs may contribute to the aggressive CMML phenotype observed upon in vivo excision. Of note, we found a striking expansion of HSPCs in the spleens of Cbl/Cbl-b-deleted mice. This extramedullary hematopoiesis, in particular the myeloid proliferation and myeloid infiltration, is consistent with clinical manifestation of CMML or JMML.
Our results show that JAK2 is a major target of CBL proteins in hematopoietic cells, in particular HSPCs. Importantly, CBL-inactivated or CBL/CBL-B-depleted cells remain sensitive to JAK inhibition, which prompted us to test JAK inhibition in vivo. We demonstrated that the JAKi ruxolitinib significantly reduced the numbers of HSPCs and myeloid-biased MPPs, reduced extramedullary hematopoiesis, and mitigated CMML development in Cbl/Cbl-b-deleted mice. Our results reveal that the efficacy of JAK inhibition is superior to FLT3 inhibition, since JAK inhibition reduces HSC and myeloid MPP expansion while sparing lymphoid MPPs. Of note, JAK2 is a major, but not the only, functionally relevant substrate for CBL proteins in hematopoietic cells. CBL has been found to promote the ubiquitination of gp130 and the GM-CSFR (Tanaka et al. 2008; Javadi et al. 2013), and Cbl-null or Cbl/Cbl-b-null HSPCs show impaired KIT and FLT3 surface down-regulation and hypersensitivity to SCF, GM-CSF, and FLT3L (Rathinam et al. 2010; An et al. 2016), all of which may synergistically promote CMML development in Cbl/Cbl-b-deleted mice.
To test the potential translational implications of our mechanistic work, we found that CBL-inactivating mutations in human AMLs closely correlate with increased amplitude and duration of JAK2/STAT5 signaling in comparison with AMLs without CBL mutations. We demonstrated that primary human CBLmut AMLs exhibit elevated JAK2 protein levels and extend the half-life of JAK2, forming the basis for the enhanced GM-CSF-induced STAT5 signaling observed in these AMLs. Strikingly, JAKis, but not FLT3is, can stratify myelomonocytic AMLs into two subgroups, with CBLmut being more sensitive than CBLWT to JAK inhibition. A biologic hallmark of JMML/CMML is their hypersensitivity to GM-CSF in both cell growth and pSTAT5 activation (Kotecha et al. 2008; Niemeyer et al. 2010; Padron et al. 2013). Among CMML patients, the pSTAT5-hypersensitive response correlated positively with high-risk disease and signaling-associated mutations (Padron et al. 2013). Our data provide mechanistic underpinnings of cytokine hyperresponsiveness in these diseases and predict particular sensitivity in those patients with dysregulated LNK, CBL, or JAK2 aberrations.
Taken together, CBL, JAK2, and LNK all play critical roles in normal and malignant hematopoiesis with overlapping and concerted functions. Our studies unravelled a novel signaling axis, CBL–LNK–JAK2, in regulating JAK2 protein turnover and signaling in HSPCs. JAKis are in clinical phase 1 studies in CMML (Padron et al. 2016). Our results implicate JAK2 as a major mediator of aberrant CBL functions in myeloid neoplasms. Given that MDS/MPN/AMLs with CBL mutations have a poor prognosis, our studies reveal a precision approach in treating these diseases.
Materials and methods
Mice, cell lines, and primary human samples
Cblf/f;Cbl-bf/f mice were generated as described (Goetz et al. 2016) and crossed to creERT2 mice from Jackson Laboratories. Lnk−/− mice were generously provided by Dr. Tony Pawson and Dr. Laura Velazquez (Samuel Lunenfeld Research Institute, Toronto, Canada). BM- or peripheral blood-derived MNCs from deidentified AML patients were obtained from the Stem Cell and Xenograft Core Facility at University of Pennsylvania. TF-1 and HEL cells were purchased from American Type Culture Collection. HEL cells were cultured in RPMI with 10% bovine calf serum. TF-1 cells were maintained in RPMI supplemented with 10% CS and 2 ng/mL hGM-CSF (PeproTech).
Constructs and antibodies
MSCV-Pgk-PAC-CBL wild type and C381A mutant were kindly provided by Dr. E. Richard Stanley (Xiong et al. 2011). pOZ-hLNK wild type and R392E mutants were generated as described previously (Jiang et al. 2012). Human CBL miR30-based shRNA (CCCGTACTATCTTGTCAAG) was constructed into PIG (MSCV-puro-IRES-GFP) retroviral vector. LentiCas9-Blast vector was used for CAS9 expression. Human CBL-b gRNA (TTCCGCAAAATAGAGCCCCA) and human LNK knockout (GGTCGAAGAGCTCCAGCACG) were inserted into lentiGuide-Puro or RFP vector.
Antibodies for biochemical studies were purchased from the following vendors: anti-pY1007/1008-JAK2 (no. 3776), JAK2 (no. 3230), pY697-STAT5 (no. 9351), pS473-AKT (no. 4051), AKT (no. 9102), pT202/204-ERK1/2 (no. 9106), ERK1/2 (no. 9102), pY507-Lyn (no. 2731), Lyn (no. 2732), and pan-Ub (P4D1, no. 3936) antibodies were from Cell Signaling Technology, Inc.; STAT5 (sc-835), Actin (sc-1616), and CBL-b (sc-8006) were from Santa Cruz Biotechnology; anti-pTyr (4G10) was from Millipore; and CBL (610441) was from BC Biosciences. hLNK and MPL antibodies were generated as described previously (Bersenev et al. 2008; Jiang et al. 2012).
Western blot and half-life assays
For Western blot analysis, TF-1 cells were starved for 1–3 h in RPMI medium (Invitrogen) plus 0.5% BSA and then treated with or without hTPO. Cells were directly lysed in LDS lysis buffer (Invitrogen) and resolved in SDS-PAGE. The blots were probed with the indicated antibodies. For CHX chase assay, TF-1/MPL cells were starved and then treated with 40 µg/mL CHX in the absence or presence of 100 ng/mL hTPO for the indicated time periods. Protein half-lives were analyzed by Western blot.
Immunoprecipitation
TF-1 cells were lysed in immunoprecipitation buffer (10 mM Tris at pH 7.4, 150 mM NaCl, 0.5% NP-40, 1 mM NaF, 1 mM Na3VO4, PMSF, protease inhibitor cocktail) for 30 min at 4°C. After clarification by centrifugation, cell lysates were precleared with protein A/G beads for 1 h and then incubated with anti-Flag beads (Sigma-Aldrich, M2) for 4 h at 4°C. Beads were then washed extensively and boiled in 50 µL of loading buffer. For pTyr (4G10) immunoprecipitation, TF-1/MPL cells were starved and pretreated with DMSO or 1 µM dasatinib for 1 h and then stimulated with or without TPO for 10 min. Cell lysates were used for immunoprecipitation as described above.
Ub pull-down assay
Ub pull-down assays were performed according to the manufacturer's protocol. Briefly, TF-1/MPL cells were starved and then either left untreated or stimulated with 100 ng/mL hTPO for 10 min. Cells were lysed in the lysis buffer (100 mM Tris-HCL at pH 8.0, 150 mM NaCl, 5 mM EDTA, 1% NP-40, 0.5% Triton-X 100, protease inhibitor cocktail [Roche], 100 µM PR-619 [LifeSensors], 4 mM 1,10-phenanthroline [o-PA; Mallinckrodt Chemicals], 4 mM N-ethylmaleimide [NEM; Sigma-Aldrich], 100 nM Flag K63-TUBE [LifeSensors, UM604]) for 1 h. Cell lysates were clarified by centrifugation, and the supernatant was diluted fourfold and then incubated for 2 h at 4°C to allow for binding of Flag-K63-TUBE to poly-Ub chains. Flag M2 affinity resins were then added to cell lysates and incubated for 2 h at 4°C. Precipitates were eluted with LDS loading buffer and then subjected to Western blot analysis. For pull-down of total ubiquitinated proteins, cells were pretreated with MG132 for 100 min in starvation medium and then stimulated with or without TPO for 20 min. Protein lysates were precipitated with anti-JAK2 antibodies, subjected to Far-Western blot analysis using TUBE1-biotin (LifeSensors, UM301) followed by streptavidin-conjugated HRP, and then developed with standard ECL reagents.
In vitro ubiquitination assay
For in vitro ubiquitination assay, Sf9-purified JAK2 (amino acids 532–1132) (SRP0172, Sigma-Aldrich) was incubated with or without 20 nM CBLTKB+RING (Progenra) in ubiquitination reaction buffer (50 mM bicine at pH 8.0, 5 mM MgCl2, 0.05% CHAPS, 5 nM E1, 200 nM E2 [UBE2D3], 600 nM Ub wild type, 200 nM biotin-Ub, 400 µM ATP, 1 mM β-mercaptoethanol) for 1 h at 37°C (Riling et al. 2015). The reaction was stopped by directly adding SDS loading buffer. Samples were subjected to Western blot analysis using streptavidin-HRP and anti-JAK2 antibodies.
For TR-FRET assay, recombinant Myc-JAK2 protein was purified from insect Sf9 cells using anti-Myc (9B11 beads; Cell Signaling Technology, Inc., no. 3400). LNK (amino acids 301–end) protein was purified from Escherichia coli BL21 (DE3). TR-FRET assay was performed as described previously (Riling et al. 2015). Briefly, the reaction was reconstituted by adding CBL E3 ligase, LNK, and JAK2 on beads with ubiquitination mixture (0.2 mM ATP, 5 nM E1, 100 nM UBE2D3 E2) and TR-FRET detection mixture (biotin-TUBE1[LifeSensors], streptavidin-terbium [CisBio], and wild-type and fluorescein-labeled Ub [LifeSensors, SI270F]) in reaction buffer. The fluorescence signal was captured in real time by PerkinElmer Life Sciences Envision plate reader (excitation: 340 nm; emission 1: 520 nm; emission 2: 480 nm). The TR-FRET ratio was calculated by emission520/emission480.
Cell growth assay
Triplicates of various TF-1 cell lines were seeded in RPMI plus 10% calf serum (100 µL per well; 2 × 105/mL) in 96-well plates in the presence of different concentrations of cytokines or inhibitors. Three days after culture, 3-(4,5-dimethylthiazole-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma-Aldrich) was added to cell medium at a final concentration of 0.5 mg/mL for 4 h at 37°C. The reaction was subsequently terminated by stopping buffer, and the absorbance was measured at OD550–600 in a spectrophotometer.
HSPC purification and BM transplantation (BMT)
Lineage− cells were first isolated from BM cells with a lineage cell depletion kit (Miltenyi Biotec). LSK or LKS− cells were sorted on a FACS Aria cytometer (BD Biosciences) and then cultured in SFEM medium (StemCell Technologies, Inc.) plus 10% FBS (SAFC Biosciences) with 20 ng/mL Flt3L, 20 ng/mL IL-6, 100 ng/mL SCF, and 20 ng/mL TPO for 2 d in the presence of 200 nM 4-OHT (Sigma-Aldrich) to induce Cre expression. For BMT, 2000 LSKs were cultured in each well in a 96-well plate. The resultant cells (CD45.2) were mixed with 3 × 105 total BM cells from isogenic competitor mice (CD45.1/CD45.2) and then injected into irradiated recipient mice (CD45.1) that were irradiated with a split dose of 10 Gy using Orthovoltage precision X-ray.
For Cbl/Cbl-b excision in vivo, we transplanted 2 × 106 total BM cells from Cblf/f;Cbl-bf/f or Cblf/f;Cbl-bf/f;CreERT2 mice into lethally irradiated recipient mice. Four weeks after BMT, transplanted mice were intraperitoneally injected with TAM (40 mg per kilogram of body weight) daily for four consecutive days. Twelve days and 45 d after TAM treatment, peripheral blood was collected for CBC and flow cytometric analysis. For secondary BMT, 2 × 106 total BM cells from primary recipient mice were injected into each lethally irradiated mouse.
CBC analysis and flow cytometric analysis of HSPCs and cells of different hematopoietic lineages
Cells from peripheral blood, BM, or spleens were collected and stained with different fluorescence-conjugated antibodies to different lineages: PE-Gr1 and APC-Mac1 for myeloid cells, PE-CD4 and CD8 for T cells, and APC-B220 for B cells, along with FITC-CD45.1 and APC-Cy7-CD45.2 to differentiate donor cells from competitor/host cells. Flow cytometry was performed on a FACS Canto analyzer. CBC was measured using a Hemavet 950 (Drew Scientific, Inc.).
For HSPC analysis, BM or spleen cells were stained with FITC-CD45.1, APC-CD45.2, biotinylated lineage cocktail (B220, CD4, CD5, CD8, CD19, Gr1, Mac1, and Ter119), PerCP-Cy5.5-Sca1, APC-Cy7-Kit, PE-Cy7-CD150, FITC-CD48, and PE-Flk2 antibodies followed by streptavidin-PE-Texas Red secondary antibodies. Cells were resuspended in DAPI-containing FACS buffer and subjected to flow cytometric analysis on LSR Fortessa.
Colony assays
Mouse BM or spleen cells were plated in semisolid methylcellulose culture (Stem Cell Technologies) according to the manufacturer's recommendations with M3434 medium for CFU-G/M colonies or M3334 medium for CFU-E colonies. The colony numbers were scored 2 d (M3334) or 8–12 d (M3434) later.
Drug treatment in vivo
Secondarily transplanted mice with Cblf/f;Cbl-bf/f;CreERT2 mice treated with TAM developed CMML at ∼9 wk. Mice were divided randomly into three groups and treated by oral gavage daily with vehicle, the JAKi ruxolitinib (60 mg per kilogram body weight), or the FLT3i quizartinib (10 mg per kilogram of body weight). Ruxolitinib was dissolved in PEG300/5% glucose (1:3), while quizartinib (both from Medkoo Biosciences) was dissolved in 5% 2-hydroxypropyl-β-cyclodextrin (Sigma-Aldrich). At 6 wk, mice were sacrificed for CBC and flow analysis for HSPC and lineage distributions in the BM and spleen.
Statistics
Statistics for MTT assays were performed by two-tailed Student's t-test, and error bars indicate mean ± SD. Kaplan-Meier survival curves were graphed in GraphPad Prism software with log-rank t-test. All other continuous variables were compared using a two-sided t-test or one-way ANOVA followed by Tukey's test for multiple comparisons, and error bars represent mean ± SEM.
Supplementary Material
Acknowledgments
We thank Dr. Martin Carroll and Ms. Rachel Astles at the Human Stem Cell and Xenotransplant Core for AML samples. We are grateful to Dr. Richard Stanley for CBL constructs and insightful discussions with Dr. Roger Greenberg. W.T. is supported by National Institutes of Health grant R01HL095675, and W.Y.L. is supported by National Health and Medical Research grants 1057762 and 1101318. W.T. received awards from Gabrielle's Angel Foundation for Cancer Research and Alex's Lemonade for Childhood Cancer Research and is a Leukemia Lymphoma Society (LLS) Scholar. H.B. is supported by National Institutes of Health grants CA87986 and CA105489 and the Dr. Rafael Bonita Memorial Fund for Cancer Research.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.297135.117.
References
- Alexander WS, Starr R, Fenner JE, Scott CL, Handman E, Sprigg NS, Corbin JE, Cornish AL, Darwiche R, Owczarek CM, et al. 1999. SOCS1 is a critical inhibitor of interferon γ signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell 98: 597–608. [DOI] [PubMed] [Google Scholar]
- An W, Mohapatra BC, Zutshi N, Bielecki TA, Goez BT, Luan H, Iseka F, Mushtaq I, Storck MD, Band V, et al. 2016. VAV1-Cre mediated hematopoietic deletion of CBL and CBL-B leads to JMML-like aggressive early-neonatal myeloproliferative disease. Oncotarget 7: 59006–59016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babon JJ, Kershaw NJ, Murphy JM, Varghese LN, Laktyushin A, Young SN, Lucet IS, Norton RS, Nicola NA. 2012. Suppression of cytokine signaling by SOCS3: characterization of the mode of inhibition and the basis of its specificity. Immunity 36: 239–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, et al. 2011. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med 364: 2496–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersenev A, Wu C, Balcerek J, Tong W. 2008. Lnk controls mouse hematopoietic stem cell self-renewal and quiescence through direct interactions with JAK2. J Clin Invest 118: 2832–2844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersenev A, Wu C, Balcerek J, Jing J, Kundu M, Blobel GA, Chikwava KR, Tong W. 2010. Lnk constrains myeloproliferative diseases in mice. J Clin Invest 120: 2058–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersenev A, Rozenova K, Balcerek J, Jiang J, Wu C, Tong W. 2012. Lnk deficiency partially mitigates hematopoietic stem cell aging. Aging Cell 11: 949–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buza-Vidas N, Antonchuk J, Qian H, Mansson R, Luc S, Zandi S, Anderson K, Takaki S, Nygren JM, Jensen CT, et al. 2006. Cytokines regulate postnatal hematopoietic stem cell expansion: opposing roles of thrombopoietin and LNK. Genes Dev 20: 2018–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caligiuri MA, Briesewitz R, Yu J, Wang L, Wei M, Arnoczky KJ, Marburger TB, Wen J, Perrotti D, Bloomfield CD, et al. 2007. Novel c-CBL and CBL-b ubiquitin ligase mutations in human acute myeloid leukemia. Blood 110: 1022–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croker BA, Krebs DL, Zhang JG, Wormald S, Willson TA, Stanley EG, Robb L, Greenhalgh CJ, Forster I, Clausen BE, et al. 2003. SOCS3 negatively regulates IL-6 signaling in vivo. Nat Immunol 4: 540–545. [DOI] [PubMed] [Google Scholar]
- Croker BA, Metcalf D, Robb L, Wei W, Mifsud S, DiRago L, Cluse LA, Sutherland KD, Hartley L, Williams E, et al. 2004. SOCS3 is a critical physiological negative regulator of G-CSF signaling and emergency granulopoiesis. Immunity 20: 153–165. [DOI] [PubMed] [Google Scholar]
- Duyvestyn JM, Taylor SJ, Dagger SA, Orandle M, Morse HC III, Thien CB, Langdon WY. 2014. Dasatinib targets B-lineage cells but does not provide an effective therapy for myeloproliferative disease in c-Cbl RING finger mutant mice. PLoS One 9: e94717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ema H, Sudo K, Seita J, Matsubara A, Morita Y, Osawa M, Takatsu K, Takaki S, Nakauchi H. 2005. Quantification of self-renewal capacity in single hematopoietic stem cells from normal and Lnk-deficient mice. Dev Cell 8: 907–914. [DOI] [PubMed] [Google Scholar]
- Goetz B, An W, Mohapatra B, Zutshi N, Iseka F, Storck MD, Meza J, Sheinin Y, Band V, Band H. 2016. A novel CBL-Bflox/flox mouse model allows tissue-selective fully conditional CBL/CBL-B double-knockout: CD4-Cre mediated CBL/CBL-B deletion occurs in both T-cells and hematopoietic stem cells. Oncotarget 7: 51107–51123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjerpe R, Aillet F, Lopitz-Otsoa F, Lang V, England P, Rodriguez MS. 2009. Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep 10: 1250–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javadi M, Richmond TD, Huang K, Barber DL. 2013. CBL linker region and RING finger mutations lead to enhanced granulocyte–macrophage colony-stimulating factor (GM-CSF) signaling via elevated levels of JAK2 and LYN. J Biol Chem 288: 19459–19470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J, Balcerek J, Rozenova K, Cheng Y, Bersenev A, Wu C, Song Y, Tong W. 2012. 14-3-3 regulates the LNK/JAK2 pathway in mouse hematopoietic stem and progenitor cells. J Clin Invest 122: 2079–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushansky K. 2003. Thrombopoietin: a tool for understanding thrombopoiesis. J Thromb Haemost 1: 1587–1592. [DOI] [PubMed] [Google Scholar]
- Kershaw NJ, Murphy JM, Liau NP, Varghese LN, Laktyushin A, Whitlock EL, Lucet IS, Nicola NA, Babon JJ. 2013. SOCS3 binds specific receptor-JAK complexes to control cytokine signaling by direct kinase inhibition. Nat Struct Mol Biol 20: 469–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N, Flores NJ, Irish JM, Simonds EF, Sakai DS, Archambeault S, Diaz-Flores E, Coram M, Shannon KM, Nolan GP, et al. 2008. Single-cell profiling identifies aberrant STAT5 activation in myeloid malignancies with specific clinical and biologic correlates. Cancer Cell 14: 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine RL, Pardanani A, Tefferi A, Gilliland DG. 2007. Role of JAK2 in the pathogenesis and therapy of myeloproliferative disorders. Nat Rev Cancer 7: 673–683. [DOI] [PubMed] [Google Scholar]
- Loh ML, Sakai DS, Flotho C, Kang M, Fliegauf M, Archambeault S, Mullighan CG, Chen L, Bergstraesser E, Bueso-Ramos CE, et al. 2009. Mutations in CBL occur frequently in juvenile myelomonocytic leukemia. Blood 114: 1859–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima H, Cazzolli H, Szpurka H, Dunbar A, Tiu R, Huh J, Muramatsu H, O'Keefe C, Hsi E, Paquette RL, et al. 2009. Mutations of e3 ubiquitin ligase cbl family members constitute a novel common pathogenic lesion in myeloid malignancies. J Clin Oncol 27: 6109–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlevede J, Droin N, Qin T, Meldi K, Yoshida K, Morabito M, Chautard E, Auboeuf D, Fenaux P, Braun T, et al. 2016. Mutation allele burden remains unchanged in chronic myelomonocytic leukaemia responding to hypomethylating agents. Nat Commun 7: 10767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesa RA, Kiladjian JJ, Verstovsek S, Al-Ali HK, Gotlib J, Gisslinger H, Levy R, Siulnik A, Gupta V, Khan M, et al. 2014. Comparison of placebo and best available therapy for the treatment of myelofibrosis in the phase 3 COMFORT studies. Haematologica 99: 292–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohapatra B, Ahmad G, Nadeau S, Zutshi N, An W, Scheffe S, Dong L, Feng D, Goetz B, Arya P, et al. 2013. Protein tyrosine kinase regulation by ubiquitination: critical roles of Cbl-family ubiquitin ligases. Biochim Biophys Acta 1833: 122–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muramatsu H, Makishima H, Maciejewski JP. 2012. Chronic myelomonocytic leukemia and atypical chronic myeloid leukemia: novel pathogenetic lesions. Semin Oncol 39: 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao T, Oshikawa G, Wu N, Kurosu T, Miura O. 2011. DNA damage stress and inhibition of Jak2-V617F cause its degradation and synergistically induce apoptosis through activation of GSK3β. PLoS One 6: e27397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naramura M, Nandwani N, Gu H, Band V, Band H. 2010. Rapidly fatal myeloproliferative disorders in mice with deletion of Casitas B-cell lymphoma (Cbl) and Cbl-b in hematopoietic stem cells. Proc Natl Acad Sci 107: 16274–16279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nie L, Zhao Y, Wu W, Yang YZ, Wang HC, Sun XH. 2011. Notch-induced Asb2 expression promotes protein ubiquitination by forming non-canonical E3 ligase complexes. Cell Res 21: 754–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemeyer CM, Kang MW, Shin DH, Furlan I, Erlacher M, Bunin NJ, Bunda S, Finklestein JZ, Sakamoto KM, Gorr TA, et al. 2010. Germline CBL mutations cause developmental abnormalities and predispose to juvenile myelomonocytic leukemia. Nat Genet 42: 794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh ST, Simonds EF, Jones C, Hale MB, Goltsev Y, Gibbs KD Jr, Merker JD, Zehnder JL, Nolan GP, Gotlib J. 2010. Novel mutations in the inhibitory adaptor protein LNK drive JAK–STAT signaling in patients with myeloproliferative neoplasms. Blood 116: 988–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padron E, Painter JS, Kunigal S, Mailloux AW, McGraw K, McDaniel JM, Kim E, Bebbington C, Baer M, Yarranton G, et al. 2013. GM-CSF-dependent pSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia. Blood 121: 5068–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padron E, Dezern A, Andrade-Campos M, Vaddi K, Scherle P, Zhang Q, Ma Y, Balasis ME, Tinsley S, Ramadan H, et al. 2016. A multi-institution phase I trial of ruxolitinib in patients with chronic myelomonocytic leukemia (CMML). Clin Cancer Res 22: 3746–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parganas E, Wang D, Stravopodis D, Topham DJ, Marine JC, Teglund S, Vanin EF, Bodner S, Colamonici OR, van Deursen JM, et al. 1998. Jak2 is essential for signaling through a variety of cytokine receptors. Cell 93: 385–395. [DOI] [PubMed] [Google Scholar]
- Pietras EM, Reynaud D, Kang YA, Carlin D, Calero-Nieto FJ, Leavitt AD, Stuart JM, Gottgens B, Passegue E. 2015. Functionally distinct subsets of lineage-biased multipotent progenitors control blood production in normal and regenerative conditions. Cell Stem Cell 17: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam C, Thien CB, Langdon WY, Gu H, Flavell RA. 2008. The E3 ubiquitin ligase c-Cbl restricts development and functions of hematopoietic stem cells. Genes Dev 22: 992–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathinam C, Thien CB, Flavell RA, Langdon WY. 2010. Myeloid leukemia development in c-Cbl RING finger mutant mice is dependent on FLT3 signaling. Cancer Cell 18: 341–352. [DOI] [PubMed] [Google Scholar]
- Riling C, Kamadurai H, Kumar S, O'Leary CE, Wu KP, Manion EE, Ying M, Schulman BA, Oliver PM. 2015. Itch WW domains inhibit its E3 ubiquitin ligase activity by blocking E2-E3 ligase trans-thiolation. J Biol Chem 290: 23875–23887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RC, Sufan RI, Zhou B, Heir P, Bunda S, Sybingco SS, Greer SN, Roche O, Heathcote SA, Chow VW, et al. 2011. Loss of JAK2 regulation via a heterodimeric VHL–SOCS1 E3 ubiquitin ligase underlies Chuvash polycythemia. Nat Med 17: 845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanada M, Suzuki T, Shih LY, Otsu M, Kato M, Yamazaki S, Tamura A, Honda H, Sakata-Yanagimoto M, Kumano K, et al. 2009. Gain-of-function of mutated C-CBL tumour suppressor in myeloid neoplasms. Nature 460: 904–908. [DOI] [PubMed] [Google Scholar]
- Saur SJ, Sangkhae V, Geddis AE, Kaushansky K, Hitchcock IS. 2010. Ubiquitination and degradation of the thrombopoietin receptor c-Mpl. Blood 115: 1254–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seita J, Ema H, Ooehara J, Yamazaki S, Tadokoro Y, Yamasaki A, Eto K, Takaki S, Takatsu K, Nakauchi H. 2007. Lnk negatively regulates self-renewal of hematopoietic stem cells by modifying thrombopoietin-mediated signal transduction. Proc Natl Acad Sci 104: 2349–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JJ, Cohen RE. 2009. Linkage-specific avidity defines the lysine 63-linked polyubiquitin-binding preference of rap80. Mol Cell 33: 775–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims JJ, Scavone F, Cooper EM, Kane LA, Youle RJ, Boeke JD, Cohen RE. 2012. Polyubiquitin-sensor proteins reveal localization and linkage-type dependence of cellular ubiquitin signaling. Nat Methods 9: 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieglitz E, Taylor-Weiner AN, Chang TY, Gelston LC, Wang YD, Mazor T, Esquivel E, Yu A, Seepo S, Olsen SR, et al. 2015. The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet 47: 1326–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Tanaka N, Saeki Y, Tanaka K, Murakami M, Hirano T, Ishii N, Sugamura K. 2008. c-Cbl-dependent monoubiquitination and lysosomal degradation of gp130. Mol Cell Biol 28: 4805–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SJ, Dagger SA, Thien CB, Wikstrom ME, Langdon WY. 2012. Flt3 inhibitor AC220 is a potent therapy in a mouse model of myeloproliferative disease driven by enhanced wild-type Flt3 signaling. Blood 120: 4049–4057. [DOI] [PubMed] [Google Scholar]
- Thien CB, Langdon WY. 2005. Negative regulation of PTK signalling by Cbl proteins. Growth Factors 23: 161–167. [DOI] [PubMed] [Google Scholar]
- Tiu RV, Sekeres MA. 2014. Making sense of the myelodysplastic/myeloproliferative neoplasms overlap syndromes. Curr Opin Hematol 21: 131–140. [DOI] [PubMed] [Google Scholar]
- Tong W, Lodish HF. 2004. Lnk inhibits Tpo–mpl signaling and Tpo-mediated megakaryocytopoiesis. J Exp Med 200: 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungureanu D, Saharinen P, Junttila I, Hilton DJ, Silvennoinen O. 2002. Regulation of Jak2 through the ubiquitin-proteasome pathway involves phosphorylation of Jak2 on Y1007 and interaction with SOCS-1. Mol Cell Biol 22: 3316–3326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk SJ, Fiskin E, Putyrski M, Pampaloni F, Hou J, Wild P, Kensche T, Grecco HE, Bastiaens P, Dikic I. 2012. Fluorescence-based sensors to monitor localization and functions of linear and K63-linked ubiquitin chains in cells. Mol Cell 47: 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velazquez L, Cheng AM, Fleming HE, Furlonger C, Vesely S, Bernstein A, Paige CJ, Pawson T. 2002. Cytokine signaling and hematopoietic homeostasis are disrupted in Lnk-deficient mice. J Exp Med 195: 1599–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Song D, Cai Y, Yu W, Yeung YG, Stanley ER. 2011. A CSF-1 receptor phosphotyrosine 559 signaling pathway regulates receptor ubiquitination and tyrosine phosphorylation. J Biol Chem 286: 952–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.