

In the Outlook from Laptenko and Prives, the authors discuss the study published in this issue by Tovy et al. regarding the control of DNA methylation homeostasis by p53 in embryonic stem cells.
Keywords: DNA methylation, p53, stem cells
Abstract
Long understood as a bona fide tumor suppressor that safeguards the integrity of the genome via regulating numerous cellular outcomes, p53 may also exert its decisive and versatile functions by controlling DNA methylation. In this issue of Genes & Development, Tovy and colleagues (pp. 959–972) report that, in naïve mouse embryonic stem cells (ESCs), p53 controls DNA methylation homeostasis by regulating the expression of key counteracting components of the DNA methylation machinery. Their findings indicate that p53 may exert its “guardian of genome” duties at least in part via safeguarding the epigenome of ESCs.
The tumor suppressor protein p53 has been a major focus of cancer research for more than three decades. Most well studied has been the ability of p53 to induce different biological outcomes, such as transient or permanent cell cycle arrest or cell death in response to diverse stress-related stimuli that lead to highly expressed and extensively modified p53 protein. However, an ever-growing body of evidence suggests that p53 may exert tumor-suppressive functions by contributing to numerous biological processes that are not obviously elicited as a result of genotoxic stress signals (Vousden and Prives 2009). Indeed, p53 has been shown to perform a number of “civic” and not yet fully appreciated duties in the regulation of stem cell renewal, differentiation, and development (Vousden and Prives 2009). Notably, in embryonic stem cells (ESCs), both differentiation and development programs demonstrate a significant degree of dependence on the correct maintenance of the epigenetic state in general and on DNA hypomethylation in particular (Cedar and Bergman 2012). In this issue of Genes & Development, Tovy et al. (2017) report that, in naïve mouse ESCs, p53 controls DNA methylation homeostasis by regulating the expression of a set of components of the DNA methylation machinery (Fig. 1). In an apparently fine-tuned balancing act, p53 represses transcription of two genes, Dnmt3a and Dnmt3b, which encode DNA methyltransferases (DNMTs) and are responsible for de novo cytosine methylation (5-methylcytosine [5mC]) of genomic DNA, while directly activating expression of Tet1 and Tet2, members of the ten-eleven translocation (TET) family of 2OG-Fe(II) dioxygenases, which are necessary for the conversion of 5mC into 5-hydroxymethylcytosine (5hmC). Remarkably, in p53-null mouse ESCs, this situation is reversed: The expression of the TET enzymes is drastically reduced, while both DNMTs undergo significant up-regulation. Consequently, p53-deficient ESC populations differ in their degree of DNA methylation, demonstrating a substantial stochastic increase in universal 5mC content, markedly reduced levels of 5hmC, and overall heterogeneity of their genomic methylation landscape. Hence, the absence of p53 in ESCs results in unbalanced DNA methylation that favors hypermethylation of genomic DNA.
Figure 1.
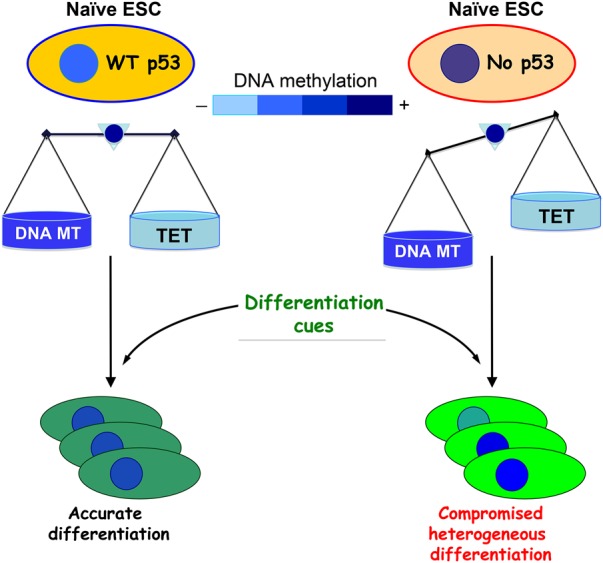
p53 keeps DNA methylation in balance in naïve ESCs. The relative degree of DNA methylation in nuclei (shown schematically as an inner circle inside the bigger oval representing the cell) is kept under control by p53 that down-regulates Dnmt3a and Dnmt3b DNMTs responsible for de novo DNA methylation and up-regulates Tet1 and Tet2 components of demethylation machinery. This balance is perturbed by loss of functional p53 in ESCs with the subsequent stochastic increase in DNA methylation and clonal heterogeneity in both naïve ESCs and the differentiated progenitors.
ESCs possess an intrinsic ability to preserve pluripotency and self-renewal over many generations. This unique and defining feature is determined by a core transcriptional program that is regulated by a complex dynamic network built of interdependent modules, of which DNA methylation is an important component (Tollervey and Lunyak 2012). The DNA methylation pattern of pluripotent ESCs is distinct from that seen in either normal differentiated cells or cancer cells (Bibikova et al. 2006). Previous observations have suggested the existence of a functional link between p53 status and DNA methylation in normal cells as well as cancer cells (Levine and Greenbaum 2012). For example, it has been reported that, prior to developing tumors, p53-null mice demonstrate an increase in global DNA methylation in the thymus and liver due to the up-regulation of the corresponding DNMTs (Park et al. 2005). Here, the work of Tovy et al. (2017) has revealed a direct connection between genomic DNA methylation and p53 transcriptional activity. Their findings identify p53 as a key regulator of the DNA methylation epigenetic branch within the transcription regulation network of ESCs. This implies that p53 may exert its “guardian of genome” duties at least in part via safeguarding the epigenome of ESCs as proposed previously (Levine and Greenbaum 2012).
Given the colossal therapeutic potential of ESCs, defining the molecular mechanisms responsible for their unique biological properties is of paramount importance. A few years ago, several independent studies reported the potential benefits of p53 inactivation for the efficient production of induced pluripotent stem cells (iPSCs) (for review, see Krizhanovsky and Lowe 2009). However, despite the promises stemming from these findings, these reports raised safety issues regarding the clinical applications of iPSCs. While the original concerns were related to the potential tumorigenic consequences associated with the loss of p53, the study by Tovy et al. (2017) has uncovered yet another problem related to the quality of such cells. Specifically, the absence of p53 in ESCs results in a marked increase in their intraclonal heterogeneity, suggesting a stochastic decrease in their “stemness.” In addition, the investigators showed that differentiated cells originating from p53-null naïve ESCs display higher heterogeneity as well. Hence, while p53 inactivation may to some degree ease the production of iPSCs, it may negatively impact the intrinsic properties of ESCs and their differentiation potential. Of course, this observation needs to be verified by future experiments using different types of stem cells and different conditions.
The study by Tovy et al. (2017) poses intriguing questions about p53 and its roles in stem cell biology. First, cell fate is known to be controlled by a collaborative action of stochastic and deterministic regulatory processes. The dynamic methylation equilibrium characteristic of the epigenome of pluripotent stem cells, but not of somatic cells, efficiently counteracts transcription noise (Shipony et al. 2014). Does p53's control over the epigenome lessen during cell differentiation? Second, the presence or absence of p53 correlates with dramatic differences in the expression of genes that regulate DNA methylation. Mechanistically, how does p53 switch from being an activator of Tet gene expression in naïve ESCs to repressing their expression in differentiated cells? Third, does p53 control its own transcriptional program in naïve ESCs via its functions in coordinating DNA methylation? In this regard, Kribelbauer et al. (2017) have just reported a role of 5mC in stabilizing p53 binding to a subset of DNA sequences and, correspondingly, increased in vivo occupancy at genomic regions marked by inactive histone modification. As has often been the case with p53, the more we learn about this fascinating transcription factor, the more is left for future studies to unravel.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.302364.117.
References
- Bibikova M, Chudin E, Wu B, Zhou L, Garcia EW, Liu Y, Shin S, Plaia TW, Auerbach JM, Arking DE, et al. 2006. Human embryonic stem cells have a unique epigenetic signature. Genome Res 16: 1075–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedar H, Bergman Y. 2012. Programming of DNA methylation patterns. Annu Rev Biochem 81: 97–117. [DOI] [PubMed] [Google Scholar]
- Kribelbauer JF, Laptenko O, Chen S, Martini GD, Freed-Pastor W, Prives C, Mann RS, Bussemaker HJ. 2017. Quantitative analysis of the DNA methylation sensitivity of transcription factor complexes. Cell Rep 10.1016/j.celrep.2017.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Lowe SW. 2009. Stem cells: The promises and perils of p53. Nature 460: 1085–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine AJ, Greenbaum B. 2012. The maintenance of epigenetic states by p53: the guardian of the epigenome. Oncotarget 3: 1503–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park IY, Sohn BH, Choo JH, Joe CO, Seong JK, Lee YI, Chung JH. 2005. Deregulation of DNA methyltransferases and loss of parental methylation at the insulin-like growth factor II (Igf2)/H19 loci in p53 knockout mice prior to tumor development. J Cell Biochem 94: 585–596. [DOI] [PubMed] [Google Scholar]
- Shipony Z, Mukamel Z, Cohen NM, Landan G, Chomsky E, Zeliger SR, Fried YC, Ainbinder E, Friedman N, Tanay A. 2014. Dynamic and static maintenance of epigenetic memory in pluripotent and somatic cells. Nature 513: 115–119. [DOI] [PubMed] [Google Scholar]
- Tollervey JR, Lunyak VV. 2012. Epigenetics: judge, jury and executioner of stem cell fate. Epigenetics 7: 823–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovy A, Spiro A, McCarthy R, Shipony Z, Aylon Y, Alton K, Ainbinder E, Furth N, Tanay A, Barton M, et al. 2017. p53 is essential for DNA methylation fidelity in naïve embryonic stem cells, and its loss promotes clonal heterogeneity. Genes Dev (this issue) 10.1101/gad.299198.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vousden KH, Prives C. 2009. Blinded by the light: the growing complexity of p53. Cell 137: 413–431. [DOI] [PubMed] [Google Scholar]