

In this study, Soto-Feliciano et al. describe the function of the plant homeodomain finger 6 (PHF6) protein in leukemia and define its role in regulating chromatin accessibility to lineage-specific transcription factors. Their findings suggest that active maintenance of a precise chromatin landscape is essential for sustaining proper leukemia cell identity and that loss of a single factor (PHF6) can cause focal changes in chromatin accessibility and nucleosome positioning that render cells susceptible to lineage transition.
Keywords: PHF6, phenotypic plasticity, chromatin regulation, leukemia, lineage maintenance, nucleosome positioning
Abstract
Developmental and lineage plasticity have been observed in numerous malignancies and have been correlated with tumor progression and drug resistance. However, little is known about the molecular mechanisms that enable such plasticity to occur. Here, we describe the function of the plant homeodomain finger protein 6 (PHF6) in leukemia and define its role in regulating chromatin accessibility to lineage-specific transcription factors. We show that loss of Phf6 in B-cell leukemia results in systematic changes in gene expression via alteration of the chromatin landscape at the transcriptional start sites of B-cell- and T-cell-specific factors. Additionally, Phf6KO cells show significant down-regulation of genes involved in the development and function of normal B cells, show up-regulation of genes involved in T-cell signaling, and give rise to mixed-lineage lymphoma in vivo. Engagement of divergent transcriptional programs results in phenotypic plasticity that leads to altered disease presentation in vivo, tolerance of aberrant oncogenic signaling, and differential sensitivity to frontline and targeted therapies. These findings suggest that active maintenance of a precise chromatin landscape is essential for sustaining proper leukemia cell identity and that loss of a single factor (PHF6) can cause focal changes in chromatin accessibility and nucleosome positioning that render cells susceptible to lineage transition.
Lymphopoiesis is a carefully orchestrated process in which hematopoietic stem cells differentiate into B cells and T cells through the activation of precise gene expression programs governed by well-described transcription factors. During the process of lymphocyte differentiation, gene expression must be activated, silenced, or maintained. In B-cell development, the accessibility of specific genomic loci for binding of transcription factors (e.g., PU.1, E2A, EBF1, and PAX5) is vital for transcriptional regulation. However, the critical mechanisms regulating the chromatin landscape and transcription factor access have yet to be elucidated (Schebesta et al. 2002).
Lymphoid cells are terminally differentiated but still possess the capacity to transdifferentiate into distinct lineages when subjected to specific genetic perturbations. For example, loss of Pax5 in mature B cells can produce functional T cells in immune-deficient mice (Cobaleda et al. 2007). Similarly, Pax5 deletion in pro-B cells allows for transdifferentiation into macrophages, granulocytes, osteoclasts, dendritic cells, and natural killer cells (Nutt et al. 1999). Overexpression of CEBPα/β can transform mature B and T cells into macrophages (Xie et al. 2004; Laiosa et al. 2006). In addition, Ebf1 loss converts pro-B cells into innate lymphoid cells and T cells (Nechanitzky et al. 2013). Interestingly, these lineage-specific transcription factors are often found to be altered in B-cell acute lymphoblastic leukemia (B-ALL). These findings highlight the plasticity of leukemia cells and how aberrant lymphoid developmental programs can favor leukemogenesis (Horcher et al. 2001; Rathert et al. 2015; Somasundaram and Sigvardsson 2015).
Plant homeodomain finger protein 6 (PHF6) was first described as the single gene mutated in the X-linked intellectual disability called Börjeson-Forssman-Lehmann syndrome (Lower et al. 2002). Inactivating mutations in PHF6 were subsequently identified in human leukemia of the T and myeloid lineages, highlighting its role as a tumor suppressor gene in these malignancies (Van Vlierberghe et al. 2010, 2011). Our group recently described a tumor-promoting role for Phf6 in a murine model of BCR–ABL1+ B-ALL (Williams et al. 2006; Meacham et al. 2015). In this study, we found that hairpin-mediated knockdown of Phf6 leads to impaired growth of B-ALL cells in vivo. Altogether, these observations suggest that PHF6 can act as a tumor suppressor or an oncogene in a lineage-dependent manner. However, the molecular mechanisms underlying PHF6's function in hematological malignancies remain entirely unknown (Fig. 1A).
Figure 1.

Phf6 loss decreases the leukemogenic potential of cells in vivo and triggers a change in disease presentation. (A) PHF6 is a lineage-specific regulator of tumor growth in B-ALL and T-cell acute lymphoblastic leukemia (T-ALL). (B). Kaplan-Meier survival analysis of mice injected with either 103 (dotted) or 106 (solid) Phf6WT (blue) and Phf6KO (red) B-ALL cells. The number (n) of mice per genotype analyzed is shown. Statistical analysis (log-rank test, Mantel-Cox) was performed for the different groups in comparison with mice injected with Phf6WT cells. P-values are shown for the comparisons. (C) Representative hematoxylin and eosin (H&E) (top) and immunohistochemistry (bottom) staining of serial sections from lymph nodes (LNs) and lymphoma (mass) of recipient mice injected with Phf6WT, shPhf6, and Phf6KO cells. mCherry immunochemistry demarcates tumor cells. Bars, 600 µm. (D, top) Size comparison of representative LNs from Phf6WT (left) and Phf6KO (right) recipient mice. (Bottom) Quantification of combined LN weight of Phf6WT (blue; n = 5) and Phf6KO (red; n = 5) recipients. (E) Tumor burden in the blood of Phf6WT (blue; n = 7) and Phf6KO (red; n = 8) recipient mice. mCherry demarcates tumor cells. (F) Bar graphs showing the percentage of the CD4+ fraction among mCherry+ cells isolated from Phf6WT (blue; n = 9) and Phf6KO (red; n = 5) tumors in bone marrow (left) and LNs (right). Data represent the mean ± standard deviation (SD) in D–F. Statistics were calculated with two-sided Student's t-test. (***) P < 0.001; (****) P < 0.0001.
Despite the knowledge obtained through sequencing studies, only a handful of functions have been described for PHF6. The protein contains two atypical PHD-like zinc finger domains, implying the capacity to bind modified histones similar to canonical PHD domains (Wysocka et al. 2006). However, PHF6 has only been shown to bind dsDNA in vitro (Liu et al. 2014). In addition, it has been shown to interact with transcriptional regulatory factors such as the nucleosome remodeling and deacetylation (NuRD) complex, the RNA polymerase II-associated factor 1 (PAF1) transcription elongation complex, and the rRNA transcriptional activator UBF (Todd and Picketts 2012; Wang et al. 2013; Zhang et al. 2013). To better understand the function of PHF6 as a potential chromatin regulator and examine its lineage-specific roles in hematological malignancies, we decided to thoroughly investigate its role in B-ALL. Here, through integrated genomics and in vivo studies, we show that PHF6 regulates the chromatin landscape of B-ALL cells, where it is responsible for maintaining a chromatin state that enables a transformed pre-B-cell identity. PHF6 controls the transcription of target genes by supporting a chromatin configuration that permits or blocks the binding of lineage-specific transcription factors. Furthermore, we show that the associated transcriptional and chromatin state changes that occur in the absence of PHF6 contribute to an emerging mechanism of drug resistance, termed pathway indifference (Cooley et al. 2015). Loss of PHF6 results in chromatin instability and genomic plasticity, which allows malignant cells to reprogram transcriptional outputs and tolerate aberrant lineage signaling.
Results
Loss of Phf6 decreases the leukemogenic potential of B-ALL cells and results in the development of mixed-lineage lymphoma in vivo
Recent studies suggest that PHF6 can act as a lineage-specific regulator of tumor growth. However, the molecular mechanisms underlying PHF6's function in hematological malignancies remain widely unclear (Fig. 1A). To evaluate the effects of complete loss of Phf6 on B-ALL growth, we engineered isogenic Phf6 knockout (Phf6KO) B-ALL cells using CRISPR–Cas9 (Supplemental Fig. S1A–D; Ran et al. 2013; Sánchez-Rivera and Jacks 2015). To maximize the chance of functionally inactivating the PHF6 protein, we used a domain-focused targeting strategy (Shi et al. 2015) by using a single-guide RNA (sgRNA) designed to target the second conserved plant homeodomain (ePHD2), which has been identified as a mutational hot spot in T-cell acute lymphoblastic leukemia (T-ALL) (Van Vlierberghe et al. 2010) and acute myeloid leukemia (AML) (Van Vlierberghe et al. 2011) (Supplemental Fig. S1B–D). The Phf6KO cells were characterized extensively with regard to their in vitro growth properties, showing no significant differences in their proliferation rates and cell cycle profiles when compared with their wild-type counterparts (Supplemental Fig. S1E,F).
To determine the effects of complete loss of Phf6 on B-ALL growth in vivo, we performed syngeneic transplants into immunocompetent recipient mice (Fig. 1B–F). Tumor formation in mice injected with 106 Phf6KO cells was significantly delayed compared with mice transplanted with (1) Phf6WT cells, (2) cells expressing an shRNA targeting Phf6 (shPhf6), and (3) Phf6KO cells rescued with the Phf6 cDNA (Fig. 1B; Supplemental Figs. S1G,H, S2A). When the number of transplanted cells was reduced 1000-fold, Phf6-deficient cells failed to develop detectable leukemia in recipient animals. Thus, complete loss of Phf6 reduces the fitness and leukemia-initiating capacity of B-ALL cells in vivo.
Notably, mice transplanted with Phf6WT and Phf6KO cells developed malignancies with pronounced differences. BCR–ABL1+ B-cell leukemia reproducibly presents with an enlarged spleen and disseminated disease that infiltrates the bone marrow (BM) and peripheral blood, as can be seen upon transplantation with Phf6WT cells (Williams et al. 2006). However, tumors that developed from Phf6KO cells pathologically resembled lymphoma, showing prominent lymphadenopathy with marked blood vessels rooting from and to the lymph nodes (LNs), which completely effaced the normal LN histology in recipient mice (Fig. 1C,D). These tumors resulted in size and combined organ weights that were >200% greater than those of wild-type B-ALL recipients (Fig. 1D). Conversely, tumor burden in the blood and the spleen size of mice injected with knockout cells were significantly reduced relative to those from mice injected with wild-type cells (Fig. 1E; Supplemental Fig. S2B). These changes were confirmed in a second isogenic Phf6KO B-ALL cell line (Supplemental Fig. S3A,B). Moreover, reintroduction of Phf6 cDNA into Phf6KO B-ALL cells resulted in leukemia that was indistinguishable in latency from that produced by Phf6WT B-ALL cells, ruling out off-target effects (Supplemental Figs. S1G,H, S2A). Altogether, the complete loss of Phf6 in B-ALL results in a drastic alteration of the natural ontology of the malignancy (changing from leukemic/disseminated disease to lymphoma) and a consequential extension in survival of recipient mice.
To further investigate the marked differences in disease presentation between Phf6KO and Phf6WT cells, we performed immunophenotypic analyses on tumor cells isolated from the LNs and BM of recipient mice. As expected, Phf6WT tumor cells isolated from the BM and LNs expressed the B-cell markers CD19 and B220 and lacked IgM (Williams et al. 2006). In contrast, tumor cells isolated from Phf6KO B-ALL recipient mice showed a modest but significant reduction of CD19 and B220 levels (Supplemental Fig. S2C–F). Strikingly, these tumor cells also had high levels of CD4 on the cell surface, indicating that a significant proportion of Phf6KO B-ALL cells aberrantly expressed a surface marker characteristic of the T-cell lineage (Fig. 1F; Supplemental Fig. S3C; Janeway et al. 1988; Zhu et al. 2010; Rothenberg et al. 2011). Additional immunophenotypic profiling demonstrated that leukemia cells from both genotypes did not express markers of additional lineages, including surface or intracellular CD3 (Borst et al. 1987) or the myeloid/dendritic cell marker CD11b (Supplemental Fig. S4A,B; Bell and Bhandoola 2008; Wada et al. 2008). As observed previously, loss of Phf6 expression in leukemia cells is detrimental exclusively in the in vivo setting (Meacham et al. 2015). In addition, Phf6KO cells cultured in vitro did not express CD4 (Supplemental Fig. S4C). Importantly, the changes in disease presentation and aberrant expression of CD4 require the complete loss of function of Phf6, as they cannot be recapitulated via potent RNAi-mediated suppression of Phf6 (Supplemental Figs. S2A, S4D–F; Meacham et al. 2015).
To rule out potential effects associated with single-cell cloning, we generated a clonal line from the Phf6WT population and performed similar survival and immunophenotyping experiments (Supplemental Fig. S4G,H). As expected, mice transplanted with the Phf6WT clone did not show a significantly different median survival compared with Phf6WT nonclonal cells. In addition, we did not observe CD4 overexpression in cells isolated from the LNs and BM of mice injected with the wild-type clone, mirroring the disease established by Phf6WT nonclonal cells. Altogether, these results indicate that PHF6 is critical for growth in vivo as a disseminated disease and suggest a role for PHF6 in maintaining a stable B-cell identity in B-ALL.
Phf6-deficient B-ALL cells have altered expression of genes involved in lineage specificity
Intrigued by the underlying phenotypic plasticity observed in Phf6KO B-ALL tumors, we sought to explore any potential transcriptional changes behind this phenotype by performing RNA sequencing (RNA-seq) (Fig. 2; Supplemental Fig. S5). Pairwise differential expression analysis between wild-type and knockout transcriptome profiles showed that Phf6KO cells had a markedly distinct gene expression pattern when compared with Phf6WT cells (Fig. 2A; Supplemental Fig. S5A; Supplemental Table 2). Differentially expressed genes between Phf6KO and Phf6WT cells were significantly enriched in pathways involved in cellular and developmental processes of lymphocytes (Fig. 2B), suggesting that complete loss of Phf6 in B-ALL cells selectively affects the expression of genes involved in lymphoid differentiation and function. Gene set enrichment analysis (GSEA) (Subramanian et al. 2005) of pairwise comparisons revealed that Phf6KO cells had a significant decrease in the expression of genes associated with B-cell development and function (Fig. 2C; Supplemental Fig. S5B, top; Supplemental Table 2; Hoffmann et al. 2002; Mori et al. 2008). Importantly, the expression of many of the genes that formed the leading-edge cluster of this B-cell signature (Cd74, IL4ra, Lyn, Ly86, and Blk) was restored by introduction of the Phf6 cDNA, indicating that these changes in gene expression are caused by the absence of Phf6 in the knockout cells and not an off-target effect (Fig. 2D). Using independent component analysis (ICA), we identified a statistically significant gene signature specific to Phf6KO replicate samples (Fig. 2E; Supplemental Table 2). GSEA of the Phf6KO-specific signature revealed that Phf6KO cells have a notable enrichment of gene sets associated with T-cell signal transduction and function, including CD4+ T-cell-specific pathways (Fig. 2F; Supplemental Fig. S5B, bottom; Supplemental Table 2; Hahtola et al. 2006; Jeffrey et al. 2006; Hutcheson et al. 2008; Abbas et al. 2009; Tokoyoda et al. 2009). These analyses suggest that Phf6KO B-ALL cells adopt a transcriptional program similar to CD4+ T cells upon complete loss of Phf6.
Figure 2.

The absence of Phf6 promotes an altered gene expression program in B-cell leukemia. (A) Heat map showing differentially expressed genes (fold change >4, false discovery rate [FDR] <0.05) in pairwise comparisons between Phf6WT (left) and Phf6KO (right) cells as determined by RNA-seq. Each column represents a replicate sample. The scale corresponds to row-wise standardized log2-transformed expression values for each gene. (B) The top gene ontology (GO) and PANTHER terms found to be enriched in Phf6KO cells. The P-value for each term is plotted as −log10(P-value). (C) GSEA plot depicting significant (P < 0.001) changes in pre-B lymphocyte signature genes upon Phf6 deletion, as compared with Phf6WT cells. (NES) Normalized enrichment score. (D) Quantitative PCR (qPCR) analysis of Phf6WT (blue) and Phf6KO (red) cells transduced with empty vector (EV; solid) or a vector expressing Phf6 cDNA (cDNA; dotted). Relative mRNA levels for B-cell-associated genes are shown: Phf6, Cd22, Cd74, Il4ra, Lyn, Ly86, and Blk. (E) Schematic representation of ICA used to identify differential expression signatures (independent components [ICs]) in the integrated RNA-seq data set comprised of Phf6WT, shPhf6, and Phf6KO cells. Hinton diagram representation of ICA-derived signatures. Columns denote signatures, and rows denote samples. Colors denote relative directionality of gene expression ([red] up-regulation; [green] down-regulation), and the size of each square represents the magnitude of the contribution of each sample to the respective IC. Each signature is two-sided. Vertical boxes denote statistically significant (P = 0.01, Mann-Whitney test) independent components. IC2 identified a Phf6KO-specific gene signature. (F) GSEA plot depicting (P = 0.08) enrichment in T-cell signal transduction signature upon Phf6 deletion, as compared with Phf6WT cells. (NES) Normalized enrichment score. Data represent the mean ± SD. Statistics for these data were calculated with two-sided Student's t-test. (**) P < 0.01; (***) P < 0.001; (****) P < 0.0001; (n.s.) not significant.
To directly compare murine CD4+ T cells and Phf6KO B-ALL cells, we applied ICA signature analysis to RNA-seq data from Phf6WT, shPhf6, and Phf6KO B-ALL cells and murine CD4+-single-positive (SP) T cells (Supplemental Fig. S5C; Miyazaki et al. 2015). This analysis unveiled a comparable expression profile between Phf6KO B-ALL cells and CD4+ T cells that was markedly distinct from shPhf6 and Phf6WT cells (Supplemental Fig. S5C,D). These results suggest that complete loss of Phf6 in B-ALL cells promotes a transcriptional program that partially resembles CD4+ T cells. It should be noted that the shPhf6 used in our previous study affects in vivo proliferation of B-ALL cells but is unable to recapitulate the breadth of transcriptional changes observed upon complete loss of Phf6 (Supplemental Figs. S4D,E, S5A,D; Meacham et al. 2015). Altogether, these findings further suggest that PHF6 is critical for the maintenance of B-cell identity through the positive transcriptional regulation of genes important for the B-cell state.
Global genomic binding profiles suggest that PHF6 interacts with chromatin
We next sought to determine whether the marked differences in gene expression profiles between knockout and wild-type cells were due to chromatin regulation by PHF6. To gain insight into the distribution of PHF6-bound sites across the B-ALL genome, we performed ChIP-seq (chromatin immunoprecipitation [ChIP] combined with high-throughput sequencing) (Lee et al. 2006) with an antibody against PHF6 (Fig. 3A). We found that PHF6 binds to both gene bodies and proximal promoter/enhancer regions of many annotated genes (Fig. 3A; Supplemental Table 3). We observed that genes that are differentially expressed in knockout cells had more than twofold enrichment of PHF6 binding at their transcriptional start sites (TSSs) (Supplemental Fig. S6A). In addition, we found that there is enrichment of PHF6 binding within the gene body of lowly expressed genes in Phf6WT cells (Supplemental Fig. S6B). Together, these data suggest that the genomic location of PHF6 binding might influence the transcriptional output in B-ALL cells.
Figure 3.

PHF6 exerts transcriptional regulation by interacting with histones rather than binding sequence-specific DNA sites. (A) Pie chart showing the distribution of 77,749 PHF6-binding sites across genomic regions in B-ALL cells. (TTS) Transcription termination site; (UTR) untranslated region. (B) De novo DNA sequence motifs identified in PHF6-bound regions at promoters of differentially expressed genes with their associated P-values. Shown are sequence logos of de novo position-weight matrices found by the MEME motif discovery tool (left) or those of known transcription factors whose motifs are found to be most similar to the de novo motif discovery results by Tomtom software (right). (C) Endogenous coimmunoprecipitation (co-IP) assay of TCF12, NF-κB, and PHF6 in the absence (top) or presence (bottom) of ethidium bromide (EtBr). Input is 7% of immunoprecipitation lysate. (D, left) Metagene tracks of PHF6 ChIP-seq signal averaged over all promoter–TSS tracks grouped by relative expression levels. (Red) High; (green) genomic; (purple) low. (Right) Metagene track of H3K27ac ChIP-seq signal averaged over all promoter–TSS regions. Shaded regions around average tracks denote estimates of 95% confidence interval (CI) of the metagene average signals based on resampling. (E) Metagene tracks of PHF6 ChIP-seq signal and correlation with histone marks: H3K27ac (blue), H3K4me3 (yellow; GSE66234), H3K27me3 (green). Pearson correlation of PHF6 and histone ChIP-seq signals across 10-kb regions spanning the TSS. Differentially expressed genes (solid line) and genome-wide genes (dotted line) are shown. (F) Endogenous co-IP assay of histone H3 and PHF6 in the absence (top) or presence (bottom) of EtBr. Input is 7% of immunoprecipitation lysate.
Previous studies have proposed that PHF6 has the ability to bind both DNA and histone proteins (Todd and Picketts 2012; Liu et al. 2014). Given these contrasting ideas, we decided to examine both modalities, first asking whether PHF6 has any DNA sequence-specific binding properties in B-ALL cells. To do this, we performed motif enrichment analysis (Bailey and Elkan 1994) of the DNA sequences surrounding the PHF6-binding peak summits near promoters of all genes and differentially expressed genes (Fig. 3B; Supplemental Fig. S6C). We found that no significant de novo motifs could be identified for PHF6 itself (Fig. 3B, left column). The motifs that passed the significance threshold bear a resemblance to motifs bound by known transcription factors (Fig. 3B, right column). Similar results were obtained from a motif search of genome-wide PHF6-binding peaks (Supplemental Fig. S6C). Since PHF6 lacked any sequence-specific binding properties, we next investigated whether PHF6 is part of a transcription factor complex that could potentially cobind and coregulate gene expression in B-ALL cells. To do this, we obtained mouse sequences for the ±1-kb regions adjacent to the TSSs of differentially expressed genes and assessed the enrichment of known motifs (those with P-value <0.01 that also have a strong PHF6 signal in ChIP-seq) (https://rdrr.io/bioc/PWMEnrich). Ultimately, 28 motifs (corresponding to 24 unique transcription factors) were identified as binding sites of putative interacting partners of PHF6 (Supplemental Table 3). These motifs were enriched for transcription factor-binding sites important during hematopoiesis and lymphopoiesis, including sites bound by PU.1, NF-kB, EGR1, EBF1, TCF3 (E2A), and TCF12 (HEB) (Supplemental Fig. S6D; Supplemental Table 3; Schebesta et al. 2002; Luo et al. 2004; Carlson et al. 2006; Wildt et al. 2007, Wang et al. 2008; Arenzana et al. 2009). Through endogenous coimmunoprecipitation (co-IP) experiments, we confirmed that PHF6 interacts with TCF12 and NF-kB in B-ALL cells (Fig. 3C, top). However, these interactions are mediated in most part via binding to DNA rather than protein–protein interactions, as demonstrated by a notable reduction in immunoprecipitated protein upon addition of the DNA-intercalating agent ethidium bromide (EtBr) (Fig. 3C, bottom). These results show that PHF6 is unable to recognize specific DNA-binding motifs/sequences and therefore is not acting as a canonical transcription factor in B-ALL cells.
We then tested whether PHF6 can regulate gene expression by interacting with histones and subsequently altering the chromatin state. In fact, we observed that the PHF6-binding profile is positively correlated with gene expression (Fig. 3D, left). Specifically, at highly expressed genes, PHF6 binding is enriched at regions flanking the TSS, similar to H3K27ac signal location (Fig. 3D). However, PHF6 signals tend to be more centered at the TSSs of genes with lower expression (Fig. 3D, left). In addition, we found that PHF6-binding enrichment is positively correlated with the presence of activating histone post-translational modifications, including H3K27ac and H3K4me3, whereas low correlation is observed with the repressive mark H3K27me3 (Fig. 3D [right], E). It had been demonstrated previously that PHF6 can interact with components of the NuRD complex as well several histone proteins (H1.2, H2B.1, H2A.Z, and H3.1) (Todd and Picketts 2012). Therefore, we decided to investigate whether these interactions also occur in B-ALL cells. Through endogenous co-IP experiments, we could not confirm interactions between PHF6 and components of the NuRD complex—RBBP4, HDAC1, or HDAC2—in B-ALL cells (data not shown). However, we were able to confirm a strong interaction between PHF6 and histone H3 (Fig. 3F, top). Importantly, this interaction is independent of the presence of DNA, as treatment of cell lysates with EtBr did not interfere with it (Fig. 3F, bottom). Altogether, these results show that PHF6 exerts transcriptional control of target genes via protein–protein interactions with histones. We conclude that the large transcriptional changes and the associated phenotypic plasticity observed in vivo are caused by loss of the PHF6–chromatin interaction that occurs upon genetic deletion of Phf6 in B-ALL cells.
PHF6 is necessary for the maintenance of chromatin organization at lineage-specific genes
Given the lineage promiscuity observed upon loss of Phf6 in vivo, we decided to further compare the transcriptional states of Phf6WT and Phf6KO cells with those of B and T cells. We used precurated lists of genes whose expression is unique and distinguishing to CD19+ B cells and CD4+ T cells (Supplemental Table 2; Painter et al. 2011). As expected, most of the B-cell signature genes are down-regulated in Phf6KO cells, whereas the T-cell signature genes showed bidirectional changes (Fig. 4A). Importantly, this observation is consistent with the mixed-lineage malignancy that arises from syngeneic transplantation of Phf6KO cells in vivo (Fig. 1F; Supplemental Fig. S2C–F).
Figure 4.

PHF6 has distinct binding patterns within lineage-specific genes that result in drastic changes in nucleosome occupancy upon genetic deletion. (A) Heat maps comparing gene expression of curated CD19+ B-cell (left) and CD4+ T-cell (right) gene sets between Phf6WT (left) and Phf6KO (right) cells as determined by RNA-seq. Scale corresponds to row-wise standardized log2-transformed expression values for each gene. Each column represents a replicate sample. (B) Motif enrichment analysis for DNA regions that undergo significant changes in chromatin accessibility in Phf6KO cells. (Top) DNA motifs with decreased chromatin accessibility upon loss of Phf6. (Bottom) DNA motifs with increased chromatin accessibility upon loss of Phf6. (C) Metagene analysis of nucleosome positions (top) and fold enrichment of PHF6 binding (bottom) plotted for global analysis (left), the CD19+ B-cell gene set (middle), and the CD4+ T-cell gene set (right) assessed at TSSs ±1-kb genomic regions. (Top) Nucleosome positions are shown for Phf6WT (solid line) and Phf6KO (dotted line) cells, called by the NucleoATAC algorithm and normalized for batch effects using the chromVAR package. (Bottom) Metagene tracks of PHF6 ChIP-seq signal averaged over the TSSs ±1 kb in Phf6WT cells. (−1/+1) +1/−1 nucleosomes flanking the TSS; (NFR) nucleosome-free region. Gray bars indicate major changes in nucleosome positioning that correspond to enriched PHF6 binding. Shaded regions around average tracks denote estimates of 95% CI of the metagene average signals based on resampling.
Lineage-specific transcriptional programs are often characterized by differential chromatin accessibility (Lara-Astiaso et al. 2014). To determine whether the changes observed in the B and T gene sets are the result of altered chromatin organization, we performed ATAC-seq (assay for transposase-accessible chromatin [ATAC] with high-throughput sequencing) (Buenrostro et al. 2013) on Phf6WT and Phf6KO B-ALL cells (Supplemental Fig. S7A–F). Phf6 loss results in focal changes in chromatin accessibility, with ∼700 regions in the genome showing statistically significant differences (Supplemental Fig. S7B). These changes are even more pronounced in the promoter regions of genes associated with B and T cells (Supplemental Fig. S7C,E,F). This analysis also demonstrated that changes in chromatin accessibility at promoter regions are predictive of changes in gene expression (Supplemental Fig. S7D,E). Consistent with our phenotypic data, differential chromatin accessibility analysis revealed that regions that become less accessible upon Phf6 loss are enriched for binding motifs of transcription factors required for B-cell development (Fig. 4B, top; Schebesta et al. 2002; Dai et al. 2007; Niebuhr et al. 2013) . Conversely, regions that became more accessible upon Phf6 loss are enriched for motifs of transcription factors associated with the development of T-ALL and AML (Fig. 4B, bottom; Eyquem et al. 2004; Thoms et al. 2011; Kiaii et al. 2013; Smeets et al. 2013). Thus, genomic regions important for B and T lineage genes as well as their regulatory factor-binding sites undergo significant changes in chromatin accessibility upon complete loss of Phf6 in B-ALL cells.
We next asked whether nucleosome positions, in addition to chromatin accessibility, were undergoing significant changes dependent on Phf6 status. To determine nucleosome positioning with high precision and resolution, we applied the NucleoATAC algorithm to our data set (Fig. 4C; Schep et al. 2015). Globally, there are few differences in nucleosome occupancy between Phf6WT (Fig. 4C, left, solid line) and Phf6KO (Fig. 4C, left, dashed line) cells. However, drastic changes in nucleosome occupancy are observed in the lineage-specific gene sets. Focusing on the CD19+ B-cell gene set, we observed an increase in nucleosome occupancy at the TSS and within the gene body upon Phf6 loss (Fig. 4C, middle). When aligned with the promoter–TSS-binding profile of PHF6 in Phf6WT cells, we observed a large overlap between regions with enriched PHF6 binding and major shifts in nucleosome occupancy (Fig. 4C, gray bar). This finding suggests that PHF6 coordinates nucleosome architecture at B-cell genes in a way that is favorable for transcriptional activity. When PHF6 is lost, nucleosome occupancy is enriched and gene expression decreases (Fig. 4A,C, middle). Focusing on the CD4+ T-cell gene subset, we observed decreased nucleosome occupancy surrounding the TSS in Phf6KO cells (Fig. 4C, right, dashed line), with preferential depletion at the −1 nucleosome (Fig. 4C, right). Our data are consistent with promoters undergoing increased chromatin accessibility, where the −1 nucleosome undergoes more pronounced depletion (Schep et al. 2015). Furthermore, PHF6 binding is enriched at sites that are undergoing global shifts in nucleosome occupancy (Fig. 4C, right). The chromatin landscape surrounding the TSSs of genes important for CD4+ T cells demonstrates fluidity in Phf6KO cells, leading to increased gene expression in a subset of T-cell genes (Fig. 4A) and presentation as a mixed-lineage lymphoma in vivo (Fig. 1). The shifts in nucleosome occupancy that we observed upon complete loss of Phf6 indicate that B-ALL cells are undergoing lasting and stable transcriptional changes resulting from alterations of the chromatin architecture, allowing for genomic (as well as phenotypic) plasticity. Specific genes undergoing changes in chromatin accessibility at regulatory elements include B-cell-specific genes Btg2 and Cd74 (both of which become less accessible upon Phf6 loss) and T-cell lineage genes Cd4 (distal enhancer) and Gata3 (which become more accessible upon Phf6 loss) (Supplemental Fig. S8A,B). Importantly, we confirmed enrichment of PHF6 binding at these target genes through ChIP combined with quantitative PCR (ChIP-qPCR) analysis (Supplemental Fig. S8C,D), suggesting that the observed shifts in chromatin accessibility are dependent on the presence of PHF6. In conclusion, we show that PHF6 is necessary for the maintenance of a chromatin state that is instrumental to B-cell identity and that Phf6 loss leads to the acquisition of a permissive/open state at loci of alternate lineages, allowing for the engagement of aberrant transcriptional programs that direct the resulting phenotypic plasticity.
Instability in the chromatin landscape allows for aberrant lineage signaling
Given the genomic plasticity observed in our Phf6KO B-ALL cells and the changes in chromatin accessibility detected at regions of T-cell transcription factor-binding sites upon Phf6 loss, we hypothesized that the genome of Phf6KO cells may now be permissive to aberrant oncogenic signaling emanating from T-cell lineage factors, such as NOTCH1. Activating mutations in NOTCH1 are well documented in >60% of cases of T-ALL (Belver and Ferrando 2016), where PHF6 is often found to be comutated with NOTCH1 (Wang et al. 2011; Li et al. 2016). Therefore, we tested the effects of overexpression of the NOTCH1 intracellular domain (NICD) in Phf6WT and Phf6KO B-ALL cells. As expected, we observed that NOTCH1 signaling dramatically inhibits the growth of Phf6WT B-ALL cells (Fig. 5A; Rothenberg 2014). However, Phf6KO cells were viable upon NICD overexpression, with growth rates equivalent to Phf6WT and Phf6KO vector control cell lines (Fig. 5A). Transplantation of Phf6KO + NICD cells into immunocompetent syngeneic mice gives rise to a latent lymphoma with extensive thymic infiltration and elevated levels of CD4 expression characteristic of T-cell malignancies (Fig. 5B–D). In conclusion, PHF6 deficiency in B-ALL cells creates a permissive chromatin state that allows transcription factors from alternate lineages to bind to their respective genomic loci, which are normally in a closed conformation in wild-type cells. Enforced expression of NOTCH1 (T-cell transcription factor) takes advantage of the chromatin fluidity characteristic of knockout cells, where Phf6KO tumors are driven more toward a “T-cell-like” state instead of a mixed-lineage phenotype.
Figure 5.

Chromatin instability allows for aberrant T-cell transcription factor signaling. (A) Cell proliferation assay comparing Phf6WT (blue; up arrow), Phf6KO (red; up arrow), Phf6WT + control vector (blue; circle-dotted), Phf6KO + control vector (red; circle-dotted), Phf6WT + NICD cDNA (blue; down triangle), and Phf6KO + NICD cDNA (red; down triangle) cells. n = 3. (B) Kaplan-Meier survival analysis of mice injected with 106 B-ALL cells of the indicated genotypes infected with control vector or activated NICD vector. The number (n) of mice per genotype analyzed is shown. Statistical analysis (log-rank test, Mantel-Cox) was performed for the different groups in comparison with mice injected with Phf6WT + control vector cells. P-values are shown for the comparisons. (C) Quantification of the combined thymus weight of Phf6WT + control vector (blue; n = 4), Phf6KO + control vector (red unfilled; n = 6), and Phf6KO + NICD cDNA (red patterned; n = 7) recipients. (D) Bar graphs showing the percentage of the CD4+ fraction among mCherry+ cells isolated from Phf6WT + control vector (blue; n = 5), Phf6KO + control vector (red unfilled; n = 4), and Phf6KO + NICD cDNA vector (red patterned; n = 6) tumors from LNs. Data represent the mean ± SD in C and D. Statistics were calculated with two-sided Student's t-test. (*) P < 0.05; (**) P < 0.01; (****) P < 0.0001.
Phenotypic plasticity underlies differential responses to anti-cancer treatments in vivo
Clinically, patients with B-cell and T-cell leukemia and lymphoma undergo different treatment strategies. BCR–ABL1-driven B-cell leukemia is often treated with targeted kinase inhibitors, whereas T-cell leukemia and lymphoma are treated with intensive chemotherapy regimens (Schultz et al. 2009; Litzow and Ferrando 2015). To further understand the shift from a disseminated leukemia to a mixed-phenotype solid lymphoma that results from complete loss of Phf6 in B-ALL cells, we examined the response of these tumors to targeted therapy (ponatinib) and frontline chemotherapy (doxorubicin).
Unbiased transcriptome analysis shows decreased dependency on BCR–ABL1 signaling pathways, the driving oncogene in this B-ALL model (Fig. 6A; Williams et al. 2006; Shamroe and Comeau 2013). Consistent with this, we observed a marked difference in the response to ponatinib (third-generation BCR–ABL1 inhibitor) between Phf6WT and Phf6KO tumors in vivo but not in vitro (Fig. 6B; Supplemental Fig. S9A). Mice transplanted with Phf6KO cells and treated with ponatinib have a median survival of 15 d, while their wild-type counterparts survived >60 d after treatment (Fig. 6B). Thus, the lineage plasticity that arises in vivo upon Phf6 loss drives a significantly decreased response to ponatinib. Furthermore, we examined the response of Phf6WT and Phf6KO tumors to the chemotherapeutic agent doxorubicin. We found that mice transplanted with Phf6KO cells are exquisitely sensitive to doxorubicin treatment in vivo, as demonstrated by an additional 13-d extension in median survival (essentially doubling their life span) when compared with mice injected with Phf6WT cells (Fig. 6C; Supplemental Fig. S9B,C). In conclusion, we found that complete loss of Phf6 in B-ALL cells changes not only the chromatin landscape of these tumors but also the phenotypic state, which is reflected in the formation of a different disease with vastly distinct responses to anti-cancer therapies. These findings support a possible underlying mechanism for the examples of lineage plasticity and drug resistance observed in a variety of malignancies and demonstrate that modulation of the chromatin landscape through the loss of regulation by a single factor (such as PHF6) may play a crucial role in acquired resistance.
Figure 6.

Loss of Phf6 results in decreased dependence on the driving oncogene BCR–ABL1 in vivo. (A) GSEA plots depicting significant changes in the targets of the BCR–ABL1 fusion signature (P = 0.0129) and dasatinib resistance signature (P = 0.0056) upon Phf6 deletion, as compared with Phf6WT cells. (NES) Normalized enrichment score. (B) Kaplan-Meier survival analysis of mice injected with 106 B-ALL cells of the indicated genotypes with subsequent mock or ponatinib treatment (30 mg/kg daily for four consecutive days). The number (n) of mice per genotype analyzed is shown. Statistical analysis (log-rank test, Mantel-Cox) was performed for the different groups in the indicated comparisons. P-value is shown for the comparison. (C) Kaplan-Meier survival analysis of mice injected with 106 B-ALL cells of the indicated genotypes with mock or 10 mg/kg doxorubicin (single immunoprecipitation dose) treatment. The number (n) of mice per genotype analyzed is shown. Statistical analysis (log-rank test, Mantel-Cox) was performed for the different groups in the indicated comparisons. P-value is shown for the comparison.
Discussion
The transcriptional regulation of lymphocyte development via lineage-specific factors is well characterized, and the perturbation of this process can promote leukemogenesis (Mullighan et al. 2007). However, the dynamic regulation of chromatin states underlying transcriptional activity and its contribution to cancer are yet to be elucidated. In this study, we show that PHF6 is a novel chromatin state regulator important for the maintenance of a chromatin landscape conducive to B-cell identity in B-ALL. Through integrated genomics (RNA-seq, ChIP-seq, and ATAC-seq) and in vivo studies, we demonstrate the crucial role that PHF6-mediated chromatin modulation plays in genomic plasticity, cell identity, leukemogenesis, and response to therapy. Our data convincingly show that Phf6 loss leads to a chromatin environment that tolerates aberrant lineage and oncogenic signaling. Moreover, we demonstrate how malignant cells can hijack chromatin instability to reprogram transcriptional output and cell identity. These findings propose the first mechanistic insights into PHF6's role in a lymphoid malignancy. We show that PHF6 binds to nucleosomes at specific genomic loci and orchestrates distinct transcriptional programs. It stabilizes chromatin accessibility at B-cell-specific genes, while T-cell transcription factor-binding sites are concealed in compacted chromatin (Fig. 7A). Upon loss of this single factor, chromatin architecture around B-cell- and T-cell-specific genes undergoes dramatic remodeling, resulting in focal genomic plasticity and acquisition of T-cell lineage markers in B-ALL cells (Fig. 7B). Given that PHF6 is frequently inactivated in T-ALL (∼30%) (Van Vlierberghe et al. 2010), we speculate that T-cell malignancies benefit from the permissive chromatin landscape that results upon PHF6 loss. Our data further support the possibility that a significant portion of T-ALLs (∼30%) (Van Vlierberghe et al. 2010) may have once started as a B-cell progenitor/precursor that acquired a mutation in PHF6 during early stages of leukemogenesis and consequently forced the cell of origin into a “pseudo-T-cell state” that then presented as a T-cell malignancy.
Figure 7.
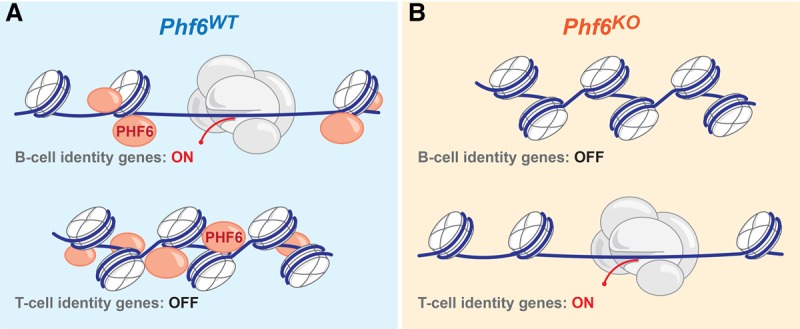
Model of PHF6 as a chromatin state regulator, permitting transcription factor binding through chromatin accessibility. (A) In wild-type cells, PHF6 binds to the ±1 nucleosome flanking the open TSSs of genes, allowing B-cell-specific transcription factors to bind, drive gene expression, and maintain B-cell identity. Conversely, PHF6 binds nucleosomes surrounding the TSSs of T-cell-specific genes, coordinating chromatin compaction and thus blocking the binding of T-cell-specific transcription factors. (B) In the absence of PHF6, chromatin is no longer maintained in an open state, B-cell transcription factors cannot bind, and expression of B-cell identity genes is down-regulated. However, T-cell identity genes are no longer inaccessible, allowing T-cell-specific transcription factors to bind and activate aberrant transcriptional programs.
Lineage switching is a rapidly emerging mechanism of resistance to targeted therapies. A variety of cancers (including lung cancer, prostate cancer, and B-cell leukemia) can acquire resistance to therapy by relapsing as a histologically distinct malignancy (Sequist et al. 2011; Yu et al. 2013; Oser et al. 2015; Gardner et al. 2016; Jacoby et al. 2016; Ku et al. 2017; Mu et al. 2017). The first functional studies investigating this process as a driver of drug resistance in prostate cancer identified epigenetic reprogramming proteins (SOX2 and EZH2) as key effectors of this phenomenon (Ku et al. 2017; Mu et al. 2017). In our study, we further demonstrate that active maintenance of chromatin architecture (by PHF6, a novel chromatin regulator) is also critical in the response to targeted therapy in B-cell leukemia. We postulate that the chromatin landscape is a pivotal component in the response of many cancer types to targeted treatments.
Here we identifed the function of PHF6 in B-ALL and ascribed its role in regulating chromatin accessibility to lineage-specific transcription factors. We showed that loss of Phf6 results in disruption of lineage differentiation via gross changes in chromatin accessibility, engagement of alternative transcriptional programs, altered disease presentation in vivo, and tolerance of aberrant oncogenic signaling. We established that the proper chromatin landscape is essential for maintaining cell identity and that loss of Phf6 can cause changes in chromatin accessibility and nucleosome positioning that result in lineage promiscuity. These findings highlight a mechanism of gene expression maintenance via chromatin landscape modulation that underlies the developmental plasticity characteristic of hematopoietic malignancies.
Materials and methods
Plasmids and cloning
A mouse Phf6 sgRNA (5′-GTACTTCAGGAGATTAAGCG-3′) was cloned into pX458 vector [pSpCas9(BB)-2A-GFP; Addgen, 48138] (Ran et al. 2013). This sgRNA was designed using the Broad Institute sgRNA Designer (Doench et al. 2014). A mouse Phf6 cDNA was obtained from OriGene (MC203493) and subcloned into pMSCV-PGK-GFP. The following NOTCH1-ICD overexpression vectors were obtained from Addgene: pLPCX-NICD (Addgene, 44471) and pLPCX-IRES-GFP (Addgene, 65436). The Phf6 shRNA (5′-GCAAGGGATTTACATGGTTTA-3′) was cloned into the mir30 backbone (Supplemental Table 1; Dickins et al. 2005; Meacham et al. 2015).
Antibodies
The antibodies used for Western blotting were anti-HSP90 (BD Biosciences, 610419), anti-PHF6 N terminus (Bethyl Laboratories, A301-450A), anti-PHF6 C terminus (Bethyl Laboratories, A301-452A), anti-Histone H3 (C-16) (Santa Cruz Biotechnology, sc-8654), and anti-PHF6 (Novus, 68262). Secondary antibodies were anti-rabbit IgG, HRP-linked (Cell Signaling Technology, 7074); anti-mouse IgG, HRP-linked (Cell Signaling Technology, 7076); and anti-goat IgG, HRP-linked (Santa Cruz Biotechnology, sc-2350). Antibodies used for immunoprecipitation were anti-PHF6 (Novus, 68262), anti-TCF12 (Cell Signaling Technology, 11825), anti-NF-kB (p65) (Santa Cruz Biotechnology, sc-372), anti-Histone H3 (Bethyl Laboratories, A300-823A), and normal rabbit IgG (Santa Cruz Biotechnology, sc-2027). Antibodies used for flow cytometry were APC anti-mouse B220 (clone RA3-6B2; BD Biosciences, 553092), V450 anti-mouse IgM (clone R6-60.2; BD Biosciences, 560575), FITC anti-mouse CD19 (clone eBio1D3; eBiosciences, 11-0193-81), APC anti-CD4 (clone RM4-5; BD Biosciences, 553051), V450 anti-CD4 (clone RM4-5; BD Biosciences, 560468), FITC anti-CD3 (clone 17A2; eBiosciences, 11-0032), and APC anti-CD11b (clone M1/70; Biolegend, 101212). Isotype controls used for flow cytometry were V450 anti-IgG2a, κ (clone R35-95; BD Biosciences, 560377); FITC anti-IgG2b, κ (clone eB149/10H5; eBiosciences, 11-4031-82); APC anti-IgG2a, κ (clone R35-95; BD Biosciences, 553932); FITC anti-IgG2a, κ (clone eBR2a; eBiosciences, 11-4321); and APC anti-IgG2b, κ (clone RTK4530; Biolegend, 400612). The antibody used for immunohistochemistry staining was anti-RFP (Rockland, 600-401-379). The antibodies used for ChIP experiments were anti-PHF6 (Bethyl Laboratories, A301-451A), anti-H3K27ac (Abcam, ab4729), anti-H3K27me3 (Abcam, ab6002), anti-PHF6 (Novus, 68262), and normal rabbit IgG (Santa Cruz Biotechnology, sc-2027).
Cell culture
Murine BCR–ABL1+; p19−/−; mCherry+ B-ALL cells (Williams et al. 2006) were cultured in RPMI-1640 (Corning) supplemented with 10% FBS, 4 mM L-glutamine, 50 µM 2-mercaptoethanol, 100 U/mL penicillin, and 100 µg/mL streptomycin. Retroviral particles were generated and packaged using the Phoenix cell system (G. Nolan, Stanford University). Briefly, a calcium phosphate transfection of 10 µg of plasmid DNA and 5 µg of helper plasmid (pCMV-Gag-Pol) was used. Transduction efficiencies were assessed 48 h after infection by flow cytometry. To generate Phf6KO cells, sgPhf6-pX458 plasmid was transfected (Lipofectamine 3000, Life Technologies) into B-ALL cells. Transfected cells were single-cell-sorted into 96-well plates based on GFP expression. Cell proliferation assay was performed by plating B-ALL cells in triplicate (1 million cells per well in 3 mL of medium in a six-well plate), counting every 24 h by Trypan blue exclusion, and replating the initial density. This was performed for 7 d.
Drug response analysis
Cells (10,000 per well) were plated in 96-well plates, and medium containing ponatinib (LC Laboratories, AP24534) or doxorubicin (Sigma-Aldrich, D1515) was added to achieve the indicated final concentrations. Cell viability was assessed 48 h after treatment by propidium iodide (Sigma-Aldrich, 81845) exclusion and flow cytometry. Inhibitor dilutions were made in cell medium immediately before use. The relative viability compared with controls diluted in equivalent medium + DMSO is shown.
Western blotting
Cells were lysed with RIPA buffer (Boston BioProducts, BP-115) supplemented with 1× protease inhibitor solution (Complete EDTA-free, Roche, 11873580001). Protein concentration of cell lysates was determined by Pierce BCA protein assay (ThermoFisher Scientific, 23225). Fifty micrograms to 70 µg of total protein was separated on 4%–12% Bis-Tris gradient SDS-PAGE gels (Life Technologies) and then transferred to PVDF membranes (EMD Millipore, IPVH00010) for blotting.
Co-IP
Antibodies were precleaned before attaching to magnetic beads using the Pierce antibody cleanup kit (ThermoFisher Scientific, 44600). A total of 10 µg of antibody was covalently attached to magnetic beads (ThermoFisher Scientific, 88828) and incubated with 1 mg of protein for 2 h at room temperature while rotating (or overnight at 4°C). Immunoprecipitation was performed according to the manufacturer's directions (Pierce Direct magnetic immunoprecipitation/co-IP kit, Thermo Fisher Scientific). For DNA-dependent interactions, 50 µg/mL EtBr was added, and lysates were incubated for 30 min on ice. Samples were centrifuged, and the supernatant was used for immunoprecipitations.
qPCR
RNA from 2 × 106 B-ALL cells was prepared using RNeasy minikit (Qiagen). Synthesis of cDNA was performed using M-MLV reverse transcriptase (Life Technologies, 28025) with oligo(dT)20 primer. qPCR was done in an Applied Biosystems StepOnePlus machine with TaqMan fast universal PCR master mix (Life Technologies, 4352042). Data were analyzed using the comparative ΔCT method and normalized to the levels of Gapdh. The TaqMan gene expression assays (Life Technologies) used were Phf6 (Mm00804415_m1), Blk (Mm00432077_m1), Cd74 (Mm00658576_m1), IL4ra (Mm01275139_m1), Ly86 (Mm00440240_m1), Lyn (Mm01217488_m1), Gapdh (Mm99999915_g1), and Cd22 (Mm00515432_m1).
Genomic DNA isolation and sequencing
Genomic DNA from B-ALL cells was isolated using the blood and cell culture DNA minikit (Qiagen) following the manufacturer's guidelines. PCR products for sequencing were amplified using HerculaseII Fusion DNA polymerase (Agilent) (see Supplemental Table 1 for primers used), gel-purified, cloned into pCR4-TOPO TA vector (Life Technologies), and subsequently sequenced.
Animal experiments
All animal studies described in this study were approved by the Massachusetts Institute of Technology Institutional Animal Care and Use Committee. For syngeneic transplants, 103 or 106 murine BCR–ABL1+p19−/− B-ALL cells (Williams et al. 2006) were injected via tail vein into syngeneic C57BL/6J (The Jackson Laboratory, 000664) recipient mice at the age of 6–8 wk. Treatment with 30 mg/kg ponatinib (administered via oral gavage, treated once daily for four consecutive days), 10 mg/kg doxorubicin (administered via immunoprecipitation, treated once; NovaPlus), or vehicle (25 mM citric acid buffer) was initiated upon significant disease presentation. Mice were observed daily and sacrificed when moribund (dehydration, ruffled fur, respiratory distress, and poor motility). Survival curves were generated using GraphPad Prism version 6.0 software, and the Mantel-Cox test was applied to pairwise comparisons of survival data. No statistical methods were used to predetermine sample size. Investigators were not blinded to group allocation during experiments and outcome assessment. Randomization was not used because all animals used in this study were of similar sex, age, and strain background.
Organ processing and cell preparation for flow cytometry
When mice reached morbidity, they were sacrificed, and lymphoid organs were collected. Briefly, cells derived from the BM were obtained by flushing the femora and tibiae with B-ALL medium. LNs, spleens, and thymi were isolated; weighed for mass measurements; ground; and filtered to obtain single-cell suspension. Subsequently, red blood cells were lysed with ACK lysing buffer (Life Technologies, A10492-01).
Immunostaining
For cell surface immunophenotyping, leukemia cell suspensions (cultured and isolated from mice) were collected and treated with the indicated antibodies. Briefly, 1 × 106 to 2 × 106 cells in 100 µL of 10% FBS in PBS were incubated with antibody for 1 h at room temperature in the dark. Antibodies were used at a 1:100 dilution. Stained samples were analyzed by flow cytometry.
For intracellular immunophenotyping, leukemia cell suspensions (cultured and isolated from mice) were collected, resuspended in PBS, and fixed with 4% formaldehyde (Tousimis, 1008A) solution for 10 min at 37°C. Cells were permeabilized with ice-cold methanol for 30 min on ice.
Cell cycle analysis
FITC BrdU flow kit (BD Biosciences, 559619) was used for cell cycle analysis. Briefly, 1 × 106 B-ALL cells plated in triplicate were labeled with 10 µM BrdU for 30 min. Cells were subsequently fixed and stained with anti-BrdU and 7-AAD and analyzed by flow cytometry (LSRFortessa, BD Biosciences).
Flow cytometry
FACSAria (BD Biosciences) was used for fluorescence-activated cell sorting, and LSRFortessa (BD Biosciences) was used for flow cytometry analysis. Data were analyzed with FlowJo software (TreeStar version 10).
Histology and immunohistochemistry
For histological and immunohistochemistry analyses, mice were euthanized by carbon dioxide asphyxiation. Lymphoid tissues were harvested, fixed overnight with 10% neutral buffered formalin (VWR), transferred to 70% ethanol solution, and subsequently embedded in parafilm. Sections were cut at a thickness of 10 µm and stained with hematoxylin and eosin (H&E) for pathological assessment. Immunohistochemistry was performed on a Thermo Autostainer 360 machine. Slides were antigen-retrieved using Thermo citrate buffer (pH 6.0) in the pretreatment module. Sections were treated with Biocare rodent block, primary antibody, and anti-rabbit HRP-polymer (Vector Laboratories). The slides were developed with Thermo Ultra DAB and counterstained with hematoxylin in a Thermo Gemini stainer, and coverslips were added using the Thermo Consul coverslipper.
RNA-seq library preparation
cDNA for RNA-seq libraries was prepared using TruSeq mRNA library preparation kit (Illumina). Illumina libraries were produced using SPRIworks (Beckman-Coulter Genomics) with a 200- to 400-base-pair (bp) size selection and enriched with 14 cycles of PCR. Library quality was determined by qPCR and on the fragment analyzer (Advanced Analytical) and loaded onto two lanes of 40-nucleotide (nt) single-end sequencing on a HiSeq2000 system (Illumina).
RNA-seq data analysis
Illumina HiSeq2000 40-nt single-end reads were mapped to the University of California at Santa Cruz (UCSC) mm9 mouse genome build (http://genome.ucsc.edu) using RSEM (Li and Dewey 2011). Raw estimated expression counts were upper-quartile-normalized to a count of 1000 (Bullard et al. 2010). Given the complexity of the data set in terms of a mixture of different conditions, a high-resolution signature discovery approach was used to characterize global gene expression profiles. ICA, an unsupervised blind source separation technique, was used on this discrete count-based expression data set to elucidate statistically independent and biologically relevant signatures. ICA is a signal processing and multivariate data analysis technique in the category of unsupervised matrix factorization methods. Conceptually, ICA decomposes the overall expression data set into independent signals (gene expression patterns) that represent distinct signatures. High-ranking positively and negatively correlated genes in each signature represent gene sets that drive the corresponding expression pattern (in either direction). Signatures were visualized using the sample to signature correspondence schematic afforded by Hinton plots, where colors represent directionality of gene expression (red indicates up-regulated, and green indicates down-regulated), and the size of each rectangle quantifies the strength of a signature (column) in a given sample (row). Each signature is two-sided, allowing for identification of up-regulated and down-regulated genes for each signature within each sample. Using input data consisting of a gene–sample matrix, ICA uses higher-order moments to characterize the data set as a linear combination of statistically independent latent variables. These latent variables represent independent components based on maximizing non-Gaussianity and can be interpreted as independent source signals that have been mixed together to form the data set under consideration. Each component includes a weight assignment to each gene that quantifies its contribution to that component. Additionally, ICA derives a mixing matrix that describes the contribution of each sample toward the signal embodied in each component. This mixing matrix can be used to select signatures among components with distinct gene expression profiles across the set of samples. The R implementation of the core JADE (Joint Approximate Diagonalization of Eigenmatrices) algorithm (Rutledge and Bouveresse 2013; https://cran.r-project.org/web/packages/JADE/index.htm; https://www.bioconductor.org/packages/release/bioc/html/MineICA.html) was used along with custom R utilities. The P-value for the knockout-specific (IC2) signature was P = 0.0119 (Mann-Whitney U-test directional P-value). Targeted pairwise differential expression analyses were conducted using EBSeq version 1.4.0 (Leng et al. 2013). Differentially expressed genes from the pairwise comparison between Phf6KO and Phf6WT B-ALL cells were determined using a significance level of false discovery rate (FDR) 0.05 and fold change >1.5. All RNA-seq analyses were conducted in the R statistical programming language (http://www.r-project.org). GSEA was carried out using the preranked mode with default settings (Subramanian et al. 2005). Heat maps were generated using the Heatplus package in R. Integrated RNA-seq signature analysis of B-ALL cells and a data set from CD4+ SP cells (Gene Expression Omnibus [GEO] accession code GSE64779, samples GSM1580493–GSM1580497) (Edgar et al. 2002; Barrett et al. 2012; Miyazaki et al. 2015) was performed using ICA. All gene lists, signatures, and sets used in this study are in Supplemental Table S2. A two-dimensional expression principal component (principal component analysis [PCA]) plot for each group of cells was generated by plotting the first two principal components of expression (explaining 75% and 11% of expression variance, respectively). Gene ontology (GO) analysis was done using DAVID, and top GO and PANTHER terms were plotted against their corresponding P-values (log-scale).
ChIP-seq
ChIP was performed as described before (Lee et al. 2006) with a few adaptations. A total of 150 × 106 cells, grown to a density of 1 × 106 cells per milliliter, was cross-linked for 10 min at room temperature by the addition of one-tenth of the volume of 11% formaldehyde solution (11% formaldehyde, 50 mM HEPES at pH 7.3, 100 mM NaCl, 1 mM EDTA at pH 8.0, 0.5 mM EGTA at pH 8.0) to the growth medium followed by 5 min of quenching with 125 mM glycine. Cells were washed twice with PBS, the supernatant was aspirated, and the cell pellet was flash-frozen in liquid nitrogen. Frozen cross-linked cells were stored at −80°C. One-hundred microliters of Protein G Dynabeads (Life Technologies) was blocked with 0.5% (w/v) BSA in PBS. Magnetic beads were bound with 10 µg of anti-H3K27ac (Abcam, ab4729), anti-H3K27me3 (Abcam, ab6002), and anti-PHF6 (Bethyl Laboratories, A301-451A). Nuclei were isolated as described previously (Lee et al. 2006) and sonicated in lysis buffer (20 mM Tris-HCl at pH 8.0, 150 mM NaCl, 2 mM EDTA at pH 8.0, 0.1% SDS, 1% Triton X-100) on a Misonix 3000 sonicator for 10 cycles of 30 sec each on ice (18–21 W) with 60 sec on ice between cycles. Sonicated lysates were cleared once by centrifugation and incubated overnight at 4°C with magnetic beads bound with antibody to enrich for DNA fragments bound by the indicated factor. Beads were washed with wash buffer A (50 mM HEPES-KOH at pH 7.9, 140 mM NaCl, 1 mM EDTA at pH 8.0, 0.1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS), buffer B (50 mM HEPES-KOH at pH 7.9, 500 mM NaCl, 1 mM EDTA at pH 8.0, 0.1% sodium deoxycholate, 1% Triton X-100, 0.1% SDS), buffer C (20 mM Tris-HCl at pH 8.0, 250 mM LiCl, 1 mM EDTA at pH 8.0, 0.5% sodium deoxycholate, 0.5% IGEPAL CA-630, 0.1% SDS), and buffer D (TE with 50 mM NaCl) sequentially. DNA was eluted in elution buffer (50 mM Tris-HCl at pH 8.0, 10 mM EDTA, 1% SDS). Cross-links were reversed overnight. RNA and protein were digested using RNase A and Proteinase K, respectively, and DNA was purified with phenol-chloroform extraction and ethanol precipitation. Purified ChIP DNA was used to prepare Illumina multiplexed sequencing libraries. Libraries were prepared following the TruSeq DNA sample preparation kit version 2 (Illumina). Amplified libraries were size-selected using a 2% gel cassette in the Pippin preparation system (Sage Science) set to capture fragments between 200 and 400 bp. Libraries were quantified by qPCR using the Illumina library quantification kit (KAPA Biosystems) according to the manufacturer's guidelines. Libraries were sequenced on the Illumina HiSeq2500 for 40 nt in single-read mode.
ChIP-seq data analysis
To compare RNA-seq with ChIP-seq data sets, normalized RNA-seq tag counts for each gene were used to plot a heat map showing the expression levels of these genes ordered by fold change values. Among these genes, 1127 symbols were mappable to ENSEMBL mouse gene IDs (unmappable gene symbols corresponded mostly to those that are not well characterized) and were used for mapping to genomic coordinates. Sequencing reads were mapped to the UCSC mm9 mouse genome using Bowtie2 (Langmead et al. 2009; Langmead and Salzberg 2012) with at most one mismatch allowed in seed alignments. Unmapped reads were removed, and alignments were output in BAM files for peak calling with the MACS software (Zhang et al. 2008). PHF6 and histone modification peaks were called using a P-value threshold of 1 × 10−6. The fraction of reads falling within the called peaks (FRiP) for PHF6 ChIP-seq data was 25.9% (8,333,082 out of 32,143,465 uniquely mappable reads), showing high ChIP quality. To examine whether PHF6-binding strength correlates with gene expression levels, RNA-seq data from Phf6WT cells were sorted and grouped by expression into three bins corresponding to high, medium, and low expression by separating into three equally sized bins. Average log2 fold change values over input signals were plotted as a metagene track for each gene set. Plots for H3K27ac signal enrichment were also generated to confirm promoter activity for each gene set. To determine PHF6 binding at promoter/enhancer regions of the differentially expressed genes, genomic sequences were obtained for the ±5-kb region surrounding the TSSs for these genes from the UCSC mm9 genome assembly and binned into 50-bp bins. ChIP-seq RPM (reads per million) values for PHF6, H3K27ac, H3K27me3, and H3K4me3 (publicly available ChIP-seq data sets were obtained as Sequence Read Archive-lite files, GEO accession code GSE66234, data sets GSM1617788–GSM1617789) (Edgar et al. 2002; Barrett et al. 2012; Wang et al. 2015) at these promoter bins were divided by the corresponding ChIP input (control) reads with a pseudocount of one and then log2-transformed to show fold enrichment. To show correlation between PHF6 and histone modification signals, Pearson's correlation coefficients between corresponding pairs of columns in the heat maps were calculated. The MEME suite (Bailey and Elkan 1994) was used for de novo motif discovery of sequences surrounding PHF6-binding peak summits. A 100-nt window upstream of and downstream from the peak summits was set to obtain sequences from the UCSC mm9 genome assembly. Among all 77,749 PHF6 summits called by the MACS pipeline (Zhang et al. 2008), 6037 fell within the proximal regulatory regions (±5 kb surrounding the TSS) of differentially expressed genes, covering a total of 1104 (out of 1123) unique promoters. For each promoter region, the PHF6 summit was selected with the highest average peak signal for motif analysis, resulting in 1044 unique summits and their surrounding sequences as input for the MEME motif discovery tool (Bailey and Elkan 1994). To see whether PHF6 displays any bias in sequence specificity at the differentially expressed gene promoters, the top 1044 among all PHF6 summits were selected and run in motif discovery in parallel. A motif length of eight was used and selected the top 10 significant motifs returned by MEME (Bailey and Elkan 1994). The motifs thus discovered were then tested for similarity to known motifs (as in the JASPAR CORE 2014 vertebrates [Mathelier et al. 2013], UniProbe mouse [Hume et al. 2015], and other human and mouse motifs [Jolma et al. 2013]) using the Tomtom tool (Gupta et al. 2007) with a minimum motif overlap of one and Pearson distance as the distance metric for motif similarity. To identify other transcription factors that cobind with PHF6, mm9 sequences were obtained for ±1-kb regions around the TSSs of differentially expressed genes and tested for known motif enrichment using the Bioconductor PWMEnrich package (https://bioconductor.org/packages/release/bioc/html/PWMEnrich.html). A default Mus musculus genomic background and the MotifDb database of motifs (https://rdrr.io/bioc/MotifDb) included in the package were used, and a P-value threshold of 0.01 was set for reporting significant transcription factors whose motifs were enriched in the selected regions. Input sequences were randomly shuffled 100 times, motif enrichment was performed for each shuffled sequence set, and motifs that came up as significant (P < 0.001) in >5% of the shuffled sets were considered to be false positive enrichments. A total of 28 motifs corresponding to 24 unique transcription factors passed the filtering step and were reported as potential cobinding partners of PHF6 at the promoters of differentially expressed genes (Supplemental Table 3). Gene functions were determined by using the PANTHER classification system (Thomas et al. 2003; Mi et al. 2004).
ChIP-qPCR
ChIP was performed as above. qPCR reactions were performed as described above. Quantities were normalized to input, calculated using the ΔΔCT method, and expressed as fold enrichment relative to a negative binding control (Pchd10). The primer sequences (forward/reverse) used were Btg2 (5′-GACACTGACAGAGCCGTTCA-3′; 5′ACACTCCTCCCACCAAACAG-3′), Cd74 (5′-GATGGCTACTCCCTTGCTGA-3′; 5′-GATGGCTACTCCCTTGCTGA-3′), Cd4 (5′-TCTCCTGCTTCAGGGTCAGT-3′; 5′-AGGGACACCCTGTTTCTGTG-3′), Gata3 (5′-AGCCCAGGACTGACTAAGCA-3′; 5′-TGTTTGGGGGTTTGTTTGTT-3′), and Pchd10 (5′-CCCTGGACTTGCTGACTAGC-3′; 5′-GTGCAAACCAGAACATGGTG-3′).
Chromatin accessibility by ATAC-seq
ATAC-seq libraries from Phf6WT and Phf6KO B-ALL cells were constructed as reported previously (Buenrostro et al. 2013). For the ATAC-seq data analysis, mapped ATAC reads were summarized into 500-bp bins. The DESeq2 package in R (Love et al. 2014) was then applied to the binned reads to analyze for differential chromatin accessibility between Phf6 wild-type and knockout cells. To call differentially accessible chromatin regions, we used a fold change cutoff of >2 and a FDR cutoff of <0.001. GREAT (genomic regions of enrichment annotations tool) (McLean et al. 2010) was used to analyze for enriched gene sets and other genomic features in the regions that were more or less accessible in Phf6KO cells separately. To test enrichment of known and de novo sequence motifs within these differentially accessible chromatin regions, we used the HOMER motif analysis toolkit (Heinz et al. 2010) on these regions using all mappable ATAC regions as background and the UCSC mm9 genome, with all other parameters set to default values.
NucleoATAC software was used with default parameters to call nucleosome signals from raw ATAC-seq signals in Phf6WT cells (Schep et al. 2015, 2017). Background-corrected nucleosome signals ±1 kb from TSSs annotated in the mm9 genome were then stacked into a heat map with rows corresponding to individual TSSs ordered by gene expression in Phf6WT cells. To visualize PHF6 occupancy at these sites, a heat map of normalized PHF6 signals in the same row order was also generated. ATAC-seq raw data and processed TSS nucleosome signals are available in the GEO database with accession number GSE98716 under superseries GSE77457.
Statistical analysis
Results are expressed as means ± standard deviation. If not stated otherwise, statistical significance was determined with Prism software (GraphPad version 6.0) by comparison of mean values by the two-tailed unpaired Student's t-test on two experimental conditions, with P < 0.05 considered statistically significant. Statistical significance levels are designated as follows: P < 0.05 (*), P < 0.01 (**), P < 0.001 (***), P < 0.0001 (****), and not significant (n.s.). Survival curves were generated using Prism software (GraphPad version 6.0), and the Mantel-Cox test was applied to pairwise comparisons of survival data. No statistical methods were used to predetermine sample size.
Accession numbers
All RNA-seq and ChIP-seq data sets have been deposited into the NCBI GEO repository under BioProjectID GSE74878. The ATAC-seq data set and processed TSS nucleosome signals are available in the GEO database with accession number GSE98716 under superseries GSE77457.
Supplementary Material
Acknowledgments
We thank Alexey Soshnev (Allis laboratory) for help in the preparation of the model figure. We thank S. Levine and S. Motola for RNA-seq support, and T. Volkert and S. Gupta for ChIP-seq support. This work was performed with the support of the Koch Institute's Robert A. Swanson (1969) Biotechnology Center. We thank K. Cormier from the Hope Babette Tang (1983) Histology Facility, and G. Paradis, M.Griffin, M. Jennings, and M. Saturno-Condón from the Flow Cytometry Core Facility. Y.M.S.-F. was supported by the National Cancer Institute (NCI) of the National Institutes of Health (NIH) under award number F31-CA183405. J.M.E.B. was supported by the National Science Foundation Graduate Research Fellowship under award number 1122374. Y.L. was supported by the MIT Department of Biology training grant. This work was supported in part by the Ludwig Center for Molecular Oncology at Massachusetts Institute of Technology, the Bridge Project (a collaboration between the Koch Institute and the Dana-Farber/Harvard Cancer Center), and Koch Institute Support (core) grant P30-CA14051 from the NCI. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or National Science Foundation.
Footnotes
Supplemental material is available for this article.
Article published online ahead of print. Article and publication date are online at http://www.genesdev.org/cgi/doi/10.1101/gad.295857.117.
References
- Abbas AR, Wolslegel K, Seshasayee D, Modrusan Z, Clark HF. 2009. Deconvolution of blood microarray data identifies cellular activation patterns in systemic lupus erythematosus. PLoS One 4: e6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arenzana TL, Smith-Raska MR, Reizis B. 2009. Transcription factor Zfx controls BCR-induced proliferation and survival of B lymphocytes. Blood 113: 5857–5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. 1994. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol 2: 28–36. [PubMed] [Google Scholar]
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. 2012. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 41: D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell JJ, Bhandoola A. 2008. The earliest thymic progenitors for T cells possess myeloid lineage potential. Nature 452: 764–767. [DOI] [PubMed] [Google Scholar]
- Belver L, Ferrando A. 2016. The genetics and mechanisms of T cell acute lymphoblastic leukaemia. Nat Rev Cancer 16: 494–507. [DOI] [PubMed] [Google Scholar]
- Borst J, van de Griend RJ, van Oostveen JW, Ang SL, Melief CJ, Seidman JG, Bolhuis RL. 1987. A T-cell receptor γ/CD3 complex found on cloned functional lymphocytes. Nature 325: 683–688. [DOI] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ. 2013. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10: 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bullard JH, Purdom E, Hansen KD, Dudoit S. 2010. Evaluation of statistical methods for normalization and differential expression in mRNA-seq experiments. BMC Bioinformatics 11: 94–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CM, Endrizzi BT, Wu J, Ding X, Weinreich MA, Walsh ER, Wani MA, Lingrel JB, Hogquist KA, Jameson SC. 2006. Kruppel-like factor 2 regulates thymocyte and T-cell migration. Nature 442: 299–302. [DOI] [PubMed] [Google Scholar]
- Cobaleda C, Jochum W, Busslinger M. 2007. Conversion of mature B cells into T cells by dedifferentiation to uncommitted progenitors. Nature 449: 473–477. [DOI] [PubMed] [Google Scholar]
- Cooley M, Fang P, Fang F, Nephew KP, Chien J. 2015. Molecular determinants of chemotherapy resistance in ovarian cancer. Pharmacogenomics 16: 1763–1767. [DOI] [PubMed] [Google Scholar]
- Dai X, Chen Y, Di L, Podd A, Li G, Bunting KD, Hennighausen L, Wen R, Wang D. 2007. Stat5 is essential for early B cell development but not for B cell maturation and function. J Immunol 179: 1068–1079. [DOI] [PubMed] [Google Scholar]
- Dickins RA, Hemann MT, Zilfou JT, Simpson DR, Ibarra I, Hannon GJ, Lowe SW. 2005. Probing tumor phenotypes using stable and regulated synthetic microRNA precursors. Nat Genet 37: 1289–1295. [DOI] [PubMed] [Google Scholar]
- Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, Root DE. 2014. Rational design of highly active sgRNAs for CRISPR–Cas9-mediated gene inactivation. Nat Biotechnol 32: 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, Lash AE. 2002. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30: 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyquem S, Chemin K, Fasseu M, Bories J-C. 2004. The Ets-1 transcription factor is required for complete pre-T cell receptor function and allelic exclusion at the T cell receptor β locus. Proc Natl Acad Sci 101: 15712–15717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner R, Wu D, Cherian S, Fang M, Hanafi L-A, Finney O, Smithers H, Jensen MC, Riddell SR, Maloney DG, et al. 2016. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 127: 2406–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Stamatoyannopoulos JA, Bailey TL, Noble WS. 2007. Quantifying similarity between motifs. Genome Biol 8: R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahtola S, Tuomela S, Elo L, Häkkinen T, Karenko L, Nedoszytko B, Heikkilä H, Saarialho-Kere U, Roszkiewicz J, Aittokallio T, et al. 2006. Th1 response and cytotoxicity genes are down-regulated in cutaneous T-cell lymphoma. Clin Cancer Res 12: 4812–4821. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38: 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann R, Seidl T, Neeb M, Rolink A, Melchers F. 2002. Changes in gene expression profiles in developing B cells of murine bone marrow. Genome Res 12: 98–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horcher M, Souabni A, Busslinger M. 2001. Pax5/BSAP maintains the identity of B cells in late B lymphopoiesis. Immunity 14: 779–790. [DOI] [PubMed] [Google Scholar]
- Hume MA, Barrera LA, Gisselbrecht SS, Bulyk ML. 2015. UniPROBE, update 2015: new tools and content for the online database of protein-binding microarray data on protein–DNA interactions. Nucleic Acids Res 43: D117–D122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK III, Wu T, Li Q-Z, Davis LS, Mohan C, Perlman H. 2008. Combined deficiency of proapoptotic regulators Bim and Fas results in the early onset of systemic autoimmunity. Immunity 28: 206–217. [DOI] [PubMed] [Google Scholar]
- Jacoby E, Nguyen SM, Fountaine TJ, Welp K, Gryder B, Qin H, Yang Y, Chien CD, Seif AE, Lei H, et al. 2016. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun 7: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janeway CA, Carding S, Jones B, Murray J, Portoles P, Rasmussen R, Rojo J, Saizawa K, West J, Bottomly K. 1988. CD4+ T cells: specificity and function. Immunol Rev 101: 39–80. [DOI] [PubMed] [Google Scholar]
- Jeffrey KL, Brummer T, Rolph MS, Liu SM, Callejas NA, Grumont RJ, Gillieron C, Mackay F, Grey S, Camps M, et al. 2006. Positive regulation of immune cell function and inflammatory responses by phosphatase PAC-1. Nat Immunol 7: 274–283. [DOI] [PubMed] [Google Scholar]
- Jolma A, Yan J, Whitington T, Toivonen J, Nitta KR, Rastas P, Morgunova E, Enge M, Taipale M, Wei G, et al. 2013. DNA-binding specificities of human transcription factors. Cell 152: 327–339. [DOI] [PubMed] [Google Scholar]
- Kiaii S, Clear AJ, Ramsay AG, Davies D, Sangaralingam A, Lee A, Calaminici M, Neuberg DS, Gribben JG. 2013. Follicular lymphoma cells induce changes in T-cell gene expression and function: potential impact on survival and risk of transformation. J Clin Oncol 31: 2654–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku S-Y, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbé DP, Gomez EC, Wang J, et al. 2017. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355: 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laiosa CV, Stadtfeld M, Xie H, de Andres-Aguayo L, Graf T. 2006. Reprogramming of committed T cell progenitors to macrophages and dendritic cells by C/EBPα and PU.1 transcription factors. Immunity 25: 731–744. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10: R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara-Astiaso D, Weiner A, Lorenzo-Vivas E, Zaretsky I, Jaitin DA, David E, Keren-Shaul H, Mildner A, Winter D, Jung S, et al. 2014. Chromatin state dynamics during blood formation. Science 345: 943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TI, Johnstone SE, Young RA. 2006. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat Protoc 1: 729–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng N, Dawson JA, Thomson JA, Ruotti V, Rissman AI, Smits BMG, Haag JD, Gould MN, Stewart RM, Kendziorski C. 2013. EBSeq: an empirical Bayes hierarchical model for inference in RNA-seq experiments. Bioinformatics 29: 1035–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12: 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Xiao L, Xu J, Zhang R, Guo J, Olson J, Wu Y, Li J, Song C, Ge Z. 2016. Co-existence of PHF6 and NOTCH1 mutations in adult T-cell acute lymphoblastic leukemia. Oncol Lett 12: 16–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litzow MR, Ferrando AA. 2015. How I treat T-cell acute lymphoblastic leukemia in adults. Blood 126: 833–841. [DOI] [PubMed] [Google Scholar]
- Liu Z, Li F, Ruan K, Zhang J, Mei Y, Wu J, Shi Y. 2014. Structural and functional insights into the human Börjeson-Forssman-Lehmann syndrome-associated protein PHF6. J Biol Chem 289: 10069–10083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15: 31–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lower KM, Turner G, Kerr BA, Mathews KD, Shaw MA, Gedeon ÁK, Schelley S, Hoyme HE, White SM, Delatycki MB, et al. 2002. Mutations in PHF6 are associated with Börjeson-Forssman-Lehmann syndrome. Nat Genet 32: 661–665. [DOI] [PubMed] [Google Scholar]
- Luo Q, Ma X, Wahl SM, Bieker JJ, Crossley M, Montaner LJ. 2004. Activation and repression of interleukin-12 p40 transcription by erythroid Kruppel-like factor in macrophages. J Biol Chem 279: 18451–18456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathelier A, Zhao X, Zhang AW, Parcy F, Worsley-Hunt R, Arenillas DJ, Buchman S, Chen C-Y, Chou A, Ienasescu H, et al. 2013. JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res 42: D142–D147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28: 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meacham CE, Lawton LN, Soto-Feliciano YM, Pritchard JR, Joughin BA, Ehrenberger T, Fenouille N, Zuber J, Williams RT, Young RA, et al. 2015. A genome-scale in vivo loss-of-function screen identifies Phf6as a lineage-specific regulator of leukemia cell growth. Genes Dev 29: 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H. 2004. The PANTHER database of protein families, subfamilies, functions and pathways. Nucleic Acids Res 33: D284–D288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki M, Miyazaki K, Chen S, Chandra V, Wagatsuma K, Agata Y, Rodewald H-R, Saito R, Chang AN, Varki N, et al. 2015. The E–Id protein axis modulates the activities of the PI3K–AKT–mTORC1–Hif1a and c-myc/p19Arf pathways to suppress innate variant T FH cell development, thymocyte expansion, and lymphomagenesis. Genes Dev 29: 409–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori S, Rempel RE, Chang JT, Yao G, Lagoo AS, Potti A, Bild A, Nevins JR. 2008. Utilization of pathway signatures to reveal distinct types of B lymphoma in the E-myc model and human diffuse large B-cell lymphoma. Cancer Res 68: 8525–8534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu P, Zhang Z, Benelli M, Karthaus WR, Hoover E, Chen C-C, Wongvipat J, Ku S-Y, Gao D, Cao Z, et al. 2017. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 355: 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, et al. 2007. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 446: 758–764. [DOI] [PubMed] [Google Scholar]
- Nechanitzky R, Akbas D, Scherer S, Györy I, Hoyler T, Ramamoorthy S, Diefenbach A, Grosschedl R. 2013. Transcription factor EBF1 is essential for the maintenance of B cell identity and prevention of alternative fates in committed cells. Nat Immunol 14: 867–875. [DOI] [PubMed] [Google Scholar]
- Niebuhr B, Kriebitzsch N, Fischer M, Behrens K, Gunther T, Alawi M, Bergholz U, Muller U, Roscher S, Ziegler M, et al. 2013. Runx1 is essential at two stages of early murine B-cell development. Blood 122: 413–423. [DOI] [PubMed] [Google Scholar]
- Nutt SL, Heavey B, Rolink AG, Busslinger M. 1999. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature 401: 556–562. [DOI] [PubMed] [Google Scholar]
- Oser MG, Niederst MJ, Sequist LV, Engelman JA. 2015. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol 16: e165–e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Painter MW, Davis S, Hardy RR, Mathis D, Benoist C, The Immunological Genome Project Consortium. 2011. Transcriptomes of the B and T lineages compared by multiplatform microarray profiling. J Immunol 186: 3047–3057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR–Cas9 system. Nat Protoc 8: 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathert P, Roth M, Neumann T, Muerdter F, Roe J-S, Muhar M, Deswal S, Cerny-Reiterer S, Peter B, Jude J, et al. 2015. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature 525: 543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg EV. 2011. T cell lineage commitment: identity and renunciation. J Immunol 186: 6649–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberg EV. 2014. Transcriptional control of early T and B cell developmental choices. Annu Rev Immunol 32: 283–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge DN, Bouveresse DJ-R. 2013. Independent components analysis with the JADE algorithm. Trends Anal Chem 50: 22–32. [Google Scholar]
- Sánchez-Rivera FJ, Jacks T. 2015. Applications of the CRISPR–Cas9 system in cancer biology. Nat Rev Cancer 15: 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schebesta M, Heavey B, Busslinger M. 2002. Transcriptional control of B-cell development. Curr Opin Immunol 14: 216–223. [DOI] [PubMed] [Google Scholar]
- Schep AN, Buenrostro JD, Denny SK, Schwartz K, Sherlock G, Greenleaf WJ. 2015. Structured nucleosome fingerprints enable high-resolution mapping of chromatin architecture within regulatory regions. Genome Res 25: 1757–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schep AN, Wu B, Buenrostro JD, Greenleaf WJ. 2017. chromVAR: inferring transcription factor variation from single-cell epigenomic data. bioRxiv 10.1101/110346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz KR, Bowman WP, Aledo A, Slayton WB, Sather H, Devidas M, Wang C, Davies SM, Gaynon PS, Trigg M, et al. 2009. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: a children's oncology group study. J Clin Oncol 27: 5175–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequist LV, Waltman BA, Dias-Santagata D, Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger S, Cosper AK, et al. 2011. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med 3: 75ra26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamroe CL, Comeau JM. 2013. Ponatinib: a new tyrosine kinase inhibitor for the treatment of chronic myeloid leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. Ann Pharmacother 47: 1540–1546. [DOI] [PubMed] [Google Scholar]
- Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. 2015. Discovery of cancer drug targets by CRISPR–Cas9 screening of protein domains. Nat Biotechnol 33: 661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeets MFMA, Chan AC, Dagger S, Bradley CK, Wei A, Izon DJ. 2013. Fli-1 overexpression in hematopoietic progenitors deregulates T cell development and induces pre-T cell lymphoblastic leukaemia/lymphoma. PLoS One 8: e62346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaram R, Sigvardsson M. 2015. Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood 126: 144–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PD, Campbell MJ, Kejariwal A, Mi H, Karlak B, Daverman R, Diemer K, Muruganujan A, Narechania A. 2003. PANTHER: a library of protein families and subfamilies indexed by function. Genome Res 13: 2129–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoms JAI, Birger Y, Foster S, Knezevic K, Kirschenbaum Y, Chandrakanthan V, Jonquieres G, Spensberger D, Wong JW, Oram SH, et al. 2011. ERG promotes T-acute lymphoblastic leukemia and is transcriptionally regulated in leukemic cells by a stem cell enhancer. Blood 117: 7079–7089. [DOI] [PubMed] [Google Scholar]
- Todd MAM, Picketts DJ. 2012. PHF6 interacts with the nucleosome remodeling and deacetylation (NuRD) complex. J Proteome Res 11: 4326–4337. [DOI] [PubMed] [Google Scholar]
- Tokoyoda K, Zehentmeier S, Hegazy AN, Albrecht I, GrUn JR, LOhning M, Radbruch A. 2009. Professional memory CD4+ T lymphocytes preferentially reside and rest in the bone marrow. Immunity 30: 721–730. [DOI] [PubMed] [Google Scholar]
- Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N, De Moerloose B, Philippé J, González-García S, Toribio ML, et al. 2010. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet 42: 338–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vlierberghe P, Patel J, Abdel-Wahab O, Lobry C, Hedvat CV, Balbin M, Nicolas C, Payer AR, Fernandez HF, Tallman MS, et al. 2011. PHF6 mutations in adult acute myeloid leukemia. Leukemia 25: 130–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada H, Masuda K, Satoh R, Kakugawa K, Ikawa T, Katsura Y, Kawamoto H. 2008. Adult T-cell progenitors retain myeloid potential. Nature 452: 768–772. [DOI] [PubMed] [Google Scholar]
- Wang L, Wildt KF, Castro E, Xiong Y, Feigenbaum L, Tessarollo L, Bosselut R. 2008. The zinc finger transcription factor Zbtb7b represses CD8-lineage gene expression in peripheral CD4+ T cells. Immunity 29: 876–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Qiu H, Jiang H, Wu L, Dong S, Pan J, Wang W, Ping N, Xia J, Sun A, et al. 2011. Mutations of PHF6 are associated with mutations of NOTCH1, JAK1 and rearrangement of SET–NUP214 in T-cell acute lymphoblastic leukemia. Haematologica 96: 1808–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Leung JWC, Gong Z, Feng L, Shi X, Chen J. 2013. PHF6 regulates cell cycle progression by suppressing ribosomal RNA synthesis. J Biol Chem 288: 3174–3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E, Kawaoka S, Roe J-S, Shi J, Hohmann AF, Xu Y, Bhagwat AS, Suzuki Y, Kinney JB, Vakoc CR. 2015. The transcriptional cofactor TRIM33 prevents apoptosis in B lymphoblastic leukemia by deactivating a single enhancer. eLife 4: e06377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wildt KF, Sun G, Grueter B, Fischer M, Zamisch M, Ehlers M, Bosselut R. 2007. The transcription factor Zbtb7b promotes CD4 expression by antagonizing Runx-mediated activation of the CD4 silencer. J Immunol 179: 4405–4414. [DOI] [PubMed] [Google Scholar]
- Williams RT, Roussel MF, Sherr CJ. 2006. Arf gene loss enhances oncogenicity and limits imatinib response in mouse models of Bcr-Abl-induced acute lymphoblastic leukemia. Proc Natl Acad Sci 103: 6688–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, et al. 2006. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature 442: 86–90. [DOI] [PubMed] [Google Scholar]
- Xie H, Ye M, Feng R, Graf T. 2004. Stepwise reprogramming of B cells into macrophages. Cell 117: 663–676. [DOI] [PubMed] [Google Scholar]
- Yu HA, Arcila ME, Rekhtman N, Sima CS, Zakowski MF, Pao W, Kris MG, Miller VA, Ladanyi M, Riely GJ. 2013. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin Cancer Res 19: 2240–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based analysis of ChIP-seq (MACS). Genome Biol 9: R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Mejia LA, Huang J, Valnegri P, Bennett EJ, Anckar J, Jahani-Asl A, Gallardo G, Ikeuchi Y, Yamada T, et al. 2013. The X-linked intellectual disability protein PHF6 associates with the PAF1 complex and regulates neuronal migration in the mammalian brain. Neuron 78: 986–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Yamane H, Paul WE. 2010. Differentiation of effector CD4 T cell populations. Annu Rev Immunol 28: 445–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.