

Abstract
Hepatic ischemia-reperfusion injury (IRI) and associated inflammation contributes to liver dysfunction and complications after liver surgery and transplantation. Mesenchymal stem cells (MSC) have been reported to reduce hepatic IRI because of their reparative immunomodulatory effects in injured tissues. Recent studies have highlighted beneficial effects of extracellular vesicles from MSCs (MSC-EV) on tissue injury. The effects of systemically administered mouse bone marrow derived MSC-EV were evaluated in an experimental murine model of hepatic IRI induced by cross clamping the hepatic artery and portal vein for 90 minutes followed by reperfusion for periods of upto 6 hours. Compared with controls, intravenous administration of MSC-EV 30 minutes prior to IRI dramatically reduced the extent of tissue necrosis, decreased caspase-3 positive and apoptotic cells, and reduced serum aminotransferase levels. MSC-EV increased hepatic mRNA expression of NACHT, LRR and PYD domains-containing protein 12 (Nlrp12), and the chemokine (C-X-C motif) ligand 1 (CXCL1), and reduced mRNA expression of several inflammatory cytokines such as IL-6 during IRI. MSC-EV increased cell viability and suppressed both oxidative injury and NF-κB activity in AML12 murine hepatocytes in vitro. In conclusion, the administration of EV derived from bone marrow derived MSCs may ameliorate hepatic IRI by reducing hepatic injury through modulation of the inflammatory response.
Keywords: Exosomes, inflammasomes, cytokines, chemokines, liver injury
Hepatic ischemia-reperfusion injury (IRI) is an important contributor to outcomes of liver surgery and transplantation. Complications associated with IRI after liver transplantation can range from early graft dysfunction to graft non-function. Cold ischemia occurring during organ preservation may be followed by warm ischemia after liver transplantation. Ischemic injury has been implicated in poor outcomes with the use of organ donations following cardiac death, where warm ischemic injury may be an important initiating event. IRI can also contribute to outcomes following hepatic resection, and resuscitation after hemorrhagic shock (1–4). In all of these settings, hepatocellular injury and IRI can occur as a result of acute interruption of blood flow to the liver with subsequent reperfusion. The subsequent acute inflammatory cascade leads to significant damage to hepatocytes and non-parenchymal cells.
Mesenchymal stem cells (MSCs) are attractive for tissue replacement because of their regenerative ability, multipotent capability and immunoregulatory effects. Indeed, MSCs derived from bone marrow, umbilical cord or adipose tissue can alleviate hepatic ischemia reperfusion injury by inhibiting apoptosis (5–8), suppressing oxidative stress (8, 9) or immunomodulation (10). Although MSCs can differentiate and replace tissue, cells may not survive long enough to do so following their infusion (11). More likely, the therapeutic benefit observed with MSCs is associated with release of paracrine factors. Systemic injection of conditioned medium from MSCs provide a significant survival benefit in vivo, and prevent the release of liver injury biomarkers, reduce hepatocyte apoptosis and increase proliferation (12) (13) (14, 15). These paracrine effects could be mediated through secreted proteins or through cell-derived extracellular vesicles (EV). However, the contributions of the latter during hepatic IRI are not well established.
Protective effects of MSCs during ischemic injury have been reported in several animal models (5–9, 14–20). Moreover, EV have been associated with protective effects in cardiac or renal ischemia (9, 16, 19, 20). EVs are a heterogeneous population that comprise of microvesicles and exosomes (21, 22). These vesicles are enclosed by a lipid bilayer membrane and their contents include proteins, lipids, RNA or DNA molecules (23). We have recently shown that EV derived from MSC (MSC-EV) can attenuate mortality in experimental hepatic failure in mice, and further that MSCs derived EV can protect hepatocytes from apoptosis and reduce injury. In the present study, we evaluated the effects of MSC-EV on experimental hepatic IRI to ascertain their potential role in ameliorating tissue injury and inflammation.
Experimental Procedures
Cells and cell culture
AML12, mouse hepatocyte cells were obtained from American Type Culture Collection (ATCC, Rockville, MD, USA) and maintained in Dulbecco’s Modified Eagle Medium (DMEM)/F-12 + Glutamax containing 6.7 ng/mL selenium, 10μg/mL insulin, 5.5μg/mL transferrin, 40 ng/mL dexamethasone (Sigma-Aldrich, St Louis, MO), and 10% FBS. Mouse (C57BL/6) bone marrow-derived MSC were obtained from Life Technologies (Grand Island, NY) and maintained in DMEM/F-12 + Glutamax containing MSC-Qualified FBS. All media was obtained from Life Technologies (Grand Island, NY). Primary hepatocytes were isolated from 6–8 week old C57BL/6 mice (Jackson Labs, Maine USA) as described by Trump et al. with modifications (24). Under isoflurane anesthesia, an abdominal incision was made, the hepatic portal vein was cannulated and a two-step buffer liver perfusion performed. The liver was removed, and hepatocytes separated by gentle mincing and filtering using a cell strainer. Hepatocytes were plated (250,000/well) into collagen coated 6-well dishes.
Biodistribution of systemically administered MSC-EV
MSC-EV were incubated with 6 μmol/l DiR (1,1′-Dioctadecyl-3,3,3′,3′-Tetramethylindotricarbocyanine Iodide) (7) (Life Technologies, Grand Island, NY) for 30 minutes, then washed with PBS and collected after ultracentrifugation. The labeled MSC-EV (2.0×1010) were administrated via subcutaneous, intraperitoneal, intravenous or per oral to healthy mice, with control mice receiving no treatment. Organs were harvested 6 hour later and labeled EVs quantitated ex situ using an in vivo imaging system (IVIS) (Perkin Elmer, Waltham, MA).
Hepatic ischemia reperfusion
All animal studies were performed in accordance with institutional animal care and use committee approved protocols (# A11514-14). First, MSC-EV (2 ×1010 particles/body) or PBS (vehicle) in a 200 μl volume were administered via tail vein injection to 10–12-week-old male C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME). After 30 minutes, mice were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg) IP, and a midline laparotomy incision performed. The hepatic artery and the portal vein above the branching to the right lateral segment were clamped using an atraumatic clip with jaws 1mm wide × 5 mm long (Cat# 14120, World Precision Instruments, Sarasota, FL), thus resulting in partial (~70%) hepatic ischemia (Supplementary Fig 1) (25). The liver was kept moist at 37°C with gauze soaked in sterile saline, while body temperature was maintained at 37°C using a heating blanket. The clip was removed after 90 minutes. Sham surgeries were identical except that hepatic blood flow was not reduced with an atraumatic clip. After 1, 3 or 6 hour reperfusion, blood was collected by cardiac puncture and the liver removed and snap-frozen in liquid nitrogen or fixed in 4% buffered formalin.
In vitro hypoxia/normoxia studies
Primary mouse hepatocytes were cultured in 6-well dishes at 250,000 cells/well and allowed to attach overnight. Cells were then treated with 1.8 × 108 mMSC-EV particles/ml or PBS (controls), and placed in a self -contained and sealed hypoxia chamber within an incubator with 5% CO2, 21% O2, balanced with N2, and 95% humidity. For hypoxic conditions, the inlet and outlet ports were opened and purged with 5% CO2 and 95% N2 at 20 psi for 4 minutes, after which the ports were sealed for 90 minutes. For normoxic conditions, cells were placed in the chamber with clamps open for 90 minutes. After that time, chambers were incubated with ports open for a further 6 hours, cells collected, and RNA isolated.
Reactive oxygen species
AML12 were plated into 96-well plates (1×104 cells/well) and incubated overnight. 100 μl of 0.1×2′,7′-dichlorodihydrofluorescin diacetate (DCFH-DA)/media was added and cells incubated at 37℃ for 60 minutes, cultivated in 100 μl vesicle-depleted medium with 2×1010 particle/ml MSC-EV or PBS (vehicle) for 24 hours. The media was removed and replaced with media with or without 50 mM hydrogen peroxide (H2O2). Intracellular ROS concentrations were measured using OxiSelect™ Intracellular ROS Assay Kit (Cell Biolabs, San Diego, CA). Fluorescence were measured using a FLUOstar Omega reader (BMG Labtech, Cary, NC) at 480/530 nm.
Nuclear factor-kappa B (NF-κB) activity
NF-κB promoter activity was determined using a Ready-To-Glow™ secreted luciferase reporter system (Clontech, Mountain View, CA). AML12 cells were plated in 6 well plates (2×105 cells/well) and transfected with the pNF-κB-MetLuc or pMetLuc2-Control plasmids using lipofectamine 2000 (Invitrogen, Carlsbad, CA). After transfection, 5×104 cells AML12 were incubated in 24-well plates overnight and then cultured in 100 μl vesicle-depleted medium with 2×1010 particles/ml MSC-EV or PBS (vehicle) for 24 hours. Cells were then incubated with or without 50 mM H2O2 for 1 hour. 50 μl of culture media from each well was transferred into a 96-well white plate, 5μl of 1X substrate/reaction buffer added, and the luminescence was measured using a FLUOstar Omega reader (BMG Labtech, Cary, NC).
EV isolation, Histopathology, western-blot, PCR, cell proliferation and protein assays
Details are outlined in the supplemental materials.
Statistical analysis
Data were expressed as mean and standard error from at least three replicates. Comparisons between groups were performed using the two-tailed Student’s t test, and results were considered to be statistically significant when p < 0.05.
Results
EV can be isolated from mouse bone marrow derived MSC in vitro
An ultracentrifugation-based protocol was used to isolate EV released by mouse bone marrow derived MSC in vitro. Isolated vesicles had a mean size of 115 ± 48 nm and peak size 89 nm by nanoparticle tracking analysis, and expressed exosome-associated proteins by immunoblot analysis (Supplementary Fig. 2). The size and surface markers support the presence of significant enrichment of exosomes within the isolated EV pool. However, the presence of other types of vesicles cannot be excluded, and we have therefore referred to the isolated vesicles as extracellular vesicles.
Bio-distribution of systemically administered MSC-EV
In order to identify an optimal and effective method for systemic administration, we examined the uptake and bio-distribution of MSC-EV administered via different routes in two mice each. Organ uptake was assessed 6 hours after administration of labeled EVs by different routes. Intra-organ accumulation of labeled MSC-EV was greatest with either intravenous or intraperitoneal administration. In both cases, accumulation of MSC-EV was most prominent in the liver and spleen compared to other organs (Fig. 1). Thus, intravenous administration is an effective and efficient route for the delivery of EVs to the liver.
Figure 1. Biodistribution of MSC-EV.
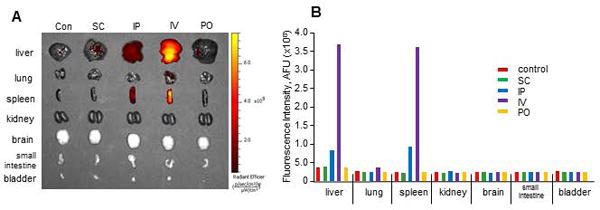
EVs labeled by DiR were administrated via subcutaneous (SC), intraperitoneal (IP), intravenous (IV) or peroral (PO) routes to healthy mice (n=2 each). (A) Accumulation of labeled EVs in these organs were detected ex vivo after 6 hours, and (B) the intensity of fluorescent signal was measured using an IVIS, and the average shown here. Hepatic uptake was noted with IP or IV, with the latter having the greatest uptake.
MSC-EV protect against experimental hepatic IRI
The effects of MSC-EV were then evaluated in a murine model of hepatic IRI. MSC-EV (2×1010 particles), or PBS (vehicle) were injected via tail vein 30 minutes prior to ischemia (Fig. 2). In the PBS control group, a temporal increase in serum alanine aminotransferase (ALT) levels was noted at 1, 3 and 6 hours of reperfusion. In contrast, serum ALT levels in the MSC-EV group reached a peak at 3 hour of reperfusion but a decrease was noted at 6 hour of reperfusion. Furthermore, reductions in aspartate aminotransferase (AST), and alkaline phosphatase (ALP), serum blood urea nitrogen (BUN), and a trend towards a reduced creatinine were also noted in mice that received MSC-EV compared with PBS controls after a 6-hour reperfusion. H&E staining of liver tissues obtained from the PBS (vehicle control) group 6 hours after reperfusion showed widespread necrosis around central veins, whereas, the extent of necrosis was significantly reduced in livers from the group that received MSC-EV (Fig. 3). Immunohistochemistry for activated caspase-3 revealed positive cell expression in many cells, and particularly around zone 1 in the PBS group. On the other hand, a significantly reduced number of caspase-3 positive cells were observed in the MSC-EV group. Thus MSC-EV can reduce biochemical and histological manifestations of hepatic injury following experimental hepatic IRI.
Figure 2. MSC-EV reduce biochemical markers of hepatic injury during hepatic ischemia reperfusion injury in vivo.
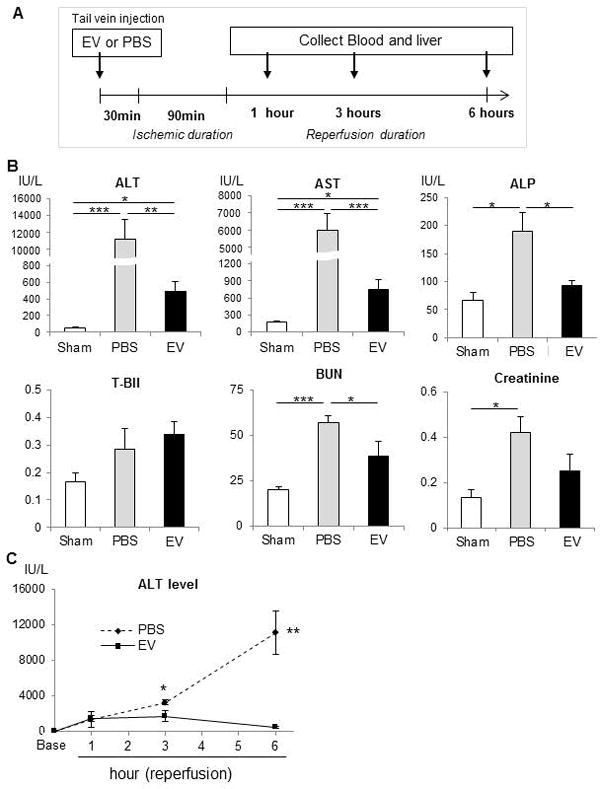
(A) Experimental schema. MSC-EV or vehicle controls (PBS) were administered intravenously 30 minutes prior to induction of hepatic ischemia for 90 minutes, followed by reperfusion for up to 6 hours. (B) Serum biochemical studies 6 hours after reperfusion in mice receiving PBS or MSC-EV (EV) or in sham operated controls. Data are expressed as the mean±SEM (n=6 mice/group). * p<0.05, ** p<0.01, *** p<0.001. (C) Time-course of serum ALT levels in mice during hepatic IRI. Data are expressed as the mean±SEM (n=3–6 mice/group). * p<0.05 or ** p<0.01in EV treated versus PBS controls at the indicated time points after reperfusion.
Figure 3. MSC-EV reduce histological features of liver injury during hepatic ischemia-reperfusion injury in vivo.
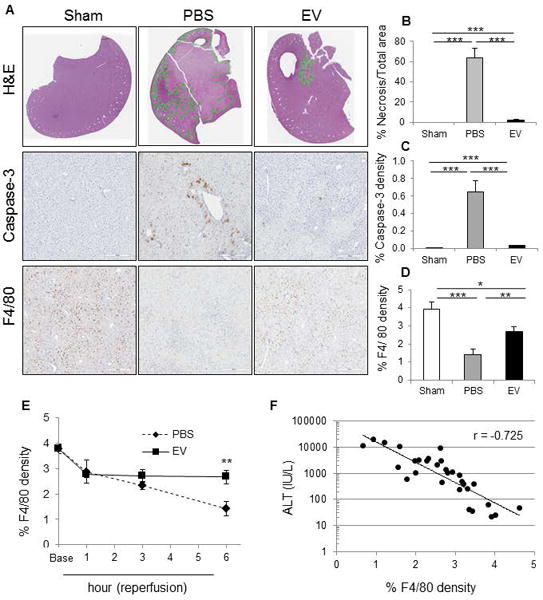
(A) H&E, activated caspase 3 and F4/80 expression in the liver of hepatic IRI mice. Caspase-3-positive cells and F4/80-positive cells are identified by brown counter-staining. Whole liver sections were scanned at ultra-resolution on the ScanScope XT, and positive staining cells were quantified using positive pixel counts by Aperio Imagescope analysis. The percentage of (B) necrosis area, (C) caspase-3-positive cells and (D) F4/80-positive cells are shown for sections from each treatment group. Data are expressed as the mean±SEM (n=6 mice/group). * p<0.05, ** p<0.01, *** p<0.001. (E) F4/80 positive cell density in liver tissues in hepatic IRI mice receiving MSC-EV (EV) or PBS control. Data are expressed as the mean±SEM (n=3–6 mice/group) of % cells expressing F4/80. ** p<0.01 between groups at at 6 hours after reperfusion. (F) Correlation between % F4/80 density in liver tissue of hepatic IRI mice and serum ALT levels in hepatic IRI mice. r = −0.73.
Within the liver, macrophages and Kupffer cells contribute to the regulation of hepatic inflammation and hepatocyte death. We observed a reduction in the number of F4/80 positive cells (macrophage/Kupffer cells) in the PBS group, with the number of cells decreasing over time. A similar reduction was also noted following sham surgery but not in MSC-EV group (Fig. 3). Indeed, in the MSC-EV treated group, the number of F4/80 positive cells increased at 1 hour of reperfusion and persisted at the same level. A strong negative correlation was observed between F4/80 positive cells and ALT level. Although the precise contribution of macrophages/Kupffer cells during liver injury is controversial, their recruitment by MSC-EV in the context of reduced injury is consistent with a protective role in this model.
MSC-EV modulate release of inflammatory mediators from hepatocytes in vitro
To understand the potential role of immune and inflammation-associated mediators, we examined the effect of MSC-EV on the secretion of cytokines and chemokines from murine hepatocytes in vitro. Assays were performed on culture supernatant obtained from AML12 cells incubated with or without MSC-EV after 24 hours. MSC-EV decreased secretion of inflammatory cytokines, such as TNF-α, IL-1α, IL-1β, IL-6, IL-12, or IFNγ compared to control. In contrast, a significant increase in CXCL1 and MCP-1 occurred with MSC-EV (Fig. 4). Next we examined effects of MSC-EV on gene expression, and observed a greater than 1.5-fold increase in mRNA expression of 18 inflammasome, chemokine or cytokine genes in vivo following administration of MSC-EV compared to PBS (vehicle control) (Supplemental Table 1). These included several inflammasome-related genes, such as Nlrp12, Pycard, Naip1, Nlrp1a or Nlrc5, and the chemokine CXCL1 mRNA (Fig 5). In contrast, a decreased mRNA expression of other chemokines such as CCL7 and CXCL3 as well as several cytokines such as IFN β1, IFNγ, IL-1β, IL-33 were observed with MSC-EV. The most prominent amongst these was a greater-than 1.5-fold decrease in IL-6 mRNA. We further examined mRNA expression in primary mouse hepatocytes after 6 hours of normoxia with or without prior exposure to 90 minutes of hypoxia (Supplemental Table 2). The potential contribution of non-parenchymal cells to changes in gene expression observed in vivo are highlighted by the differences between mRNA changes in vivo and in vitro.
Figure 4. MSC-EV alter cytokine/chemokine release.
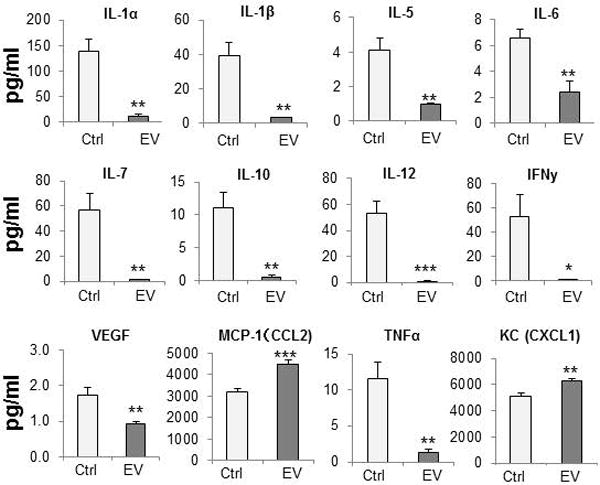
Cytokine and chemokine levels in culture supernatants from AML12 cells incubated with or without MSC-EV. Data are expressed as the mean±SEM (n=4/group) * p<0.05, ** p<0.01, *** p<0.001.
Figure 5. Expression of inflammasome and cytokine-associated genes in liver tissue of hepatic IRI mice.
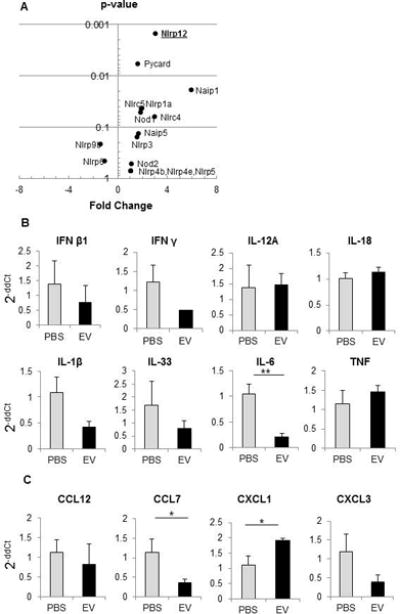
mRNA expression was assessed in liver tissue of hepatic IRI mice treated with MSC-EV or with PBS (vehicle). (A) Plot of p value versus fold-change in expression for Inflammasome-associated genes. (B,C) Alterations in expression of selected cytokine (B) or chemokine (C) genes. Data are expressed as the mean±SEM (n=3 mice/group). * p<0.05, ** p<0.01.
MSC-EV attenuate oxidative stress and NF-κB activity in hepatocytes
In order to ascertain the potential effect of MSC-EV on hepatocyte injury, we examined cell viability in AML12 murine hepatocytes incubated with MSC-EV (0–1×1010 particles/ml) for 24 hours, (Supplementary Figure 3). The results indicate that MSC-EV can directly reduce hepatocyte injury in vitro. The observations made from the in vivo studies indicated that MSC-EV could, in addition, modulate inflammatory responses during hepatic IRI. Oxidative injury associated with an increase in ROS and NF-κB activity can contribute to hepatocyte injury during hepatic IRI (26). Thus, we next evaluated the ability of MSC-EV to dampen or ameliorate the effect of MSC-EV on oxidative stress and NF-κB activation in hepatocytes in vitro. For these studies, we evaluated the effects of MSC-EV on hydrogen peroxide induced ROS generation and NF-κB activity in AML12 cells. An increase in ROS activity was observed following exposure to H2O2, for an hour. This was significantly reduced in the presence of pre-incubation with MSC-EV for 24 hrs (Fig. 6). Similarly, the increase in activity of NF-κB was also reduced by MSC-EV. Thus, MSC-EV could modulate responses to oxidative stress during hepatic IRI and dampen the inflammatory responses that contribute to injury.
Figure 6. MSC-EV modulate oxidative stress.
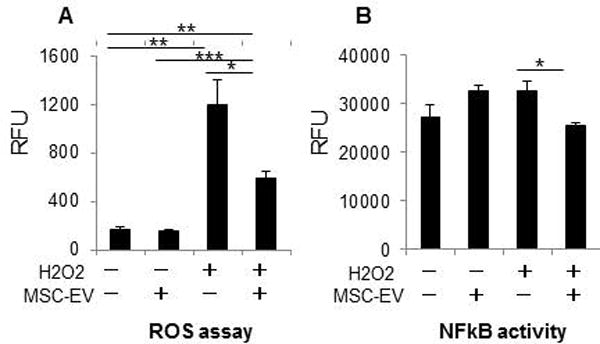
(A) ROS assay in AML12 incubated with or without 1×1010 particles/ml MSC-EV in presence or absence of hydrogen peroxide induced oxidative stress (B) NFκB activity in AML12 incubated with or without 1×1010 particles/ml MSC-EV in presence or absence of hydrogen peroxide. Data are expressed as the mean±SEM (n=3/group) * p<0.05, ** p<0.01, *** p<0.001.
MSC-EV enhance Nlrp12 and CXCL1 mRNA expression in the liver
The direct effects of MSC-EV on hepatocyte gene expression were evaluated in vitro. RNA was extracted from AML12 murine hepatocytes incubated with either 1×1010 particles/ml MSC-EV or PBS (vehicles) for 6 hours, and qRT-PCR performed. The most significant increase in expression of inflammasome genes was noted for Nlrp12, which was increased > 3-fold (p<0.001) in the MSC-EV group compared with either PBS or sham groups. Nlrp12 mRNA expression was significantly increased in vivo during hepatic IRI following administration of MSC-EV group compared to sham or PBS group. Even though an increase in Nlrp12 was only appreciated after 6 hours of reperfusion, there was no significant change in Nlrp12 mRNA expression in the PBS control group (Fig 7).
Figure 7. MSC-EV upregulate hepatic Nlrp12 mRNA in vivo and in vitro.
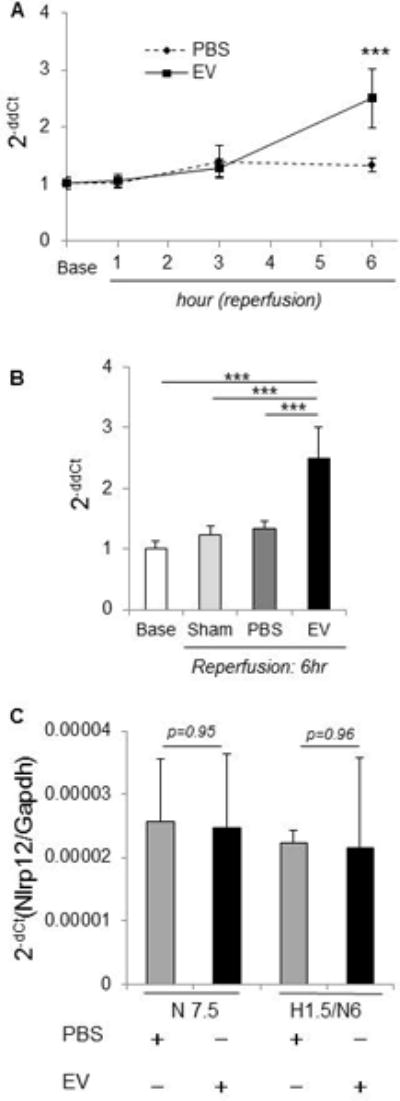
(A) The time-course of Nlrp12 mRNA level in liver tissue of IRI mice. * p<0.05, *** p<0.001 between groups at the indicated times after reperfusion. (B) Nlrp12 mRNA level in hepatic IRI mice at 6 hours after reperfusion. *** p<0.001, Data are expressed as the mean±SEM (n=3–6 mice/group). (C) Nlrp12 mRNA was assessed in primary mouse hepatocytes cultured in the presence of MSC-EV or PBS (controls) after 6 hrs of normoxia with or without prior exposure to 90 minutes hypoxia. Data represents fold-change of Nlrp12 mRNA with MSC-EV compared with PBS (controls). N7.5 = normoxia 7.5 hr, H1.5/N6 = 1.5 hr hypoxia followed by 6 hr normoxia
An increase in mRNA expression of several genes such as Nlrp3, Cxcl1, Cxcl3, Mapk13 or Ptgs2 was noted in hepatocytes incubated with MSC-EV compared to vehicle controls (Supplemental Table 3). Amongst these a > 1.5-fold increase in CXCL1 mRNA was noted in liver tissue of IRI mice receiving EV in vivo and in hepatocytes treated with MSC-EV in vitro compared with controls (Supplementary Fig. 4). Consistent with this, a significant increase in CXCL1 mRNA was also observed within liver tissue in MSC-EV group compared to PBS group after 3 hours and 6 hour of reperfusion (Fig. 8). Although an increase in CXCL1 mRNA was also noted after sham surgery and in livers from mice receiving PBS, this was further significantly increased with MSC-EV after IRI at 6 hours of reperfusion. An increase in cell viability was observed in vitro, in AML12 cells incubated with recombinant CXCL1(0–100 ng/ml) for 24 hours (Supplementary Fig 5). CXCL1 mRNA is amongst several mRNA identified as being selectively enriched within EV derived from MSC, compared to their expression in the cells of origin (Supplemental Table 4). We therefore postulate that CXCL1 mRNA is enriched within MSC-EV and the transfer of CXCL1 to hepatocytes following uptake of MSC-EV could contribute to reduction of hepatic IRI.
Figure 8. Effect of MSC-EV on CXCL1 mRNA expression in vivo and in vitro.
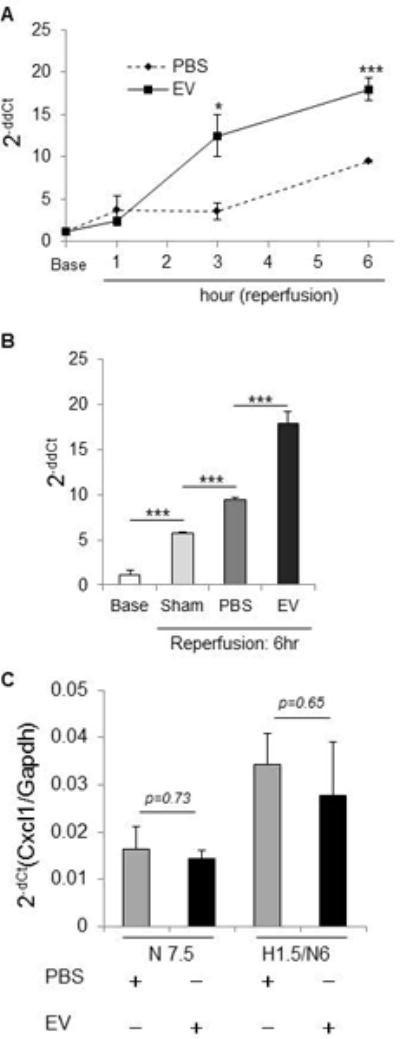
(A) The time-course of CXCL1 mRNA level in liver of hepatic IRI mice. * p<0.05 *** p<0.001 between EV treated and control groups at the indicated time points. (B) CXCL1 mRNA level in hepatic IRI mice at 6 hours after reperfusion. (C) CXCL1 mRNA was assessed in primary mouse hepatocytes cultured with MSC-EV or PBS (controls) for 6 hrs under normoxia with or without prior exposure to 90 minutes hypoxia. Data represents fold-change of Nlrp12 mRNA with MSC-EV compared with PBS (controls). *** p<0.001. Data are expressed as the mean±SEM (n=3–6 mice/group). N7.5 = normoxia 7.5 hr, H1.5/N6 = 1.5 hr hypoxia followed by 6 hr normoxia
Discussion
The multipotent capacity of MSC is an attractive property that supports their use as therapeutic agents to enhance tissue repair and regeneration (27, 28). In addition to an ability to de-differentiate, MSC are capable of modulating inflammatory and immune responses (6, 29, 30). Amelioration of experimental hepatic IRI by bone marrow or adipose derived MSC has been observed in several studies in rodents refs (5, 6, 31–35). These effects have been presumed to reflect the regenerative properties of MSC, but occur rapidly and inconsistent with the time required for tissue replacement. Moreover, MSC are short-lived when administered intravenously (36, 37). Thus, we and others have postulated that the observed beneficial effects are mediated by soluble or acellular factors such as EV released by MSC. We have identified that MSC-EV can localize to the injured liver after systemic administration. MSC-EV have advantages over the use of MSC for therapeutic use by avoiding undesirable effects associated with use of cellular material. The size differences between MSCs (~15–19 μm) (38) and MSC-EV (115±48 nm) also provide further advantages. Thus, while intravenously administered MSCs may become trapped in the lungs (38, 39) and form potentially lethal microemboli(40), we did not observe any similar effects following IV administration of MSC-EV in our studies.
MSC-EV reduced the expression of inflammatory mediators both in vivo and in vitro. The rapid initiation of a sterile inflammatory response could contribute to hepatic IRI. Such responses could be mediated through the release of endogenous danger signals that activate the innate immune system. The NOD-like receptors (NLR) are a major class of cytosolic pattern recognition receptors (PRR). Activation of some family members such as Nlrp1, Nlrp3 and Nlrc4 in response to recognition of intracellular danger associated molecular patterns (DAMPs) and pathogen associated molecular patterns (PAMPs) can result in inflammasome formation. Subsequently caspase-1 activation results in post-translational cleavage of pro-IL-1β and pro-IL-18 into their active forms. IL-1β and IL-18 are important mediators of the inflammatory and cell death response (41, 42). Caspase-1, and inflammasome-dependent pathways may contribute to hepatic IRI. However, even though MSC-EV increased expression of several NLRs, hepatic IL-1β expression was decreased during IRI by MSC-EV. Rather than through direct effects on pro-inflammatory NLR expression, our observations imply that the effects of MSC-EV involve attenuation of inflammation through Nlrp12. Nlrp12 is a negative regulator of inflammatory activity in vitro in the immune system and other settings, through attenuation of non-canonical NF-kB signaling (43, 44). MSC-EV suppressed both experimentally induced oxidative stress and NF-κB activity in hepatocytes, and expression of several inflammation-associated cytokines such as IL-6 and IL-1β that are transcriptionally regulated by NF-κB activation (44). These effects could serve to protect the liver during inflammatory stress. Consequently, attenuation of inflammatory responses associated with tissue injury as a mechanism for the beneficial effects of MSC-EV during hepatic IRI. Thus, targeting Nlrp12 mediated anti-inflammatory responses to modulate hepatic IRI warrants further evaluation as a therapeutic strategy.
In addition to ameliorating the sterile inflammatory response, MSC-EV can directly modulate inflammatory responses such as through CXCL1, a secreted chemokine which binds the G-protein coupled receptor chemokine (C-X-C motif) receptor 2 (CXCR2) to recruit and activate leukocytes. In mice CXCL1 deficiency is associated with colitis and defects in immune cell recruitment to the lung. An increased expression of CXCR2 on hepatocytes has been reported during hepatic injury in several studies, and receptor engagement altering hepatocyte survival and proliferation in a dose dependent mechanism (45). CXCL1 is upregulated in liver allografts that experience early allograft dysfunction, the clinical sequelae of IRI, following transplantation (46). MSC-EV increased CXCL1 release from AML12 hepatocytes in vitro. CXCL1 mRNA expression was increased during hepatic IRI in vivo, with expression further enhanced by MSC-EV. Furthermore CXCL1 directly increased hepatocyte proliferation, consistent with other reports (47). Thus, we speculate that CXCL1 may participate in tissue restoration responses to injury. Although not conclusively demonstrated, increased hepatic expression of CXCL1 could contribute to recruitment of cells that release NLRP12. The number of F4/80 positive cells in liver were significantly increased in MSC-EV or sham group compared with PBS (vehicle) after reperfusion, and negatively correlated with serum ALT levels. A contribution of F4/80 expressing cells with attenuation of injury is consistent with other reports of hepatic protection linked to Kupffer cell-dependent expression of anti-inflammatory cytokines such as interleukin-10 (48). The precise involvement of Kupffer cells needs to be examined because their functional inactivation has been reported to attenuate injury during reperfusion (49, 50).
In conclusion, these studies support the use of EVs derived from MSCs as a therapeutic strategy to alleviate hepatic IRI following hepatic surgery or liver transplantation. The regenerative capacity of donor organs is reduced by hepatic IRI that accompanies liver transplantation. Reducing this injury could increase the use of marginal livers that are obtained from extended criteria donors such as older donors or following cardiac death for transplantation. Therapeutic interventions that alleviate hepatic IRI will increase the donor organ pool available for transplantation and thereby reduce mortality from advanced liver disease.
Supplementary Material
Acknowledgments
Grants and Financial Support: Supported in part by Grant UH3TR000884 from the National Institutes of Health
Abbreviations
- CXCL1
Chemokine (C-X-C motif) ligand 1
- EV
Extracellular vesicles
- H2O2
Hydrogen peroxide
- IRI
Ischemia-reperfusion injury
- MSC
Mesenchymal stem cells
- Nlrp12
NACHT, LRR and PYD domains-containing protein 12
- PBS
Phosphate-buffered saline
Footnotes
Disclosures: There are no potential conflicts relevant to the manuscript
References
- 1.Lentsch AB, Kato A, Yoshidome H, McMasters KM, Edwards MJ. Inflammatory mechanisms and therapeutic strategies for warm hepatic ischemia/reperfusion injury. Hepatology. 2000;32:169–173. doi: 10.1053/jhep.2000.9323. [DOI] [PubMed] [Google Scholar]
- 2.Huguet C, Gavelli A, Bona S. Hepatic resection with ischemia of the liver exceeding one hour. J Am Coll Surg. 1994;178:454–458. [PubMed] [Google Scholar]
- 3.Lemasters JJ, Thurman RG. Reperfusion injury after liver preservation for transplantation. Annu Rev Pharmacol Toxicol. 1997;37:327–338. doi: 10.1146/annurev.pharmtox.37.1.327. [DOI] [PubMed] [Google Scholar]
- 4.Vedder NB, Fouty BW, Winn RK, Harlan JM, Rice CL. Role of neutrophils in generalized reperfusion injury associated with resuscitation from shock. Surgery. 1989;106:509–516. [PubMed] [Google Scholar]
- 5.Kanazawa H, Fujimoto Y, Teratani T, Iwasaki J, Kasahara N, Negishi K, Tsuruyama T, et al. Bone marrow-derived mesenchymal stem cells ameliorate hepatic ischemia reperfusion injury in a rat model. PLoS One. 2011;6:e19195. doi: 10.1371/journal.pone.0019195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saidi RF, Rajeshkumar B, Shariftabrizi A, Bogdanov AA, Zheng S, Dresser K, Walter O. Human adipose-derived mesenchymal stem cells attenuate liver ischemia-reperfusion injury and promote liver regeneration. Surgery. 2014;156:1225–1231. doi: 10.1016/j.surg.2014.05.008. [DOI] [PubMed] [Google Scholar]
- 7.Sun CK, Chang CL, Lin YC, Kao YH, Chang LT, Yen CH, Shao PL, et al. Systemic administration of autologous adipose-derived mesenchymal stem cells alleviates hepatic ischemia-reperfusion injury in rats. Crit Care Med. 2012;40:1279–1290. doi: 10.1097/CCM.0b013e31823dae23. [DOI] [PubMed] [Google Scholar]
- 8.Jin G, Qiu G, Wu D, Hu Y, Qiao P, Fan C, Gao F. Allogeneic bone marrow-derived mesenchymal stem cells attenuate hepatic ischemia-reperfusion injury by suppressing oxidative stress and inhibiting apoptosis in rats. Int J Mol Med. 2013;31:1395–1401. doi: 10.3892/ijmm.2013.1340. [DOI] [PubMed] [Google Scholar]
- 9.Arslan F, Lai RC, Smeets MB, Akeroyd L, Choo A, Aguor EN, Timmers L, et al. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res. 2013;10:301–312. doi: 10.1016/j.scr.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 10.Wan CD, Cheng R, Wang HB, Liu T. Immunomodulatory effects of mesenchymal stem cells derived from adipose tissues in a rat orthotopic liver transplantation model. Hepatobiliary Pancreat Dis Int. 2008;7:29–33. [PubMed] [Google Scholar]
- 11.Mummery CL, Davis RP, Krieger JE. Challenges in Using Stem Cells for Cardiac Repair. Science Translational Medicine. 2010;2 doi: 10.1126/scitranslmed.3000558. [DOI] [PubMed] [Google Scholar]
- 12.van Poll D, Parekkadan B, Cho CH, Berthiaume F, Nahmias Y, Tilles AW, Yarmush ML. Mesenchymal stem cell-derived molecules directly modulate hepatocellular death and regeneration in vitro and in vivo. Hepatology. 2008;47:1634–1643. doi: 10.1002/hep.22236. [DOI] [PubMed] [Google Scholar]
- 13.Parekkadan B, van Poll D, Suganuma K, Carter EA, Berthiaume F, Tilles AW, Yarmush ML. Mesenchymal stem cell-derived molecules reverse fulminant hepatic failure. PLoS One. 2007;2:e941. doi: 10.1371/journal.pone.0000941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pan GZ, Yang Y, Zhang J, Liu W, Wang GY, Zhang YC, Yang Q, et al. Bone marrow mesenchymal stem cells ameliorate hepatic ischemia/reperfusion injuries via inactivation of the MEK/ERK signaling pathway in rats. J Surg Res. 2012;178:935–948. doi: 10.1016/j.jss.2012.04.070. [DOI] [PubMed] [Google Scholar]
- 15.Fu J, Zhang H, Zhuang Y, Liu H, Shi Q, Li D, Ju X. The role of N-acetyltransferase 8 in mesenchymal stem cell-based therapy for liver ischemia/reperfusion injury in rats. PLoS One. 2014;9:e103355. doi: 10.1371/journal.pone.0103355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai RC, Arslan F, Lee MM, Sze NS, Choo A, Chen TS, Salto-Tellez M, et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010;4:214–222. doi: 10.1016/j.scr.2009.12.003. [DOI] [PubMed] [Google Scholar]
- 17.Zhang J, Chen GH, Wang YW, Zhao J, Duan HF, Liao LM, Zhang XZ, et al. Hydrogen peroxide preconditioning enhances the therapeutic efficacy of Wharton’s Jelly mesenchymal stem cells after myocardial infarction. Chin Med J (Engl) 2012;125:3472–3478. [PubMed] [Google Scholar]
- 18.Hagiwara M, Shen B, Chao L, Chao J. Kallikrein-modified mesenchymal stem cell implantation provides enhanced protection against acute ischemic kidney injury by inhibiting apoptosis and inflammation. Hum Gene Ther. 2008;19:807–819. doi: 10.1089/hum.2008.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gatti S, Bruno S, Deregibus MC, Sordi A, Cantaluppi V, Tetta C, Camussi G. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol Dial Transplant. 2011;26:1474–1483. doi: 10.1093/ndt/gfr015. [DOI] [PubMed] [Google Scholar]
- 20.Bruno S, Grange C, Deregibus MC, Calogero RA, Saviozzi S, Collino F, Morando L, et al. Mesenchymal stem cell-derived microvesicles protect against acute tubular injury. J Am Soc Nephrol. 2009;20:1053–1067. doi: 10.1681/ASN.2008070798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruno S, Grange C, Collino F, Deregibus MC, Cantaluppi V, Biancone L, Tetta C, et al. Microvesicles derived from mesenchymal stem cells enhance survival in a lethal model of acute kidney injury. PLoS One. 2012;7:e33115. doi: 10.1371/journal.pone.0033115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.He J, Wang Y, Sun S, Yu M, Wang C, Pei X, Zhu B, et al. Bone marrow stem cells-derived microvesicles protect against renal injury in the mouse remnant kidney model. Nephrology (Carlton) 2012;17:493–500. doi: 10.1111/j.1440-1797.2012.01589.x. [DOI] [PubMed] [Google Scholar]
- 23.Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383. doi: 10.1083/jcb.201211138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klaunig JE, Goldblatt PJ, Hinton DE, Lipsky MM, Chacko J, Trump BF. Mouse liver cell culture. I. Hepatocyte isolation. In vitro. 1981;17:913–925. doi: 10.1007/BF02618288. [DOI] [PubMed] [Google Scholar]
- 25.Abe Y, Hines IN, Zibari G, Pavlick K, Gray L, Kitagawa Y, Grisham MB. Mouse model of liver ischemia and reperfusion injury: method for studying reactive oxygen and nitrogen metabolites in vivo. Free radical biology & medicine. 2009;46:1–7. doi: 10.1016/j.freeradbiomed.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teoh NC, Farrell GC. Hepatic ischemia reperfusion injury: pathogenic mechanisms and basis for hepatoprotection. J Gastroenterol Hepatol. 2003;18:891–902. doi: 10.1046/j.1440-1746.2003.03056.x. [DOI] [PubMed] [Google Scholar]
- 27.Almeida-Porada G, Zanjani ED, Porada CD. Bone marrow stem cells and liver regeneration. Exp Hematol. 2010;38:574–580. doi: 10.1016/j.exphem.2010.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ishikawa T, Banas A, Hagiwara K, Iwaguro H, Ochiya T. Stem cells for hepatic regeneration: the role of adipose tissue derived mesenchymal stem cells. Curr Stem Cell Res Ther. 2010;5:182–189. doi: 10.2174/157488810791268636. [DOI] [PubMed] [Google Scholar]
- 29.Ortiz LA, Dutreil M, Fattman C, Pandey AC, Torres G, Go K, Phinney DG. Interleukin 1 receptor antagonist mediates the antiinflammatory and antifibrotic effect of mesenchymal stem cells during lung injury. Proc Natl Acad Sci U S A. 2007;104:11002–11007. doi: 10.1073/pnas.0704421104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zappia E, Casazza S, Pedemonte E, Benvenuto F, Bonanni I, Gerdoni E, Giunti D, et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood. 2005;106:1755–1761. doi: 10.1182/blood-2005-04-1496. [DOI] [PubMed] [Google Scholar]
- 31.Nowacki M, Nazarewski L, Pokrywczynska M, Kloskowski T, Tyloch D, Pietkun K, Jundzill A, et al. Long-term influence of bone marrow-derived mesenchymal stem cells on liver ischemia-reperfusion injury in a rat model. Annals of transplantation. 2015;20:132–140. doi: 10.12659/AOT.892364. [DOI] [PubMed] [Google Scholar]
- 32.Jin G, Qiu G, Wu D, Hu Y, Qiao P, Fan C, Gao F. Allogeneic bone marrow-derived mesenchymal stem cells attenuate hepatic ischemia-reperfusion injury by suppressing oxidative stress and inhibiting apoptosis in rats. International journal of molecular medicine. 2013;31:1395–1401. doi: 10.3892/ijmm.2013.1340. [DOI] [PubMed] [Google Scholar]
- 33.Pan GZ, Yang Y, Zhang J, Liu W, Wang GY, Zhang YC, Yang Q, et al. Bone marrow mesenchymal stem cells ameliorate hepatic ischemia/reperfusion injuries via inactivation of the MEK/ERK signaling pathway in rats. The Journal of surgical research. 2012;178:935–948. doi: 10.1016/j.jss.2012.04.070. [DOI] [PubMed] [Google Scholar]
- 34.Seki T, Yokoyama Y, Nagasaki H, Kokuryo T, Nagino M. Adipose tissue-derived mesenchymal stem cell transplantation promotes hepatic regeneration after hepatic ischemia-reperfusion and subsequent hepatectomy in rats. The Journal of surgical research. 2012;178:63–70. doi: 10.1016/j.jss.2012.02.014. [DOI] [PubMed] [Google Scholar]
- 35.Sun CK, Chang CL, Lin YC, Kao YH, Chang LT, Yen CH, Shao PL, et al. Systemic administration of autologous adipose-derived mesenchymal stem cells alleviates hepatic ischemia-reperfusion injury in rats. Critical care medicine. 2012;40:1279–1290. doi: 10.1097/CCM.0b013e31823dae23. [DOI] [PubMed] [Google Scholar]
- 36.Saat TC, van den Engel S, Bijman-Lachger W, Korevaar SS, Hoogduijn MJ, JN IJ, de Bruin RW. Fate and Effect of Intravenously Infused Mesenchymal Stem Cells in a Mouse Model of Hepatic Ischemia Reperfusion Injury and Resection. Stem cells international. 2016;2016:5761487. doi: 10.1155/2016/5761487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eggenhofer E, Benseler V, Kroemer A, Popp FC, Geissler EK, Schlitt HJ, Baan CC, et al. Mesenchymal stem cells are short-lived and do not migrate beyond the lungs after intravenous infusion. Frontiers in immunology. 2012;3:297. doi: 10.3389/fimmu.2012.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schrepfer S, Deuse T, Reichenspurner H, Fischbein MP, Robbins RC, Pelletier MP. Stem cell transplantation: the lung barrier. Transplant Proc. 2007;39:573–576. doi: 10.1016/j.transproceed.2006.12.019. [DOI] [PubMed] [Google Scholar]
- 39.Furlani D, Ugurlucan M, Ong L, Bieback K, Pittermann E, Westien I, Wang WW, et al. Is the intravascular administration of mesenchymal stem cells safe? Mesenchymal stem cells and intravital microscopy. Microvascular Research. 2009;77:370–376. doi: 10.1016/j.mvr.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 40.Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, Semprun-Prieto L, et al. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009;5:54–63. doi: 10.1016/j.stem.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Franchi L, Munoz-Planillo R, Nunez G. Sensing and reacting to microbes through the inflammasomes. Nature Immunology. 2012;13:325–332. doi: 10.1038/ni.2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 43.Allen IC, Wilson JE, Schneider M, Lich JD, Roberts RA, Arthur JC, Woodford RM, et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity. 2012;36:742–754. doi: 10.1016/j.immuni.2012.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zaki MH, Man SM, Vogel P, Lamkanfi M, Kanneganti TD. Salmonella exploits NLRP12-dependent innate immune signaling to suppress host defenses during infection. Proc Natl Acad Sci U S A. 2014;111:385–390. doi: 10.1073/pnas.1317643111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saiman Y, Friedman SL. The role of chemokines in acute liver injury. Front Physiol. 2012;3:213. doi: 10.3389/fphys.2012.00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurian SM, Fouraschen SM, Langfelder P, Horvath S, Shaked A, Salomon DR, Olthoff KM. Genomic profiles and predictors of early allograft dysfunction after human liver transplantation. Am J Transplant. 2015;15:1605–1614. doi: 10.1111/ajt.13145. [DOI] [PubMed] [Google Scholar]
- 47.Vansaun MN, Mendonsa AM, Lee Gorden D. Hepatocellular proliferation correlates with inflammatory cell and cytokine changes in a murine model of nonalchoholic fatty liver disease. PLoS One. 2013;8:e73054. doi: 10.1371/journal.pone.0073054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sutter AG, Palanisamy AP, Ellet JD, Schmidt MG, Schnellmann RG, Chavin KD. Intereukin-10 and Kupffer cells protect steatotic mice livers from ischemia-reperfusion injury. Eur Cytokine Netw. 2014;25:69–76. doi: 10.1684/ecn.2015.0359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology. 2003;125:1246–1257. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- 50.Lee YG, Lee SH, Lee SM. Role of Kupffer cells in cold/warm ischemia-reperfusion injury of rat liver. Arch Pharm Res. 2000;23:620–625. doi: 10.1007/BF02975251. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.