

Significance
Nature produces a variety of proteins that could be tapped for therapeutic applications. This paper focuses on the bacterial enzyme β-lactamase, a component of antibody-directed enzyme prodrug therapies designed to activate cytotoxic prodrugs selectively at sites of malignancy. Unfortunately, like many other nonhuman proteins, β-lactamase evokes an antidrug immune response that limits its clinical potential. This paper demonstrates that a multiobjective library-design method enables incorporation of mutations throughout the protein, modifying portions that trigger immune recognition while simultaneously preserving stability and catalytic activity. The libraries were inherently enriched in beneficial variants, and they produced numerous candidates that were both highly functional and immunologically stealthy. The method is general purpose and could enable functional deimmunization of other biotherapeutic agents.
Keywords: deimmunization, computational protein design, immunogenicity, T-cell epitope, combinatorial library
Abstract
Therapeutic proteins of wide-ranging function hold great promise for treating disease, but immune surveillance of these macromolecules can drive an antidrug immune response that compromises efficacy and even undermines safety. To eliminate widespread T-cell epitopes in any biotherapeutic and thereby mitigate this key source of detrimental immune recognition, we developed a Pareto optimal deimmunization library design algorithm that optimizes protein libraries to account for the simultaneous effects of combinations of mutations on both molecular function and epitope content. Active variants identified by high-throughput screening are thus inherently likely to be deimmunized. Functional screening of an optimized 10-site library (1,536 variants) of P99 β-lactamase (P99βL), a component of ADEPT cancer therapies, revealed that the population possessed high overall fitness, and comprehensive analysis of peptide–MHC II immunoreactivity showed the population possessed lower average immunogenic potential than the wild-type enzyme. Although similar functional screening of an optimized 30-site library (2.15 × 109 variants) revealed reduced population-wide fitness, numerous individual variants were found to have activity and stability better than the wild type despite bearing 13 or more deimmunizing mutations per enzyme. The immunogenic potential of one highly active and stable 14-mutation variant was assessed further using ex vivo cellular immunoassays, and the variant was found to silence T-cell activation in seven of the eight blood donors who responded strongly to wild-type P99βL. In summary, our multiobjective library-design process readily identified large and mutually compatible sets of epitope-deleting mutations and produced highly active but aggressively deimmunized constructs in only one round of library screening.
Protein engineering techniques have enabled targeted modification of biotherapeutics to mitigate the T-cell–mediated immune response that otherwise can undermine clinical applications (1). These approaches mutate specific amino acids in a therapeutic candidate so as to disrupt MHC II binding of constituent peptides processed from the protein following uptake by antigen-presenting cells (APCs). The amino acid substitutions prevent APCs from displaying peptide epitopes to cognate CD4+ T cells, thereby short-circuiting T-cell–driven antidrug antibody responses. Mutagenic deletion of T-cell epitopes has been demonstrated for a number of therapeutic proteins (2–7), and in some cases a correlation with reduced antidrug antibody titers has been shown (8–11). More recently, a direct tripartite link from elimination of MHC II-binding peptides to reduction in antidrug antibodies to improvement in therapeutic efficacy was demonstrated for a reengineered antibacterial enzyme designed to treat methicillin-resistant Staphylococcus aureus infections (11). Given that antibody humanization methods likewise reduce T-cell recognition, there is mounting evidence that T-cell epitope deletion is a clinically useful strategy for addressing biotherapeutic immunogenicity.
In pursuing deimmunization by epitope deletion, the key challenge is to incorporate mutations that dampen immune recognition sufficiently while not undermining therapeutic function. A successful outcome requires satisfying both of these challenging objectives, but testing them simultaneously involves difficult and expensive in vivo studies that are necessarily limited in scope. Lead compounds therefore are developed with extensive in vitro work, usually casting reduction of immunogenicity and maintenance of therapeutic function as separate goals to be addressed sequentially and perhaps even iteratively.
Typical approaches are immunogenicity driven (2, 3, 5–7), starting with experimental mapping of epitopes via in vitro peptide binding or ex vivo cellular immunoassays. Verified immunogenic peptides are subjected to alanine scanning or similar trial-and-error mutagenesis to find mutations yielding reduced immune recognition. Deimmunizing mutations then are engineered into full-length protein variants, which are subsequently tested for expression, stability, and molecular function. Although immunogenicity-driven efforts benefit from early confirmation of bona fide epitopes, they are time, labor, and resource intensive. Moreover the “hit rate” for identifying deimmunizing but function-preserving mutations is low, further exacerbating the arduous nature of these efforts.
In contrast, function-driven approaches can leverage high-throughput screens to reduce the burden of producing and testing individual variants and increase the probability of identifying a highly active design. However, mutations selected for function are unlikely to reduce immunogenicity by chance, and there is currently no high-throughput screen to readily quantify immunogenicity in large protein libraries. In function-driven approaches, therefore, libraries must be biased to focus on potentially deimmunizing mutations. Cantor et al. (9) pioneered a “neutral drift” approach in which predicted epitopes were sequentially targeted for elimination. In each round, key MHC-binding positions of one predicted epitope were subjected to combinatorial saturation mutagenesis, and an ultra-high-throughput screen was used to select functional variants. Highly active variants predicted to have reduced MHC II molecular recognition were used as templates for subsequent rounds of mutagenesis and screening.
These immunogenicity- and function-driven approaches target epitopes one by one, either independently or sequentially. In contrast, Pareto optimal deimmunization strategies explicitly design for the trade-offs between immunogenicity and function, seeking to delete multiple epitopes simultaneously throughout the protein while maintaining overall stability and function (12–16). In these approaches, in silico methods predict the effects of mutations on both epitope content, by any of a variety of epitope predictors, and on molecular function, by sequence and/or structural analysis. Computational protein design frameworks then optimize individual variants against both criteria. Pareto optimal deimmunization manifests two important characteristics: designs are global, simultaneously accounting for the effects of widespread mutations on overall immunogenicity and function (in contrast to epitope-by-epitope and peptide-based approaches), and they are globally optimal, provably making the best trade-offs between the modeled criteria (in contrast to approaches reliant on computational or experimental sampling). Pareto optimal deimmunization thus can be seen as a special case of provably optimal multistate protein design (17) in which the positive design goal seeks to maintain therapeutic function and the negative design goal seeks to reduce immune recognition. Pareto optimal designs encode combinations of epitope-deleting, functionality-preserving mutations that should “play well together” in a given variant, facilitating the rapid advance of promising candidates to experimental evaluation (18–20).
Here we use a computational library optimization methodology that combines the principles of Pareto optimal deimmunization of individual variants with the advantages of library-based function-driven deimmunization. Integrating an in silico T-cell epitope predictor with statistical sequence potentials of residue plasticity, libraries are optimized so that the constituent populations are biased toward members predicted to exhibit both low epitope content and high function (Fig. 1). This strategy accounts for T-cell epitopes that may be widely distributed throughout a target protein and simultaneously ensures that the cumulative effects of nonconservative and interdependent epitope-deleting substitutions do not undermine protein stability and activity. As a consequence, screening effort is allocated to those variants most likely to possess desirable characteristics for both objectives. When applied to P99 β-lactamase (P99βL), a component of ADEPT (antibody-directed enzyme prodrug therapy) anticancer therapies (21), the resulting libraries yielded a variety of highly active and highly deimmunized variants in only one round of design and screening. We ultimately characterized a 14-mutation variant having wild-type stability, better than wild-type activity, and vastly reduced immune cell activation among genetically diverse human subjects.
Fig. 1.

Pareto optimal deimmunization libraries. (A) Combinatorial libraries comprised all combinations of mutations at selected positions; illustrated are three different libraries incorporating different substitutions (empty squares, in contrast to wild-type solid squares) at different positions. Deimmunization is inherently a multiobjective problem, because beneficial variants must simultaneously reduce immunogenicity but maintain therapeutic function. One of the three illustrated libraries (blue) focuses only on reducing immunogenicity without regard for loss of function; consequently the functional screen finds no viable variants (below the dashed line). Another library (red) focuses only on maintaining function and thus yields many functional variants but makes little progress in reducing immunogenicity. The third library (green) strikes a balance; importantly and by design, variants selected for good function are also of lower immunogenicity. (B) Pareto optimal deimmunization library design seeks to balance the properties of interest, optimizing libraries to be enriched in high-quality variants according to both function and immunogenicity. Here, SEpi assesses expected immunogenicity risk and SSeq assesses expected perturbation to function (relative to wild type) over the set of variants in a combinatorial library. Pareto optimal libraries are undominated: The score for one criterion cannot be improved without the score for the other becoming worse. Mapping the frontier of all Pareto optimal designs reveals the trade-offs and the balance that can be achieved, unknown a priori.
Results
Combinatorial Deimmunization Library Design.
Pareto optimal deimmunized libraries were designed to balance the two goals of reducing immunogenicity and maintaining high function so that variants identified by subsequent functional screening would also likely be deimmunized (Fig. 1). Libraries were designed as sets of residue positions at which substitutions and alternative amino acids for each location were introduced, so that all combinations of substitutions would be evaluated. A library’s immunogenicity score, SEpi, was computed as the expected value over all library members of the number of peptide–allele hits (lower is better) identified by a standard MHC II epitope predictor (22). A library’s function score, SSeq, was computed as the expected value over all library members of a standard sequence potential (lower is better) incorporating position-specific conservation (one-body) and positions-specific covariation (two-body) terms computed from a multiple sequence alignment of P99βL homologs (15). For wild-type P99βL (hereafter simply “P99βL”), SEpi = 123, and, by construction, SSeq = 0. The design process then optimized the set of libraries comprising the Pareto frontier, i.e., libraries making optimal trade-offs between the two scores, given that any library better for one score must be worse for the other.
In this manner, both 10- and 30-site P99βL Pareto optimal deimmunization libraries were designed (Fig. 2). As introduced intuitively in Fig. 1, we found that P99βL libraries optimized for sequence score alone contained members that were likely to maintain high function but make little progress in reducing epitope content (red contours in Fig. 2 A and B). Conversely, libraries optimized for epitope score alone were more likely to delete numerous epitopes but might do so by incorporating particularly disruptive sets of substitutions (blue contours in Fig. 2 A and B). Libraries near the “elbow” of the Pareto frontier struck a balance, and we selected one such library for each mutational load (green contours in Fig. 2 A and B; also see Table S1). Lib10 was composed of 1,536 unique designs from 10 degenerate codons encoding the wild type and one or two mutations per site. Lib30 had ∼2.15 × 109 unique members from 30 degenerate codons encoding the wild type and one to four mutations per site. Note that by allowing mutations at more sites, Lib30 is able to attain a better epitope score than Lib10 but sacrifices sequence score, because each additional mutation increases the chance of incurring a sequence penalty, relative to wild type.
Fig. 2.

Library design spaces. Libraries were designed to select residue positions and amino acid substitutions targeting either 10 sites (A and C) or 30 sites (B and D). (A and B) Libraries were optimized for enrichment in variants with a desired balance between immunogenicity, captured by an epitope score SEpi (x axis), and function, captured by a sequence score SSeq (y axis). Pareto optimal libraries, those making undominated trade-offs between the average values of the two criteria over their constituents, are marked with circles; the wild type is marked with an asterisk. The red contours represent the distributions of variant scores within a library optimized solely for sequence score, the blue contours represent the distributions of variant scores within a library optimized solely for epitope score, and the green contours represent the distributions of variant scores within a library optimized for a balanced combination (Lib10 and Lib30). Experimentally characterized library members are marked with triangles (no selective pressure) and squares (cefazolin concentration of 20 µg/mL); gray markers in A are for members of an EP library with average mutational load of 4. (C and D) Different mutations were incorporated when optimizing for sequence score only (Top), a balance (Middle, Lib10/Lib30), or epitope score only (Bottom). Boxes indicate mutations incorporated in the variants selected at a cefazolin concentration ([cefa]) of 20 μg/mL.
Table S1.
Degenerate codons for Lib10 and Lib30
| Position | Wild type | Lib10 | Lib30 | ||
| Codon | Mutations | Codon | Mutations | ||
| 13 | A | GMG | EA | GMT | DA |
| 19 | L | STT | LV | ||
| 21 | K | RAG | KE | RAG | KE |
| 28 | M | WTG | ML | ||
| 31 | A | GST | AG | ||
| 37 | K | RAG | KE | ||
| 48 | I | RTT | IV | RTT | IV |
| 49 | A | RCT | TA | ||
| 86 | D | RAT | ND | ||
| 95 | Q | SAG | QE | ||
| 103 | G | GRT | DG | GRT | DG |
| 105 | R | MGT | SR | MGT | SR |
| 133 | R | CRT | HR | ||
| 137 | N | RAT | ND | RAT | ND |
| 149 | L | CWG | QL | ||
| 172 | Q | SAG | QE | ||
| 183 | K | AAW | KN | AAW | KN |
| 193 | K | RAG | KE | ||
| 194 | A | KCT | AS | ||
| 210 | R | CRT | HR | CRT | HR |
| 220 | A | KCT | AS | ||
| 234 | V | STT | LV | STT | LV |
| 239 | A | GMT | DA | ||
| 261 | R | CRG | QR | ||
| 262 | I | AYS | TIM | RYT | TIAV |
| 285 | E | GAW | ED | ||
| 299 | A | RCT | TA | ||
| 326 | V | GYT | AV | ||
| 335 | G | GST | AG | ||
| 336 | I | WTA | IL | ||
The mutational composition of libraries optimized for SSeq alone, SEpi alone, or both scores simultaneously (i.e., the elbow plans) were analyzed, revealing clear distinctions in how the libraries achieved their design objectives (Fig. 2 C and D). The Lib10 and Lib30 elbow plans seek to strike a balance between the two objective functions and therefore draw mutations from both the SSeq-only and SEpi-only libraries (red and blue boxes, respectively, Fig. 2 C and D). These SSeq- and SEpi-biased mutations are not favorable across the full Pareto frontier, but their strong contribution to preservation of function and reduction of immunogenicity, respectively, render them suitable for the centrally located elbow plans. Moreover, the Lib10 and Lib30 elbow plans encode a large fraction of mutant codons (6 and 16, respectively) that are either common among all libraries on the Pareto frontier or, alternatively, are unique, relative to SSeq-only or SEpi-only mutations (green boxes, Fig. 2 C and D). These substitutions individually balance both evolutionary constraints for maintaining function and deimmunizing constraints for deleting epitopes. The elbow libraries guarantee an optimal mixture of aggressive SSeq mutations, aggressive SEpi mutations, and balanced mutations. Notably, the Lib10 mutation sites are a subset of Lib30, suggesting core mutational priorities that are conserved across mutational loads.
Library Population Fitness and Variant Distribution.
The overall fitness of each library was assessed by quantitative culture, plating 1 million to 3 million cells on agar containing escalating concentrations of the β-lactam antibiotic cefazolin (Fig. 3A). An error-prone (EP) library bearing approximately the same mutational load as Lib10 provided a benchmark for the fitness cost of stochastic mutations. Cultures expressing parental P99βL served as a control. The P99βL control exhibited a sharp decrease in colony counts (>90%) at cefazolin concentrations between 40 and 75 μg/mL, a result typical for clonal bacterial populations [e.g., comparable to the precipitous loss of outgrowth over a single twofold antibiotic dilution in minimal inhibitory concentration assays (23)]. In contrast, each of the libraries exhibited a more protracted loss of viable colonies spanning as many as six twofold antibiotic dilutions. This result was consistent with expectations for heterogeneous populations encoding diverse P99βL variants; low-activity clones will die at low antibiotic concentrations, but more active clones will continue to grow out at higher concentrations.
Fig. 3.

Lib10 peptide immunoreactivity and library fitness. (A) The fitness of each library is characterized by the concentration of cefazolin (x axis) vs. the relative colony outgrowth in terms of the total number of cells plated (y axis). Shown are data for Lib10 (red), Lib30 (blue), EP library (gray), and a wild-type culture (black) for reference. Neither the wild type nor Lib10 yielded outgrowth on 100 μg/mL agar, whereas EP exhibited 0.6% viability and Lib30 exhibited 0.007% viability at this top concentration. (B) For each mutation incorporated in Lib10, synthetic peptides corresponding to the wild type or variant sequences were assayed for binding against four different soluble MHC II proteins. The immunoreactivity of each variant in the library was assessed in terms of the number of moderate-to-strong MHC II-binding events (IC50 <10 μM) summed over the four alleles and the constituent peptides. Wild-type P99βL presents 10 such peptide–MHC binding events.
With respect to fractional outgrowth across the cefazolin dilution series, Lib10 exhibited the highest average fitness of any culture. There was minimal loss of Lib10 viability at antibiotic concentrations up to 30 μg/mL and more than 40% outgrowth at a concentration of 75 μg/mL. No Lib10 colonies were detected at the concentration of 100 μg/mL on agar plates. Lib30 likewise appeared to contain high-performance enzymes, as evidenced by 0.007% library outgrowth under highly stringent selection at the antibiotic concentration of 100 μg/mL, at which wild-type P99βL and Lib10 failed to grow on agar medium. However, the average population fitness of Lib30 was lower than that of Lib10, in part because ∼65% of the naive Lib30 population contained frameshift mutations and premature stop codons. The EP library manifested better average fitness than Lib30 but exhibited a similar overall trend. Specifically, the EP library suffered significant reductions in outgrowth at low cefazolin concentrations, but a small fraction of EP clones (∼0.6%) remained viable even at antibiotic concentrations of 100 μg/mL. Similar to Lib30, the average fitness of the EP library was reduced by a large fraction of clones bearing frameshifts or empty vector background (∼50%); these clones contributed to the sharp loss of viability at 5 μg/mL. In total, these preliminary screening results indicated that the deimmunization libraries contained large proportions of highly active variants and thereby achieved the functional design objective.
Randomly chosen Lib10, Lib30, and EP colonies were sequenced from the naive libraries (no cefazolin selection) and from plates containing cefazolin at a concentration of 20 μg/mL. To assess where the sequenced clones fell in the library design space, SEpi and SSeq scores were computed for each clone. The pre- and postselection Lib10 members were broadly distributed across the design space, and no observable shift in the distribution of SEpi and SSeq scores was obvious following functional selection on cefazolin (Fig. 2A). Without exception, sequenced Lib10 members had lower SEpi scores than wild-type P99βL, whereas EP variants having similar mutational loads failed to achieve any meaningful reduction in SEpi and in many cases were worse than wild-type P99βL. The sequenced Lib30 variants were also distributed broadly across the design space, and functional selection with 20 μg/mL cefazolin did not appear to skew their distribution (Fig. 2B). As per the deimmunization design objective, the Lib30 variants achieved even greater overall reductions in SEpi than the Lib10 variants. These results demonstrate that deimmunization libraries, both before and after functional selection, are indeed biased toward reduced immunogenic potential, as embodied by the SEpi score.
Library Population Relative Immunoreactivity.
To validate the above predictive assessments of reduced immunogenic potential experimentally, the relative immunoreactivities of all Lib10 members were quantified by measuring MHC II binding of constituent peptides for each target site. Peptides 15–18 amino acids in length, representing all possible mutations in Lib10 (Table S2), were synthesized, and their affinities for the soluble human MHC II proteins DRB1*0101, 0401, 0701, and 1501 were determined via competition assays against known T-cell epitopes (24). A total of nine wild-type and 12 variant peptides were assessed against each of the four alleles, for a total of 84 peptide affinity measurements in 48 matched pairs of wild-type vs. the corresponding deimmunized peptide (Fig. S1). In 24 of these cases, the variant peptide had negligible MHC II-binding affinity (IC50 ≥250 μM) or had a binding affinity reduced by more than 10-fold relative to the wild-type peptide. In 11 instances the variant affinity reductions were more modest, between two- and 10-fold. In contrast, there were only eight cases in which variant peptides increased MHC II-binding affinity, and in five of these cases the increase was negligible to small, i.e., less than threefold. Only two variant peptides, V243L and R105S, increased MHC II-binding affinity for any allele by more than fivefold relative to their wild-type counterparts, and in both cases the increased affinity of the variant peptide failed to meet the threshold for biologically relevant MHC II binding (typically <1 µM) (25–28).
Table S2.
Lib10 peptide–MHC binding
| Wild type or mutation | Peptide | Experimental IC50, μM | |||
| 0101 | 0401 | 0701 | 1501 | ||
| A13 | EKQLAEVVANTITPLM | >250 | 15.95 | 0.62 | 7.40 |
| A13E | EKQLAEVVENTITPLM | >250 | 71.12 | 74.46 | >250 |
| K21 | ITPLMKAQSVPGMAV | 3.58 | 3.623 | 41.49 | 5.378 |
| K21E | ITPLMEAQSVPGMAV | 16.7 | 11.37 | 40.43 | 16.79 |
| I48 | KPHYYTFGKADIAAN | 0.79 | 199.05 | >250 | >250 |
| I48V | KPHYYTFGKADVAAN | 0.714 | 73.46 | >250 | >250 |
| G103+R105 | GKQWQGIRMLDLATYT | 219.75 | >250 | 10.74 | 96.31 |
| G103D | GKQWQDIRMLDLATYT | >250 | >250 | 33.01 | 117.7 |
| G103D+R105S | GKQWQDISMLDLATYT | >250 | >250 | >250 | >250 |
| R105S | GKQWQGISMLDLATYT | 26.73 | >250 | >250 | 69.45 |
| N137 | SLLRFYQNWQPQWKPGTT | >250 | >250 | >250 | <0.01 |
| N137D | SLLRFYQDWQPQWKPGTT | >250 | 155.2 | >250 | 0.083 |
| K183 | PLKLDHTWINVPKAEEA | >250 | 185.5 | >250 | >250 |
| K183N | PLNLDHTWINVPKAEEA | >250 | >250 | >250 | >250 |
| R210 | GKAVRVSPGMLDAQAY | >250 | >250 | >250 | 21.18 |
| R210H | GKAVHVSPGMLDAQAY | >250 | >250 | >250 | 91.5 |
| V234 | MANWVMANMAPENVA | 12.41 | 23.64 | 16.95 | >250 |
| V234L | MANWLMANMAPENVA | 10.77 | 22.64 | 5.678 | 16.25 |
| I262 | GIALAQSRYWRIGSMYQG | 22.17 | 0.4567 | 1.675 | 0.323 |
| I262T | GIALAQSRYWRTGSMYQG | >250 | 2.185 | 4.147 | 5.725 |
| I262M | GIALAQSRYWRMGSMYQG | 12.4 | 0.2449 | 11.6 | 2.413 |
Fig. S1.

IC50 values are plotted as cognate wild type (Left) and variant (Right) pairs. Lines connect measurements for the same MHC II allele (DRB1*0101 blue; 0401 red; 0701 green; 1501 purple). Larger positive slopes of connecting lines indicate a greater fold decrease in affinity relative to the wild type; lines with negative slopes indicate a mutation that enhanced MHC II binding. Shading designates binding strength: strong (IC50 < 1 μM, dark gray), moderate (1 μM ≤ IC50 < 10 μM, medium gray), weak (10 μM ≤ IC50 < 100 μM, light gray), or nonbinding (IC50 ≥ 100 μM, white).
To facilitate protein-to-protein comparisons, each of the 84 peptide affinity measurements were classified as strong (IC50 < 1 μM), moderate (1 μM ≤ IC50 < 10 μM), weak (10 μM ≤ IC50 < 100 μM), or nonbinding (IC50 ≥ 100 μM) (Fig. S1), and whole-protein immunoreactivity scores were calculated by summing the number of strong and moderate binding peptides (IC50 <10 μM) for each individual library member. The distribution of scores for all Lib10 members shows that the overall population has substantially reduced immunoreactivity compared with wild-type P99βL (Fig. 3B). Of 1,536 library members, 79% had fewer MHC-binding peptides than wild-type P99βL, 13% possessed the same number, and only 8% had more. Importantly, one sixth of the library represented highly epitope-depleted variants in which 50% or more of the strong- to moderate-binding peptides were eliminated. Thus, protein ensembles generated with the deimmunization library algorithm manifested reduced immunoreactivity by objective experimental measures. We therefore conclude that the deimmunization design objective was achieved.
Isolation and Characterization of Highly Active Variants.
Initial efforts to isolate highly active P99βL variants used a liquid growth selection in which library members, in the form of bacterial cells, were compartmentalized in inverted emulsions containing cefazolin antibiotic. Lib10 and the EP library yielded outgrowth following compartmentalization and selection at 50 μg/mL cefazolin, but only Lib10 yielded outgrowth at the more stringent cefazolin concentration of 200 μg/mL. Sequencing of 16 Lib10 clones from the 200 μg/mL emulsion selection revealed that the enriched population generally maintained the mutational diversity seen in the naive library members. Among the 14 unique sequences, all the originally encoded mutations appeared in three or more distinct clones, with no change in the average number of amino acid substitutions per variant (naive = 5 ± 2; selected = 5 ± 1). As an aside, the observation that Lib10 exhibited outgrowth at higher cefazolin concentrations in microcompartments and Lib30 did so on agar plates (Fig. 3A) was attributed to complex interplay between factors such as enzyme activity, efficiency, stability, expression level, and differences in liquid vs. agar growth environments. Notably, individual Lib10 clones selected at 200 μg/mL cefazolin in inverted emulsions were not viable when subsequently streaked on agar plates containing either 200 or 100 μg/mL cefazolin, as was consistent with the original results of functional screening.
Five Lib10 variants from the 200 μg/mL cefazolin selection were expressed, purified, and characterized in greater detail (Table 1). We note that clinical application of a P99βL ADEPT therapy would use β-lactam half mustards or other prodrugs that release toxic reaction products, whereas here we used the colorimetric nitrocefin substrate as an experimentally tractable surrogate. Consistent with isolation from a stringent growth selection, kinetic analysis with the nitrocefin β-lactam substrate revealed that all five variants possessed high catalytic proficiency. In particular, the variants exhibited kcat values two- to 2.5-fold greater than P99βL and had similar improvements in catalytic efficiency, as measured by kcat/Km values. Although the variants’ activities were improved, their thermostabilities decreased. The Lib10 variants manifested a loss of 3–9.5 °C in thermostability, with the six- and seven-mutation variants having greater reductions than the four- and five-mutation variants. Importantly, however, the melting temperatures (Tm) of the selected Lib10 variants were generally similar to those of four- to eight-mutation P99βL variants deimmunized in earlier studies (18–20). Thus, rigorous kinetic and thermostability analysis of the selected Lib10 variants, combined with the MHC II-binding assays described above, demonstrated that the library design algorithms achieved both objectives, reducing immunoreactivity and maintaining stability and activity.
Table 1.
In vitro stability and activity measurements for selected variants
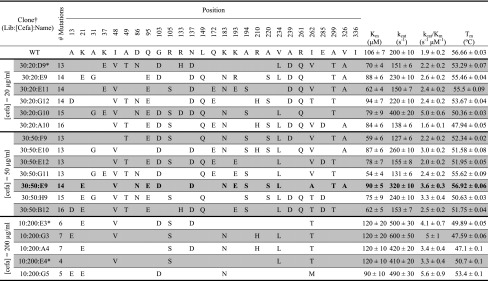 |
Bold entry indicates the variant subsequently tested in ex vivo PBMC assays.
Nomenclature is “library mutation no.:cefazolin selection concentration:clone designation,” e.g., “10:200:G3.”
Duplicate variants identified twice in the selected populations.
Although the Lib30 population failed to produce outgrowth in inverted emulsions, Lib30 colonies could be grown readily on nutrient agar plates at 20 and 50 μg/mL cefazolin (Fig. 3A). Six clones from the 20 μg/mL plates and seven clones from the 50 μg/mL plates were chosen for purification and detailed functional characterization (Table 1). The variants’ catalytic constants (kcat) and catalytic efficiencies (kcat/Km) were generally lower than those of the Lib10 variants characterized above. This observation is consistent with the higher selection stringency applied during screening of Lib10. Nonetheless, Lib30 variants selected from both cefazolin concentrations retained catalytic activity similar to that of P99βL, with one clone from each selection exhibiting substantially improved activity and efficiency (variants 30:20:G10 and 30:50:E9; Table 1). Notably, Lib30 variants trended toward higher thermostability than the Lib10 variants, with average Tm values as follows: 50 ± 2 °C for 10:200:clones, 53 ± 3 °C for 30:20:clones, and 53 ± 2 °C for 30:50:clones. Indeed, two Lib30 variants from each selection exhibited wild-type thermostability. One of these was the variant 30:50:E9 (bold text, Table 1), which also possessed better than wild-type activity. Given its superior performance in functional assays, this enzyme was chosen for more advanced immunological analysis using ex vivo cellular immunoassays.
Ex Vivo Human Peripheral Blood Mononuclear Cell Assays.
The EpiSweep-based deimmunization algorithm used here seeks to disrupt MHC II-mediated molecular recognition of immunogenic peptide fragments, and thus the peptide–MHC II binding assays described above provided a direct experimental readout of this design objective. Ultimately, however, the goal of deimmunization is to reduce immunogenicity in human subjects. Ex vivo immunoassays with human peripheral blood mononuclear cells (PBMCs) are a widely recognized means of measuring immunogenicity risk for preclinical biotherapeutic candidates (29). As implemented in various formats, PBMC assays measure immune cell activation following exposure of human immune cells to the test protein. Here, we used the methods of Sette and coworkers (30) and Pastan and coworkers (6) to quantify the activation of human PBMCs following stimulation with either P99βL or the 30:50:E9 variant. Both proteins were tested against PBMCs from 18 blood donors having distinct HLA genotypes (Table S3). Notably, the library design algorithm optimized against a limited subset of only eight DRB1 HLA alleles, which are nonetheless more broadly representative of human HLA DRB-binding specificities (27, 31). The panel of 18 PBMC donors encoded seven of the eight target alleles (lacking DRB1*0401) as well as 23 other DRB1 alleles that were not explicitly considered during the design and optimization process.
Table S3.
Human PBMC donor genotypes
| Lot no. | Donor no. | Ethnicity | Age, y | Gender | HLA Class II DRB1 | HLA Class II DQA | HLA Class II DQB | HLA Class II DP |
| LP_37 | 30 | Hispanic | 53 | Male | *1406, *1502 | *0101, *0301 | DQ3*0302, DQ5*0501 | w4*0401P, w4*0402P |
| LP_53 | 40 | Asian | 36 | Male | *0405, *1202 | *0101, *0301 | DQ3*0302, DQ5*0501 | w4*0401, w19*1901 |
| LP_83 | 61 | Caucasian | 41 | Male | *0701, *1301 | *0103, 0201 | DQ2*0202, DQ6*0603 | w2*0201, w4*0402 |
| LP_84 | 62 | Hispanic | 20 | Male | *0403, *1501 | *0101, *0501 | DQ2*0201, DQ5*0501 | w1*0101, w5*0501 |
| LP_100 | 76 | African American | 36 | Male | *0901, *1303 | *0105, *0301 | DQ3*0302, DQ5*0501 | w1*0101, w4*0402 |
| LP_106 | 82 | Hispanic | 37 | Male | *0407, *1602 | *0102, *0505 | DQ3*0301, DQ5*0502 | w135*13501, w100*10001 |
| LP_112 | 85 | Caucasian Hispanic | 44 | Female | *0801, *1101 | *0401, *0505 | DQ3*0301, DQ4*0402 | w4*0401P, w4*0402P |
| LP_127 | 99 | Hispanic | 53 | Male | *0410, *0802 | *0505, *0505 | DQ3*0301, DQ3*0319 | w2*0201, w85*8501 |
| LP_130 | 102 | African American | 54 | Male | *1454, *1503 | *0102, *0503 | DQ7*0301 | w1*0101, w4*0401 |
| LP_132 | 104 | African American | 35 | Male | *0302, *1201P | *0102, *0104 | DQ6*0602, DQ6*0602 | w1*0101, w1*0101 |
| LP_164 | 118 | Hispanic | 25 | Male | *1102, *1104 | *0105, *0401 | DQ3*0319, DQ5*0501 | w2*0201, w13*1301 |
| LP_149 | 121 | Hispanic | 46 | Male | *0101, *0407P | *0103, *0301 | DQ3*0302, DQ6*0603 | w4*0401P, w4*0402P |
| LP_151 | 123 | Hispanic | 20 | Male | *1001, *1402 | *0102, *0301 | DQ3*0302, DQ6*0602 | w4*0401, w11*1101 |
| LP_154 | 126 | Caucasian | 29 | Male | *0701, *1302 | *0303, *0601 | DQ3*0301, DQ4*0402 | not tested |
| LP_155 | 127 | Hispanic | 31 | Male | *0402, *1301P | *0301, *0505 | DQ3*0301, DQ3*0302 | w4*0402, w4*0402 |
| LP_173 | 142 | Filipino | 28 | Female | *1101P, *1502 | *0301, *0401 | DQ3*0302, DQ4*0402 | w4*0401P, w4*0402P |
| LP_175 | 144 | Hispanic | 22 | Male | *0102, *0301 | *0201, *0505 | DQ3*0301, DQ3*0303 | w4*0401, w11*1101 |
| LP_176 | 145 | Caucasian | 22 | Male | *0101P, *0404 | *0102, *0201 | DQ2*0202, DQ6*0604 | w4*0402, w4*0402 |
| LP_190 | 155 | Caucasian | 50 | Female | *0701, *1103 | *0201, *0303 | DQ2*0202, DQ2*0202 | w1*0101, w2*0201 |
Replicate aliquots of donor cells were expanded in the presence of either P99βL or the 30:50:E9 variant so as to drive the proliferation of T cells that recognized epitopes in the respective protein. Subsequently, the expanded cell populations were restimulated with one of two separate pools of 12 synthetic peptides, corresponding to either the wild-type or the 30:50:E9 sequence at all 30 target sites (Fig. S2). The number of activated immune cells was then quantified by IL-2 ELISpot. Ten donors exhibited little to no response against either protein, one donor exhibited a strong response against both proteins, and the remaining seven donors showed a strong response against P99βL but not the 30:50:E9 variant (Fig. 4). Thus, the 30:50:E9 variant proved to be broadly deimmunized for genetically diverse subjects, although the data suggest that DRB1*0801 may still restrict one or more immunogenic epitopes in the variant. Importantly, the mutations engineered into the 30:50:E9 variant did not result in the generation of neo-epitopes, nor did they reveal cryptic epitopes, at least among the patient genotypes examined here. These results demonstrate that deimmunization for genetically diverse patient populations may be achieved by explicit optimization against a smaller and more tractable subset of representative HLA alleles.
Fig. S2.

Alignment of wild-type and 30:50:E9 peptides with the P99βL protein sequence. The amino acids highlighted in yellow are mutated in 30:50:E9.
Fig. 4.

Human PBMC cellular immunoassays. PBMCs from 18 donors were expanded in the presence of either wild-type P99βL or the 30:50:E9 variant, driving the proliferation of cells that recognized the respective enzyme. Activation of the proliferated cell populations was then quantified by ELISpot following restimulation with wild-type or variant peptides, respectively. The seven donors to the left of the vertical dashed lines exhibited a strong response to the wild type but minimal response to variant enzymes. One donor shown between the vertical dashed lines exhibited a strong response to both the wild type and variant enzymes. The remaining 10 donors at the right of the vertical dashed lines exhibited minimal responses to both enzymes.
Discussion
The studies described here demonstrate a Pareto optimal library approach to biotherapeutic deimmunization. By extending the EpiSweep algorithm to design combinatorial protein libraries, as opposed to individual protein designs, exceptionally high mutational loads were brought to bear on the deimmunization problem. Such high mutational loads may be important for achieving deimmunization against genetically diverse patient populations or when seeking to deimmunize proteins replete with immunogenic epitopes. In prior studies, smaller numbers of P99βL T-cell epitopes had been deleted by the introduction of two to eight mutations (3, 18–20, 32). Here, deimmunized P99βL libraries produced highly active variants bearing as many as 16 mutations, and in several cases the engineered enzymes possessed equivalent stability and higher catalytic activity than the parental P99βL protein.
Functional deimmunization necessitates balancing two objectives that potentially conflict with each other, because mutations made to eliminate epitopes may detrimentally impact function. As such, functional deimmunization is an instance of multistate design, seeking simultaneously to preserve therapeutic function and eliminate MHC II binding of constituent peptides. Multistate design is particularly challenging when designing positively for some criteria but negatively against others (33), requiring that the goals be treated separately instead of simply combined into a single goal. A variety of algorithmic approaches, based on explicit structural modeling (34–37), sequence potentials derived from the evolutionary record (15), and sequence potentials derived from structural modeling (38), have driven applications such as modifying protein interaction specificity (39–41), designing peptide inhibitors (42–44), altering substrate specificity (45–47), characterizing resistance mechanisms (48), and enabling a single protein to adopt multiple folds (49). We have focused on a Pareto optimization framework (50) as the basis for elucidating and explicitly optimizing trade-offs between criteria and thereby providing the recently well-discussed advantages of provable optimality guarantees, such as enabling more direct interpretation of experimental data according to the driving models without concern for algorithmic bias or sampling failure (17). We here pursue sequence-based design, relying on mutations observed in evolutionarily related proteins (but now in combinations not observed in databases) to provide sufficient flexibility to achieve our goals. Although structure-based design allows stepping outside the confines of evolutionarily accepted mutations, we conjecture that such measures were not necessary here because of the richness of the sequence record, the size of the feasible design space, and the do-no-harm context of deimmunization, in which function is to be preserved rather than altered. Nonetheless, we have recently extended the combinatorial library design to support structure-based modeling in cases where that is advantageous (51).
Although deimmunization via combinatorial libraries necessitates functional library screening, a key advantage of computationally optimized libraries is the ability to control library diversity and size so as to best match available screening technology. The cefazolin selection for P99βL enabled successful screening of the large and diverse Lib30 population (2 × 109 members), although the more constrained Lib10 population (only 1,536 unique variants) could have been screened readily using microtiter plates. Despite Lib10’s constrained diversity and small size, it also yielded highly active and highly engineered variants. The isolation of deimmunized and functionally enhanced candidates from both the large and small libraries highlights the utility of the optimization algorithms: Productive libraries can be designed for and enriched with screening technologies of widely variable throughput.
In other work, a library-based neutral drift strategy was used to delete T-cell epitopes from asparaginase (9), an important therapeutic agent for acute lymphoblastic leukemia. Although successful, this effort used an iterative strategy in which T-cell epitopes were deleted via the synthesis and screening of large, combinatorial libraries that targeted three putative epitopes in a sequential fashion. Thus, the approach sought to delete a given epitope individually without consideration of any remaining epitopes. Although successful in this case, such “greedy” experimental algorithms run the risk of overlooking globally optimal combinations of mutations. In contrast, the EpiSweep library design strategy seeks globally optimal protein designs by simultaneously mutating all target epitopes, thereby considering all combinations of the chosen deimmunization mutations. In the case of optimized P99βL libraries, highly active, highly stable, and aggressively deimmunized variants were isolated in a single round of screening. We therefore conclude that computationally optimized deimmunization libraries represent a broadly configurable and effective strategy for creating high-performance biotherapeutic agents that evade immune surveillance.
Materials and Methods
Degenerate codon libraries were synthesized by GenScript. Plasmid purification kits were purchased from Qiagen, and DNA purification kits were from Zymo Research. Oligonucleotides were purchased from Integrated DNA Technology. All sequencing was performed by Genewiz. All restriction enzymes and PCR reagents were purchased from New England BioLabs. Growth medium was purchased from Becton Dickinson. Ni-NTA resin was purchased from Qiagen. Nitrocefin was purchased from Oxoid. SYPRO Orange was purchased from Sigma. MicroAmp Fast Optical 0.1 mL 96-well plates and MicroAmp Fast Optical Adhesive Film were purchased from Applied Biosystems. Cefazolin was purchased from MP Biomedical. Isopropyl β-d-1-thiogalactopyranoside (IPTG) and kanamycin were purchased from Research Products International. Pico-Surf1 (2% in Novec 7500) for inverted emulsions was purchased from Sphere Fluidics and 1H,1H,2H,2H-Perfluoro-1-octanol was from Sigma. The microfluidics device was manufactured by the Stanford foundry. Peptides were ordered from GenScript and were greater than 85% pure. Biotinylated tracer peptides were purchased from 21st Century Biochemicals. MHC II DR molecules were from Benaroya Research Institute. Anti-MHC II-DR antibody was purchased from BioLegend. DELFIA Eu-labeled streptavidin was from PerkinElmer. Human donor PBMCs were purchased from Cellular Technology Limited, IL-2 was sourced from PeproTech, and IL-2 ELISpot assay kits were from Mabtech. Unless noted, all other chemicals and reagents were from VWR.
Library Design.
EpiSweep is an integrated deimmunization software suite that is generic to the epitope score, supports both sequence- and structure-based analysis of mutational effects on function, and designs either individual variants or combinatorial libraries (52). Although structure-based library designs have been described elsewhere (11, 51), the results presented here demonstrate the use of a sequence score to drive the design of a combinatorial deimmunization library and demonstrate the scaling of the approach to a massive library size. In short, the sequence score is derived from a multiple sequence alignment of P99βL homologs obtained by PSI-BLAST; it includes both one-body (conservation) and two-body (coupling) terms (15). The epitope score used here is based on ProPred (22) predictions for eight HLA-DRB1 MHC alleles (DRB1*0101, 0301, 0401, 0701, 0801, 1101, 1301 and 1501) that are more broadly representative of human genetic diversity (27, 31). The epitope score counts the number of peptide–allele hits at a standard 5% threshold. Libraries are designed by simultaneously selecting residue positions and degenerate oligonucleotides from a set of allowed positions and position-specific mutational choices derived from the sequence analysis (excluding mutations to/from proline and cysteine) (53).
The evaluation of a design is based on average scores over its constituent members, which can be evaluated in terms of precomputed position-specific average scores without having to enumerate the variants (53). Here we use SEpi and SSeq to denote these library-averaged scores, i.e., expected values over the variants in a library. EpiSweep generates an entire Pareto frontier of designs (and successively suboptimal frontiers if desired), yielding the designs making the best trade-offs between the two scores. This combinatorial optimization problem is specified as an integer linear program and solved with the IBM ILOG CPLEX software.
During optimization, libraries are evaluated based on scores averaged across the entire population, but for visualization purposes 10-site library score distributions were computed by enumerating all variants and 30-site library score distributions were computed from randomly selected sets of 3,000 variants.
Library Construction.
The general structure of all P99βL genes and the overall cloning strategy have been described in detail elsewhere (32). Lib10 and Lib30 were constructed by GenScript and delivered as pooled plasmid in a pET26b backbone. Degenerate codons comprising Lib10 and Lib30 are detailed in Table S1. The EP gene library was synthesized using error-prone PCR conditions (54), and the amplified genes were cloned into the XbaI and HindIII sites of pET26b. One hundred nanograms of each plasmid library was electroporated separately into five freshly prepared aliquots of BL21(DE3) cells [F− ompT hsdSB (rB− mB−) gal dcm (DE3)], yielding 5 × 106 to 1.2 × 107, 8 × 106 to 1.5 × 107, and 1.3 × 106 to 1.8 × 106 transformants per 100 ng of DNA for the Lib10, Lib30, and EP libraries, respectively.
Inverted Emulsion Selections.
Selections in inverted microemulsions used the microfluidic-based encapsulation technique of Scanlon et al. (55), with the modification that compartments were generated at room temperature and contained liquid growth medium as described below, as opposed to agarose hydrogel growth medium. Libraries were first expanded from glycerol stocks by 6–8 h of growth with shaking at 37 °C in Luria-Bertani broth containing 30 µg/mL kanamycin (LB-Kan30). Cultures were diluted 1:100 in fresh LB-Kan30 and were grown for 1.75 h at 37 °C. Cultures were temperature-shifted to 16 °C and induced for 8 h with 1 mM IPTG. Following low-temperature induction in bulk medium, cultures were diluted to an OD600 = 0.1 in fresh LB-Kan30 medium containing 1 mM IPTG and cefazolin at the desired selection concentration. Emulsions were incubated without shaking overnight at 37 °C and then were broken by extraction with 1H,1H,2H,2H-Perfluoro-1-octanol as described (55). The aqueous phase of the emulsion extract was incubated at 37 °C without shaking for 4 h, and the expansion, subculture, induction, and encapsulation/selection process was repeated with increasing concentrations of cefazolin until the selected cultures yielded no visible outgrowth during the subsequent 8-h expansion step.
Agar Plate Selections.
Libraries were expanded from glycerol stocks, subcultured, and induced at 16 °C as described above for the inverted emulsion selections. Following 8 h of low-temperature induction in bulk medium, replicate 100-μL aliquots of neat culture and two dilutions (1:103 and 1:106) were plated either on LB-Kan30 agar containing 1 mM IPTG (no selection) or on LB-Kan30 agar containing 1 mM IPTG and increasing concentrations of cefazolin (0, 1, 5, 10, 20, 30, 40, 50, 75, 100, 200, 300, 400, 500, 750, and 1,000 μg/mL). Plates were incubated at 37 °C for 20 h; then colonies were counted from both the control and selection plates, and fitness was calculated as the ratio of colony counts.
Enzyme Expression and Purification.
Expression and immobilized metal ion affinity purification of individual enzyme variants were performed as described elsewhere without modification (32). Analytical size exclusion chromatography of small-scale purifications showed a peak consistent with monomeric P99βL molecular mass with no evidence of dimers, trimers, tetramers, or other discrete oligomers. Protein preparations used in kinetic and thermostability analyses were ≥95% pure.
Enzyme Functional Analysis.
Kinetic and thermostability analysis of individual enzyme variants was performed as described elsewhere without modification (32). Each of two independent protein preparations was assayed in triplicate, and average values ± SD are reported.
Peptide–MHC II Binding Assays.
Peptide–MHC II affinity measurements were determined as described elsewhere without modification (24). Synthetic peptides used in the binding studies are shown in Table S2.
Immune Cell Activation Assays.
Protein immunogenic potential was quantified by measuring the activation of human immune cells from 19 donors (Table S3) using an established protocol (6, 30). Briefly, 2 × 106 live PBMCs were cultured in 24-well plates using 1 mL of RPMI 1640 medium supplemented with 5% human AB serum and either 5 µg/mL wild-type P99βL or 5 µg/mL 30:50:E9 deimmunized β-lactamase. Before cell culture, endotoxin was removed from the protein preparations by Triton X-114 extraction (56), and endotoxin levels were verified to be less than 0.1 endotoxin units per milligram of protein. Cells were incubated at 37 °C, 5% CO2, and on days 4, 7, and 11 half of the culture medium volume was replaced with RPMI 1640 supplemented with 5% human AB serum and 10 U/mL IL-2. On day 14, cells were harvested, washed, counted, and screened for antigen-specific reactivity by IL-2 ELISpot according to the manufacturer’s protocol. For each donor, aliquots of both the P99βL and 30:50:E9 expanded immune cells (200,000 cells per well) were rechallenged with a corresponding pool of 12 synthetic peptides that covered all 30 target sites of the respective protein antigen (each peptide at a final concentration of 1 µM; see Table S2). As controls, cells were also rechallenged with an equivalent volume of DMSO vehicle (negative control) or 2 µg/mL phytohemagglutinin (PHA) (positive control). Each rechallenge condition was measured in triplicate. Donor cells were accepted for analysis if the following criteria, defined before the experiment, were met: (i) greater than 80% viability of the expanded cells on day 14; (ii) at least 60% of the expanded cells were CD3+/CD4+; and (iii) the PHA rechallenge control produced at least 200 IL-2–producing cells. Donor DRB1*0405/1202 failed the PHA criterion and was excluded. All other donors passed the inclusion criteria. Donors were considered positive for a protein antigen if they yielded at least 60 spot-forming cells after background subtraction (i.e., the number of peptide-stimulated spots minus the number of DMSO-stimulated spots).
Acknowledgments
We thank Prof. Margaret Ackerman for reviewing this work. This work was supported by NIH Grant R01-GM-098977 (to C.B.-K. and K.E.G.). R.S.S. was supported in part by a Luce Foundation Fellowship and in part by a Thayer Innovation Program Fellowship from the Thayer School of Engineering. J.R.K. was supported in part by a Mazilu Fellowship. Computational resources were provided by National Science Foundation Grant CNS-1205521.
Footnotes
Conflict of interest statement: K.E.G. and C.B.-K. are Dartmouth faculty and co-members of Stealth Biologics, LLC, a Delaware biotechnology company. This work has been reviewed and approved as specified in K.E.G.’s and C.B.-K.’s Dartmouth conflict-of-interest management plans.
This article is a PNAS Direct Submission. C.D.S. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1621233114/-/DCSupplemental.
References
- 1.Griswold KE, Bailey-Kellogg C. Design and engineering of deimmunized biotherapeutics. Curr Opin Struct Biol. 2016;39:79–88. doi: 10.1016/j.sbi.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cizeau J, Grenkow DM, Brown JG, Entwistle J, MacDonald GC. Engineering and biological characterization of VB6-845, an anti-EpCAM immunotoxin containing a T-cell epitope-depleted variant of the plant toxin bouganin. J Immunother. 2009;32:574–584. doi: 10.1097/CJI.0b013e3181a6981c. [DOI] [PubMed] [Google Scholar]
- 3.Harding FA, et al. A beta-lactamase with reduced immunogenicity for the targeted delivery of chemotherapeutics using antibody-directed enzyme prodrug therapy. Mol Cancer Ther. 2005;4:1791–1800. doi: 10.1158/1535-7163.MCT-05-0189. [DOI] [PubMed] [Google Scholar]
- 4.Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the CDR regions. MAbs. 2010;2:256–265. doi: 10.4161/mabs.2.3.11641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jones TD, et al. Identification and removal of a promiscuous CD4+ T cell epitope from the C1 domain of factor VIII. J Thromb Haemost. 2005;3:991–1000. doi: 10.1111/j.1538-7836.2005.01309.x. [DOI] [PubMed] [Google Scholar]
- 6.Mazor R, et al. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc Natl Acad Sci USA. 2014;111:8571–8576. doi: 10.1073/pnas.1405153111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warmerdam PAM, et al. Elimination of a human T-cell region in staphylokinase by T-cell screening and computer modeling. Thromb Haemost. 2002;87:666–673. [PubMed] [Google Scholar]
- 8.Yeung VP, et al. Elimination of an immunodominant CD4+ T cell epitope in human IFN-β does not result in an in vivo response directed at the subdominant epitope. J Immunol. 2004;172:6658–6665. doi: 10.4049/jimmunol.172.11.6658. [DOI] [PubMed] [Google Scholar]
- 9.Cantor JR, et al. Therapeutic enzyme deimmunization by combinatorial T-cell epitope removal using neutral drift. Proc Natl Acad Sci USA. 2011;108:1272–1277. doi: 10.1073/pnas.1014739108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mazor R, et al. Elimination of murine and human T-cell epitopes in recombinant immunotoxin eliminates neutralizing and anti-drug antibodies in vivo. Cell Mol Immunol. 2017;14:432–442. doi: 10.1038/cmi.2015.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao H, et al. Depletion of T cell epitopes in lysostaphin mitigates anti-drug antibody response and enhances antibacterial efficacy in vivo. Chem Biol. 2015;22:629–639. doi: 10.1016/j.chembiol.2015.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi Y, Griswold KE, Bailey-Kellogg C. Structure-based redesign of proteins for minimal T-cell epitope content. J Comput Chem. 2013;34:879–891. doi: 10.1002/jcc.23213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.King C, et al. Removing T-cell epitopes with computational protein design. Proc Natl Acad Sci USA. 2014;111:8577–8582. doi: 10.1073/pnas.1321126111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parker AS, Choi Y, Griswold KE, Bailey-Kellogg C. Structure-guided deimmunization of therapeutic proteins. J Comput Biol. 2013;20:152–165. doi: 10.1089/cmb.2012.0251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parker AS, Griswold KE, Bailey-Kellogg C. Optimization of therapeutic proteins to delete T-cell epitopes while maintaining beneficial residue interactions. J Bioinform Comput Biol. 2011;9:207–229. doi: 10.1142/s0219720011005471. [DOI] [PubMed] [Google Scholar]
- 16.Parker AS, Zheng W, Griswold KE, Bailey-Kellogg C. Optimization algorithms for functional deimmunization of therapeutic proteins. BMC Bioinformatics. 2010;11:180. doi: 10.1186/1471-2105-11-180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gainza P, Nisonoff HM, Donald BR. Algorithms for protein design. Curr Opin Struct Biol. 2016;39:16–26. doi: 10.1016/j.sbi.2016.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Salvat RS, Choi Y, Bishop A, Bailey-Kellogg C, Griswold KE. Protein deimmunization via structure-based design enables efficient epitope deletion at high mutational loads. Biotechnol Bioeng. 2015;112:1306–1318. doi: 10.1002/bit.25554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Salvat RS, et al. Computationally driven deletion of broadly distributed T cell epitopes in a biotherapeutic candidate. Cell Mol Life Sci. 2014;71:4869–4880. doi: 10.1007/s00018-014-1652-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salvat RS, Parker AS, Choi Y, Bailey-Kellogg C, Griswold KE. Mapping the Pareto optimal design space for a functionally deimmunized biotherapeutic candidate. PLoS Comput Biol. 2015;11:e1003988. doi: 10.1371/journal.pcbi.1003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bagshawe KD. Antibody-directed enzyme prodrug therapy (ADEPT) for cancer. Expert Rev Anticancer Ther. 2006;6:1421–1431. doi: 10.1586/14737140.6.10.1421. [DOI] [PubMed] [Google Scholar]
- 22.Singh H, Raghava GPS. ProPred: Prediction of HLA-DR binding sites. Bioinformatics. 2001;17:1236–1237. doi: 10.1093/bioinformatics/17.12.1236. [DOI] [PubMed] [Google Scholar]
- 23.Jenkins SG, Schuetz AN. Current concepts in laboratory testing to guide antimicrobial therapy. Mayo Clin Proc. 2012;87:290–308. doi: 10.1016/j.mayocp.2012.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salvat R, Moise L, Bailey-Kellogg C, Griswold KE. A high throughput MHC II binding assay for quantitative analysis of peptide epitopes. J Vis Exp. 2014;85:e51308. doi: 10.3791/51308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hill JA, Wang D, Jevnikar AM, Cairns E, Bell DA. The relationship between predicted peptide-MHC class II affinity and T-cell activation in a HLA-DRbeta1*0401 transgenic mouse model. Arthritis Res Ther. 2003;5:R40–R48. doi: 10.1186/ar605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sidney J, del Guercio M-F, Southwood S, Sette A. The HLA molecules DQA1*0501/B1*0201 and DQA1*0301/B1*0302 share an extensive overlap in peptide binding specificity. J Immunol. 2002;169:5098–5108. doi: 10.4049/jimmunol.169.9.5098. [DOI] [PubMed] [Google Scholar]
- 27.Southwood S, et al. Several common HLA-DR types share largely overlapping peptide binding repertoires. J Immunol. 1998;160:3363–3373. [PubMed] [Google Scholar]
- 28.Wang P, et al. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput Biol. 2008;4:e1000048. doi: 10.1371/journal.pcbi.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jawa V, et al. T-cell dependent immunogenicity of protein therapeutics: Preclinical assessment and mitigation. Clin Immunol. 2013;149:534–555. doi: 10.1016/j.clim.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 30.Oseroff C, et al. Molecular determinants of T cell epitope recognition to the common Timothy grass allergen. J Immunol. 2010;185:943–955. doi: 10.4049/jimmunol.1000405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Greenbaum J, et al. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics. 2011;63:325–335. doi: 10.1007/s00251-011-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Osipovitch DC, et al. Design and analysis of immune-evading enzymes for ADEPT therapy. Protein Eng Des Sel. 2012;25:613–623. doi: 10.1093/protein/gzs044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davey JA, Chica RA. Multistate approaches in computational protein design. Protein Sci. 2012;21:1241–1252. doi: 10.1002/pro.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allen BD, Mayo SL. An efficient algorithm for multistate protein design based on FASTER. J Comput Chem. 2010;31:904–916. doi: 10.1002/jcc.21375. [DOI] [PubMed] [Google Scholar]
- 35.Hallen MA, Donald BR. Comets (Constrained Optimization of Multistate Energies by Tree Search): A provable and efficient protein design algorithm to optimize binding affinity and specificity with respect to sequence. J Comput Biol. 2016;23:311–321. doi: 10.1089/cmb.2015.0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Leaver-Fay A, Jacak R, Stranges PB, Kuhlman B. A generic program for multistate protein design. PLoS One. 2011;6:e20937. doi: 10.1371/journal.pone.0020937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yanover C, Fromer M, Shifman JM. Dead-end elimination for multistate protein design. J Comput Chem. 2007;28:2122–2129. doi: 10.1002/jcc.20661. [DOI] [PubMed] [Google Scholar]
- 38.Negron C, Keating AE. Multistate protein design using CLEVER and CLASSY. Methods Enzymol. 2013;523:171–190. doi: 10.1016/B978-0-12-394292-0.00008-4. [DOI] [PubMed] [Google Scholar]
- 39.Grigoryan G, Reinke AW, Keating AE. Design of protein-interaction specificity gives selective bZIP-binding peptides. Nature. 2009;458:859–864. doi: 10.1038/nature07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Havranek JJ, Harbury PB. Automated design of specificity in molecular recognition. Nat Struct Biol. 2003;10:45–52. doi: 10.1038/nsb877. [DOI] [PubMed] [Google Scholar]
- 41.Rudicell RS, et al. NISC Comparative Sequencing Program Enhanced potency of a broadly neutralizing HIV-1 antibody in vitro improves protection against lentiviral infection in vivo. J Virol. 2014;88:12669–12682. doi: 10.1128/JVI.02213-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foight GW, Ryan JA, Gullá SV, Letai A, Keating AE. Designed BH3 peptides with high affinity and specificity for targeting Mcl-1 in cells. ACS Chem Biol. 2014;9:1962–1968. doi: 10.1021/cb500340w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roberts KE, Cushing PR, Boisguerin P, Madden DR, Donald BR. Computational design of a PDZ domain peptide inhibitor that rescues CFTR activity. PLoS Comput Biol. 2012;8:e1002477. doi: 10.1371/journal.pcbi.1002477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zheng F, et al. Computational design of selective peptides to discriminate between similar PDZ domains in an oncogenic pathway. J Mol Biol. 2015;427:491–510. doi: 10.1016/j.jmb.2014.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen CY, Georgiev I, Anderson AC, Donald BR. Computational structure-based redesign of enzyme activity. Proc Natl Acad Sci USA. 2009;106:3764–3769. doi: 10.1073/pnas.0900266106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lippow SM, et al. Engineering enzyme specificity using computational design of a defined-sequence library. Chem Biol. 2010;17:1306–1315. doi: 10.1016/j.chembiol.2010.10.012. [DOI] [PubMed] [Google Scholar]
- 47.Murphy PM, Bolduc JM, Gallaher JL, Stoddard BL, Baker D. Alteration of enzyme specificity by computational loop remodeling and design. Proc Natl Acad Sci USA. 2009;106:9215–9220. doi: 10.1073/pnas.0811070106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reeve SM, et al. Protein design algorithms predict viable resistance to an experimental antifolate. Proc Natl Acad Sci USA. 2015;112:749–754. doi: 10.1073/pnas.1411548112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ambroggio XI, Kuhlman B. Computational design of a single amino acid sequence that can switch between two distinct protein folds. J Am Chem Soc. 2006;128:1154–1161. doi: 10.1021/ja054718w. [DOI] [PubMed] [Google Scholar]
- 50.He L, Friedman AM, Bailey-Kellogg C. A divide-and-conquer approach to determine the Pareto frontier for optimization of protein engineering experiments. Proteins. 2012;80:790–806. doi: 10.1002/prot.23237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Verma D, Grigoryan G, Bailey-Kellogg C. Structure-based design of combinatorial mutagenesis libraries. Protein Sci. 2015;24:895–908. doi: 10.1002/pro.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Choi Y, Verma D, Griswold KE, Bailey-Kellogg C. EpiSweep: Computationally driven reengineering of therapeutic proteins to reduce immunogenicity while maintaining function. Methods Mol Biol. 2017;1529:375–398. doi: 10.1007/978-1-4939-6637-0_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Parker AS, Griswold KE, Bailey-Kellogg C. Optimization of combinatorial mutagenesis. J Comput Biol. 2011;18:1743–1756. doi: 10.1089/cmb.2011.0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fromant M, Blanquet S, Plateau P. Direct random mutagenesis of gene-sized DNA fragments using polymerase chain reaction. Anal Biochem. 1995;224:347–353. doi: 10.1006/abio.1995.1050. [DOI] [PubMed] [Google Scholar]
- 55.Scanlon TC, Dostal SM, Griswold KE. A high-throughput screen for antibiotic drug discovery. Biotechnol Bioeng. 2014;111:232–243. doi: 10.1002/bit.25019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu S, et al. Removal of endotoxin from recombinant protein preparations. Clin Biochem. 1997;30:455–463. doi: 10.1016/s0009-9120(97)00049-0. [DOI] [PubMed] [Google Scholar]