

Significance
Nav1.1 is a potential therapeutic target for brain disorders, such as epilepsy, Alzheimer's disease, and autism. Moreover, this voltage-gated sodium (Nav) channel subtype contributes to mechanical pain by regulating excitability in a specific subset of sensory neurons within the peripheral nervous system. The results of our study shed light on the molecular mechanisms underlying Nav1.1 inactivation and suggest pharmacologic approaches to address relevant disease-related phenotypes.
Keywords: Nav1.1, fast inactivation, slow inactivation, Hm1a spider toxin, ICA-121431
Abstract
The Nav1.1 voltage-gated sodium channel is a critical contributor to excitability in the brain, where pathological loss of function leads to such disorders as epilepsy, Alzheimer’s disease, and autism. This voltage-gated sodium (Nav) channel subtype also plays an important role in mechanical pain signaling by primary afferent somatosensory neurons. Therefore, pharmacologic modulation of Nav1.1 represents a potential strategy for treating excitability disorders of the brain and periphery. Inactivation is a complex aspect of Nav channel gating and consists of fast and slow components, each of which may involve a contribution from one or more voltage-sensing domains. Here, we exploit the Hm1a spider toxin, a Nav1.1-selective modulator, to better understand the relationship between these temporally distinct modes of inactivation and ask whether they can be distinguished pharmacologically. We show that Hm1a inhibits the gating movement of the domain IV voltage sensor (VSDIV), hindering both fast and slow inactivation and leading to an increase in Nav1.1 availability during high-frequency stimulation. In contrast, ICA-121431, a small-molecule Nav1.1 inhibitor, accelerates a subsequent VSDIV gating transition to accelerate entry into the slow inactivated state, resulting in use-dependent block. Further evidence for functional coupling between fast and slow inactivation is provided by a Nav1.1 mutant in which fast inactivation removal has complex effects on slow inactivation. Taken together, our data substantiate the key role of VSDIV in Nav channel fast and slow inactivation and demonstrate that these gating processes are sequential and coupled through VSDIV. These findings provide insight into a pharmacophore on VSDIV through which modulation of inactivation gating can inhibit or facilitate Nav1.1 function.
Proper electrical signaling within neuronal systems requires an exquisite balance between excitation and inhibition at the molecular, cellular, and network levels. Among the most robust loci for regulating excitability are voltage-gated sodium (Nav) channels, a protein family consisting of nine subtypes (Nav1.1–1.9) that initiate and propagate action potentials throughout the human body (1). Loss-of-function mutations in Nav1.1 produce epilepsy syndromes, likely reflecting the critical importance of this Nav channel subtype in controlling the excitability of inhibitory interneurons in the brain (2–4). In contrast, gain-of-function mutations that diminish Nav1.1 fast inactivation are linked to familial hemiplegic migraine type 3 (5–9). Therefore, Nav1.1 has emerged as an important therapeutic target for brain disorders and, more recently, mechanical pain (10).
A high degree of amino acid sequence similarity among Nav channels complicates the development of subtype-selective pharmacologic agents, however (11). In addition, an efficacious Nav1.1 compound should target the appropriate gating transition to restore normal excitability patterns within a particular tissue. For example, neurons with a reduced complement of functional Nav1.1 channels will be most prone to dysfunction during elevated or sustained neuronal firing, because the pool of available channels will be further reduced by use-dependent accumulation of channels in inactivated states (12).
In this context, a compound that inhibits channel inactivation would be particularly beneficial. However, Nav channel inactivation is a complex process that includes binding of an inactivation particle to the intracellular side of the pore (fast inactivation), as well as a structurally and temporally distinct process whereby the selectivity filter becomes nonconducting after prolonged depolarization (slow inactivation) (1, 13, 14). Mammalian Nav channels are large proteins composed of four homologous domains that form a pseudo-fourfold symmetric channel comprising a central pore surrounded by four voltage sensors, one from each domain (1). Accumulating evidence suggests that activation gate opening is associated with movement of voltage sensors in domains I–III (VSDI–III), whereas fast inactivation is initiated by subsequent movement of the voltage sensor in domain IV (VSDIV) (1, 15, 16); however, the role of VSDIV in slow inactivation and the possibility of functional coupling between fast and slow inactivation remain matters of debate (17–19).
In the present study, we probed the molecular details of channel inactivation in relation to VSDIV using Hm1a, a recently discovered spider toxin that selectively enhances the Nav1.1 current through a two-point interaction with VSDIV (10). We found that Hm1a inhibits fast and slow inactivation, whereas a small-molecule inhibitor, ICA-121431 (20), acts solely on slow inactivation through interactions with the same voltage sensor, thus supporting a role for VSDIV in both fast and slow inactivation. Moreover, we show that a Nav1.1 mutant lacking fast inactivation has dramatic effects on slow inactivation, further suggesting coupling between these processes (21). Finally, we found that inhibition of fast and slow inactivation increases Nav1.1 availability in a frequency-dependent manner, reminiscent of migraine-linked mutations in Nav1.1 (5–9). These effects highlight the potential for pharmacologically targeting the VSDIV region as a therapeutic strategy for managing neurologic disorders in which Nav1.1 function is diminished (2, 3).
Results
Hm1a Binds to VSDIV to Alter Nav1.1 Fast Inactivation and Amplify Window Current.
We previously reported that Hm1a interacts with extracellular loops connecting transmembrane segments 1-2 and 3-4 (S1-S2/S3-S4) in VSDIV to inhibit Nav1.1 fast inactivation (10). Application of Hm1a also increases peak Na+ currents and produces a considerable persistent current at depolarized potentials (10). To further probe the underlying mechanism(s) of these actions, we first studied the kinetic and steady-state effects of Hm1a on fast inactivation. Facilitation of peak Na+ current can result from increased Nav1.1 availability. Indeed, Hm1a accelerates recovery from fast inactivation at very short time intervals, and shifts the voltage dependence of recovery into the physiological voltage range (Fig. 1 A and B). These effects can account for the observed toxin-dependent increase in peak Na+ current in cells heterologously expressing Nav1.1, as well as the increased action potential firing rate in mechanosensitive primary afferent fibers (10). These properties are similar to those described for other toxins from arachnids that interact with the S3b-S4 motif in VSDIV (1).
Fig. 1.

Hm1a affects Nav1.1 inactivation and creates a window current. (A) Recovery of Nav1.1 currents after fast and slow inactivation. Oocytes were depolarized to elicit peak conductance, then rested for a variable interval before a second depolarization to determine the fraction of channels recovered from inactivation. The duration of the interval ranged from 0 to 50 ms in 1-ms steps and from 50 ms to 3,000 ms in 50-ms steps to examine recovery from fast and slow inactivation, respectively. Black circles depict Nav1.1 before the application of 100 nM toxin application, and red circles depict Nav1.1 after this application. (B) Voltage dependence of recovery from fast inactivation in the presence (red) or absence (black) of 100 nM Hm1a. (C) Representative current traces showing SSI at a constant test voltage after prepulses at different voltages. (Top Left) In the absence of toxin, Nav1.1 is completely inactivated after prepulse potentials ≥−40 mV (test pulse, −15 mV, prepulse for 50 ms, −90 mV to 5 mV in 5-mV steps). (Top Right) In the presence of toxin, the same protocol produces a plateau at depolarized potentials. (Bottom Left) However, pulsing to very depolarize potentials (up to +60 mV) forces channels to completely fast inactivate. (Bottom Right) Switching to low-Na+ buffer enables measurement of outward currents at a +40-mV test potential, allowing for a more reliable measurement of steady-state inhibition. (D) Normalized conductance-voltage (G/Gmax, filled circles) and SSI (I/Imax, open circles) relationships of Nav1.1 before (black) and after (red) treatment with 1 μM Hm1a. The SSI relationship was obtained using the protocol shown in C. (E) Representative TTX-subtracted window currents from Nav1.1 in the presence (red) or absence (black) of 1 μM Hm1a, obtained by applying a voltage ramp from −70 to +70 mV over 1 s. (Inset) A nearly identical trace obtained when reversing the protocol from +70 to −70 mV. (F) Representative current traces elicited at the indicated voltages displaying different levels of sustained current in the presence of 1 μM Hm1a. Values are presented as mean ± SEM with n = 4–6. (Scale bars: horizontal, 100 ms; vertical, as indicated.)
Although kinetic changes to fast inactivation are often described for VSDIV toxins, their effects on steady-state inactivation (SSI) are less well defined. We previously found that Hm1a introduced a plateau in the SSI curve around the test potential, such that inactivation appeared incomplete (10) (Fig. 1C, Top). By extending the range of depolarized potentials in our SSI protocol, we discovered that more channels are forced into the inactivated state after depolarization to voltages >0 mV (Fig. 1C, Bottom Left). To record an SSI curve using a more depolarized test potential, we switched to low external Na+, enabling us to measure outward currents at +40 mV (Fig. 1C, Bottom Right). Under these conditions, the SSI curve in the presence of Hm1a was substantially right-shifted (V1/2 = −35 ± 2 mV vs −60 ± 1 mV in the presence or absence of toxin, respectively (Fig. 1D). Importantly, SSI in the absence of Hm1a was unchanged when current was measured at a +40-mV test potential in low external Na+ (Fig. S1A).
Fig. S1.

Effect of LqqIV on Nav1.1 SSI and voltage-dependence of slow inactivation. (A) Normalized steady-state inactivation (I/Imax, open circles) relationships of Nav1.1 before (black) and after (red) treatment with 100 nM LqqIV, measured at +40 mV in low external Na+. The SSI relationship was obtained using the protocol shown in Fig. 1D. Also shown is the SSI under normal saline conditions (black filled circles). (B) Voltage dependence of slow inactivation in the absence (black) or presence (red) of 100 nM LqqIV (open circles). Oocytes were held at −90 mV and exposed to the different conditioning voltages for 10 s, followed by a −100-mV pulse for 50 ms to remove fast inactivation and then a test pulse to −20 mV. Values are shown as mean ± SEM with n = 5. Boltzmann fit values are reported in the text.
To test whether shifts in SSI are a common feature of VSDIV-binding toxins, we examined the effects of LqqIV, a VSDIV-binding α-scorpion toxin (22, 23) using this same protocol. Similar to Hm1a, LqqIV shifted the SSI curve to more positive potentials (V1/2 = 67 ± 3 mV) (Fig. S1A). Our results with Hm1a predict a range of voltages over which Nav1.1 channels will activate but then incompletely inactivate, thereby producing a sustained “window current.” Indeed, such a current developed in the presence of Hm1a during a slow voltage ramp from −70 to +70 mV (Fig. 1E). This window current was due to steady-state effects rather than kinetic effects on fast inactivation, because indistinguishable window currents were observed when the ramp was reversed (+70 mV to −70 mV over 1 s) (Fig. 1F). Importantly, a small but mostly noninactivating current produced at ∼ −40 mV may decrease the threshold for action potential firing (24), as is seen in sensory neurons after application of Hm1a (10).
VSDIV Plays a Role in Slow Inactivation.
Having established the VSDIV-mediated effect of Hm1a on Nav1.1 fast inactivation and window current, we next investigated the influence of toxins on slow inactivation, which develops over seconds to tens of seconds. To assess this gating parameter, we held oocytes at various potentials for 10 s to monitor the voltage dependence and entry into the slow inactivated state. We found that Hm1a (1 μM) substantially shifted the voltage dependence of slow inactivation (V1/2) from −68 ± 2 mV in control conditions to −42 ± 1 mV after toxin application (ΔV1/2 ∼26 mV) (Fig. 2A). LqqIV (100 nM) had a comparable effect, with a V1/2 of −65 ± 1 mV in control and −44 ± 1 mV on toxin application (Fig. S1B). The time course of entry into the slow inactivated state was slightly slower in the presence of Hm1a across a range of test pulse durations, although the effect on kinetics was less pronounced compared with the effect on voltage dependence (Fig. 2B). In contrast, Hm1a did not influence the voltage dependence of recovery from slow inactivation (Fig. 2C). Indeed, by monitoring sustained current at −30 mV during this protocol, we found that sustained current was reduced, as if Hm1a unbound during prolonged depolarizations (Fig. 2D). Taken together, these data demonstrate that Hm1a influences entry into slow inactivation by interacting with VSDIV, indicating a role for this voltage sensor in Nav1.1 slow inactivation.
Fig. 2.

Hm1a inhibits entry into the slow inactivated state. (A) Voltage dependence of slow inactivation in the absence (black) or presence (red) of 1 μM Hm1a (filled circles). Oocytes were held at −90 mV and exposed to the different conditioning voltages for 10 s, followed by a −100-mV pulse for 50 ms to remove fast inactivation and then a test pulse to −20 mV. V1/2, control, −68 ± 2 mV; V1/2, Hm1a, −42 ± 1 mV for Hm1a. Hm1a significantly shifts the V1/2 of slow inactivation. Values are shown as mean ± SEM with n = 5. P < 0.001, Student’s paired two-tailed t test. (B) Kinetics of entry into the slow inactivated state in the absence (black) or presence (red) of Hm1a (1 μM). Oocytes were held at −90 mV and pulsed to −20 mV for various durations, followed by a 50-ms pulse to −100 mV to recover channels from fast inactivation and a test pulse at −20 mV for 50 ms. Entry into slow inactivation is significantly slower in the presence of Hm1a across a range of durations (300 ms–10 s; indicated with an asterisk). Values are shown as mean ± SEM with n = 5. P < 0.01, two-way ANOVA with Tukey’s post hoc test. (C) Recovery from slow inactivation in the absence (black) or presence (red) of Hm1a (100nM). Oocytes were held at −90 mV and exposed to −20 mV for 10 s, pulsed to −100 mV for 200 ms, then held at various recovery voltages for 10 s before a test pulse at −30 mV. Data are normalized to the peak current at −30 mV assayed before recovery from the slow inactivation protocol. Values are shown as mean ± SEM with n = 5. (D) Representative current traces before (Left) and after (Right) recovery from the slow inactivation protocol as in C.
Hm1a Increases Nav1.1 Availability and Supports Elevated Action Potential Firing.
The combined effect of Hm1a on fast and slow inactivation is sufficient to explain the previously reported increased excitability of Nav1.1-expressing neurons in the presence of the toxin (10). Indeed, the effects on excitability would be particularly noticeable during rapid neuronal firing, when fast and slow inactivation limit firing rate and duration, respectively. To replicate these effects in a heterologous system, we exposed Nav1.1-expressing oocytes to prepulse trains of simulated action potentials and measured currents without and with 100 nM Hm1a (Fig. 3A). We found that Nav1.1 availability dramatically increased with the toxin, by 3- to 10-fold depending on the stimulation rate and resting membrane potential (Fig. 3B). Strikingly similar phenotypes have been observed in studies of migraine mutations associated with Nav1.1, including impaired fast and slow inactivation and increased channel availability in response to rapid channel stimulation (5–9).
Fig. 3.

Hm1a prevents frequency-dependent current reduction in available channels. (A) Representative current traces from oocytes expressing Nav1.1 before stimulation (control, gray trace), and after 50-Hz simulated action potentials for 1 s at a holding potential (HP) of −90 mV in the absence (black trace) or presence (red trace) of 100 nM Hm1a. (B) Group data showing the availability of Nav1.1 following the simulated action potentials at the rate and holding potential indicated. Hm1a significantly increases Nav1.1 availability over a range of conditions. Values are shown as mean ± SEM with n = 5. P < 0.001, two-way ANOVA.
A Nav1.1 Inhibitor Promotes Slow Inactivation Through VSDIV.
ICA-121431 (ICA) is a small molecule inhibitor of Nav1.1 and Nav1.3 that has been shown to target extracellular amino acids within S2 and S3 in VSDIV, a locus also required for Hm1a efficacy (10, 20). Indeed, we found that Nav1.4 can be rendered ICA-sensitive by replacing the S1-S2 and S3-S4 loops of VSDIV with those from Nav1.1 (Fig. 4A), demonstrating that ICA and Hm1a sensitivity is specified by the same regions. (The Nav1.4 S1-S2/S3-S4 chimera does not show use-dependent inhibition by itself; Fig. S2.) In accordance with previous results (20), we observed strong use dependence for ICA (500 nM), such that Nav1.1 currents were fully inhibited at a stimulation rate of 5 Hz but unaffected at lower (0.2 Hz) rates (Fig. 4B). To better understand how ICA affects Nav1.1 gating, we measured activation–voltage and SSI relationships using prolonged intersweep intervals (10 s) to prevent use-dependent effects from ICA. Under these conditions, ICA did not significantly affect Nav1.1 activation (V1/2 = −38 ± 1 mV in control and −37 ± 1 mV with compound) or SSI (V1/2 = −42 ± 1 mV in control and −42 ± 2 mV with compound) (Fig. 4C). In contrast, the same concentration of ICA significantly shifted the voltage dependence of slow inactivation to more negative voltages (V1/2 = −68 ± 1 mV in control and −78 ± 1 mV with compound) and markedly accelerated entry into slow inactivation (Fig. 4D). Because both ICA and Hm1a interact with the same region in VSDIV, we coapplied ICA and Hm1a (500 nM each) to Nav1.1-expressing oocytes and exposed them to different stimulation rates. At high stimulation rates (5 Hz), Hm1a was unable to overcome ICA-induced inhibition, consistent with the idea that the toxin likely dissociates when channels are exposed to prolonged depolarization (Fig. 2D). During low-frequency stimulation at 0.2 Hz, ICA did not affect the ability of Hm1a to inhibit fast inactivation, thus resulting in channels that inactivated incompletely (Fig. 4E). Taken together, these results show that ICA inhibits Nav1.1 currents by binding to VSDIV to specifically target the slow inactivation process.
Fig. 4.

ICA accelerates entry into slow inactivation without affecting fast inactivation. (A, Left) Representative current traces from oocytes expressing Nav1.4 or Nav1.4 containing the S1-S2/S3-S4 VSDIV loop of Nav1.1 (black, control) in the presence (cyan) of 500 nM ICA-121431 during repeated stimulations from −90 mV to −10 mV at 5 Hz. (A, Right) Summary of current inhibition at −10 mV (holding −90 mV) as a result of applying 500 nM ICA-121431 on five oocytes expressing Nav1.4 or Nav1.4 containing the S1-S2/S3-S4 VSDIV loop of Nav1.1. (B) Representative current traces from oocytes expressing Nav1.1 in the presence of 500 nM ICA-121431 during repeated stimulation from a −90 mV holding potential to −30 mV at the indicated rate. (C, Left) Voltage-activation relationship for Nav1.1 in the absence (black) or presence (blue) of ICA (500 nM). The protocol was as shown in Fig. 3, except cells were held for 10 s at −90 mV between sweeps. The V1/2 of activation was −37 ± 1 mV in the presence of ICA vs −38 ± 1 mV in the absence of ICA. This shift did not reach statistical significance (P > 0.05, Student’s t test). (C, Right) SSI curve for which cells were held for 10s at −90 mV between sweeps. Values are shown as mean ± SEM with n = 6. (D, Left) Voltage dependence of slow inactivation in the absence and presence of ICA (500 nM) using a protocol similar to that shown in Fig. 2. (D, Right) Time-dependence of entry into slow inactivation according to the protocol in Fig. 2. Values are shown as mean ± SEM with n = 6. (E) Currents elicited during stimulation as indicated in the presence of both ICA (500 nM) and Hm1a (500 nM).
Fig. S2.

Nav1.4 VSDIV S1-S2/S3-S4 does not show use-dependent inhibition. Shown is a representative experiment demonstrating that before the addition of ICA-121431, peak current from the Nav1.4 VSDIV S1-S2/S3-S4 chimera reaches steady state during 1-Hz pulses to −30 mV. The ICA-121431-induced block washes out, and peak current returns to the steady-state level before drug application.
Disruption of Nav1.1 Fast Inactivation Influences Slow Inactivation.
Up to this point, our data suggest that VSDIV is involved in both fast and slow inactivation and that these two processes can be separated pharmacologically. To further investigate the nature of their relationship, we constructed the Nav1.1 L417W/L419C/A420W (WCW) triple mutant and measured its gating properties. These three residues are located at the intracellular end of the DI S6 helix, where mutations have been shown to remove fast inactivation of Nav1.4, Nav1.5, and Nav1.7 (25-27). Likewise, we found that fast inactivation was largely abolished in the Nav1.1 WCW mutant (Fig. 5 A and B). In addition, the voltage dependence of activation was shifted to more negative potentials [V1/2 of −23 ± 2 mV for wild-type (WT) vs −36 ± 1 mV for WCW mutant channels] (Fig. 5B). Monitoring slow inactivation in this mutant revealed that significantly stronger depolarization was required to drive the channel into the slow inactivated state (V1/2 = −68 ± 2 mV for WT vs −46 ± 1 mV for the WCW mutant) (Fig. 5C); however, the time-dependence of entry into the slow inactivated state at −20 mV was increased in the mutant (Fig. 5D). This gating behavior is similar to that observed in the Nav1.4 WCW mutant, where fast and slow inactivation display an inverse relationship, suggesting functional coupling (27). Such a correlation also has been detected in other fast inactivation-deficient Nav channels, including the QQQ mutant (28, 29) and chloramine T- or pronase-treated channels (30, 31), as well as, to a certain extent, Shaker Kv channels, in which experiments have shown that N-type inactivation (fast) is at least partially coupled to C-type inactivation (slow) (21).
Fig. 5.

Nav1.1 WCW affects fast and slow inactivation. (A) Representative family of current traces from oocytes expressing the Nav1.1 WCW mutant exposed to various activating voltages from −100 to 0 mV. As illustrated, fast inactivation is removed. (B) Activation–voltage (solid symbols, calculated G/Gmax) and SSI (open symbols, I/Imax) relationships of Nav1.1 WCW. Also shown in gray is the activation–voltage relationship obtained from measuring tail currents (I/Imax). The V1/2 for this curve (−39 ± 1 mV) is similar to that obtained from the calculated activation–voltage relationship. Values are shown as mean ± SEM with n = 4. (C) Voltage dependence of slow inactivation for Nav1.1 WCW (filled circles) compared with WT Nav1.1 (open circles). Values are shown as mean ± SEM with n = 4. (D) Entry into slow inactivation for Nav1.1 WCW (filled circles) compared with WT Nav1.1 (open circles). Values are shown as mean ± SEM with n = 4.
The gating characteristics of the Nav1.1 WCW channel can be explained if these mutations destabilize the VSDIV-activated state, thereby inhibiting the resting-to-activated step while promoting the activated-to-immobilized transition. Although we cannot exclude the effects of mutating three Nav1.1 pore residues on other channel regions, selective destabilization of the VSDIV-activated state is a likely consequence, because it prevents binding of the inactivation particle to the intracellular pore, a gating process known to be coupled to the activated state of VSDIV (1). Of note, Hm1b, a close relative of Hm1a (10), does not affect the activation–voltage and SSI relationships of Nav1.1 WCW (Fig. S3A). Moreover, the voltage dependence of slow inactivation is much less affected compared with the effects of Hm1a on WT Nav1.1 (Fig. S3B), further supporting the idea that the effect of Hm1a/b on slow inactivation is likely indirect and mediated primarily through the inhibition of fast inactivation.
Fig. S3.

Effect of Hm1b on Nav1.1 WCW. (A) Activation–voltage (solid symbols, calculated G/Gmax) and steady-state inactivation (open symbols, I/Imax) relationships of Nav1.1 WCW before (black) and after (red) the addition of 500 nM Hm1b. The V1/2, control value of −33 ± 1 mV does not differ significantly from the V1/2, Hm1b value of −32 ± 1 mV. Steady-state inactivation curves were not fitted with the Boltzmann function. (B) Voltage dependence of slow inactivation for Nav1.1 WCW before (black) and after (red) the addition of 500 nM Hm1b toxin. V1/2, control = −45 ± 1 mV; V1/2, Hm1b = −38 ± 1 mV. Values are shown as mean ± SEM with n = 4.
A Model of Sequential Nav1.1 VSDIV Movements.
In considering models that can account for our observations, we propose that VSDIV undergoes sequential movements that couple fast and slow inactivation (Fig. 6A). The first step initiates fast inactivation and corresponds to the transition of VSDIV from the resting state to the activated state, a step that accounts for the majority of gating charge movement (15, 32–35). The second, more weakly voltage-dependent, transition moves the activated VSDIV to an immobilized state that is coupled to selectivity filter collapse and slow inactivation, as has been proposed for other voltage-gated channels (14, 17, 36). The effect of Hm1a (10) on Nav1.1 (Figs. 1, 2, 3, and 5) can be explained primarily as inhibition of the first VSDIV transition, thereby resulting in direct effects on fast inactivation and indirect effects on slow inactivation, particularly with respect to voltage dependence. Conversely, ICA accelerates entry into slow inactivation by promoting the activated-to-immobilized transition of VSDIV, with more modest effects on the voltage dependence of slow inactivation and no effect on the voltage dependence of activation or SSI.
Fig. 6.

Model for VSDIV-controlled inactivation gating in Nav1.1. (A) Sequential states of VSDIV are shown with the functional state of the ion channel coupled to each position shown above. In this model, Hm1a stabilizes the resting state of the voltage sensor, whereas ICA exclusively promotes movement from the activated to the immobilized state. (B) A three-state model of sequential VSDIV movements with simple rate changes recapitulating the effects of Hm1a and ICA on Nav1.1 (Table S1). Simulated SSI (Left), voltage dependence of slow inactivation (Middle), and time dependence of entry into slow inactivation (Right) are shown. WT, Hm1a, and ICA models are shown in black, red, and blue, respectively.
To test our hypotheses, we constructed a three-state model of sequential inactivation in which the first transition represents VSDIV activation (coupled to channel inactivation) and the second transition represents VSDIV immobilization (coupled to channel slow inactivation) (Materials and Methods and Fig. 6B). Original data were fit to reproduce slow and fast SSI curves, along with the time course of entry into the slow inactivated state (10-s pulses to −20 mV). As shown in Fig. 6B, the model accurately reproduces the inactivation properties obtained from experimental data (WT model). Remarkably, all of the effects of Hm1a on Nav1.1 gating can be simulated with a minimal number of perturbations to the fast inactivation transition of the WT model; i.e., slowing the forward step into the fast inactivated state and accelerating the reverse step (Fig. 6B and Table S1). Similarly, a simple acceleration of the second transition by a factor of 3 compared with the WT model reproduced the observed effects of ICA on the time course of entry into the slow inactivated state, as well as the leftward shift in the slow inactivated state availability curve (Fig. 6B and Table S1). Taken together, these simulations support a sequential inactivation scheme in which the initial, fast-inactivation coupled movement of VSDIV is followed by a slower, less voltage-dependent transition to the immobilized, slow-inactivation–coupled state.
Table S1.
Values for the computational simulations shown in Fig. 6B
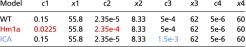 |
Colored numbers indicate altered parameters as mentioned in the text.
Discussion
Nav channel inactivation is a complex process consisting of fast and slow components that may involve a contribution from one or more voltage sensor domains (1, 13, 15, 16, 19). Understanding these processes and elucidating strategies for their pharmacologic modulation may aid drug discovery efforts aimed at treating Nav1.1-associated disorders, such as epilepsy, migraine, and mechanical pain (2, 3, 5–8, 10, 11). Here we exploited two pharmacologic agents to probe the role of VSDIV in Nav1.1 inactivation. Our data suggest that the VSDIV-targeting spider toxins Hm1a/b (10) increase Nav1.1 currents by inhibiting the initial voltage-sensor movement to hamper fast inactivation (Fig. 1). In addition, these toxins shift voltage dependence of slow inactivation to more positive potentials and impede entry into the slow inactivated state (Figs. 2 and 3). We further show that the small-molecule inhibitor ICA-121431 (20) exclusively influences slow inactivation by binding to VSDIV, and that a mutant (WCW) that destabilizes fast inactivation also inhibits the voltage dependence of slow inactivation, but enhances the kinetics of entry into this state (Figs. 4 and 5).
Although these data do not rule out contributions of VSDI–III or other regions to macroscopic fast or slow inactivation (19, 37–39), they do suggest a prominent role for VSDIV in slow inactivation, especially because this process can be promoted (ICA) or inhibited (Hm1a) through pharmacologic agents that target this particular voltage sensor domain. Notably, the concept that slow inactivation is tied to a VSDIV transition subsequent to activation supports previous models proposing that Nav channel voltage sensor immobilization and slow inactivation are coupled (1, 15–18, 36). Taken together, our results provide support for a VSDIV-centric model of Nav1.1 inactivation in which both fast and slow inactivation processes are coupled to sequential movements of this voltage sensor (Fig. 6).
Partial loss of Nav1.1 function in parvalbumin-positive interneurons in the brain is sufficient to cause epilepsy (12, 40). These neurons display high-frequency firing properties that exacerbate an Nav1.1 deficit by driving remaining channels into the inactivated state. Thus, certain aspects of Hm1a action, including its subtype selectivity, may be well suited to treat epilepsy associated with reduced Nav1.1 function (11). However, the introduction of a large window current by the toxin is a complicating factor, given that this current also has been linked to seizures (41). In fact, injection of high concentrations of Hm1a into the mouse brain was shown to be lethal (42), an observation consistent with the idea that elevated toxin concentrations can introduce a dangerous level of persistent Nav1.1 current. Moreover, Nav1.1 expression in other regions of the brain may contribute to Hm1a toxicity (4). Nonetheless, our Nav1.1 inactivation gating model provides a rationale for developing new Nav1.1-selective agents with a greater safety margin (11). For example, a VSDIV-binding compound that equally favors the resting and activated voltage sensor over a subsequent slow-inactivation–coupled state should prevent Nav1.1 accumulation in the slow inactivated state without introducing persistent current. In contrast to pharmacologically activating Nav1.1 by targeting VSDIV, blocking channel currents by promoting entry into the slow inactivated state (ICA) may be efficacious in treating such disorders as irritable bowel syndrome, where enhanced Nav1.1 function or expression may dysregulate the excitability of sensory nerve fibers that mediate mechanical pain (10).
Finally, our data show that Hm1a inhibits fast and slow inactivation to increase available channels during periods of rapid stimulation. Strikingly similar phenotypes are exhibited by Nav1.1 mutants linked to migraine (5–7, 9). Of note, migraine mutations tend to cluster around Nav1.1 VSDIV, lending further support to the utility of Hm1a for phenocopying FHM3. As such, Hm1a could represent a new tool with which to probe the contributions of central or peripheral Nav1.1-expressing neurons to migraine pain.
Materials and Methods
Reagents and Buffers.
Hm1a was purified from Heteroscodra maculata venom as described previously (10). LqqIV was provided by Martin-Eauclaire and Pierre Bougis, Aix-Marseille Université (22). ICA-121431 was purchased from Tocris. Human Nav1.1 was obtained from Origene Technologies and modified as reported previously (11). Rat Nav1.4 was a gift from Baron Chanda, University of Wisconsin–Madison. cRNA was synthesized in vitro using T7 polymerase (mMessage mMachine Kit; Life Technologies), preceded by enzyme linearization of sequenced DNA. Nav1.1 DNA was amplified using CopyCutter EPI400 Escherichia coli (Epicentre), whereas Nav1.4 was propagated in JM-109 E. coli (Promega). Expanded DNA stocks were checked for rearrangements using restriction mapping with appropriate enzymes (New England BioLabs). All other chemicals were obtained from Sigma-Aldrich.
Xenopus laevis Oocyte Electrophysiology.
Nav1.1, Nav1.4, and the PCR-generated Nav1.1 WCW mutant were expressed in Xenopus laevis oocytes (animals from Xenopus 1 or ovaries acquired from Nasco) that had been incubated in Barth’s medium [88 mM NaCl, 1 mM KCl, 0.33 mM Ca(NO3)2, 0.41 mM CaCl2, 0.82 mM MgSO4, 2.4 mM NaHCO3, 5 mM Hepes, and 0.1 mg/mL gentamycin, pH 7.6 with NaOH] at 17 °C for 1–3 d after cRNA injection, and then studied using a two-electrode voltage-clamp recorder (OC-725C; Warner Instruments or GeneClamp 500B; Axon Instruments) with a 150-μL recording chamber or a small-volume (<20 μL) oocyte perfusion chamber (AutoMate Scientific). Data were filtered at 4 kHz and digitized at 20 kHz using pClamp 10 software (Molecular Devices). Microelectrode resistance was 0.5–1 MΩ when filled with 3 M KCl. The external recording solution contained 100 mM NaCl, 5 mM Hepes, 1 mM MgCl2, and 1.8 mM CaCl2, pH 7.6 with NaOH, except for the low-Na+ solution, which contained 5 mM NaCl, 93 mM KCl, 1 mM MgCl2, and 5 mM Hepes, pH 7.6 with NaOH. All experiments were performed at ∼22 °C, and toxin samples were diluted in recording solution with 0.1% BSA. Off-line data analyses were performed using Clampfit 10 (Molecular Devices), GraphPad Prism 6, and Origin 7.5 (Originlab). Statistical analysis was performed as appropriate, and data are reported in the figure legends or text. To avoid functional interference of auxiliary proteins (23), Nav channel gating properties were investigated in oocytes without the presence of β subunits.
Computational Modeling.
Computation results were obtained by fitting the rate constants of a three-state model with Arrhenius equations of the form a = c1⋅exp(v/x1) and b = c2⋅exp(−v/x2). Simulations were performed in MATLAB (MathWorks), with a deterministic solution (using forward Kolmogorov equations) for the linear system under imposed voltage clamp. For initial conditions, the channel was assumed to be available when held at the holding potential (−140 mV). For the simulations shown in Fig. 6B, the forward (a) and reverse (b) rates between states are governed by the following equations, where the values used for the constants in the equations are given in Table S1: fast inactivation transition, a1 = c1⋅exp(v/x1) and b1 = c2⋅exp(−v/x2); slow inactivation transition, a2 = c3⋅exp(v/x3) and b2 = c4⋅exp(−v/x4).
Acknowledgments
We thank Glenn King (Institute for Molecular Bioscience, University of Queensland) for sharing the Heteroscodra maculata spider venom and John Gilchrist for generating the Nav1.1 WCW mutant. This work was supported by National Institutes of Health Ruth Kirschstein Postdoctoral Fellowship F32NS081907 (to J.D.O.) and National Institutes of Health Grants R37NS065071 (to D.J.), R01 NS081115 (to D.J.), and R01 NS091352 (to F.B.).
Footnotes
Conflict of interest statement: J.D.O. is currently an employee of Vertex Pharmaceuticals. His contributions to this study were made while at University of California, San Francisco. This work was neither carried out nor supported by Vertex Pharmaceuticals.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1621263114/-/DCSupplemental.
References
- 1.Ahern CA, Payandeh J, Bosmans F, Chanda B. The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J Gen Physiol. 2016;147:1–24. doi: 10.1085/jgp.201511492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Catterall WA, Kalume F, Oakley JC. NaV1.1 channels and epilepsy. J Physiol. 2010;588:1849–1859. doi: 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Escayg A, Goldin AL. Sodium channel SCN1A and epilepsy: Mutations and mechanisms. Epilepsia. 2010;51:1650–1658. doi: 10.1111/j.1528-1167.2010.02640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trimmer JS, Rhodes KJ. Localization of voltage-gated ion channels in mammalian brain. Annu Rev Physiol. 2004;66:477–519. doi: 10.1146/annurev.physiol.66.032102.113328. [DOI] [PubMed] [Google Scholar]
- 5.Cestèle S, et al. Self-limited hyperexcitability: Functional effect of a familial hemiplegic migraine mutation of the Nav1.1 (SCN1A) Na+ channel. J Neurosci. 2008;28:7273–7283. doi: 10.1523/JNEUROSCI.4453-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cestèle S, Schiavon E, Rusconi R, Franceschetti S, Mantegazza M. Nonfunctional NaV1.1 familial hemiplegic migraine mutant transformed into gain of function by partial rescue of folding defects. Proc Natl Acad Sci USA. 2013;110:17546–17551. doi: 10.1073/pnas.1309827110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fan C, et al. Early-onset familial hemiplegic migraine due to a novel SCN1A mutation. Cephalalgia. 2016;36:1238–1247. doi: 10.1177/0333102415608360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gargus JJ, Tournay A. Novel mutation confirms seizure locus SCN1A is also familial hemiplegic migraine locus FHM3. Pediatr Neurol. 2007;37:407–410. doi: 10.1016/j.pediatrneurol.2007.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kahlig KM, et al. Divergent sodium channel defects in familial hemiplegic migraine. Proc Natl Acad Sci USA. 2008;105:9799–9804. doi: 10.1073/pnas.0711717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Osteen JD, et al. Selective spider toxins reveal a role for the Nav1.1 channel in mechanical pain. Nature. 2016;534:494–499. doi: 10.1038/nature17976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gilchrist J, et al. Nav1.1 modulation by a novel triazole compound attenuates epileptic seizures in rodents. ACS Chem Biol. 2014;9:1204–1212. doi: 10.1021/cb500108p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tai C, Abe Y, Westenbroek RE, Scheuer T, Catterall WA. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc Natl Acad Sci USA. 2014;111:E3139–E3148. doi: 10.1073/pnas.1411131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hille B. 2001. Ion Channels of Excitable Membranes (Sinauer Associates, Sunderland. MA), 3rd Ed, p 814.
- 14.Silva J. Slow inactivation of Na+ channels. Handb Exp Pharmacol. 2014;221:33–49. doi: 10.1007/978-3-642-41588-3_3. [DOI] [PubMed] [Google Scholar]
- 15.Capes DL, Goldschen-Ohm MP, Arcisio-Miranda M, Bezanilla F, Chanda B. Domain IV voltage-sensor movement is both sufficient and rate-limiting for fast inactivation in sodium channels. J Gen Physiol. 2013;142:101–112. doi: 10.1085/jgp.201310998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bezanilla F. How membrane proteins sense voltage. Nat Rev Mol Cell Biol. 2008;9:323–332. doi: 10.1038/nrm2376. [DOI] [PubMed] [Google Scholar]
- 17.Capes DL, Arcisio-Miranda M, Jarecki BW, French RJ, Chanda B. Gating transitions in the selectivity filter region of a sodium channel are coupled to the domain IV voltage sensor. Proc Natl Acad Sci USA. 2012;109:2648–2653. doi: 10.1073/pnas.1115575109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitrovic N, George AL, Jr, Horn R. Role of domain 4 in sodium channel slow inactivation. J Gen Physiol. 2000;115:707–718. doi: 10.1085/jgp.115.6.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silva JR, Goldstein SAN. Voltage-sensor movements describe slow inactivation of voltage-gated sodium channels, I: Wild-type skeletal muscle NaV1.4. J Gen Physiol. 2013;141:309–321. doi: 10.1085/jgp.201210909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCormack K, et al. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc Natl Acad Sci USA. 2013;110:E2724–E2732. doi: 10.1073/pnas.1220844110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: Effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- 22.Kopeyan C, Martinez G, Rochat H. Primary structure of toxin IV of Leiurus quinquestriatus quinquestriatus: Characterization of a new group of scorpion toxins. FEBS Lett. 1985;181:211–217. [Google Scholar]
- 23.Gilchrist J, Das S, Van Petegem F, Bosmans F. Crystallographic insights into sodium-channel modulation by the β4 subunit. Proc Natl Acad Sci USA. 2013;110:E5016–E5024. doi: 10.1073/pnas.1314557110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carter BC, Giessel AJ, Sabatini BL, Bean BP. Transient sodium current at subthreshold voltages: Activation by EPSP waveforms. Neuron. 2012;75:1081–1093. doi: 10.1016/j.neuron.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edrich T, Wang SY, Wang GK. State-dependent block of human cardiac hNav1.5 sodium channels by propafenone. J Membr Biol. 2005;207:35–43. doi: 10.1007/s00232-005-0801-4. [DOI] [PubMed] [Google Scholar]
- 26.Wang GK, Calderon J, Wang SY. State- and use-dependent block of muscle Nav1.4 and neuronal Nav1.7 voltage-gated Na+ channel isoforms by ranolazine. Mol Pharmacol. 2008;73:940–948. doi: 10.1124/mol.107.041541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang SY, Bonner K, Russell C, Wang GK. Tryptophan scanning of D1S6 and D4S6 C-termini in voltage-gated sodium channels. Biophys J. 2003;85:911–920. doi: 10.1016/S0006-3495(03)74530-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hilber K, et al. Interaction between fast and ultra-slow inactivation in the voltage-gated sodium channel: Does the inactivation gate stabilize the channel structure? J Biol Chem. 2002;277:37105–37115. doi: 10.1074/jbc.M205661200. [DOI] [PubMed] [Google Scholar]
- 29.West JW, et al. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc Natl Acad Sci USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rudy B. Slow inactivation of the sodium conductance in squid giant axons: Pronase resistance. J Physiol. 1978;283:1–21. doi: 10.1113/jphysiol.1978.sp012485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang SY, Wang GK. A mutation in segment I-S6 alters slow inactivation of sodium channels. Biophys J. 1997;72:1633–1640. doi: 10.1016/S0006-3495(97)78809-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chanda B, Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J Gen Physiol. 2002;120:629–645. doi: 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen LQ, Santarelli V, Horn R, Kallen RG. A unique role for the S4 segment of domain 4 in the inactivation of sodium channels. J Gen Physiol. 1996;108:549–556. doi: 10.1085/jgp.108.6.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kühn FJ, Greeff NG. Movement of voltage sensor S4 in domain 4 is tightly coupled to sodium channel fast inactivation and gating charge immobilization. J Gen Physiol. 1999;114:167–183. doi: 10.1085/jgp.114.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheets MF, Kyle JW, Kallen RG, Hanck DA. The Na channel voltage sensor associated with inactivation is localized to the external charged residues of domain IV, S4. Biophys J. 1999;77:747–757. doi: 10.1016/S0006-3495(99)76929-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ghovanloo MR, Aimar K, Ghadiry-Tavi R, Yu A, Ruben PC. Physiology and pathophysiology of sodium channel inactivation. Curr Top Membr. 2016;78:479–509. doi: 10.1016/bs.ctm.2016.04.001. [DOI] [PubMed] [Google Scholar]
- 37.Jurkat-Rott K, et al. Voltage-sensor sodium channel mutations cause hypokalemic periodic paralysis type 2 by enhanced inactivation and reduced current. Proc Natl Acad Sci USA. 2000;97:9549–9554. doi: 10.1073/pnas.97.17.9549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kontis KJ, Goldin AL. Sodium channel inactivation is altered by substitution of voltage sensor positive charges. J Gen Physiol. 1997;110:403–413. doi: 10.1085/jgp.110.4.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cha A, Ruben PC, George AL, Jr, Fujimoto E, Bezanilla F. Voltage sensors in domains III and IV, but not I and II, are immobilized by Na+ channel fast inactivation. Neuron. 1999;22:73–87. doi: 10.1016/s0896-6273(00)80680-7. [DOI] [PubMed] [Google Scholar]
- 40.Ogiwara I, et al. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stafstrom CE. Persistent sodium current and its role in epilepsy. Epilepsy Curr. 2007;7:15–22. doi: 10.1111/j.1535-7511.2007.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Escoubas P, Diochot S, Célérier ML, Nakajima T, Lazdunski M. Novel tarantula toxins for subtypes of voltage-dependent potassium channels in the Kv2 and Kv4 subfamilies. Mol Pharmacol. 2002;62:48–57. doi: 10.1124/mol.62.1.48. [DOI] [PubMed] [Google Scholar]