

Significance
Muscle contraction is required for critical physiological functions. It relies on the interaction of myosin motors with the thin filament (TF), which is regulated through a translocation of tropomyosin on the surface of F-actin by the troponin complex in response to Ca2+. The lack of high-resolution structure of the TF under relaxing (low-Ca2+) and activating (high-Ca2+) conditions impairs our understanding of the mechanism of cardiac muscle regulation. Here we report high-resolution structures of the native cardiac TF under relaxing and activating conditions. Our data lead to a model for cardiac TF regulation by Ca2+ levels that is an important step in understanding how the components of cardiac muscle work in concert to maintain healthy heart functions.
Keywords: thin filament, cardiac muscle regulation, cryoelectron microscopy
Abstract
Muscle contraction relies on the interaction of myosin motors with F-actin, which is regulated through a translocation of tropomyosin by the troponin complex in response to Ca2+. The current model of muscle regulation holds that at relaxing (low-Ca2+) conditions tropomyosin blocks myosin binding sites on F-actin, whereas at activating (high-Ca2+) conditions tropomyosin translocation only partially exposes myosin binding sites on F-actin so that binding of rigor myosin is required to fully activate the thin filament (TF). Here we used a single-particle approach to helical reconstruction of frozen hydrated native cardiac TFs under relaxing and activating conditions to reveal the azimuthal movement of the tropomyosin on the surface of the native cardiac TF upon Ca2+ activation. We demonstrate that at either relaxing or activating conditions tropomyosin is not constrained in one structural state, but rather is distributed between three structural positions on the surface of the TF. We show that two of these tropomyosin positions restrain actomyosin interactions, whereas in the third position, which is significantly enhanced at high Ca2+, tropomyosin does not block myosin binding sites on F-actin. Our data provide a structural framework for the enhanced activation of the cardiac TF over the skeletal TF by Ca2+ and lead to a mechanistic model for the regulation of the cardiac TF.
Muscle contraction is based on the interaction between two filamentous systems: the thick (myosin) and the thin (actin) filaments. Thin filaments (TFs) are comprised of F-actin, tropomyosin (Tpm), and troponin (Tn) complex. Tpm is a coiled-coil protein that geometrically complements F-actin by binding to seven successive actin subunits (1). The Tn complex alters the position of the Tpm cable upon binding Ca2+ (2, 3). The steric blocking model of muscle regulation holds that at relaxing (low Ca2+) conditions Tpm blocks myosin binding sites on the surface of F-actin, whereas Tpm moves away from the myosin binding sites at activating (high Ca2+) conditions (2, 3). Kinetics, biochemical, and structural studies have suggested that there are three structural states for Tpm, termed “blocked,” “closed,” and “myosin” (“open”), which correspond to relaxing, activating, and myosin-bound conditions, respectively (4–6). The current model of the muscle regulation is based on the analysis of negatively stained TFs and suggests a two-step muscle activation (3, 7). At relaxing conditions Tn keeps Tpm constrained in the blocked state. Binding of Ca2+ to the Tn complex releases Tpm from the blocked state so that Tpm swings by ∼25° from the blocked to the closed state, partially exposing myosin binding sites on F-actin. Because the myosin binding sites are not fully exposed and actomyosin interactions are constrained, the model advocates that under activating conditions a small number of myosin molecules should strongly bind to F-actin to complete activation of the TF by swinging Tpm by an additional ∼10° from its closed into the myosin state.
There are significant differences in the mechanisms of regulation of cardiac and skeletal TFs. Ca2+ alone produces only ∼20% of maximal activation of skeletal TF and rigor-bound myosin is required to complete activation (8, 9), whereas Ca2+ alone activates cardiac TFs by ∼70% (10). Presumably, significant differences between the TF proteins in cardiac muscle and skeletal muscle (11–13) are responsible for their different response to Ca2+ levels.
All our current structural knowledge on the Ca2+ activation of the TF relies on the analysis of negatively stained TFs (3, 5, 14). Recent studies on the interaction of F-actin with Tpm demonstrated that the position of Tpm on the surface of F-actin is different in negatively stained and frozen hydrated samples (15). Therefore, it has been suggested that negative staining induces repulsions at the Tpm–F-actin interface which are responsible for the leftward shift of Tpm on the surface of F-actin (15).
Here we used a single-particle approach to helical reconstruction (16) of frozen hydrated native cardiac TFs under relaxing and activating conditions to reveal the azimuthal movement of the Tpm cable on the surface of F-actin upon Ca2+ activation.
Results
The Canonical-Blocked Structural State of the TF Is Not Required for the Cardiac TF Inhibition at low Ca2+.
Because most of the structural studies on the Ca2+-dependent regulation of the TF were done using negative staining (3, 5), we performed 3D reconstructions of negatively stained native cardiac TFs (SI Appendix, Fig. S1). In the negatively stained samples the Tn densities were readily visible (SI Appendix, Fig. S1A, red arrowheads). We generated 3D reconstructions of segments of TFs that possessed well-defined Tn lumps at both low (pCa > 8) and high (pCa = 4) Ca2+ levels (SI Appendix, Fig. S1 B and C, respectively). In agreement with previously published data (3, 5, 14) we found that at low Ca2+ Tpm was in the “canonical-blocked” state (SI Appendix, Fig. S1B, red ribbons), whereas at pCa = 4 the Tpm cable was shifted leftward into its closed state (SI Appendix, Fig. S1C, green ribbons). It has been shown that in the negatively stained Tpm–F-actin complex cross-linking of Tpm to F-actin shifts the position of Tpm from the canonical blocked to the closed structural state (15). We therefore performed 3D reconstructions of cardiac TFs cross-linked with glutaraldehyde (CL-TF) at low and high Ca2+ (SI Appendix, Fig. S1 D and E, respectively). Only segments with well-defined Tn lumps were chosen for 3D reconstruction (SI Appendix, Fig. S1A, red arrowheads). We found that in contrast to the native TFs the position of Tpm in the CL-TFs at low Ca2+ was shifted leftward from the canonical blocked to the closed structural state (SI Appendix, Fig. S1D, green ribbons). At high Ca2+ the Tpm cable was shifted leftward even further toward the myosin structural state (SI Appendix, Fig. S1E, yellow ribbons).
To determine whether the glutaraldehyde cross-linking alters the activation state of the native cardiac TFs in solution, we measured myosin-S1 TF-dependent activation of product dissociation from myosin-ADP-Pi at low and high Ca2+ for both native and CL-TFs (SI Appendix, Fig. S2). We found very little effect of glutaraldehyde on product activation kinetics by F-actin (SI Appendix, Fig. S2A), but the effect of glutaraldehyde on the TF-dependent product dissociation was more pronounced (SI Appendix, Fig. S2B). At pCa > 8, CL-TFs were partially activated, whereas at pCa = 4 CL-TFs were somewhat inhibited in comparison with their native counterparts. Nevertheless, CL-TFs retained a significant difference in their activation state at low and high Ca2+ levels, suggesting that the canonical blocked structural state which was absent in the cross-linked sample was not required per se for the inhibition of the native cardiac TF at low Ca2+ conditions.
Cryoelectron Microscopy Defines Three Structural States of Tpm on the Surface of F-Actin.
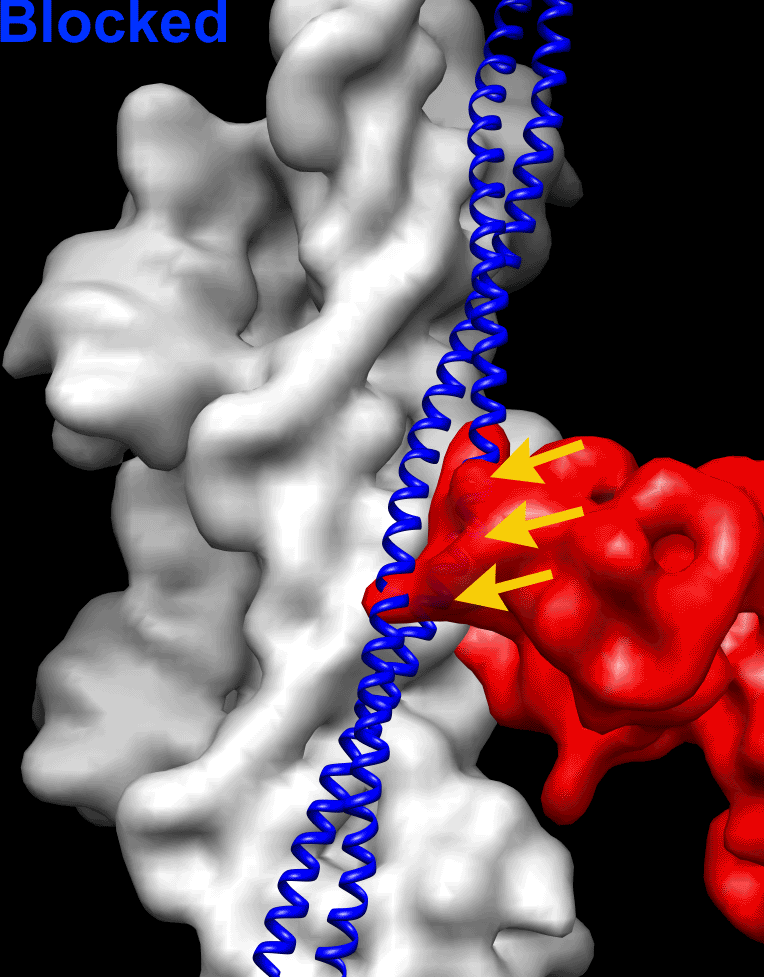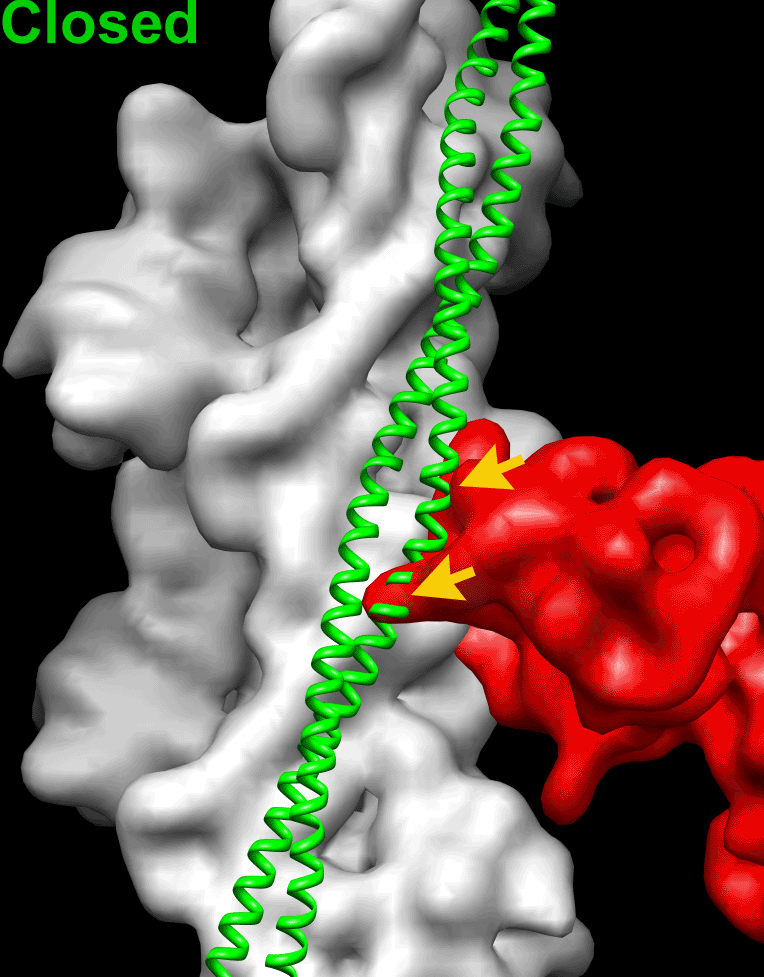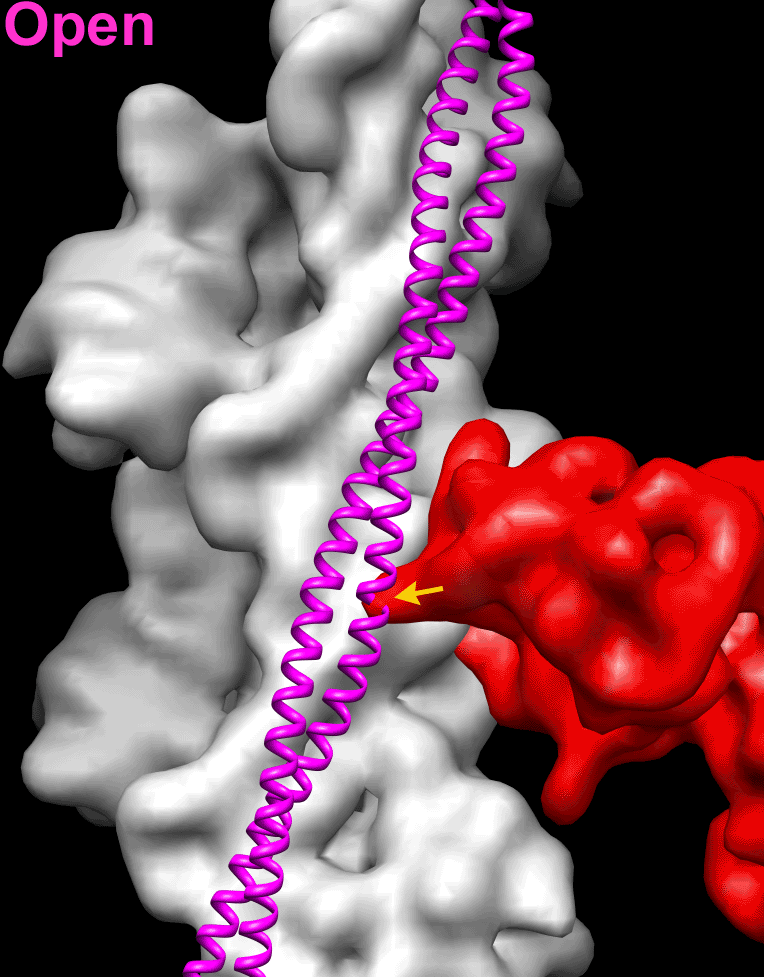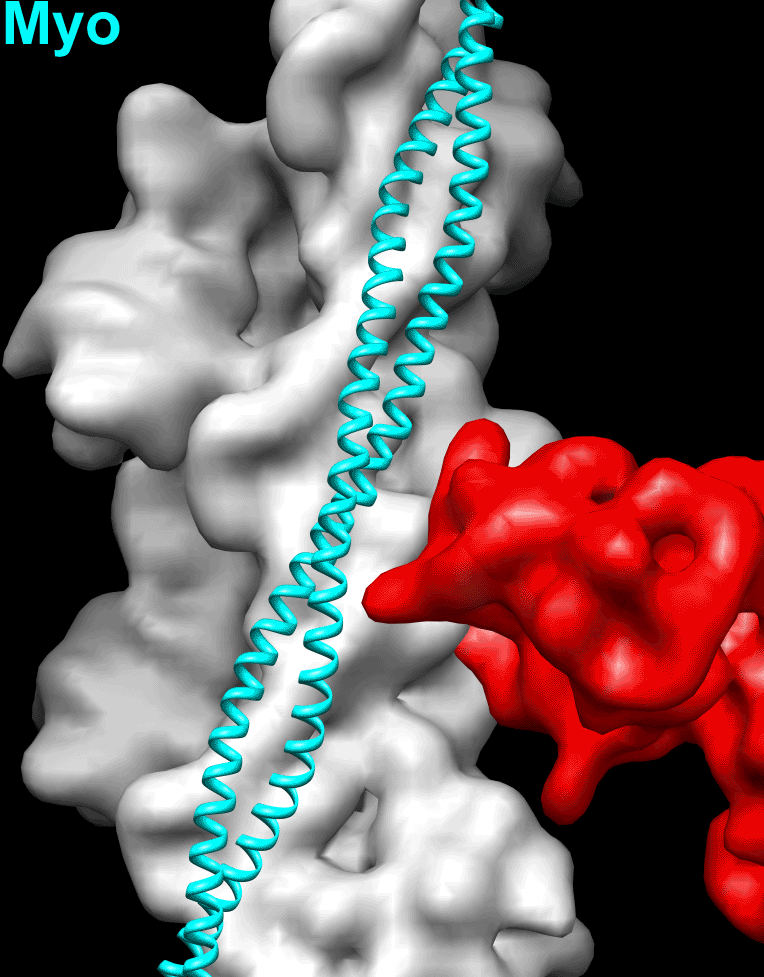
Because the position of Tpm on the surface of the actin filament is altered by negative staining (SI Appendix, Fig. S1), we used frozen hydrated native cardiac TFs to study the movement of the Tpm cable upon TF activation by Ca2+. Frozen hydrated native TFs possessed well-defined Tn densities (Fig. 1A, Inset). Because it has been shown that TFs are heterogeneous at either low or high Ca2+ concentrations (5), we used cross-correlation sorting of the TF segments using three positions of Tpm on the surface of F-actin, namely canonical blocked (14), “apo” (15), and myosin (17) as reference structures. These structural states of Tpm were chosen because they apparently cover the entire swing of the Tpm cable on the surface of F-actin. The resultant frequency distribution (Fig. 1B) shows that the population of each class depends on both the activation state of the TF and on the presence of glutaraldehyde. Each class was independently reconstructed (SI Appendix, Fig. S3A). The corresponding 3D reconstructions of the apo classes and myosin classes derived from native TFs imaged at either activating or relaxing conditions were indistinguishable (SI Appendix, Fig. S3A). Therefore, these classes were combined (SI Appendix, Fig. S3A, black boxes) to yield better 3D maps and more reliable corresponding pseudoatomic models (Fig. 1C, c-closed and c-open). Similarly, apo and myosin classes derived from the cross-linked TFs imaged under activating or relaxing conditions were combined (SI Appendix, Fig. S3B, black boxes) and the resultant maps are shown in Fig. 1D (c-closed and c-open). Segments of TFs at pCa = 4 assigned to the canonical blocked structural class (∼9%) gave a poor 3D reconstruction that possessed Tpm in the apo structural state (SI Appendix, Fig. S3A, black X) and therefore were discarded. The position of the Tpm in the 3D reconstruction of native TF segments assigned to the canonical blocked structural class (∼18%) (Fig. 1C, c-blocked and Fig. 1E, blue surface) was inconsistent with the canonical blocked state (14) (Fig. 1E, red surface), due to ∼15° leftward azimuthal rotation from the canonical blocked state (Fig. 1E, red surface) toward the apo Tpm state (Fig. 1E, green surface). Because in this structural state Tpm has severe clashes with myosin head (see below in Fig. 2 A and E), we termed it a “cardiac-blocked” (c-blocked) state. The 3D map calculated from segments selected as being in the apo structural state (Fig. 1C, c-closed) yielded a perfect match with the proposed model of the apo state (15, 18). We found that the azimuthal position of Tpm on F-actin in the apo state (15, 18) was very similar to the azimuthal position of Tpm on F-actin in the closed structural state obtained by the negative staining protocol here (SI Appendix, Fig. S1C) and by others (3, 5). Therefore, we termed this state a “cardiac-closed” (c-closed) state. Finally, segments that yielded the best correlation with the reference having Tpm in the myosin state yielded a 3D reconstruction (Fig. 1C, c-open and Fig. 3F, magenta surface) where the Tpm cable was between the myosin (17) (Fig. 1F, cyan surface) and the closed (Fig. 1F, green surface) positions of Tpm on the surface of F-actin (Fig. 1F). In this structural state myosin binding sites were exposed (see below in Fig. 2 C and G). Therefore, we termed this structural state a “cardiac open” (c-open) state. Therefore, the cardiac TF open and myosin structural states are not the same and differ by 5° azimuthal rotation (Fig. 1F). To summarize, we found three structural states of Tpm on the surface of F-actin in the frozen hydrated cardiac TFs (c-blocked, c-closed, and c-open).
Fig. 1.

Three-dimensional reconstruction of frozen hydrated native and cross-linked cardiac TFs. (A) Electron micrograph of frozen hydrated cardiac TFs (pCa = 4) shows that some filaments possess well-defined Tn densities (black arrows), whereas others do not (white arrow). (Inset) Examples of segments of native and cross-linked (CL) TFs at low (pCa > 8) and high (pCa = 4) Ca2+ used for image analysis. Tn complexes are marked with black arrowheads. (B) Pseudoatomic models of the canonical-blocked (14), apo (15), and myosin (17) structural states of Tpm were used as reference structures in cross-correlation sorting of native and CL TFs at low (pCa > 8) and high (pCa = 4) Ca2+. (C and D) Three-dimensional reconstructions of native (C) and CL (D) cardiac TFs possessing Tpm in the c-blocked (blue ribbons), c-closed (green ribbons), or c-open (magenta ribbons) structural states. Density maps are shown as transparent gray surfaces, whereas actin subunits are tan ribbons. (E) Position of Tpm in the c-blocked state (blue surface) is compared with the previously observed canonical-blocked (14) (red surface) and apo (15) (green surface) positions of Tpm on F-actin. The top view shows an ∼10° swing of Tpm in the c-blocked state from its apo state. (F) Tpm position in the c-open state (magenta surface) is compared with the previously defined apo (15) (green surface) and myosin (17) (cyan surface) positions on F-actin. The top view shows that the c-open state is located between the apo and the myosin positions of Tpm on F-actin.
Fig. 2.

Superimposition of the c-blocked, c-closed, and c-open structural states with the Pi-release (Pi-R) (A–D) and rigor (E–H) states of myosin. (A–D) Tpm in either c-blocked (A) or c-closed (B) positions has a severe sterical clash with myosin in the Pi-R state (large and medium red arrowheads, respectively), whereas in the c-open state only a minor sterical hindrance is present (C, small red arrowhead). In the c-myosin state myosin and Tpm have no clashes (D). (E–H) In the c-blocked and c-closed structural states Tpm yielded an overwhelming sterical clash with the rigor-bound myosin head (E and F, large and medium red arrowheads, respectively). In the c-open state only a small portion of loop-4 of the myosin head clashes with Tpm (G, small red arrowhead). (H) Atomic model of rigor myosin–Tpm–F-actin complex (17).
Fig. 3.

Contacts of Tpm with F-actin in the c-blocked (A and B), c-closed (C and D), and c-open (E and F) states of the native (A, C, and E) and CL (B, D, and F) cardiac TFs. Residues of F-actin involved in the interaction with Tpm in native TF are marked in blue (A), green (D), and magenta (E), whereas actin residues presumably CL to Tpm by glutaraldehyde (B, D, and F) are marked in red. (G) In the native cardiac TF the Tpm interface on actin comprises residues located in SD3 of actin: 333/335 for the c-blocked state (blue spheres), 326/328/311 for the closed state (green spheres), and 307/311 for the c-open state (magenta spheres). (H) The transition of the inner Tpm strand, which interacts with F-actin, between the c-blocked (blue ribbons) and c-closed (green ribbons) state requires a 10° azimuthal movement of Tpm (black arrow), whereas the transition between the c-closed (green ribbons), c-open (magenta ribbons), and c-myosin (cyan ribbons) states involves rocking movement around a common anchor point (red arrow). (I) The outer Tpm strand, which is distal from F-actin, makes an ∼20° swing upon transition from the c-closed (green ribbons) to the c-myosin (cyan ribbons) structural state (red arrow). Actual density maps are shown as transparent gray surfaces, whereas actin molecules are tan ribbons.
To elucidate the influence of the cross-linking on the position of the Tpm on F-actin we compared 3D reconstructions of the c-blocked, c-closed, and c-open structural classes obtained from native TFs (Fig. 1C) and CL-TFs (Fig. 1D). For either c-closed or c-open classes we found very little difference in the position of Tpm in the native or cross-linked TFs (SI Appendix, Fig. S4A), whereas a small leftward swing of the Tpm cable was detected upon cross-linking in the c-blocked state (SI Appendix, Fig. S4A, black arrow). This leftward movement is consistent with the partial activation of the TFs upon cross-linking (SI Appendix, Fig. S2B) and will be discussed below.
Finally, we compared the frequencies of the three determined structural states (e.g., c-blocked, c-closed, and c-open) found within the native (Fig. 1B, Left) and cross-linked (Fig. 1B, Right) TFs under relaxed (Fig. 1B, light-gray bins) and activated (Fig. 1B, dark-gray bins) conditions. At low Ca2+ we found the majority of native TF segments (∼75%) in either c-blocked or c-closed structural states (Fig. 1B, Left). At high Ca2+ we did not detect any segments that yielded a reasonable 3D reconstruction of the c-blocked state (see above). At the same time high Ca2+ gave an ∼1.8-fold boost in the frequency of the c-open structural state compared with the relaxing conditions (Fig. 1B, Left). We did not find a significant difference between the frequency distributions of the three structural states of the native and cross-linked TFs at relaxing conditions (Fig. 1B, compare left and right panels). In contrast to that, we found that at activating conditions ∼15% of CL-TFs were found in the c-blocked state, whereas in the absence of glutaraldehyde the c-blocked state was nonexistent (Fig. 1B). This suggests that at high Ca2+ Tpm is not rigidly trapped in either c-closed or c-open structural states, but rather may still fluctuate. Therefore, a small fraction of the TFs in the c-blocked structural state can be trapped by glutaraldehyde so that the frequency of this otherwise rare structural state is enhanced over the time of the cross-linking. Because in the c-blocked state Tpm interferes with the actomyosin interactions (see below), elevated levels of the c-blocked state explain the slightly slower kinetics of product dissociation from myosin-S1-mdADP-Pi after cross-linking with glutaraldehyde (SI Appendix, Fig. S2B).
The Interface Between Tpm and F-Actin Suggests the Mechanism of Azimuthal Movement of Tpm upon TF Activation.
We examined the contacts of Tpm with F-actin in the c-blocked, c-closed, and c-open structural states in the presence and absence of glutaraldehyde. Because Tpm is comprised of seven pseudorepeats that overlap on the surface of F-actin, helical averaging eliminates any information on the location of these pseudorepeat units on the surface of F-actin. Therefore, we replaced actual Tpm residues with a corresponding pseudoatomic model. Relatively high resolution in our cryoelectron microscopy (cryoEM) maps allowed us to determine which actin residues may be involved in the interaction with the Tpm (Fig. 3). In the 3D map of the native TFs in the c-blocked state we found a bridge of density that involves Arg335 and Pro333 (Fig. 3A, blue spheres). In the 3D map of the same structural state derived from the CL-TFs the bridge of density formed by the Pro333/Arg335 contact (Fig. 3B, blue spheres) was more prominent, suggesting that Lys336 located next to Pro333 and Arg335 was cross-linked to Tpm by glutaraldehyde (Fig. 3B, red spheres). We also found a contact that was absent in the native TFs presumably formed by Lys328 cross-linked to the Tpm cable (Fig. 3B, red spheres). In the c-closed state of the native TFs we found two contacts between the Tpm and F-actin––one involved Asp311, and the other comprised Lys326 and Lys328 (Fig. 3C, green spheres). In the 3D reconstruction of the c-closed state derived from the CL-TFs we found two bridges of density between Tpm and F-actin––one involved Lys328 (Fig. 3D, green spheres), whereas the other contact was apparently produced by the Lys238 cross-linked to Tpm (Fig. 3D, red spheres). The comparison of the 3D reconstructions of the c-open state in the absence and presence of glutaraldehyde revealed a significant difference in the contacts between the Tpm and F-actin in the native and cross-linked TFs (Fig. 3 E and F). In the native TFs we found a bridge of density presumably comprised of Asp311 and Pro307 interacting with Tpm (Fig. 3E, magenta spheres). A set of very different contacts seemingly involving Lys238, Lys326, and Lys328 cross-linked to the Tpm cable was found in the presence of glutaraldehyde (Fig. 3F, red spheres).
The difference in the interface between the native and the CL TFs explains a leftward shift of Tpm in the c-blocked state in the presence of glutaraldehyde (SI Appendix, Fig. S4A, black arrow). Presumably, a contact introduced by glutaraldehyde between Lys328 and Tpm pulls Tpm leftward. Such a movement of Tpm is consistent with the gradual activation of the TF by glutaraldehyde at relaxing conditions (SI Appendix, Fig. S2B). Despite a significant difference in the interface between the native and the cross-linked TFs (Fig. 3 C–F) the positions of the Tpm cable on the surface of F-actin in either c-closed or c-open structural states are very similar (SI Appendix, Fig. S4A). The only rational explanation for this finding is that despite the difference in the contacts between Tpm and F-actin in the native and CL TFs, the Tn complex oversees the position of the Tpm cable on the surface of F-actin.
The actin residues that we found to be involved in the interaction with Tpm are in good agreement with previously published data (14, 15, 17). In the c-blocked state Pro333 and Arg335 are at the interface with Tpm, confirming computational modeling results (14). In the c-closed structural state Lys326, Lys328, and Asp311 interaction with Tpm is in excellent agreement with the structural data obtained by cryoEM (15). Asp311, which is at the Tpm interface with F-actin in the c-open state, has been found to bind to Tpm in cryoEM structure of myosin–Tpm–F-actin complex (17). Because we have not found a canonical-blocked structural state (14) in our frozen hydrated TFs, Asp25, Arg28, and Arg147 are not involved in Tpm binding to F-actin in the native cardiac TF.
Next, we analyzed the movement of Tpm cable between the three observed structural states. Each actin protomer within the TF interacts with one of the two Tpm ɑ-helical strands (the “inner” strand), whereas the other strand is distal from the actin surface (the “outer” strand). First, we calculated the trajectory of the Tmp cable in the c-blocked → c-closed transition (Fig. 3H, black arrow; Movie S1) which appeared to be an ∼10° leftward azimuthal movement of both inner and outer Tpm strands. Such a movement can be achieved by either an azimuthal rotation (roll) or by an upward movement (shift) of Tpm on the surface of F-actin. In contrast to the c-blocked → c-closed Tpm movement, the trajectory of the inner and outer strands of Tpm upon c-closed → c-open and c-open → myosin transitions were different (Movie S1). First, we traced the movement of the inner Tpm strand upon the c-blocked → c-open → myosin transition, which was found to be a rocking movement around the anchor point (Fig. 3H, red arrow; SI Appendix, Movie S1) originally predicted by Sousa et al. (18). Next, we compared the successive movement of the outer Tpm ɑ-helical strand. We found that upon c-closed → c-open → myosin transition, the rocking movement of the inner Tpm strand around the anchor point (Fig. 3H, red arrow) results in a significant ∼20° azimuthal movement of the distal outer Tpm strand (Fig. 3I, red arrow). Therefore, the minor repositioning of the inner Tpm strand on the surface of F-actin is greatly amplified by the dramatic movement of the outer strand.
The position of the actin residues involved in the interaction with Tpm forms a compact interface on the surface of subdomain 3 (SD3) of the actin molecule (Fig. 3G). Interestingly, Asp311 is involved in all of the structural states of interaction of actin with Tpm (e.g., c-closed, c-open, and myosin), except the c-blocked structural state. This suggests that residue 311 comprises the interface for a rocking movement of Tpm on F-actin. The importance of the Asp311 for the Tpm interaction with F-actin has been previously reported (19, 20).
The Three Structural States of the TF Revealed by cryoEM Suggest a Novel Mechanism of TF Activation.
Next, we sought to evaluate whether the c-blocked, c-closed, and c-open structural states interfere with the actomyosin interactions. It has been suggested that loop-2 of myosin initiates weak binding of myosin to the TF (21, 22). Subsequent binding of the lower L50 domain of myosin to F-actin through the helix-turn-helix motif (23) promotes Pi release (24) and completes the initial strong binding of myosin to F-actin. We approximated the initial strong binding mode of myosin using the same approach described by von der Ecken et al. (23) (SI Appendix, Fig. S5). Superimposition of the c-blocked, c-closed, and c-open structural states with myosin in the Pi-R state bound to F-actin (Fig. 2) revealed that Tpm in either c-blocked (Fig. 2A) or c-closed (Fig. 2B) positions had severe sterical clashes with myosin (Fig. 2 A and B, red arrowheads). At the same time, in the c-open state only a minor sterical hindrance with the tip of the myosin loop-4 was observed (Fig. 2C, small red arrowhead). Next, we sought to evaluate whether the c-blocked, c-closed, and c-open structural states interfere with the rigor binding of myosin (Movie S2). We therefore superimposed the pseudoatomic model of rigor actomyosin complex (23) with the models of the three structural states of the native cardiac TF (Fig. 2 E–H). In the c-blocked and the c-closed structural states Tpm yielded overwhelming sterical clashes with the myosin head (Fig. 2 E and F, red arrowheads). In the c-open state we found that only a small portion of loop-4 of myosin clashed with Tpm (Fig. 2G, small red arrowhead). Therefore, we conclude that either c-closed or c-blocked structural states of Tpm should inhibit actomyosin interactions. In either Pi-R or the rigor state myosin has no clashes with Tpm in the c-open structural state, except the tip of the myosin loop-4 which is very minor. Therefore, Tpm in the c-open state is permissive for both the initial and the rigor myosin binding. In the atomic model of rigor actomyosin Asp378 of loop-4 makes a contact with Lys325 and Lys327 of actin involved in the interaction of actin with Tpm in the c-closed structural state (23). In the c-open structural state Lys325 and Lys327 of actin are free to interact with myosin Asp378 because they are not making contact with the Tpm cable anymore. Presumably, the formation of this contact between actin and myosin moves Tpm from the c-open to the myosin state (∼5° swing) and completes the proposed isomerization from the weak to the strong actomyosin bound state (4).
Stopped-Flow Kinetic Measurements of Product Dissociation from TF-Myosin-ADP-Pi.
To evaluate whether enhanced frequency of the c-open state leads to the enhanced activation of the cardiac TF we used double-mixing stopped flow to determine the effect of Ca2+ on the rates of Pi dissociation from the cardiac TF-myosin-ADP-Pi under conditions similar to those used in cryoEM experiments (SI Appendix, Figs. S6 and S7). These data show that at physiological ionic strength cardiac TF are activated to ∼70% of the maximal level by Ca2+ in the absence of rigor-bound myosin, so that there is a relatively small additional ∼30% activation of the TF by addition of rigor-bound myosin. Therefore, the enhanced frequency of the c-open structural state at high Ca2+ (Fig. 1B) corroborates with the kinetic data and provides a structural framework for enhanced activation of the cardiac TF by Ca2+.
Discussion
The mechanism of the TF activation by Ca2+ has been studied for decades, but the lack of a high-resolution structure of the TF under relaxing and activating conditions is one of the main drawbacks in our understanding of the mechanism of muscle contraction. Almost all of the structural data on the regulation of the TF were obtained using negative staining (3, 5), which is incapable of yielding high-resolution density maps. Here we report high-resolution structures of the frozen hydrated native cardiac TF under relaxing and activating conditions. We found that in cardiac TFs Tpm can exist in three structural states on the surface of F-actin, two of which, namely c-blocked and c-open, have not been observed in negatively stained TFs. Importantly, cardiac-open (c-open) structural state is different from the canonical-open or myosin state, which represents the position of Tpm in the presence of rigor-bound myosin (Fig. 1F). In the c-open state the myosin binding sites are fully accessible except for the tip of myosin loop-4 (Fig. 2 C and G). Therefore, we distinguish between the c-open and the myosin states of Tpm in cardiac TF.
In the original model derived from the 3D reconstructions of the negatively stained TFs (Fig. 4A), the Ca2+-induced swing of the Tpm cable upon activation of the TF is ∼25°, whereas rigor-bound myosin induces an additional ∼10° azimuthal rotation of Tpm (5). Therefore, the overall swing of the Tpm cable is predicted to be ∼35°. Our structural data derived from the frozen hydrated native cardiac TFs show that the maximal swing of the Tpm upon Ca2+-induced activation of the TF is ∼15°, whereas rigor-bound myosin adds an additional ∼5° azimuthal rotation of Tpm (Fig. 4B). Intensities measured from the low-angle X-ray fiber diagrams obtained from rabbit psoas muscle fibers showed ∼15° azimuthal movement of Tpm upon Ca2+-induced muscle activation, and ∼7° movement upon binding of active cross-bridges (6). Therefore, cryoEM data are in excellent agreement with the X-ray fiber diffraction experiments. An ∼2° difference in the overall Tpm swing between our data obtained from cardiac TFs and X-ray fiber diagrams obtained from skeletal muscles may reflect the difference in the activating potential between cardiac and skeletal striated muscles (10).
Fig. 4.

Model for the Ca2+-dependent activation of the TF. (A) In the canonical model derived from the negatively stained TFs the swing of the Tpm cable from the canonical-blocked to the closed structural state upon Ca2+-induced activation of the TF is ∼25° (red arrow), whereas rigor-bound myosin adds an additional ∼10° azimuthal rotation of Tpm (black arrow) (5). (B) CryoEM-based model for the activation of the cardiac TF shows an ∼15° swing of Tpm upon Ca2+-induced activation of the cardiac TF (red arrow), whereas rigor-bound myosin adds an additional ∼5° azimuthal rotation of the Tpm cable toward subdomain 4 of actin (black arrow). (C–I) Comparison of the canonical model for the TF activation (C–F) with the model for cardiac TF activation (G–I). (C) At low Ca2+ Tpm in the canonical-blocked state (red) blocks myosin heads (yellow) binding to F-actin (red arrows). (D) Upon Ca2+-induced activation of the TF, Tpm moves to the closed structural state (green) partially exposing myosin binding sites so that a few myosin molecules (yellow) can bind to the TF (green arrow). (E) Strongly bound myosin heads locally activate the TF by switching Tpm from its closed (green) to myosin (cyan) state. (F) The activated myosin Tpm state cooperatively propagates along the TF. (G) At low Ca2+ Tpm in the cardiac TF is in either c-blocked (blue) or c-closed (green) inhibitive states which block myosin (yellow) binding to F-actin (red arrows). (H) Upon Ca2+-induced activation of the cardiac TF Tpm occupies either c-closed (green) or c-open structural state (magenta). In c-open structural state myosin binding sites are almost fully exposed and myosin molecules (yellow) freely bind to the TF (green arrows). (I) Strongly bound myosin heads shift Tpm further from either c-closed or c-open into the myosin (cyan) structural state.
It has been shown that negative staining biases the position of Tpm in the Tpm–F-actin complex so that instead of being in the apo state, as observed in frozen hydrated samples, Tpm is found in the canonical-blocked state (15). We show here (SI Appendix, Fig. S1) that the native cardiac TFs behave similarly to the Tpm–F-actin complex upon negative staining and that cross-linking by glutaraldehyde prevents the movement of Tpm into the canonical-blocked state. Consistently, our kinetic measurements (SI Appendix, Fig. S2B) show that the canonical-blocked state is not required per se for the cardiac TF inhibition, because cross-linked cardiac TFs that do not possess the canonical-blocked state in either negatively stained (SI Appendix, Fig. S1 D and E) or frozen hydrated (Fig. 1D) samples retain the inhibited state in the ATPase assay. Therefore, we conclude that in native cardiac TFs the canonical-blocked structural state is induced by the negative staining protocol.
At either relaxing or activating conditions Tpm is not fixed in one particular state (Fig. 1B). At relaxing conditions ∼75% of the TFs are in the inhibited c-blocked and c-closed structural states, whereas ∼25% TF segments are in the activated c-open state. This is consistent with the diversity of the structural composition of the TF found using biochemical approaches (25) and explains how at low Ca2+ conditions rigor myosin can bind and activate the TF (26, 27). The frequency of the activated c-open state at low Ca2+ is higher than expected from the kinetic measurements (SI Appendix, Figs. S6 and S7) and is similar to what has been observed in the analysis of negatively stained TFs (5). This discrepancy between kinetic and EM data has yet to be resolved. At activating conditions TFs are comprised of the permissive c-open and inhibitory c-closed structural states (Fig. 1B). Presumably, at activating conditions regions of the TFs in the inhibitory c-closed structural state become fully activated by the rigor-bound myosins through a positive cooperativity in cross-bridge binding (28).
The existing steric blocking model of muscle regulation holds that at relaxing conditions Tpm in the canonical-blocked state blocks myosin binding sites on F-actin (Fig. 4C), whereas at activating conditions the swing of Tpm from the canonical-blocked to the closed state partially exposes myosin binding sites on F-actin, so that binding of a small number of rigor myosins (Fig. 4D, green arrow) fully activates the TF (Fig. 4 E and F). Therefore, the existing model advocates for a two-step activation of the TF which relies on a subsequent binding of myosin to the Ca2+-activated TF. This model is in agreement with the observed activation of the skeletal TFs where Ca2+ on its own activates the TF by ∼20% and rigor-bound myosin is crucial in achieving a full TF activation (8, 9). In contrast to the skeletal TF, Ca2+ induces ∼70% activation of the cardiac TF (SI Appendix, Figs. S6 and S7) and therefore the role of the rigor-bound cross-bridges is not as significant. In our model for the cardiac TF activation Ca2+-enhanced c-open structural state allows immediate binding of a large number of myosin cross-bridges (Fig. 4H), which complete the activation of the cardiac TF (Fig. 4I). Therefore, in the model for the cardiac TF regulation, the first step of TF activation by Ca2+ overwhelms the second myosin-dependent activation step. Our data provide a structural framework for the enhanced contribution of Ca2+ to the overall activation of the cardiac TF in comparison with the skeletal TF. Because there are significant differences between the TF proteins in cardiac muscle and skeletal muscle (11, 12, 29, 30), it is not surprising that the regulation of the skeletal and cardiac TFs is different. Analysis of frozen hydrated skeletal TFs is required to mechanistically explain the difference in the activation of skeletal and cardiac striated muscles.
Materials and Methods
Proteins and Buffers.
Native porcine cardiac thin filaments were purified as described previously (10). A-buffer was used for cryoEM experiments: 50 mM KAc, 10 mM Mops, 3 mM MgCl2, pH 7.0.
Cross-Linking Experiments.
TF in A-buffer in activating [0.2 mM Ca2+] or relaxing [2 mM EGTA] conditions were incubated with 0.25% (vol) of glutaraldehyde for 5 min at 20 °C. To stop reaction 1 M Tris⋅HCl, pH 7.0 buffer was added into the mixture and allowed to react for an additional 5 min.
Kinetic Measurements.
TF activation of product dissociation from myosin-ADP-Pi by TFs was measured using double-mixing stopped-flow fluorescence as described previously (10) but under the conditions similar to those used in the cryoEM experiments (SI Appendix).
CryoEM.
Sample preparation.
Samples (1.8 µL) of [1 μM] native or cross-linked TFs in A-buffer in activating [0.2 mM Ca2+] or relaxing [2 mM EGTA] conditions were applied to lacey carbon grids, blotted with Whatman #1 filter paper for 3 s, and vitrified in a Vitrobot Mark IV (FEI, Inc.).
Three-dimensional reconstruction.
Samples were imaged with a Titan Krios at 300 keV using the Falcon II direct electron detector. The SPIDER software package (31) was used for image processing; in addition, CTFFIND3 software (32) was used to determine the defocus values in the micrographs. The EMAN package (33) was used to extract filament images from micrographs. Fourier shell correlation was used to estimate the resolution (SI Appendix, Figs. S2B and S3B). Image analysis is explained in detail in SI Appendix.
Modeling.
The model of the Tpm–F-actin complex extracted from the myosin-Tpm-F-actin complex (23) (Protein Data Bank ID code 5JLH) was used as the starting model for the refinement against the cryoEM density maps. The program DireX (34) was used for the model refinement. The density maps were masked around the rigid-body docked starting model with a radius of 8 Å using a smooth edge. H bonds present in the starting structure were restrained during the refinement. All models were refined for 50 steps against a target map filtered to 8 Å, whereas the Fourier interval 6–8 Å was used for cross-validation to prevent overfitting. The DireX refinement was followed by 50 steps of minimization with PHENIX (35).
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
We thank Kelly Dryden for assistance with the microscopy at University of Virginia. This work was supported by American Heart Association Grant in Aid 560851 (V.E.G.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank and Electron Microscopy Data Bank (accession nos. 5NOG, EMD-3665, 5NOL, EMD-3667, 5NOJ, EMD-3666).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1700868114/-/DCSupplemental.
References
- 1.Brown JH, Cohen C. Regulation of muscle contraction by tropomyosin and troponin: How structure illuminates function. Adv Protein Chem. 2005;71:121–159. doi: 10.1016/S0065-3233(04)71004-9. [DOI] [PubMed] [Google Scholar]
- 2.Spudich JA, Huxley HE, Finch JT. Regulation of skeletal muscle contraction. II. Structural studies of the interaction of the tropomyosin-troponin complex with actin. J Mol Biol. 1972;72:619–632. doi: 10.1016/0022-2836(72)90180-5. [DOI] [PubMed] [Google Scholar]
- 3.Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. J Mol Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- 4.McKillop DFA, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: Evidence for three states of the thin filament. Biophys J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pirani A, et al. Single particle analysis of relaxed and activated muscle thin filaments. J Mol Biol. 2005;346:761–772. doi: 10.1016/j.jmb.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 6.Poole KJ, et al. A comparison of muscle thin filament models obtained from electron microscopy reconstructions and low-angle X-ray fibre diagrams from non-overlap muscle. J Struct Biol. 2006;155:273–284. doi: 10.1016/j.jsb.2006.02.020. [DOI] [PubMed] [Google Scholar]
- 7.Lehman W, Orzechowski M, Li XE, Fischer S, Raunser S. Gestalt-binding of tropomyosin on actin during thin filament activation. J Muscle Res Cell Motil. 2013;34:155–163. doi: 10.1007/s10974-013-9342-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heeley DH, Belknap B, White HD. Mechanism of regulation of phosphate dissociation from actomyosin-ADP-Pi by thin filament proteins. Proc Natl Acad Sci USA. 2002;99:16731–16736. doi: 10.1073/pnas.252236399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heeley DH, Belknap B, White HD. Maximal activation of skeletal muscle thin filaments requires both rigor myosin S1 and calcium. J Biol Chem. 2006;281:668–676. doi: 10.1074/jbc.M505549200. [DOI] [PubMed] [Google Scholar]
- 10.Houmeida A, Heeley DH, Belknap B, White HD. Mechanism of regulation of native cardiac muscle thin filaments by rigor cardiac myosin-S1 and calcium. J Biol Chem. 2010;285:32760–32769. doi: 10.1074/jbc.M109.098228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wilkinson JM, Grand RJ. Comparison of amino acid sequence of troponin I from different striated muscles. Nature. 1978;271:31–35. doi: 10.1038/271031a0. [DOI] [PubMed] [Google Scholar]
- 12.van Eerd JP, Takahshi K. Determination of the complete amino acid sequence of bovine cardiac troponin C. Biochemistry. 1976;15:1171–1180. doi: 10.1021/bi00650a033. [DOI] [PubMed] [Google Scholar]
- 13.Pearlstone JR, Carpenter MR, Smillie LB. Amino acid sequence of rabbit cardiac troponin T. J Biol Chem. 1986;261:16795–16810. [PubMed] [Google Scholar]
- 14.Li XE, et al. Tropomyosin position on F-actin revealed by EM reconstruction and computational chemistry. Biophys J. 2011;100:1005–1013. doi: 10.1016/j.bpj.2010.12.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.von der Ecken J, et al. Structure of the F-actin-tropomyosin complex. Nature. 2015;519:114–117. doi: 10.1038/nature14033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Egelman EH. A robust algorithm for the reconstruction of helical filaments using single-particle methods. Ultramicroscopy. 2000;85:225–234. doi: 10.1016/s0304-3991(00)00062-0. [DOI] [PubMed] [Google Scholar]
- 17.Behrmann E, et al. Structure of the rigor actin-tropomyosin-myosin complex. Cell. 2012;150:327–338. doi: 10.1016/j.cell.2012.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sousa DR, Stagg SM, Stroupe ME. Cryo-EM structures of the actin:tropomyosin filament reveal the mechanism for the transition from C- to M-state. J Mol Biol. 2013;425:4544–4555. doi: 10.1016/j.jmb.2013.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Debold EP, et al. Human actin mutations associated with hypertrophic and dilated cardiomyopathies demonstrate distinct thin filament regulatory properties in vitro. J Mol Cell Cardiol. 2010;48:286–292. doi: 10.1016/j.yjmcc.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerson JH, Bobkova E, Homsher E, Reisler E. Role of residues 311/312 in actin-tropomyosin interaction. In vitro motility study using yeast actin mutant e311a/r312a. J Biol Chem. 1999;274:17545–17550. doi: 10.1074/jbc.274.25.17545. [DOI] [PubMed] [Google Scholar]
- 21.Joel PB, Trybus KM, Sweeney HL. Two conserved lysines at the 50/20-kDa junction of myosin are necessary for triggering actin activation. J Biol Chem. 2001;276:2998–3003. doi: 10.1074/jbc.M006930200. [DOI] [PubMed] [Google Scholar]
- 22.Murphy CT, Spudich JA. The sequence of the myosin 50-20K loop affects Myosin’s affinity for actin throughout the actin-myosin ATPase cycle and its maximum ATPase activity. Biochemistry. 1999;38:3785–3792. doi: 10.1021/bi9826815. [DOI] [PubMed] [Google Scholar]
- 23.von der Ecken J, Heissler SM, Pathan-Chhatbar S, Manstein DJ, Raunser S. Cryo-EM structure of a human cytoplasmic actomyosin complex at near-atomic resolution. Nature. 2016;534:724–728. doi: 10.1038/nature18295. [DOI] [PubMed] [Google Scholar]
- 24.Llinas P, et al. How actin initiates the motor activity of Myosin. Dev Cell. 2015;33:401–412. doi: 10.1016/j.devcel.2015.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Maytum R, Westerdorf B, Jaquet K, Geeves MA. Differential regulation of the actomyosin interaction by skeletal and cardiac troponin isoforms. J Biol Chem. 2003;278:6696–6701. doi: 10.1074/jbc.M210690200. [DOI] [PubMed] [Google Scholar]
- 26.Swartz DR, Moss RL. Influence of a strong-binding myosin analogue on calcium-sensitive mechanical properties of skinned skeletal muscle fibers. J Biol Chem. 1992;267:20497–20506. [PubMed] [Google Scholar]
- 27.Hill TL, Eisenberg E, Greene L. Theoretical model for the cooperative equilibrium binding of myosin subfragment 1 to the actin-troponin-tropomyosin complex. Proc Natl Acad Sci USA. 1980;77:3186–3190. doi: 10.1073/pnas.77.6.3186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bremel RD, Weber A. Cooperation within actin filament in vertebrate skeletal muscle. Nat New Biol. 1972;238:97–101. doi: 10.1038/newbio238097a0. [DOI] [PubMed] [Google Scholar]
- 29.Solaro RJ, Moir AJ, Perry SV. Phosphorylation of troponin I and the inotropic effect of adrenaline in the perfused rabbit heart. Nature. 1976;262:615–617. doi: 10.1038/262615a0. [DOI] [PubMed] [Google Scholar]
- 30.Collins JH, Potter JD, Horn MJ, Wilshire G, Jackman N. The amino acid sequence of rabbit skeletal muscle troponin C: Gene replication and homology with calcium-binding proteins from carp and hake muscle. FEBS Lett. 1973;36:268–272. doi: 10.1016/0014-5793(73)80388-6. [DOI] [PubMed] [Google Scholar]
- 31.Frank J, et al. SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 32.Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 33.Ludtke SJ, Baldwin PR, Chiu W. EMAN: Semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- 34.Schröder GF, Brunger AT, Levitt M. Combining efficient conformational sampling with a deformable elastic network model facilitates structure refinement at low resolution. Structure. 2007;15:1630–1641. doi: 10.1016/j.str.2007.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Adams PD, et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.