

Abstract
D-penicillamine (DPEN), a copper chelator, has been used in the treatment of Wilson's disease, cystinuria, and rheumatoid arthritis. Recent evidence suggests that DPEN in combination with biologically relevant copper (Cu) concentrations generates H2O2 in cancer cell cultures, but the effects of this on cancer cell responses to ionizing radiation and chemotherapy are unknown. Increased steady-state levels of H2O2 were detected in MB231 breast and H1299 lung cancer cells following treatment with DPEN (100 μM) and copper sulfate (15 μM). Clonogenic survival demonstrated that DPEN-induced cancer cell toxicity was dependent on Cu and was significantly enhanced by depletion of glutathione [using buthionine sulfoximine (BSO)] as well as inhibition of thioredoxin reductase [using Auranofin (Au)] prior to exposure. Treatment with catalase inhibited DPEN toxicity confirming H2O2 as the toxic species. Furthermore, pretreating cancer cells with iron sucrose enhanced DPEN toxicity while treating with deferoxamine, an Fe chelator that inhibits redox cycling, inhibited DPEN toxicity. Importantly, DPEN also demonstrated selective toxicity in human breast and lung cancer cells, relative to normal untransformed human lung or mammary epithelial cells and enhanced cancer cell killing when combined with ionizing radiation or carboplatin. Consistent with the selective cancer cell toxicity, normal untransformed human lung epithelial cells had significantly lower labile iron pools than lung cancer cells. These results support the hypothesis that DPEN mediates selective cancer cell killing as well as radio-chemo-sensitization by a mechanism involving metal ion catalyzed H2O2-mediated oxidative stress and suggest that DPEN could be repurposed as an adjuvant in conventional cancer therapy.
Keywords: D-penicillamine, transition metal ions, breast cancer, lung cancer, oxidative metabolism, hydrogen peroxide
Graphical abstract
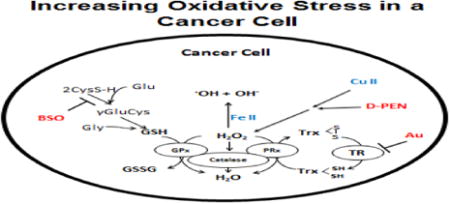
Introduction
The most successful adjuvants to cancer therapy are selectively cytotoxic to malignant cells while sparing normal tissues. Recently there has been a great deal of interest in repurposing redox active drugs that have been used in humans as adjuvants to cancer therapy based on fundamental differences in oxidative metabolism between normal and cancer cells leading to increased steady-state levels of O2•- and H2O2 [1-3]. This paradigm of drug repurposing is based on the in depth assessment of biochemical differences between tumor and normal cell redox metabolism targetable for therapy development. Recent evidence continues to support the hypothesis that cancer cells have aberrant mitochondrial function, leading to increased steady state levels of O2•- and H2O2, compared to normal cells [4-6]. Because of the elevated baseline levels of reactive oxygen species (ROS) in cancer cells, it has also been proposed that using agents that further increase ROS levels will lead to selective cell death in cancer cells while leaving normal cells, relatively unaffected [1, 6-8].
Recent data also suggests that metabolism of redox active metal ions such as Cu and Fe is also dysregulated in tumor versus normal cells. For example, breast and lung cancer patients contain elevated serum copper levels when compared to cancer-free patients [8]. Breast and lung tumors also contain markedly increased copper when compared to their matched normal tissue [9-11]. Labile iron concentrations are also increased in both serum and tumor tissue from cancer patients [12]. Currently, there is also promising research focusing on cancer treatment strategies based on copper and iron chelation [13, 14].
D-penicillamine (DPEN) is an aminothiol and copper chelator used in the treatment of Wilson's disease, cystinuria, and rheumatoid arthritis. When DPEN chelates Cu(II) in either the labile or ceruloplasmin-bound form, it is reduced to Cu(I) and can generate ROS via one electron reductions of O2 to form O2•- that rapidly dismutes to form H2O2 [15] [16]. In addition, increased levels of O2•- in cancer cells could lead to increased labile Fe(II) that could react with H2O2 to form •OH, creating a selectively cytotoxic environment in cancer cells treated with DPEN [17].
In the current report increased steady-state levels of H2O2 were quantified in lung and breast cancer cells in vitro when treated with DPEN and physiologically relevant concentrations of copper. DPEN also induced clonogenic cell killing in cancer cells that was inhibited by catalase demonstrating the H2O2-dependence of this biological response. DPEN-induced clonogenic cell killing was further enhanced via inhibition hydroperoxide metabolism in both lung and breast cancer cells using the thioredoxin reductase inhibitor auranofin (Au) and the glutathione synthesis inhibitor buthionine sulfoximine (BSO). In addition, the importance of redox active Fe in DPEN-induced effects was apparent when cancer cells were sensitized to DPEN-induced killing by pre-treatment with Fe-sucrose and protected by deferoxamine. Importantly the toxicity of DPEN was significantly greater in human breast and lung cancer cells as compared to non-immortalized primary human mammary and bronchial epithelial cells possibly because of the higher baseline labile iron pools in cancer cells compared to normal cells. DPEN also enhanced the cytotoxicity of carboplatin and radiation in human cancer cells. These results support the hypothesis that DPEN-induced cytotoxicity and radio-chemosensitization are mediated by H2O2 and redox active metals as well as supporting the speculation that DPEN could be repurposed as an adjuvant in cancer therapy.
Methods
Cells and Culture Conditions
MB231 human breast carcinoma cells, H1299 human lung carcinoma cells and H292 human lung carcinoma cells were obtained from ATCC and maintained in RPMI 1640 (Mediatec) with 10% fetal bovine serum (FBS; HyClone). Non-immortalized primary human bronchial epithelial cells (HBEpC) were obtained and maintained as suggested in bronchial/tracheal epithelial cell growth media from Cell Applications, Inc. Non-immortalized primary human mammary epithelial cells (HMEC) were obtained and maintained in mammary epithelial growth media as suggested by the vendor (Lonza). Experiments with non-transformed cells (HBEpC and HMEC) were performed between 3-10 population doublings from receipt of the cells from the company. In our experience, the doubling time of the cells did not change until after 10 population doublings. These cells maintained a healthy replicative lifespan in culture which was verified in each experiment by measuring plating efficiency.
Clonogenic Cell Survival Assay
120,000 H1299 cells, 125,000 MB231 or 150,000 H292 cells were plated in 60-mm dishes in 21% O2 and 37°C and treated during exponential growth using D-penicillamine (DPEN; Sigma-Aldrich; 100 μM), auranofin (Au; Enzo Life Science; 0.5 μM), buthionine sulfoximine (BSO; Sigma-Aldrich; 1000 μM) bovine catalase (cat; Sigma-Aldrich; 50 U/mL), and/or carboplatin (Hospira, Inc.) for 24 hours. Unless otherwise stated, all cell groups were dosed with copper (II) sulfate pentahydrate (CuSO4; Sigma-Aldrich; 15 μM) at t0. In experiments using deferoxamine (DFO; Sigma-Aldrich; 40 μM) and iron sucrose (Fe sucrose; Venofer; 250 μM) indicated dishes were pretreated for 2 hours. Dishes were washed twice with media to remove extracellular DFO and iron sucrose, and CuSO4 was reapplied to dishes after washing prior to DPEN exposure for 2 hours. In experiments with radiation, cells were irradiated with a dose of 1 Gy, 2 Gy, or 4 Gy (dose rate, 0.365 Gy/min), using a 37Cs source (JL Shepherd, San Fernando, CA). Cells were returned to the incubator for 2 hours followed by analysis with the clonogenic survival assay as described [18]. Media containing any floating cells was removed from the treatment dish. Attached cells were trypsinized with 0.25% trypsin-EDTA and trypsin was inactivated by re-combining cells with the media from the same treatment dish containing 10% FBS. Samples were centrifuged, re-suspended in fresh media and the resulting total cell population counted using a Beckman Coulter Counter. The cells were then plated 60-mm dishes at a variety of densities ranging from 200-2000 cells per dish. Clones were grown for 10-12 days in complete media with 0.1% gentamycin. Cells were fixed with 70% ethanol, stained with Coomassie blue, and colonies containing > 50 cells were counted. The treatment groups for each cell line were normalized to the control group. The survival analysis was done using a minimum of 3 cloning dishes per experimental condition, and the experiments were repeated a minimum of 3 times on separate occasions.
Calcein Labile Iron Detection Assay
To visualize the intracellular labile iron pool (LIP), the fluorescent dye Calcein-AM (Molecular Probes) was used as described previously with modifications [19]. The method for assessing LIP was based on the measurement of Calcein florescence in cells in the presence and absence of the iron chelator, 100 μM 2′,2′-bipyridyl (BIP-Sigma) [19]. 150,000 H292 and 250,000 HBEpC cells were plated in 60 mm dishes in their respective media and grown for 3 days at 4% O2. Media was changed to HBEpC media in all dishes then treated with either 250 μM Fe sucrose (positive control) or 250 μM DFO (negative control) for 4 hours, washed with PBS, trypsinized, counted and 106 cells each placed in 15 mL tubes. After centrifuging at 1200 rpm for 5 minutes, cells were resuspended in PBS containing 500 nM Calcein-AM incubate at 37°C for 15 minutes. A background sample was created by adding only PBS. Calcein-AM was then removed by centrifugation, and cells were resuspended in 1 mL PBS and each sample was split into two flow tubes and 2,2 bipyridyl was added to one sample in each treatment group. BIP-treated samples were run at least 15 minutes after the addition to ensure adequate time for iron chelation. Cells were analyzed using FACS on LSR Violet (Becton Dickinson LSR II) with 515/15 emission filter.
Tumor versus Normal cells Clonogenic Survival Assays
100,000 H1299T cells and 250,000 HBEC cells or 125,000 MB231 cells and 300,000 HMEC cells were plated on 60-mm dishes as before for 72 hours at 4% O2 and 37°C. All cells were then washed with warm PBS and placed in either complete Bronchial/Tracheal Epithelial Cell Basal Media (H1299T and HBEC cells) or Mammary Epithelial Cell Basal Medium (HMEC and MB231 cells)(Cell Applications, Inc.). Cells were then treated with 15 μM CuSO4 and 100 μM DPEN for 3 hours, 50 μM H2O2 for 3 hours, or 10 μM carboplatin for 24. A clonogenic cell survival assay was performed as above with the following exceptions. All cells were trypsinized using trypsin/EDTA followed by trypsin neutralizing solution (Cell Applications, Inc) instead of FBS containing media. HBEC cloning dishes contained 100,000 lethally irradiated (30 Gy) H1299T feeder cells to encourage colony formation. Lethally irradiated feeder cells were also plated in dishes without test cells to confirm the lack of colony formation.
Peroxy Orange-1 H2O2 Production Flow Cytometry Assay
Exponentially growing MB231 cells were assayed for peroxide content using PeroxyOrange-1 (PO-1; BD Biosciences) as previously described [20]. Briefly, dishes were washed with warm phosphate buffer solution (PBS) in dimly lit conditions. 10 μM PO-1 probe was added. The dishes were then incubated at 37°C for 1 hour, washed with warm PBS, and dosed with 2 mL of cell media containing 15 μM CuSO4, 100 or 500 μM DPEN, and/or 100 U/mL catalase as a negative control. A positive control was generated by adding H2O2 every 30 minutes starting at t0 to reach a steady state of 100 μM. The treated cells were allowed to incubate at 37°C for 3 hours. All subsequent steps utilizing the PO-1 probe were done on ice to preserve the analytical capabilities of the probe. The dishes were then washed with PBS and trypsinized with 0.5% trypsin for 10-15 minutes. The trypsin was then neutralized by re-suspending cells in PBS containing 10% FBS. Cells were centrifuged at 4°C, 1200 rpm for 5 minutes, resuspended in 300 μL PBS, and analyzed using flow cytometry (Becton Dickinson LSR II) with a 561 nm laser, 585/20 collection.
Hydrogen Peroxide Quantification
To quantify differences in steady-state levels of intracellular H2O2 during exposure to DPEN, we utilized a method based on the stoichiometric inactivation of catalase by 3-amino-1,2,4-triazole (3AT) in the presence of H2O2 as previously described [5, 21, 22]. One catalase active site specifically reacts with one molecule of H2O2 leading to the formation of one molecule of the Compound I intermediate [22]. Compound I can then covalently interact with one molecule of 3AT leading to the irreversible inhibition of catalase activity. The pseudo first-order rate constant for the 3AT-mediated inactivation of catalase (kAT) is then derived through kinetic analysis. Intracellular steady-state H2O2 concentrations are determined by: [H2O2] = kAT/k1, where k1 = 1.7 × 107 M-1 s-1 [21-23]. Because endogenous catalase activity in H1299T lung cancer cells is relatively low, the cells were first transduced with replication incompetent adenoviral vector AdCMV-Catalase (Viraquest). Cells were first counted to determine the quantity of virus needed. Media was changed to include 2% FBS, 25 MOI adenovirus was added, and cells incubated for 6 hours at 37°C at 21% O2. The media was then changed to 10% FBS and cells were allowed to recover for 48 hours in the absence of virus. After 48 hours, the cells were rinsed in PBS and FBS-free media was added. 50 mM 3AT was added to the cultures for 5 minutes before 15 μM CuSO4 and 100 μM DPEN was added. A positive control was generated by treating cells with 100 μM H2O2. Cells were harvested by rinsing twice in ice cold PBS, scraping into catalase buffer (50 mM K Phosphate pH 7.0) and frozen until analysis of remaining catalase activity. Control cells were harvested at 0, 15, 30, 45 and 60 minutes while the DPEN + CuSO4 and H2O2 treated dishes were harvested at 0, 5 10 and 15 minutes. Two dishes were assayed at each time point and the experiment was repeated on three separate occasions.
Catalase Activity Assay
Samples were thawed, homogenized by sonication and the proteins quantified using the Lowry assay. Catalase activity was determined on the soluble portion of the homogenates by measuring the disappearance of 10 mm hydrogen peroxide (Δ∈240 = 39.4 m−1 cm–1) in 50 mM potassium phosphate, pH 7.0, monitored at 240 nm and the units were expressed as milli-k units/mg of protein as described [24].
Xenograft Tumor Growth
Female athymic nude mice were purchased from Envigo and housed according to Office of The Institutional Animal Care and Use Committee. 106 H292 cells were injected into the right flank and when tumor volumes averaged 100-300 mm3, mice were randomized into treatment or control groups (N=6 mice per group). DPEN (150 mg/kg) was administered by IP injection twice daily in normal saline for injection. Control mice were injected with vehicle. Tumor volumes were measured using Vernier calipers, and both tumor volumes and mouse weights were measured daily.
Statistical Analysis
Unless otherwise noted in figure legend, data was analyzed for significance using unpaired t-tests in GraphPad Prism.
Results
DPEN combined with copper results in increased steady-state levels of hydrogen peroxide in cancer cells
To verify previous reports [25] of increased H2O2 in cancer cells treated with DPEN and CuSO4, the PO1 oxidation assay was used in MB231 breast cancer cells. Intracellular oxidation of the PO1 probe as detected by flow cytometry increased significantly 1.3-fold following treatment with 100 μM DPEN + 15 μM CuSO4 and 5-fold following treatment with 500 μM DPEN + 15 μM CuSO4 (Figure 1A). The highest DPEN dose resulted in a PO1 oxidation mean fluorescent intensity similar to the 100 μM H2O2 control. Pretreatment of the MB231 cells with 100 U/mL catalase prior to treatment with DPEN and CuSO4 completely inhibited the oxidation of PO1, confirming that the probe oxidation was mediated by H2O2.
Figure 1. Generation of H2O2 by DPEN and CuSO4 in breast and lung cancer cell cultures.
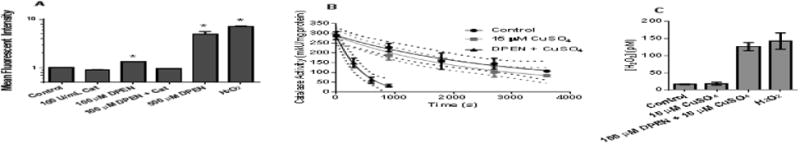
(A) MB231 cells were pre-incubated with PeroxyOrange-1 (PO-1) probe for one hour then treated with 100 μM DPEN, catalase (100 U/mL), or genuine H2O2 (100 μM) for 3 hours followed by analysis by flow cytometry. 15 μM CuSO4 was added to all groups at the time of drug treatment. *Significantly different than control; p< 0.05; n=4. (B) The rate of catalase inactivation in the presence of 3-aminotriazole was measured in H1299 lung cancer cells treated with 15 μM CuSO4 and 100 μM DPEN at various time points and (C) intracellular [H2O2] was calculated using the rates of catalase activity disappearance as previously described [23]. p<0.05; n=3. Errors represent ± 1SEM.
To quantify the increased steady-state levels of H2O2 in H1299T lung cancer cells during exposure to DPEN and CuSO4, a kinetic analysis was used based on the stoichiometric inactivation of catalase by 3-amino-1,2,4-triazole (3AT) in the presence of H2O2 (Figure 1B). 100 μM DPEN was chosen because it reflects a pharmacologically relevant level in humans [26] and 15 μM CuSO4 was used because this represents a physiological relevant tissue concentration in humans. [8]. Addition of CuSO4 did not significantly increase intracellular H2O2 from baseline (17 pM vs. 19 pM). However, the addition of the combination of DPEN and CuSO4 significantly increased the intracellular H2O2 to 127 pM, similar to the levels seen upon the addition of 100 μM genuine exogenous H2O2 (142 pM) (Figure 1C). Together this data strongly supports the hypothesis that treatment of cancer cells with physiologically relevant concentrations of DPEN and CuSO4 results in significant increases in steady-state [H2O2].
DPEN decreases breast and lung cancer cell clonogenic survival which is enhanced with Au and BSO
The lack of toxic effects of DPEN or CuSO4 individually was confirmed using clonogenic survival by treating H292 lung and MB231 breast cancer cells 24 hour with 15 μM CuSO4 or 100 μM DPEN or the combination (Figure 2A). Major cellular detoxification pathways for H2O2 include the glutathione and thioredoxin dependent cellular peroxidases. Because DPEN + CuSO4 resulted in H2O2 production, we combined DPEN with Au, a thioredoxin reductase inhibitor, and BSO, an inhibitor of glutathione synthesis. When treated with 100 μM DPEN clonogenic survival of MB231 dropped to 64%, while 1 mM BSO reduced survival to 91% (Figure 2B). When DPEN was combined with BSO significantly greater killing was noted and survival dropped to 48% (Figure 2B). Additionally, 0.5 μM Au reduced survival to 83% and when DPEN was combined with Au resulted survival dropped significantly to 42% (Figure 2B). When 50 U/mL catalase was added immediately before drug treatment with DPEN and Au, toxicity was inhibited demonstrating the H2O2 dependence of DPEN mediated cell killing (Figure 2B). When these experiments were repeated in H1299 and H292 lung cancer cells (Figure 2C) similar results were obtained showing the generality of the results with DPEN in three different human cancer cell lines. These results suggest that increased toxicity associated with inhibitors of thioredoxin and GSH metabolism are additive when combined with DPEN + CuSO4.
Figure 2. Clonogenic cell killing by DPEN is dependent on CuSO4 and enhanced by inhibitors of glutathione (BSO) and thioredoxin-dependent (Au) metabolism in human lung and breast cancer cells.
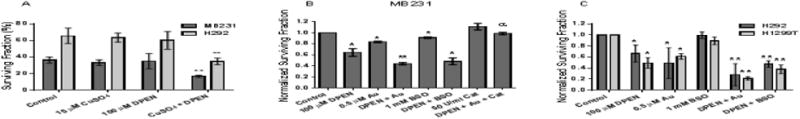
Clonogenic survival assays were performed with lung cancer cells or breast cancer cells. (A) MB231 breast and H292 lung cancer cells were treated with 15 μM CuSO4 and/ or 100 μM DPEN for 24 hours. (B) MB231 cells (C) H1299T and H292 cells were treated with the indicated concentrations of DPEN, Au, and/or BSO for 24 hours, 15 μM CuSO4 was added to all dishes at the time of treatment. 50 U/mL bovine catalase was added immediately prior to drug treatment. *Significantly different than control. **Significantly different than either drug alone. α significantly different than drugs without catalase. p< 0.05; n=3. Errors represent ± 1SEM.
Labile iron contributes to DPEN+ CuSO4 toxicity in breast and lung cancer cells
To determine if labile iron played a role in DPEN effects on clonogenic cell survival, MB231 and H292 cells were treated with 250 μM Fe sucrose or 40 μM DFO for 2 hours, then the cells were rinsed, and treated with DPEN + CuSO4 (Figure 3). DFO was selected for iron chelation because it has a binding constant for Fe3+ that is more than twice that for Cu2+ (pKs 30.6 and 14.1 respectively) [27] as well as inhibiting the redox cycling of Fe [28]. Fe sucrose was chosen because it is a clinically available source of supplemental iron that can be given to patients [29]. DFO and Fe sucrose did not cause clonogenic cell killing when used a single agents indicating these treatments were non-toxic (Figure 3AB). Interestingly pretreatment with Fe sucrose prior to DPEN + CuSO4 significantly increased clonogenic cell killing in breast (Fig 3A) and lung (Fig 3B) cancer cell lines when compared to DPEN + CuSO4 alone. Following treatment with DFO, DPEN toxicity was significantly inhibited in both breast and lung cancer cells despite the continued presence of 15 μM CuSO4 during DPEN exposure when the DFO had already been washed off (Figure 3AB). These results support the conclusion that redox active Fe as well as Cu play a significant role in the mechanism of H2O2 mediated cell killing induced by exposure to DPEN.
Figure 3. Clonogenic cell killing in cancer cells by DPEN + CuSO4 is enhanced with Fe sucrose and inhibited with Fe chelation.
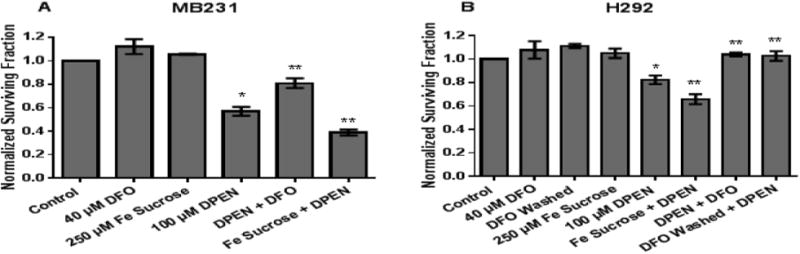
Iron chelation using 40 μM DFO for 2 hours prior to 100 μM DPEN + CuSO4 decreases clonogenic killing in MB231 cancer cells (A) and H292 cancer cells (B). In contrast, pretreatment with 250 μM Fe sucrose for 2 hours prior to addition of DPEN + CuSO4 enhances clonogenic cell death (A,B). 15 μM CuSO4 was added to all groups at time of DPEN treatment. *Significantly different than control; p< 0.05. **Significantly different than DPEN. p<0.05; n= 3. Errors represent ± 1 SEM.
DPEN + CuSO4 selectively induce clonogenic cell killing in breast and lung cancer cells compared to normal breast and lung cells
When clonogenic cell survival was measured in H1299T lung cancer cells treated with DPEN and compared to HBEpC non-transformed primary lung epithelial cells, DPEN was found to be selectively cytotoxic to the lung cancer cells (Figure 4A). Likewise when similar experiments were performed in MB231 breast cancer cells and compared to non-transformed normal HMEC breast epithelial cells, DPEN was found to be selectively cytotoxic to the breast cancer cells (Figure 4B). DPEN oxidation is accompanied by H2O2 production with a molar ratio of approximately 2:1 [16]. As expected the direct clonogenic toxic effects of 50 μM H2O2 on HMEC and MB231 cells exhibited similar results as treatment with 100 μM DPEN + Cu (Figure 4C).
Figure 4. DPEN + CuSO4 selectively induced clonogenic cell killing in human breast and lung epithelial cancer cells as compared to normal non-transformed human breast and lung epithelial cells. Intracellular labile iron pool is significantly higher in lung cancer cells compared to normal non-immortalized lung epithelial cells.
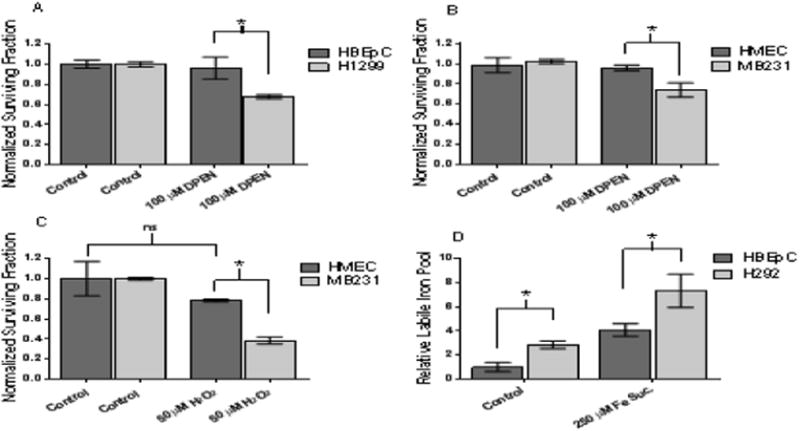
(A) Normal lung epithelial cells (HBEpC) and lung cancer cells (H1299) were treated 100 μM DPEN + 15 μM CuSO4 for 3 hours then subjected clonogenic assay. (B) Normal breast epithelial cells (HMEC) and breast cancer cells (MB231) similarly treated with 100 μM DPEN + 15 μM CuSO4 followed by clonogenic assay. (C) HMEC and MB231 cells were treated for 3 hours with 50 μM H2O2 followed by the clonogenic assay. (D) Relative intracellular labile iron pools measured using Calcein AM dye followed by flow cytometry comparing HBEpC and H292 cells. Cells were pre-treated with 250 μM Fe sucrose or 250 μM DFO as a positive and negative control. *Significantly different. p<0.05; n=3. Errors represent ± 1 SEM.
To further confirm labile Fe plays a key role in the cytotoxic effect observed with DPEN + CuSO4, intracellular LIPs were measured in H292 lung cancer cells and normal lung epithelial cells HBEpC using Calcein-AM as previously described [30] (Figure 4D). This probe binds Fe(II) rapidly, stoichiometrically, and reversibly while forming fluorescence-quenched Calcein-Fe complexes. Intracellular LIP is assessed from the relative rise in fluorescence elicited by addition of the highly permeant and high-affinity binding chelator, BIP. The results in Figure 4D demonstrate that the relative LIP of H292 lung cancer cells is 2.8 fold greater than the LIP of HBEpC. In addition pretreatment of cells with 250 μM Fe sucrose resulted in an approximate twice as much LIP in H292 cancer cells as compared to HBEpC normal cells. As expected, pretreatment with the iron chelator, DFO, resulted in a LIP below the detectible limit (data not shown). These results support the conclusion that fundamental differences in oxidative metabolism govern the selective toxicity of DPEN in cancer versus normal cells.
DPEN + CuSO4 decrease clonogenic cell survival when combined with radiation or carboplatin
Both breast and lung cancer patients are treated with radiation and/or carboplatin. To test if DPEN + CuSO4 could be combined with these standard cancer therapy modalities MB231 and H292 cancer cells with DPEN + CuSO4 followed by 2 Gy IR (Figure 5A). In both cell lines the combination of DPEN with IR resulted in significantly more cell killing than either treatment alone. These results were also recapitulated with 1 and 4 Gy IR doses with similar results (data not shown). Catalase reversed the toxicity resulting from DPEN or IR + DPEN but not IR alone in both MB231 and H292 cell lines showing that H2O2 was responsible for the effects of DPEN (Figure 5A). Similarly DPEN significantly increased clonogenic cell killing when combined with 5, 10, and 20 μM carboplatin compared to either agent alone (Figure 5B). Non-transformed human bronchial epithelial cells (HBEpC) along with H292 and H1299 lung cancer cells were treated with carboplatin, DPEN + Cu and the combination followed by clonogenic survival. The resulting data in Figure 5C continues to support the hypothesis that DPEN + Cu differentially sensitize lung cancer cells to carboplatin, relative to normal cells.
Figure 5. DPEN + Cu enhances cancer cell clonogenic cell killing when combined with ionizing radiation (IR) or Carboplatin.
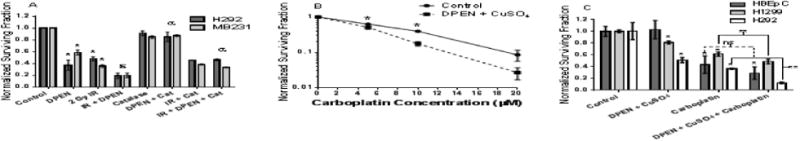
(A) H292 and MB231 cancer cells were treated with 100 μM DPEN + 15 μM CuSO4 for a total of 3 hours, with exposure to 2 Gy IR after one hour of drug treatment. Catalase protected the cancer cells from the enhanced cell killing of the combination of IR + DPEN. *-Significantly different than IR alone control; p< 0.05; n= 3. ε Significantly different than DPEN alone. a Significantly different than IR + DPEN. (B) H292 cells were treated with DPEN + CuSO4 and the indicated concentrations of carboplatin for 24 hours followed by clonogenic cell survival analysis. Survival data were normalized to the toxicity of DPEN + CuSO4 alone. *Significantly different than untreated control. (C) HBEpC (n = 2 experiments plated into at least 6 cloning dishes per treatment group), H292 (n = 3 experiments plated into at least 9 cloning dishes per treatment group), and H1299 (n = 2 experiments plated into at least 6 cloning dishes per treatment group) were treated with 10 μM carboplatin for 24 hours with DPEN added for the last 3 hours, followed by clonogenic assay. CuSO4 was present in all dishes at the beginning of treatment. ** Significantly different than either drug alone. p<0.05, Errors represent ± 1 SEM.
DPEN decreases tumor growth of H292 lung cancer xenographs
Results of in vivo studies showed that DPEN effectively decreased H292 tumor growth in mouse xenografts (Supplemental Figure 1A). Tumors were measured daily and each tumor's beginning volume was measured relative to the average volumes of tumor on the first day of treatment with DPEN (150 mg/kg 2×/day). Analysis was discontinued on the day the first mouse reached criteria for euthanasia (tumor diameter of 1.5 mm). Compared to control, DPEN alone showed a statistically significant decrease in tumor growth rate at day 9 (Supplemental Figure 1A). DPEN treatment did not result in a significant difference in mouse weight or any other observable evidence of toxicity. This in vivo data shows promise for the future exploration of DPEN in combination with standard of care chemotherapy or radiation.
Discussion
When developing cancer therapies, exploiting the fundamental biochemical differences between cancer and normal cells is essential. One such difference is alterations in oxidative metabolism that have been noted in cancer versus normal cells [4, 31, 32] as well as cancer stem cells [33]. We have recently published data demonstrating that disruption of redox balance radiosensitizes breast cancer stem cells [34]. Cancer cells appear to demonstrate both increased steady state levels of ROS (O2•- and H2O2) and abnormal metabolism of transition metals when compared to normal cells [4, 35]. It has been shown that free and bound copper and iron concentrations are higher in breast compared to the normal surrounding tissue [9, 10, 36]. These metal ions participate in many critical biological functions however the same properties that allow for the gain or loss of electrons also results in cytotoxicity through the donation of electrons to oxygen, leading to the generation of highly toxic hydroxyl radicals and other ROS. Recently, the importance of ferrous iron [Fe(II)] as the critical initiator of the Fenton reaction and the accompanying pathology in situ in carcinogenesis models was clearly demonstrated [37, 38]. Others have demonstrated that H2O2, generated via plasma activated media, induces apoptosis in lung cancer cells compared to normal cells in a Fe(II) dependent mechanism [39]. Based on alterations in oxidative metabolism as well as differences in redox active metal ions in cancer cells, agents that generate H2O2 represent new therapeutic tools for selectively killing cancer [25, 30].
D-penicillamine, a potent copper chelator, has been used in patients with rheumatic arthritis and Wilson's disease for many years. DPEN is tolerated well in humans, and when dosed at 750 mg per day, steady-state plasma levels reach 100 μM [26]. However proteinuria, thrombocytopenia, mucocutaneous lesions, and neutropenia have been reported in patients on DPEN therapy which limits its chronic use. The majority of in vivo copper is bound to either ceruloplasmin or albumin with a much smaller fraction being labile copper. Each ceruloplasmin protein has the ability to bind 8 copper atoms, however it is known that only 6 are tightly bound at physiologic conditions and at least one is exchangeable and available for chemical reaction [40]. Previous studies have shown the generation of H2O2 by DPEN in the presence of physiologically relevant concentrations of ceruloplasmin [15]. Additionally, albumin binds copper even less tightly than ceruloplasmin explaining why DPEN is effective at removing excess albumin-bound and free copper in Wilson's disease patients [25]. The resulting copper chelation and reduction produces H2O2 as previously described [15]. It has been reported that the H2O2 production from DPEN is concentration dependent up to 4 μM of copper intimating the differences between the tumor tissue and the normal surrounding tissue (1.57 μM vs 0.57 μM respectively) could result in a significantly different amount of H2O2 production in tumor tissue in vivo [10, 41]. Using PeroxyOrange-1, we have demonstrated that intracellular H2O2 increases with increasing DPEN concentration in breast cancer cells. Additionally, using the 3-amiotriazole mediated inhibition of catalase activity we quantified the intracellular H2O2 concentrations in lung cancer cells after treatment with biologically relevant concentrations of DPEN. Within minutes DPEN + CuSO4 significantly increased the intracellular [H2O2] similar to the levels seen upon exposure to 50 μM exogenous H2O2. Using the clonogenic survival assay to measure cancer cell reproductive integrity, we demonstrated that agents that inhibit H2O2 metabolism (Au and BSO) enhance cell killing when combined with DPEN + Cu, while catalase inhibited DPEN-induced cell death, clearly demonstrating that H2O2 is responsible for cell death.
To determine the role of redox active iron in DPEN-induced cancer cell toxicity, we chelated Fe using DFO and demonstrated that DPEN toxicity was inhibited. Consistent with this finding when cells were preloaded with Fe sucrose prior to treatment with DPEN + CuSO4 cancer cell death was significantly enhanced. Furthermore, the Calcein assay confirmed that the chelation and addition of Fe lead to a significant decrease and increase of the labile iron pool respectively. Previous reports have demonstrated the iron catalyzed oxidation rates of DPEN are minor at physiologic pH [25]. Although iron does not oxidize DPEN directly, we have demonstrated that its presence inside the cell is important for toxicity possibly by converting the H2O2 to more highly toxic oxidants such as •OH via the Fenton reaction. Finally we observed that DPEN was selectively toxic to breast and lung cancer cells vs normal breast and lung epithelial cells which could be the result of fundamental differences in oxidative metabolism involving either the removal of H2O2 or the formation of •OH by metal catalyzed reactions in cancer versus normal cells.
DPEN has been investigated in a phase 2 trial of 40 glioblastoma patients in combination with a low copper diet and radiation. Doses of DPEN of 2000 mg/day for several months resulted minimal side effects (mainly leukopenia and thrombocytopenia) as previously described for use in rheumatoid arthritis. DPEN did not improve patient survival; however this could be because it was combined with a low copper diet as part of an antiangiogenic strategy or issues related to crossing the blood brain barrier [42]. DPEN has safely and effectively been given alone and combined with other standard-of-care chemotherapeutics in xenograft mouse models in the treatment of multiple cancers [43-45]. Our in vitro data demonstrates that DPEN + CuSO4 contribute to enhancement of both radiation and carboplatin cell death which are considered standard of care agents for both lung and breast cancers. In aggregate, the data gathered in the current study supports the hypothesis that DPEN + Cu selectively kills cancer cells via the production of H2O2 which is also selectively enhanced in cancer cells via labile iron pools. This data supports further investigation of the combination of DPEN + Cu as an adjuvant to cancer therapy.
Supplementary Material
Supplemental Figure 1: The effect of DPEN on tumor growth rate in H292 lung cancer xenografts. 106 H292 cells were injected into the right flank of athymic nude mice. Treatments included control (n=6) or DPEN (150 mg/kg 2x/day) (n=6). Tumor volumes and mouse weights were measure daily. (A) Relative tumor volumes represent the average volumes for each day relative to the average volumes of tumor on the first day of treatment. Average tumor volume was significantly different on day 9 of treatment (day of first mouse death). (B) Average weight of mice in groups showed no significant change in weight indicating the treatments were well-tolerated. Error bars represent SEM. * indicates p < 0.05 compared to control using ANOVA with Fisher's LSD.
Highlights.
DPEN + Cu at physiologic concentrations increase H2O2 levels in cancer cells.
DPEN's clonogenic toxicity is enhanced using auranofin and buthionine sulfoximine.
DPEN + Cu treatment is more toxic to cancer cells than to normal epithelial cells.
DPEN toxicity in correlated with intracellular labile iron pools.
Labile iron pools are higher in cancer cells verses normal lung epithelial cells.
Acknowledgments
We would like to thank the Flow Cytometry Core, the Holden Comprehensive Cancer Center (HCCC), the medical student summer research program, and T35HL007485, T32CA078586, R01CA182804, P30CA086862 for support. We would also like to acknowledge the staff at the University of Iowa College of Medicine and Holden Comprehensive Cancer Center Radiation and Free Radical Research (RFRR) Core for radiation services.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wondrak GT. Redox-directed cancer therapeutics: molecular mechanisms and opportunities. Antioxid Redox Signal. 2009;11(12):3013–69. doi: 10.1089/ars.2009.2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fath MA, Ahmad IM, Smith CJ, Spence J, Spitz DR. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clin Cancer Res. 2011;17(19):6206–17. doi: 10.1158/1078-0432.CCR-11-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Du J, Cieslak JA, 3rd, Welsh JL, Sibenaller ZA, Allen BG, Wagner BA, Kalen AL, Doskey CM, Strother RK, Button AM, Mott SL, Smith B, Tsai S, Mezhir J, Goswami PC, Spitz DR, Buettner GR, Cullen JJ. Pharmacological Ascorbate Radiosensitizes Pancreatic Cancer. Cancer Res. 2015;75(16):3314–26. doi: 10.1158/0008-5472.CAN-14-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aykin-Burns N, Ahmad IM, Zhu Y, Oberley LW, Spitz DR. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J. 2009;418(1):29–37. doi: 10.1042/BJ20081258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ahmad I, A-B N, Sim JE, Walsh SA, Higashikubo R, Buettner GR, Venkataraman S, Mackey MA, Flanagan SW, Oberley LW, Spitz DR. Mitochondrial O2·- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. Journal of Biological Chemistry. 2005;280(6):4254–4263. doi: 10.1074/jbc.M411662200. [DOI] [PubMed] [Google Scholar]
- 6.Zhou D, Shao L, Spitz DR. Reactive oxygen species in normal and tumor stem cells. Adv Cancer Res. 2014;122:1–67. doi: 10.1016/B978-0-12-420117-0.00001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oberley LW, Rogers KL, Schutt L, Oberley TD, Leuthauser SW, Sorenson JR. Possible role of glutathione in the antitumor effect of a copper-containing synthetic superoxide dismutase in mice. J Natl Cancer Inst. 1983;71(5):1089–94. [PubMed] [Google Scholar]
- 8.Zowczak M, I M, Torlinski L, Cofta S. Analysis of serum copper and zinc concentrations in cancer patients. Biological Trace Element Research. 2001;82(1):1–8. doi: 10.1385/BTER:82:1-3:001. [DOI] [PubMed] [Google Scholar]
- 9.Mulware SJ. Comparative Trace Elemental Analysis in Cancerous and Noncancerous Human Tissues Using PIXE. J Biophys. 2013:192026. doi: 10.1155/2013/192026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuo HW, Chen SF, Wu CC, Chen DR, Lee JH. Serum and tissue trace elements in patients with breast cancer in Taiwan. Biol Trace Elem Res. 2002;89(1):1–11. doi: 10.1385/BTER:89:1:1. [DOI] [PubMed] [Google Scholar]
- 11.Adachi S, Takemoto K, Ohshima S, Shimizu Y, Takahama M. Metal concentrations in lung tissue of subjects suffering from lung cancer. Int Arch Occup Environ Health. 1991;63(3):193–7. doi: 10.1007/BF00381568. [DOI] [PubMed] [Google Scholar]
- 12.Farah IO, Trimble Q, Ndebele K, Mawson A. Significance of differential metal loads in normal versus cancerous cadaver tissues - biomed 2010. Biomed Sci Instrum. 2010;46:404–9. [PMC free article] [PubMed] [Google Scholar]
- 13.Garber K. Biomedicine. Targeting copper to treat breast cancer. Science. 2015;349(6244):128–9. doi: 10.1126/science.349.6244.128. [DOI] [PubMed] [Google Scholar]
- 14.Corce V, Gouin SG, Renaud S, Gaboriau F, Deniaud D. Recent advances in cancer treatment by iron chelators. Bioorganic & medicinal chemistry letters. 2016;26(2):251–6. doi: 10.1016/j.bmcl.2015.11.094. [DOI] [PubMed] [Google Scholar]
- 15.Lipsky PE. Immunosuppression by D-penicillamine in vitro. Inhibition of human T lymphocyte proliferation by copper- or ceruloplasmin-dependent generation of hydrogen peroxide and protection by monocytes. The Journal of clinical investigation. 1984;73(1):53–65. doi: 10.1172/JCI111207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Starkebaum G, R RK. D-Penicillamine: analysis of the mechanism of copper-catalyzed hydrogen peroxide generation. The Journal of Immunology. 1985;134(5):3371–3378. [PubMed] [Google Scholar]
- 17.Keyer K, Imlay JA. Superoxide accelerates DNA damage by elevating free-iron levels. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(24):13635–40. doi: 10.1073/pnas.93.24.13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Spitz DR, Malcolm RR, Roberts RJ. Cytotoxicity and metabolism of 4-hydroxy-2-nonenal and 2-nonenal in H2O2-resistant cell lines. Do aldehydic by-products of lipid peroxidation contribute to oxidative stress? Biochem J. 1990;267(2):453–9. doi: 10.1042/bj2670453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Epsztejn S, Kakhlon O, Glickstein H, Breuer W, Cabantchik I. Fluorescence analysis of the labile iron pool of mammalian cells. Anal Biochem. 1997;248(1):31–40. doi: 10.1006/abio.1997.2126. [DOI] [PubMed] [Google Scholar]
- 20.Dickinson BC, Huynh C, Chang CJ. A palette of fluorescent probes with varying emission colors for imaging hydrogen peroxide signaling in living cells. Journal of the American Chemical Society. 2010;132(16):5906–15. doi: 10.1021/ja1014103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Royall JA, Gwin PD, Parks DA, Freeman BA. Responses of vascular endothelial oxidant metabolism to lipopolysaccharide and tumor necrosis factor-alpha. Arch Biochem Biophys. 1992;294(2):686–94. doi: 10.1016/0003-9861(92)90742-f. [DOI] [PubMed] [Google Scholar]
- 22.Chance B, S H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiological Reviews. 1979;59(3):527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- 23.Ahmad I, A-B N, Sim JE, Walsh SA, Higashikubo R, Buettner GR, Venkataraman S, Mackey MA, Flanagan SW, Oberley LW, Spitz DR. Mitochondrial superoxide and H2O2 mediate glucose deprivation-induced stress in human cancer cells. Journal of Biological Chemistry. 2005;280(6):4254–4263. doi: 10.1074/jbc.M411662200. [DOI] [PubMed] [Google Scholar]
- 24.Spitz DR, Elwell JH, Sun Y, Oberley LW, Oberley TD, Sullivan SJ, Roberts RJ. Oxygen toxicity in control and H2O2-resistant Chinese hamster fibroblast cell lines. Arch Biochem Biophys. 1990;279(2):249–60. doi: 10.1016/0003-9861(90)90489-l. [DOI] [PubMed] [Google Scholar]
- 25.Gupte A, Mumper RJ. An investigation into copper catalyzed D-penicillamine oxidation and subsequent hydrogen peroxide generation. Journal of inorganic biochemistry. 2007;101(4):594–602. doi: 10.1016/j.jinorgbio.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Muijsers AO, van de Stadt RJ, Henrichs AM, Ament HJ, van der Korst JK. D-penicillamine in patients with rheumatoid arthritis. Serum levels, pharmacokinetic aspects, and correlation with clinical course and side effects. Arthritis and rheumatism. 1984;27(12):1362–9. doi: 10.1002/art.1780271206. [DOI] [PubMed] [Google Scholar]
- 27.Sillén LG, Martell AE. Supplement, Chemical Society. London: 1971. Stability constants of metal-ion complexes. [Google Scholar]
- 28.Burkitt MJ, Kadiiska MB, Hanna PM, Jordan SJ, Mason RP. Electron spin resonance spin-trapping investigation into the effects of paraquat and desferrioxamine on hydroxyl radical generation during acute iron poisoning. Mol Pharmacol. 1993;43(2):257–63. [PubMed] [Google Scholar]
- 29.Johnson AC, Becker K, Zager RA. Parenteral iron formulations differentially affect MCP-1, HO-1, and NGAL gene expression and renal responses to injury. Am J Physiol Renal Physiol. 2010;299(2):F426–35. doi: 10.1152/ajprenal.00248.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schoenfeld JD, Sibenaller ZA, Mapuskar KA, Wagner BA, Cramer-Morales KL, Furqan M, Sandhu S, Carlisle TL, Smith MC, Abu Hejleh T, Berg DJ, Zhang J, Keech J, Parekh KR, Bhatia S, Monga V, Bodeker KL, Ahmann L, Vollstedt S, Brown H, Shanahan Kauffman EP, Schall ME, Hohl RJ, Clamon GH, Greenlee JD, Howard MA, Shultz MK, Smith BJ, Riley DP, Domann FE, Cullen JJ, GR Buettner, Buatti JM, Spitz DR, Allen BA. O2·-and H2O2-Mediated Disruption of Feetabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell. 2017 doi: 10.1016/j.ccell.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 31.Spitz DR, Sim JE, Ridnour LA, Galoforo SS, Lee YJ. Glucose deprivation-induced oxidative stress in human tumor cells. A fundamental defect in metabolism? Ann N Y Acad Sci. 2000;899:349–62. doi: 10.1111/j.1749-6632.2000.tb06199.x. [DOI] [PubMed] [Google Scholar]
- 32.Fath MA, Diers AR, Aykin-Burns N, Simons AL, Hua L, Spitz DR. Mitochondrial electron transport chain blockers enhance 2-deoxy-D-glucose induced oxidative stress and cell killing in human colon carcinoma cells. Cancer biology & therapy. 2009;8(13):1228–36. doi: 10.4161/cbt.8.13.8631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, Joshua B, Kaplan MJ, Wapnir I, Dirbas FM, Somlo G, Garberoglio C, Paz B, Shen J, Lau SK, Quake SR, Brown JM, Weissman IL, Clarke MF. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458(7239):780–3. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rodman SN, Spence JM, Ronnfeldt TJ, Zhu Y, Solst SR, O'Neill RA, Allen BG, Guan X, Spitz DR, Fath MA. Enhancement of Radiation Response in Breast Cancer Stem Cells by Inhibition of Thioredoxin- and Glutathione-Dependent Metabolism. Radiation research. 2016;186(4):385–395. doi: 10.1667/RR14463.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalinowski DS, Stefani C, Toyokuni S, Ganz T, Anderson GJ, Subramaniam NV, Trinder D, Olynyk JK, Chua A, Jansson PJ, Sahni S, Lane DJ, Merlot AM, Kovacevic Z, Huang ML, Lee CS, Richardson DR. Redox cycling metals: Pedaling their roles in metabolism and their use in the development of novel therapeutics. Biochim Biophys Acta. 2016;1863(4):727–48. doi: 10.1016/j.bbamcr.2016.01.026. [DOI] [PubMed] [Google Scholar]
- 36.Rehman S, Husnain SM. A probable risk factor of female breast cancer: study on benign and malignant breast tissue samples. Biol Trace Elem Res. 2014;157(1):24–9. doi: 10.1007/s12011-013-9865-7. [DOI] [PubMed] [Google Scholar]
- 37.Ito F, Nishiyama T, Shi L, Mori M, Hirayama T, Nagasawa H, Yasui H, Toyokuni S. Contrasting intra- and extracellular distribution of catalytic ferrous iron in ovalbumin-induced peritonitis. Biochemical and biophysical research communications. 2016;476(4):600–6. doi: 10.1016/j.bbrc.2016.06.003. [DOI] [PubMed] [Google Scholar]
- 38.Mukaide T, Hattori Y, Misawa N, Funahashi S, Jiang L, Hirayama T, Nagasawa H, Toyokuni S. Histological detection of catalytic ferrous iron with the selective turn-on fluorescent probe RhoNox-1 in a Fenton reaction-based rat renal carcinogenesis model. Free Radic Res. 2014;48(9):990–5. doi: 10.3109/10715762.2014.898844. [DOI] [PubMed] [Google Scholar]
- 39.Adachi T, Tanaka H, Nonomura S, Hara H, Kondo S, Hori M. Plasma-activated medium induces A549 cell injury via a spiral apoptotic cascade involving the mitochondrial-nuclear network. Free radical biology & medicine. 2015;79:28–44. doi: 10.1016/j.freeradbiomed.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 40.Jackson GE, May PM, Williams DR. The action of chelating agents in the removal of copper from ceruloplasmin: an in vitro study. FEBS letters. 1978;90(1):173–7. doi: 10.1016/0014-5793(78)80323-8. [DOI] [PubMed] [Google Scholar]
- 41.Gupte A, Mumper RJ. Copper chelation by D-penicillamine generates reactive oxygen species that are cytotoxic to human leukemia and breast cancer cells. Free Radic Biol Med. 2007;43(9):1271–8. doi: 10.1016/j.freeradbiomed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 42.Brem S, Grossman SA, Carson KA, New P, Phuphanich S, Alavi JB, Mikkelsen T, Fisher JD. C.N.S.C. New Approaches to Brain Tumor Therapy, Phase 2 trial of copper depletion and penicillamine as antiangiogenesis therapy of glioblastoma. Neuro-oncology. 2005;7(3):246–53. doi: 10.1215/S1152851704000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen SJ, Kuo CC, Pan HY, Tsou TC, Yeh SC, Chang JY. Mechanistic basis of a combination D-penicillamine and platinum drugs synergistically inhibits tumor growth in oxaliplatin-resistant human cervical cancer cells in vitro and in vivo, Biochemical pharmacology. 2015;95(1):28–37. doi: 10.1016/j.bcp.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 44.Qiao S, Cabello CM, Lamore SD, Lesson JL, Wondrak GT. D-Penicillamine targets metastatic melanoma cells with induction of the unfolded protein response (UPR) and Noxa (PMAIP1)-dependent mitochondrial apoptosis. Apoptosis: an international journal on programmed cell death. 2012;17(10):1079–94. doi: 10.1007/s10495-012-0746-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshii J, Yoshiji H, Kuriyama S, Ikenaka Y, Noguchi R, Okuda H, Tsujinoue H, Nakatani T, Kishida H, Nakae D, Gomez DE, De Lorenzo MS, Tejera AM, Fukui H. The copper-chelating agent, trientine, suppresses tumor development and angiogenesis in the murine hepatocellular carcinoma cells. International journal of cancer. 2001;94(6):768–73. doi: 10.1002/ijc.1537. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: The effect of DPEN on tumor growth rate in H292 lung cancer xenografts. 106 H292 cells were injected into the right flank of athymic nude mice. Treatments included control (n=6) or DPEN (150 mg/kg 2x/day) (n=6). Tumor volumes and mouse weights were measure daily. (A) Relative tumor volumes represent the average volumes for each day relative to the average volumes of tumor on the first day of treatment. Average tumor volume was significantly different on day 9 of treatment (day of first mouse death). (B) Average weight of mice in groups showed no significant change in weight indicating the treatments were well-tolerated. Error bars represent SEM. * indicates p < 0.05 compared to control using ANOVA with Fisher's LSD.