

Abstract
Numerous epidemiological studies have shown a significantly higher risk for development of Alzheimer’s disease (AD) in patients affected by type 2 diabetes (T2D), but the molecular mechanism responsible for this association is presently unknown. Both diseases are considered protein misfolding disorders associated to the accumulation of protein aggregates; amyloid-beta (Aβ) and tau in the brain during AD, and islet amyloid polypeptide (IAPP) in pancreatic islets in T2D. Formation and accumulation of these proteins follows a seeding-nucleation model, where a misfolded aggregate or “seed” promotes the rapid misfolding and aggregation of the native protein. Our underlying hypothesis is that misfolded IAPP produced in T2D potentiates AD pathology by cross-seeding Aβ, providing a molecular explanation for the link between these diseases. Here, we examined how misfolded IAPP affects Aβ aggregation and AD pathology in vitro and in vivo. We observed that addition of IAPP seeds accelerates Aβ aggregation in vitro in a seeding-like manner and the resulting fibrils are composed of both peptides. Transgenic animals expressing both human proteins exhibited exacerbated AD-like pathology compared to AD transgenic mice or AD transgenic animals with type-1 diabetes (T1D). Remarkably, IAPP colocalized with amyloid plaques in brain parenchymal deposits, suggesting these peptides may directly interact and aggravate the disease. Furthermore, inoculation of pancreatic IAPP aggregates into the brains of AD transgenic mice resulted in more severe AD pathology and significantly greater memory impairments than untreated animals. This data provides a proof-of-concept for a new disease mechanism involving the interaction of misfolded proteins through cross-seeding events which may contribute to accelerate or exacerbate disease pathogenesis. Our findings could shed light on understanding the linkage between T2D and AD, two of the most prevalent protein misfolding disorders.
Introduction
Protein misfolding disorders (PMDs) are a group of diseases caused by the formation and accumulation of proteinaceous misfolded aggregates1,2. The disease-associated misfolded proteins usually organize in a β-sheet structure, leading to the formation of stable oligomeric and fibrillar species that accumulate in tissues in the form of amyloid-like deposits. In each disease the main protein component of the aggregates is different and include, for example, amyloid-beta (Aβ) and hyperphosphorylated tau (ptau) in Alzheimer’s disease (AD), alpha-synuclein (α-syn) in Parkinson’s disease, TDP-43 in amyotrophic lateral sclerosis, islet amyloid polypeptide (IAPP) in type 2 diabetes (T2D) and prion protein (PrPSc) in transmissible spongiform encephalopathies. In some cases, more than one misfolded protein can be found in a single disease, with AD as the most emblematic case, because it features the simultaneous accumulation of Aβ and pTau aggregates. In addition, it has become clear that there is a substantial overlap of pathological abnormalities, leading to the relatively frequent appearance of mixed pathologies, characterized by the presence of multiple protein aggregates in the same tissue3–8. For instance, it is common to find in AD brains accumulation of α-syn and TDP43, in addition to the expected presence of amyloid plaques and neurofibrillary tangles. Similarly, misfolded IAPP can aggregate and deposit in the brain of patients with T2D and AD9, while Aβ and ptau can be found in the pancreas of T2D patients10. The exact relationship between different protein aggregates is currently unknown, but it is possible that aggregation of one protein may induce the misfolding and aggregation of another protein.
Extensive epidemiological studies have reported an association between T2D and AD, and a higher risk for dementia has been found in individuals who suffer from diabetes and insulin resistance11–13. In fact, glucose metabolism impairment and T2D significantly increases the risk for AD14 and the development of mild cognitive impairment (MCI)15,16. Nearly 80% of all AD patients exhibit glucose tolerance impairment or have been diagnosed with diabetes17. Furthermore, IAPP levels in these patients correlate with the Aβ concentration in the plasma18. The mechanism for the risk association between AD and T2D is unknown and several hypotheses have been proposed, including: alterations in insulin signaling, oxidative stress, abnormal clearance capacity, hypercholesterolemia, and interactions at the level of protein misfolding19.
Despite the fact that each PMD is associated to the misfolding of a different protein, the process of protein misfolding and aggregation in all PMDs follows a similar mechanism -the so-called seeding-nucleation model- and results in the formation of structures rich in β-sheet conformation, partially resistant to proteolysis and with a high tendency to form larger order aggregates20,21. A seeded-nucleated aggregation starts with a slow nucleation phase followed by a rapid elongation stage20. In the nucleation phase, the rate-determining step is the formation of a stable seed or nucleus of polymerized protein. Once the first seeds are formed, a rapid and exponential recruitment of soluble protein increases the population of misfolded units. A typical feature of the seeding-nucleation model is the ability of preformed seeds to accelerate the process of conversion by recruiting the soluble normal protein into the growing aggregate20. Interestingly, the fact that the process as well as the end-products and intermediates of the seeding-nucleation mechanism are similar in all PMDs raise the possibility that seeds composed by one protein may catalyze the polymerization of other proteins22. This process of heterologous seeding, also known as “cross-seeding”, has been extensively described using pure preparations of proteins in test tube experiments, especially between proteins with some degree of sequence homology23–26. In particular, several in vitro studies have described cross-seeding between IAPP and Aβ26–28. The underlying hypothesis of the current study is that IAPP aggregates can interact with Aβ, inducing cross-seeding of amyloid aggregation and thus, accelerating or exacerbating the pathological features of AD. The main goal of this study was to investigate a possible molecular interaction through cross-seeding between Aβ and IAPP proteins in vitro, the effect of the coexistence of both misfolded proteins in vivo, and the acceleration of cerebral amyloid deposition by exogenous administration of IAPP aggregates into a transgenic mouse model of AD. Our findings may contribute to understand the molecular mechanism responsible for the linkage between AD and T2D observed in epidemiological studies.
Materials and Methods
Peptide synthesis
Aβ40, Aβ42 and IAPP peptides were produced by solid-phase synthesis by Dr. James I Elliott (The ERI Amyloid Laboratory, LLC) and purified (>95% purity) by reverse-phase chromatography. Peptides were dissolved in 50% acetonitrile, frozen, and lyophilized overnight. Lyophilized Aβ and IAPP were then dissolved at a concentration of 2.5 mg/mL in either 10mM NaOH (pH 12) or 2 mM HCl (pH 2), respectively. The material was centrifuged in a 30 kDa cut-off filter for 12 min at 14,000 × g at 4°C to obtain the “seed-free” solution.
In vitro aggregation assay
Aβ seed-free solution was diluted to a final concentration of 0.05 μg/μL and incubated with shaking (450 rpm) at 25°C, for 5 h to produce oligomeric seeds. For IAPP oligomer preparation, IAPP seed-free solution was diluted to a final concentration of 0.04 μg/μL and incubated at room temperature, with shaking (450 rpm) for 90 min. For the seeding assays, 2 μM seed-free Aβ1–40 was mixed in 100 μl of Tris-HCl buffer in the presence of 1% of IAPP oligomers (heterologous seeding), or 1% of Aβ oligomers (homologous seeding). The same volume of Tris-HCl buffer was added for negative controls. Then, 20 μl of 50 μM Thioflavin T (ThT) was added so aggregation could be measured by fluorescence at 485 nm over time.
Electron microscopy
Amyloid fibrils were visualized by transmission electron microscopy (TEM), using negative staining, as previously described29. To better visualize fibrils, Aβ1–42 peptide was used at a concentration of 37 μM, which was incubated in the presence of 10% molar ratio of IAPP. For immuno-gold labeling, 10 μL of the IAPP-Aβ fibrils were added to Formvar/carbon coated 200 mesh copper TEM support grids (EMS) and blocked with normal goat serum. After glutaraldehyde fixation, the grid was incubated with a mixture of primary antibodies (mouse anti-Aβ 4G8 and rabbit anti-IAPP). After rinsing, the grid was incubated in a mixture of secondary colloidal gold antibodies – 6 nm goat anti-mouse IgG and 12 nm goat anti-rabbit IgG (Jackson Immuno Research). 2% uranyl acetate solution was placed on the grid, rinsed, and allowed to air-dry. Grids were imaged in a JEOL 1200 transmission electron microscope at 60 kv, and images were captured with a 2k×2k Gatan Orius CCD camera.
Transgenic mice
APP (Tg2576 on a 129S2 background, The Jackson Laboratory) transgenic mice overexpress a mutant form of APP bearing the Swedish mutation. Heterozygous APP animals begin to develop amyloid deposits by 9–10 months in the hippocampus and cortex, numerous parenchymal Aβ plaques appear by 11–13 months, and behavioral impairments are displayed by 12 months of age30. As a model for IAPP aggregation, we used transgenic mice over-expressing human IAPP (FVB background; a kind gift from Dr. Peter C. Butler, UCLA)31. In homozygosity, transgenic mice spontaneously develop IAPP amyloid deposits in the pancreas, which are associated with selective β-cell death, impaired insulin secretion and hyperglycemia31. We bred these colonies in our facilities to obtain double transgenic animals with two (IAPP+/+ × APP) or one (IAPP+/− × APP) copy of the human IAPP, and selected for the animals having one copy of the human, mutant APP gene. Appropriate controls were produced by breeding APP mice with the same mixed background (FVB/129S2) of the IAPP transgenic mice. Mice were sacrificed by CO2 inhalation at 8.5 months old and perfused transcardially. One hemisphere was frozen for biochemical studies, and the other half was fixed for histological analysis. All animal experiments were carried out in accordance with the NIH regulations and approved by the committee of animal use for research at the University of Texas Health Science Center at Houston Medical School.
Induction of diabetes by streptozotocin administration
As a model of type 1 diabetes (T1D), mice were treated with streptozotocin (STZ) as previously described32. STZ is a diabetogenic drug that specifically destroys pancreatic β-cells. Briefly, STZ (Sigma-Aldrich) was diluted in sodium citrate buffer and injected intraperitoneally at 110 mg/kg per day for two consecutive days. Mice were fasted for 16 h before collecting blood from the tail vein and measuring glucose levels with a Contour Blood Glucose Monitoring System (Bayer). Animals were considered to have T1D when their blood glucose levels were higher than 500 mg/dL. Pancreases from positive animals were harvested and stored at −80°C.
Intracerebral inoculation of pancreas extracts
Pancreas specimens from IAPP+/+ animals showing extensive IAPP deposition and STZ-injected WT animals were homogenized at 10% (w/v) in PBS containing protease inhibitors and stored at −80°C. 2-month-old APP mice received a stereotaxic intracerebral injection in the right hippocampus with 10 μL of 10% pancreas homogenate.
Behavioral testing
At 8 months old, experimental animals were assayed in the Barnes maze to examine hippocampal/medial temporal lobe function related to spatial reference memory. The Barnes maze is a circular platform with 40 holes and is surrounded by black curtains with visual clues to enable the animal to navigate and reach the escape hole. We used negative stimuli (buzz and light) to stimulate the animal to enter into the escape hole. On day 1, the mouse is familiarized and adapted to the task. Spatial acquisition was done during days 2–5, where the animal is placed in the center of the platform and allowed to explore the maze for a maximum of 3 min with 4 trials each day. The animal’s latency to the escape hole is averaged over training trials in blocked days, as well as short term memory is designated as the first trial on day 5. Trials were recorded and tracking analyzed by TopScan 2.0. Any animal demonstrating motor impairments was not used in analyses.
Biochemical extraction and quantification of Aβ aggregates
Mouse brain hemispheres were homogenized at 10% w/v in PBS containing protease inhibitors and centrifuged at 32,600 rpm for 1 h at 4°C in an ultracentrifuge (Beckman-Coulter). The supernatant was collected to measure the concentration of soluble Aβ containing a mixture of monomers and low molecular weight oligomers. The pellets were resuspended in 200 μL of 70% formic acid followed by sonication. Samples were centrifuged for 30 min under the same conditions, and the supernatant was collected and neutralized in 1 M Tris buffer, pH 11, to detect the fraction of insoluble Aβ, corresponding mostly to fibrillary aggregates. Levels of Aβ42 were measured using Human Aβ ELISA Kits (Invitrogen) on an ELISA plate reader (EL800 BIO-TEK).
Histological staining
Brains and pancreases were post-fixed into formalin and embedded in paraffin. Serial 10-μm-thick sections from all animal groups (n=5–10/group; 5 sections/stain/animal) were processed in parallel for immuno-staining. After blocking the endogenous peroxidase activity with 3% H2O2 and 10% methanol for 20 min, sections were incubated overnight at room temperature in one of the following primary antibodies: 82E1 (1:1,000 IBL America), 4G8 (1:1,000 Covance) monoclonal anti-Aβ antibodies; or rabbit anti-hIAPP (1:1,000 Peninsula Laboratories). Sections stained for Aβ were pretreated with 85% formic acid. Primary antibody was detected by incubating 1 h with the corresponding Alexa Fluor linked secondary antibody. For Thioflavin-S (ThS) staining, tissue slices were incubated in 0.025% ThS solution for 8 min. Finally, all sections were cover slipped with mounting medium containing DAPI.
Image analysis
Photomicrographs of samples examined under an epifluorescent microscope (DMI6000B, Leica) were taken with a digital camera (DFC310 FX Leica), imported into ImageJ 1.45s software (NIH), and converted to black and white images. Threshold intensity was manually set and kept constant, and the number of pixels was determined for 82E1, 4G8, or hIAPP immunostained sections to quantify the amyloid load in hippocampal and cortical areas of the brain as well as in the Islets of Langerham. Burden was defined as the area labeled per total area analyzed. Amyloid plaque quantification was performed manually using the same software by calculating the area and the number of plaques per total analyzed area.
Statistical analysis
Graphs are expressed as means ± standard error (SEM). After confirming normal distribution with Skewness and Kurtosis statistic test, one way analysis of variance (ANOVA) followed by a post-hoc Tukey’s multiple comparisons test were used to analyze differences among groups. Two-ways ANOVA followed by a post-hoc Tukey’s multiple comparisons test and Bonferroni post-test were used to analyze the Barnes maze data. Statistical analyses were performed using GraphPad Prism 5.0 software (GraphPad Software Inc). Statistical differences for all tests were considered significant at the p < 0.05 level.
Results
In vitro aggregation of Aβ is accelerated by IAPP seeds
First, we wanted to determine if IAPP oligomeric species could cross-seed Aβ misfolding and aggregation in vitro. Under the conditions the experiments were carried out, Aβ1–40 monomers do not substantially aggregate until 144 h of incubation in the absence of pre-formed seeds (Fig. 1A, circles). Addition of homologous Aβ seeds accelerated misfolding and aggregation of Aβ, reaching maximal aggregation at approximately 96 h (Fig. 1A, diamonds). When the reaction was seeded with the same concentration of IAPP aggregates, Aβ aggregation showed an intermediate kinetic, reaching maximal aggregation by 144 h (Fig. 1A, triangles). These results indicate that IAPP aggregates can serve as a nucleus to cross-seed Aβ aggregation in vitro, confirming previous reports26–28. To further study whether Aβ and IAPP can directly interact within the aggregates, we visualized the aggregates produced in the presence of IAPP seeds by transmission electron microscopy with immunogold to specifically label the presence of Aβ or IAPP in the fibrils. As shown in Fig. 1B, in many instances, Aβ and IAPP are in close proximity (see red arrows in Fig. 1B), although it is clear also that homologous labeling is much more frequent. These results suggest that Aβ and IAPP may interact by a cross-seeding mechanism. However, we cannot rule out that the co-localization of both proteins in the same aggregates may in part be due to IAPP aggregates, or even monomers present in the preparation, contributing to fibril extension initiated by Aβ seeds.
Figure 1. In vitro Aβ aggregation is enhanced by IAPP heterologous seeding.
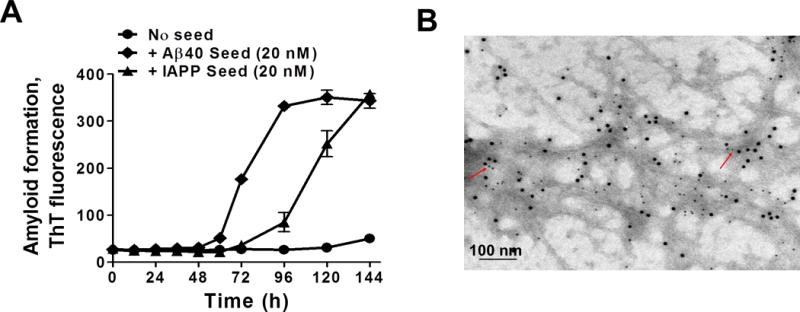
A: Soluble seed-free Aβ40 (2000 nM) in 100 mM Tris-HCl pH 7.4 (200 μl) containing 5 μM thioflavin T (ThT) was allowed to aggregate at 22°C with intermittent shaking at 500 rpm in the absence or in the presence of either 20 nM Aβ40 seeds (homologous seeding) or 20 nM IAPP seeds (heterologous seeding). The extent of aggregation was periodically monitored by measuring the ThT fluorescence at excitation of 435 nm and emission of 485 nm. Error bars indicate standard deviation (S.D.) B: Transmission electron microscopy microphotography of double immuno-gold labeling of fibrils formed by the addition of IAPP seeds (10% with respect to Aβ) into seed-free Aβ1–42 (37 μM) aggregation solution. After in vitro cross-seeding, fibrils were stained using antibodies coupled to gold nanoparticles. IAPP was detected with an antibody coupled to 12 nm gold particles while Aβ was labeled with 6 nm gold particles. Scale bar: 100 nm. Red arrows point to aggregates showing staining for both peptides.
Transgenic animals expressing IAPP and APP human genes displayed higher Aβ deposition in the brain
Next, we wanted to analyze the possibility that IAPP aggregates may also seed Aβ aggregation in vivo. For this purpose, we bred transgenic mouse models for amyloidosis associated to T2D and AD to generate animals with two (IAPP+/+) or one (IAPP+/−) copies of the human IAPP in the context of one copy of mutant APP (APPSwe+/−), termed IAPP+/+ × APP and IAPP+/− × APP, respectively. As controls, APP transgenic mice were bred with animals having the same genetic background (FVB/129S2) of the IAPP transgenic mouse. As a further control and to rule out an effect caused by glucose impairment instead of IAPP aggregates, we used a model of T1D induced by administration of STZ in APP animals (Tg2576 transgenic mice) having the same background and age than experimental animals. All animals were sacrificed at 8.5 months when standard APP animals start to display the first amyloid plaques. As illustrated in Fig. 2A–B, IAPP+/+ × APP animals demonstrated a statistically significant difference in Aβ burden in the hippocampus and the cortex (p=0.0021 and p<0.0001, respectively), in comparison to heterozygous IAPP+/− × APP, APP STZ-injected, and untreated APP animals. Moreover, the number of plaques found in IAPP+/+ × APP animals was also higher than the other control groups (Fig. 2C–D), indicating that the presence of IAPP in AD animals can increase both Aβ burden and plaque density in the brain (p=0.0012 and p<0.0001, respectively). We also detected that a significant percentage of the deposits were bigger (more than 500 μm2) in IAPP+/+ × APP compared to APP or STZ-injected animals (Fig. 2E–F; p=0.0074), suggesting that the presence of IAPP induced Aβ plaques to grow faster. Interestingly, double transgenic mice harboring one copy of the IAPP gene have an intermediate phenotype, showing significantly higher amyloid pathology than the control groups in some, but not all measurements (Fig. 2). To evaluate a possible interaction between IAPP and Aβ aggregates, we double-stained brain samples using antibodies that recognize human IAPP and Aβ, and the results clearly showed that both proteins were detectable in amyloid plaques (Fig. 2G). Interestingly, when pancreas tissue from these animals was examined, we could observe that the load of IAPP aggregates was substantially higher in animals over-expressing the human mutant APP gene (Supplementary Fig 1), suggesting that Aβ pathology may also promote pancreatic IAPP aggregation.
Figure 2. IAPP+/+ × APP mice display increased Aβ-immunopositive deposition in the brain.
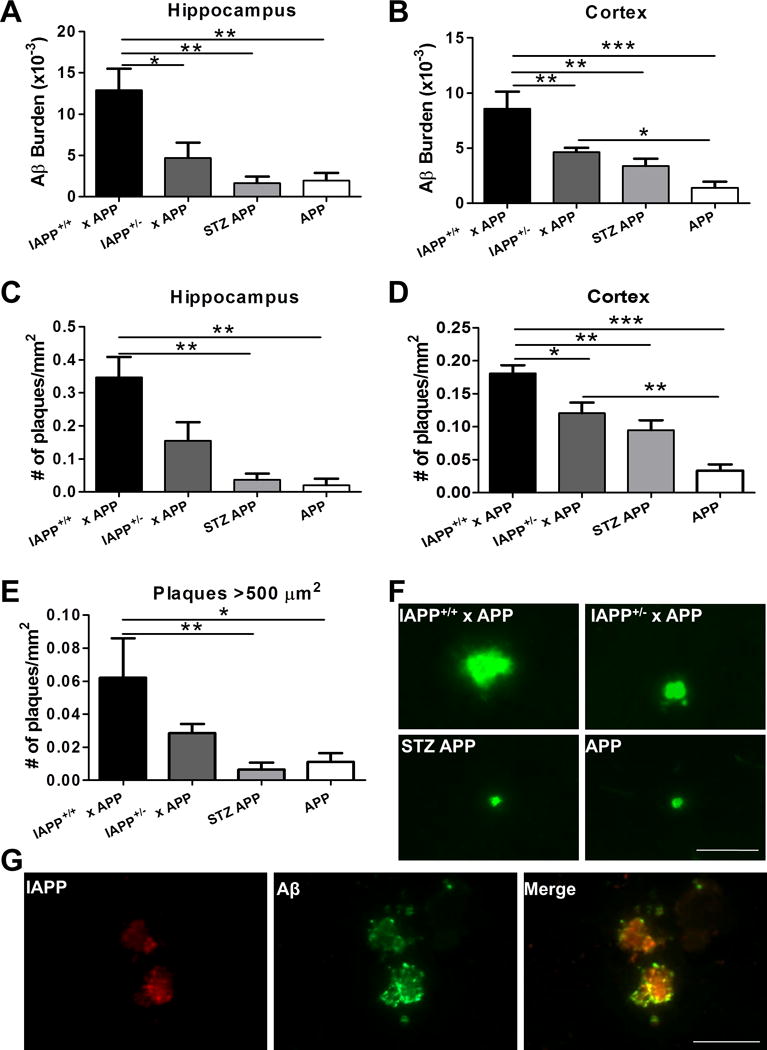
Aβ deposition was analyzed in the brain of transgenic animals over-expressing human IAPP and APP and control animals by immunohistological staining using 4G8 antibody. n=5–10 animals/group; 5 sections/animal. A–B: Brain Aβ burden was quantified as the immune-reactive area per total area analyzed in IAPP+/+ × APP, IAPP+/− × APP, APP animals injected with STZ, and untreated APP mice in hippocampal and cortical areas. C–D: Amyloid plaque density was measured as the number of plaques per mm2 in the hippocampus and cortices. E: Plaques bigger than 500 μm2 were quantified in the brain of experimental and control groups. Data in panel A–E was analyzed by one-way ANOVA, followed by the Tukey’s multiple comparison post-hoc test. *p<0.05; **p<0.01; ***p<0.001. F: Representative pictures of amyloid plaques in the cortical area reactive to human anti-Aβ antibody 82E1 in analyzed groups. G: Double immune-staining of IAPP (in red) and Aβ (in green), and co-localization (yellow) in the cortical area of IAPP+/+ × APP transgenic mice. Scale bar: 50 μm.
Intracerebral inoculation of IAPP aggregates promotes Aβ aggregation
To study in a more direct manner if the presence of IAPP aggregates was indeed responsible for the enhanced amyloid pathology observed in our double transgenic animals, we intracerebrally injected APP mice with pancreas homogenate (PH) from IAPP transgenic mice harboring extensive amounts of IAPP aggregates. For this experiment, the following groups of intracerebral inoculations were done: 1) APP transgenic mice inoculated with PH from IAPP+/+ animals containing high levels of ThS-positive IAPP aggregates (Suppl. Fig. 2), 2) APP mice inoculated with PH from WT mice with STZ-induced T1D and lacking IAPP aggregates, and 3) WT animals inoculated with PH containing aggregated IAPP. In addition, we also analyzed untreated APP animals.
As before, animals were sacrificed at 8.5 months of age and accumulation of Aβ deposits was evaluated by histological and biochemical techniques. Analysis of Aβ deposition showed that the APP animals inoculated with IAPP-containing PH displayed a significantly higher Aβ burden in both the cortex and the hippocampus with respect to controls (Fig. 3A–C; p=0.0001 and p=0.0054, respectively). This increase was also observed in the number (Fig. 3D–E; p=0.0002 and p=0.013, respectively) and the size of plaques (Fig. 3F; p=0.0002 and p=0.036, respectively; ˃500 μm2 in the cortex). To measure the effect of injecting pancreas extract containing IAPP aggregates by a different procedure, we performed biochemical studies to quantify the levels of soluble and insoluble Aβ using a sequential extraction procedure that enables to separate low molecular weight aggregates (soluble material) and large fibrillar aggregates (insoluble material). As shown in figure 3G and H, the amount of soluble and insoluble Aβ measured in the brain of APP transgenic mice injected with PH from IAPP mice was significantly higher compared with that from untreated animals.
Figure 3. Aβ pathology is aggravated by exogenous addition of pancreas homogenates containing IAPP aggregates.
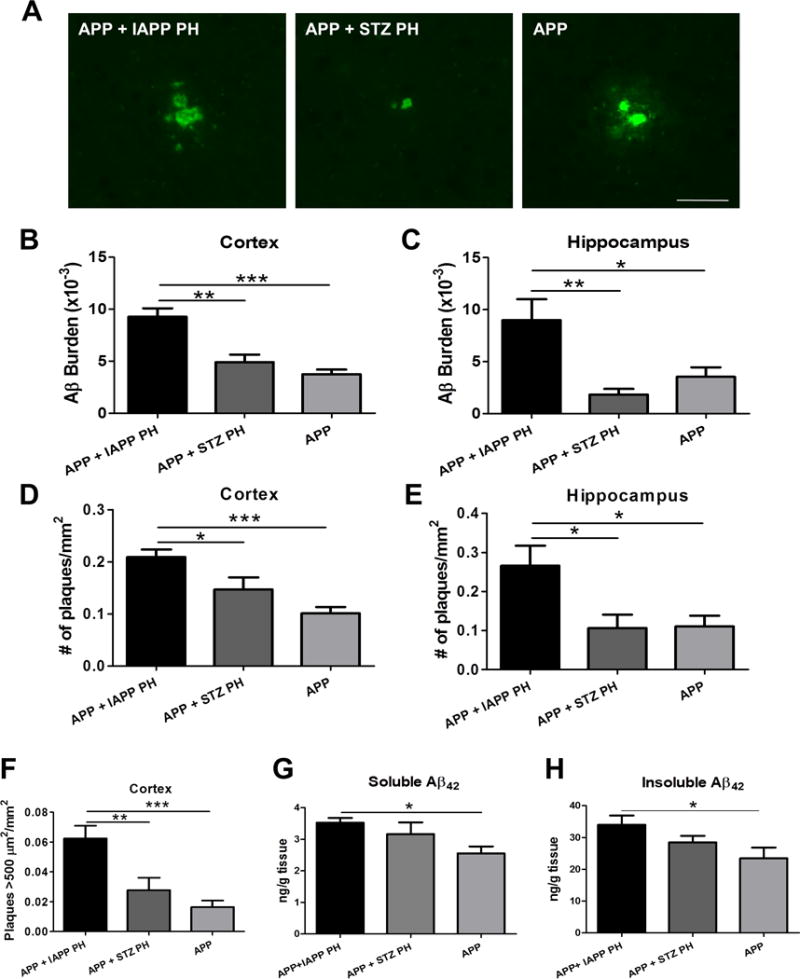
A: Representative microphotography of fluorescent immuno-labeled Aβ in the brain of APP animals intracerebrally inoculated with IAPP-PH, WT STZ-injected PH and untreated APP animals. Scale bar: 25 μm. B–C: Immunohistochemical quantification of Aβ burden (immune-reactive area per total area analyzed) in cortical and hippocampal brain areas in experimental and control groups. D–E: The number of plaques per mm2 was quantified to analyze differences in the amyloid plaque density in the brain of APP mice inoculated with IAPP-PH, WT STZ-injected PH and untreated APP animals. F: The density of plaques bigger than 500 μm2 per mm2 was quantified in the cortical area of experimental and control groups. G-H: Whole brain homogenate of APP mice intracerebrally inoculated with IAPP-PH, WT STZ-injected PH, and untreated APP animals was fractionated by ultracentrifugation. PBS and formic acid soluble fractions containing either oligomeric soluble (panel G) or fibrillar insoluble Aβ (panel H) were extracted and quantified using an ELISA kit to measure Aβ42 protein concentration per gram of tissue. Data in panels B–H was statistically analyzed by One-way ANOVA, followed by the Tukey’s multiple comparison test, *p<0.05; **p<0.01; ***p<0.001. n=5–10 animals/group; 5 sections/animal.
Intracerebral inoculation of IAPP aggregates enhances memory impairment
To study whether the exacerbated amyloid deposition observed after injection of PH containing IAPP aggregates resulted in cognitive impairment, we measured memory performance in the same group of animals. For this purpose, animals from each experimental and control group were subjected to the Barnes maze test at the age of 8 months. In comparison to the control groups, animals injected with IAPP-containing PH took more time to find the escape box throughout the entire learning period (Fig. 4A; p<0.0001) and performed worse when evaluated for short term memory (Fig. 4B; p=0.0015).
Figure 4. APP animals inoculated with pancreas homogenate containing aggregated IAPP display learning and memory deficits.
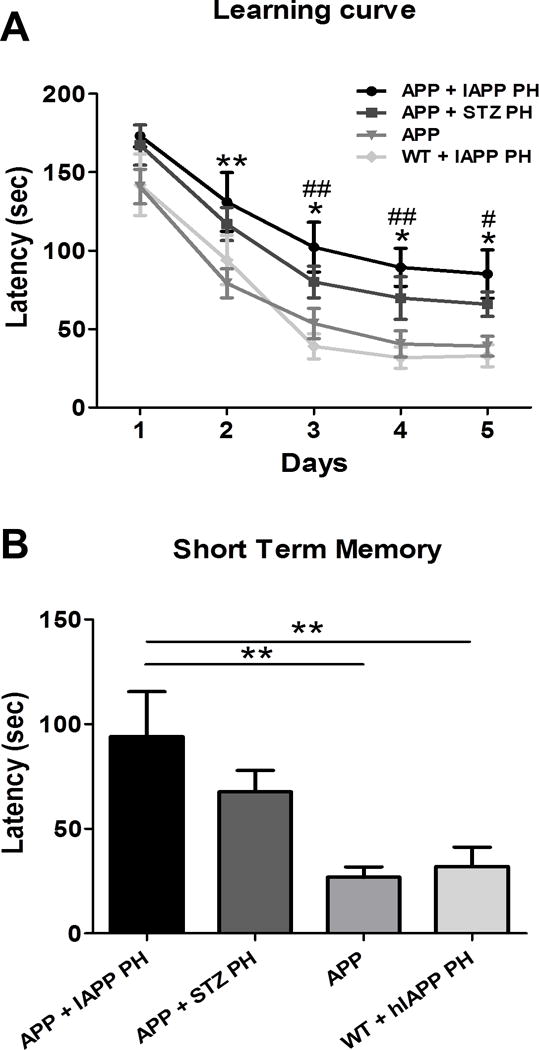
Learning and memory was measured by Barnes maze, a spatial working memory, hippocampal dependent task that measures the spatial navigation and memory of an animal. A: Learning was measured in experimental and control groups at 8 months old in 5–10 animals for five consecutive days as the primary latency in seconds. Two-way ANOVA, Tukey’s: *p<0.05, **p<0.01 compared to untreated APP; #p<0.05, ##p<0.01 compared to inoculated WT. B: Short term memory was measured as the time in seconds the animals take to reach the escaping box (latency) in the day 5 after the learning period. One-way ANOVA, Tukey’s: **p<0.01.
Collectively, these results suggest that intracerebral inoculation of PH containing IAPP aggregates accelerates Aβ deposition and enhances memory impairment.
Discussion
Numerous epidemiological and experimental studies have indicated that individuals with diabetes have a higher risk to develop dementia and AD, providing a robust link between T2D and AD11,19,33. Insulin resistance has been shown to be associated with acceleration of Aβ fibrillogenesis34 and cognitive impairment35. Insulin administration resulted in a reduction of amyloid accumulation, and improved cognitive performance in mice and humans affected by AD36,37. Experimental induction of diabetes has also been shown to promote the accumulation of ptau38,39. Moreover, treatment with anti-diabetic drugs (including roziglitazone, pioglitazone, and liraglutide) was reported to reduce Aβ and tau deposition and prevent memory impairment in animal models of AD40,41 as well as to provide some cognitive improvement in patients affected by AD42–44. It is often assumed that reduced glucose metabolism in the brain of diabetic patients or a negative effect of the lack of insulin or insulin resistance might be the main culprit of increasing the risk for AD development. Since both AD and T2D are protein misfolding disorders characterized by the accumulation of protein aggregates in the respective target organs, in this study we analyzed a putative alternative mechanism for the association, based on the direct interaction between IAPP and Aβ aggregates. In this study we show that synthetic IAPP aggregates are able to seed and enhance Aβ aggregation in vitro and both peptides appear to be part of the same fibrillar aggregates. The ability to cross-seed may reside in the shared characteristics between Aβ and IAPP misfolded aggregates. Both Aβ and IAPP are peptides of similar length produced by the proteolytic processing of a larger protein. Moreover, they have a 25% amino acid sequence identity, particularly in regions responsible for misfolding and aggregation45. Finally, both peptides are highly amyloidogenic and considered to be cytotoxic in their oligomeric conformation46,47.
Interestingly, it has been reported that IAPP aggregates can be found in the brain of patients affected simultaneously by T2D and AD9. Some of the IAPP aggregates appear associated with Aβ deposits, suggesting that IAPP and Aβ may also interrelate in vivo in human patients9,45. A similar result was found in transgenic rats over-expressing human IAPP in the pancreas, which also display IAPP deposits in the brain parenchyma48. In order to study the potential of misfolded IAPP to reach the brain, interact with Aβ, and modify AD pathology, we generated a double transgenic animal model that develops simultaneously pancreatic IAPP and cerebral Aβ deposits. In these mice, IAPP over-expression in the pancreatic β-cells triggers the accumulation of IAPP aggregates in the islets of Langerham and glucose impairment to precede Aβ plaque formation in the brain. This transgenic model resembles the onset and development of both diseases, since T2D pathology likely precedes AD brain alterations in human patients. We hypothesize that IAPP aggregates generated in the pancreas circulate through the blood, get into the brain, and can nucleate the misfolding and aggregation of the Aβ peptides that are locally produced. Consistent with our hypothesis, over-production of human IAPP in APP mice promotes higher accumulation of Aβ in the brain, and IAPP staining is readily detectable in the core of senile plaques. Furthermore, we can observe a dose-dependency in the Aβ burden, plaque density, and size of Aβ plaques in relation to the expression levels of IAPP in our double transgenic animals. IAPP+/+ × APP mice developed more and bigger plaques than IAPP+/− × APP animals, indicating that the amount of IAPP produced in the pancreas regulates Aβ deposition in the brain in a dose-dependent manner. Interestingly, the load of IAPP aggregates in the pancreas of the double transgenic mice was also increased compared with animals expressing only IAPP, suggesting that Aβ aggregates may also promote T2D abnormalities. Of note, epidemiological studies have shown an increase risk for T2D in patients affected by AD17.
Our results are supported by a previous observation in monkeys that spontaneously develop both AD and T2D pathologies, which showed accelerated Aβ pathology compared to non-diabetic monkeys49. Although previous studies have reported that induction of T1D leads to accelerated brain pathology in transgenic animal models by increasing levels of Aβ4250,51, under our experimental conditions, there was not significant difference in Aβ deposition in animals with diabetes induced by streptozotocin administration compared to untreated APP mice. This result indicates that increased Aβ deposition is likely not produced by glucose metabolism impairment.
To further characterize the effect of IAPP in Aβ pathology, we exogenously inoculated APP animals with PH containing aggregated IAPP. As a control, PH without IAPP aggregates was used. In addition, WT animals were inoculated with IAPP-containing PH. Our results showed that APP mice injected intra-cerebrally with PH harboring IAPP aggregates have increased Aβ deposition compared to control groups. Strikingly, this group of animals displayed significant problems in learning and memory, likely as a consequence of the exacerbated brain pathology. This data goes along with a recent study from Westermark and colleagues describing that synthetic IAPP or Aβ aggregates intravenously injected into transgenic mice over-expressing IAPP, accelerated pancreatic pathology, providing further support for cross-seeding interaction between these peptides45. Unfortunately, in this study, the effect of IAPP aggregates in cerebral Aβ deposition was not analyzed.
In conclusion, our findings suggest that IAPP and Aβ may interact by cross-seeding, providing a potential new mechanistic explanation for the higher risk of AD in people affected by T2D. Obviously, our findings do not rule out the implication of other pathways in the linkage, such as glucose metabolism, insulin resistance, oxidative or endoplasmic reticulum stress, insulin-induced amyloid pathology, or impairments of clearance pathways, among others. Further studies need to be conducted to explore in more detail a cross-seeding interaction between IAPP and Aβ and its role in disease induction and progression.
Supplementary Material
Acknowledgments
We thank Dr. Peter C. Butler (University of California at Los Angeles) for the donation of the IAPP diabetic colony and Dr. Charles Mays (University of Texas Medical School at Houston) for extensive editing of the manuscript. This work was partially funded by NIH grant R01GM100453 to CS.
Footnotes
Conflict of interests
The authors declare no competing financial interests.
References
- 1.Chiti F, Dobson CM. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 2.Soto C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat Rev Neurosci. 2003;4:49–60. doi: 10.1038/nrn1007. [DOI] [PubMed] [Google Scholar]
- 3.Clinton LK, Blurton-Jones M, Myczek K, Trojanowski JQ, LaFerla FM. Synergistic Interactions between Abeta, tau, and alpha-synuclein: acceleration of neuropathology and cognitive decline. J Neurosci. 2010;30:7281–7289. doi: 10.1523/JNEUROSCI.0490-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giasson BI, Lee VM, Trojanowski JQ. Interactions of amyloidogenic proteins. Neuromolecular Med. 2003;4:49–58. doi: 10.1385/NMM:4:1-2:49. [DOI] [PubMed] [Google Scholar]
- 5.Walker L, McAleese KE, Thomas AJ, Johnson M, Martin-Ruiz C, Parker C, et al. Neuropathologically mixed Alzheimer’s and Lewy body disease: burden of pathological protein aggregates differs between clinical phenotypes. Acta Neuropathol. 2015;129:729–748. doi: 10.1007/s00401-015-1406-3. [DOI] [PubMed] [Google Scholar]
- 6.Kovacs GG, Alafuzoff I, Al-Sarraj S, Arzberger T, Bogdanovic N, Capellari S, et al. Mixed brain pathologies in dementia: the BrainNet Europe consortium experience. Dement Geriatr Cogn Disord. 2008;26:343–350. doi: 10.1159/000161560. [DOI] [PubMed] [Google Scholar]
- 7.Barker WW, Luis CA, Kashuba A, Luis M, Harwood DG, Loewenstein D, et al. Relative frequencies of Alzheimer disease, Lewy body, vascular and frontotemporal dementia, and hippocampal sclerosis in the State of Florida Brain Bank. Alzheimer Dis Assoc Disord. 2002;16:203–212. doi: 10.1097/00002093-200210000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Guerrero-Munoz MJ, Castillo-Carranza DL, Krishnamurthy S, Paulucci-Holthauzen AA, Sengupta U, Lasagna-Reeves CA, et al. Amyloid-beta oligomers as a template for secondary amyloidosis in Alzheimer’s disease. Neurobiol Dis. 2014;71:14–23. doi: 10.1016/j.nbd.2014.08.008. [DOI] [PubMed] [Google Scholar]
- 9.Jackson K, Barisone GA, Diaz E, Jin LW, Decarli C, Despa F. Amylin deposition in the brain: A second amyloid in Alzheimer disease? Ann Neurol. 2013;74:517–526. doi: 10.1002/ana.23956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miklossy J, Qing H, Radenovic A, Kis A, Vileno B, Laszlo F, et al. Beta amyloid and hyperphosphorylated tau deposits in the pancreas in type 2 diabetes. Neurobiol Aging. 2010;31:1503–1515. doi: 10.1016/j.neurobiolaging.2008.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Biessels GJ, Staekenborg S, Brunner E, Brayne C, Scheltens P. Risk of dementia in diabetes mellitus: a systematic review. Lancet Neurol. 2006;5:64–74. doi: 10.1016/S1474-4422(05)70284-2. [DOI] [PubMed] [Google Scholar]
- 12.Luchsinger JA, Reitz C, Patel B, Tang MX, Manly JJ, Mayeux R. Relation of diabetes to mild cognitive impairment. Arch Neurol. 2007;64:570–575. doi: 10.1001/archneur.64.4.570. [DOI] [PubMed] [Google Scholar]
- 13.Kopf D, Frolich L. Risk of incident Alzheimer’s disease in diabetic patients: a systematic review of prospective trials. J Alzheimers Dis. 2009;16:677–685. doi: 10.3233/JAD-2009-1011. [DOI] [PubMed] [Google Scholar]
- 14.Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology. 1999;53:1937–1942. doi: 10.1212/wnl.53.9.1937. [DOI] [PubMed] [Google Scholar]
- 15.Velayudhan L, Poppe M, Archer N, Proitsi P, Brown RG, Lovestone S. Risk of developing dementia in people with diabetes and mild cognitive impairment. Br J Psychiatry. 2010;196:36–40. doi: 10.1192/bjp.bp.109.067942. [DOI] [PubMed] [Google Scholar]
- 16.Morris JK, Vidoni ED, Honea RA, Burns JM. Impaired glycemia increases disease progression in mild cognitive impairment. Neurobiol Aging. 2014;35:585–589. doi: 10.1016/j.neurobiolaging.2013.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC. Increased risk of type 2 diabetes in Alzheimer disease. Diabetes. 2004;53:474–481. doi: 10.2337/diabetes.53.2.474. [DOI] [PubMed] [Google Scholar]
- 18.Qiu WQ, Wallack M, Dean M, Liebson E, Mwamburi M, Zhu H. Association between amylin and amyloid-beta peptides in plasma in the context of apolipoprotein E4 allele. PLoS One. 2014;9:e88063. doi: 10.1371/journal.pone.0088063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Biessels GJ, Kappelle LJ. Increased risk of Alzheimer’s disease in Type II diabetes: insulin resistance of the brain or insulin-induced amyloid pathology? Biochem Soc Trans. 2005;33:1041–1044. doi: 10.1042/BST0331041. [DOI] [PubMed] [Google Scholar]
- 20.Jarrett JT, Lansbury PT., Jr Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer’s disease and scrapie? Cell. 1993;73:1055–1058. doi: 10.1016/0092-8674(93)90635-4. [DOI] [PubMed] [Google Scholar]
- 21.Soto C, Estrada L, Castilla J. Amyloids, prions and the inherent infectious nature of misfolded protein aggregates. Trends Biochem Sci. 2006;31:150–155. doi: 10.1016/j.tibs.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 22.Morales R, Moreno-Gonzalez I, Soto C. Cross-seeding of misfolded proteins: implications for etiology and pathogenesis of protein misfolding diseases. PLoS Pathog. 2013;9:e1003537. doi: 10.1371/journal.ppat.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan J, Fu X, Ge F, Zhang B, Yao J, Zhang H, et al. Cross-seeding and cross-competition in mouse apolipoprotein A-II amyloid fibrils and protein A amyloid fibrils. Am J Pathol. 2007;171:172–180. doi: 10.2353/ajpath.2007.060576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vitrenko YA, Gracheva EO, Richmond JE, Liebman SW. Visualization of aggregation of the Rnq1 prion domain and cross-seeding interactions with Sup35NM. J Biol Chem. 2007;282:1779–1787. doi: 10.1074/jbc.M609269200. [DOI] [PubMed] [Google Scholar]
- 25.Krebs MR, Morozova-Roche LA, Daniel K, Robinson CV, Dobson CM. Observation of sequence specificity in the seeding of protein amyloid fibrils. Protein Sci. 2004;13:1933–1938. doi: 10.1110/ps.04707004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O’Nuallain B, Williams AD, Westermark P, Wetzel R. Seeding specificity in amyloid growth induced by heterologous fibrils. J Biol Chem. 2004;279:17490–17499. doi: 10.1074/jbc.M311300200. [DOI] [PubMed] [Google Scholar]
- 27.Ono K, Takahashi R, Ikeda T, Mizuguchi M, Hamaguchi T, Yamada M. Exogenous amyloidogenic proteins function as seeds in amyloid beta-protein aggregation. Biochim Biophys Acta. 2014;1842:646–653. doi: 10.1016/j.bbadis.2014.01.002. [DOI] [PubMed] [Google Scholar]
- 28.Andreetto E, Yan LM, Tatarek-Nossol M, Velkova A, Frank R, Kapurniotu A. Identification of hot regions of the Abeta-IAPP interaction interface as high-affinity binding sites in both cross- and self-association. Angew Chem Int Ed Engl. 2010;49:3081–3085. doi: 10.1002/anie.200904902. [DOI] [PubMed] [Google Scholar]
- 29.Shahnawaz M, Soto C. Microcin amyloid fibrils are a reservoir of toxic oligomeric species. J Biol Chem. 2012 doi: 10.1074/jbc.M111.282533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, et al. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 31.Janson J, Soeller WC, Roche PC, Nelson RT, Torchia AJ, Kreutter DK, et al. Spontaneous diabetes mellitus in transgenic mice expressing human islet amyloid polypeptide. Proc Natl Acad Sci U S A. 1996;93:7283–7288. doi: 10.1073/pnas.93.14.7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tesch GH, Allen TJ. Rodent models of streptozotocin-induced diabetic nephropathy. Nephrology (Carlton) 2007;12:261–266. doi: 10.1111/j.1440-1797.2007.00796.x. [DOI] [PubMed] [Google Scholar]
- 33.Luchsinger JA. Type 2 diabetes and cognitive impairment: linking mechanisms. J Alzheimers Dis. 2012;30(Suppl 2):S185–S198. doi: 10.3233/JAD-2012-111433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yamamoto N, Matsubara T, Sobue K, Tanida M, Kasahara R, Naruse K, et al. Brain insulin resistance accelerates Abeta fibrillogenesis by inducing GM1 ganglioside clustering in the presynaptic membranes. J Neurochem. 2012;121:619–628. doi: 10.1111/j.1471-4159.2012.07668.x. [DOI] [PubMed] [Google Scholar]
- 35.Bourdel-Marchasson I, Lapre E, Laksir H, Puget E. Insulin resistance, diabetes and cognitive function: consequences for preventative strategies. Diabetes Metab. 2010;36:173–181. doi: 10.1016/j.diabet.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 36.Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B, et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology. 2008;70:440–448. doi: 10.1212/01.WNL.0000265401.62434.36. [DOI] [PubMed] [Google Scholar]
- 37.Haj-ali V, Mohaddes G, Babri SH. Intracerebroventricular insulin improves spatial learning and memory in male Wistar rats. Behav Neurosci. 2009;123:1309–1314. doi: 10.1037/a0017722. [DOI] [PubMed] [Google Scholar]
- 38.Kim B, Backus C, Oh S, Hayes JM, Feldman EL. Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology. 2009;150:5294–5301. doi: 10.1210/en.2009-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ke YD, Delerue F, Gladbach A, Gotz J, Ittner LM. Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One. 2009;4:e7917. doi: 10.1371/journal.pone.0007917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Escribano L, Simon AM, Gimeno E, Cuadrado-Tejedor M, Lopez de MR, Garcia-Osta A, et al. Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology. 2010;35:1593–1604. doi: 10.1038/npp.2010.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McClean PL, Parthsarathy V, Faivre E, Holscher C. The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci. 2011;31:6587–6594. doi: 10.1523/JNEUROSCI.0529-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate SR, Asthana S, et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005;13:950–958. doi: 10.1176/appi.ajgp.13.11.950. [DOI] [PubMed] [Google Scholar]
- 43.Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, et al. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006;6:246–254. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 44.Hanyu H, Sato T, Kiuchi A, Sakurai H, Iwamoto T. Pioglitazone improved cognition in a pilot study on patients with Alzheimer’s disease and mild cognitive impairment with diabetes mellitus. J Am Geriatr Soc. 2009;57:177–179. doi: 10.1111/j.1532-5415.2009.02067.x. [DOI] [PubMed] [Google Scholar]
- 45.Oskarsson ME, Paulsson JF, Schultz SW, Ingelsson M, Westermark P, Westermark GT. In vivo seeding and cross-seeding of localized amyloidosis: a molecular link between type 2 diabetes and Alzheimer disease. Am J Pathol. 2015;185:834–846. doi: 10.1016/j.ajpath.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 46.Janson J, Ashley RH, Harrison D, McIntyre S, Butler PC. The mechanism of islet amyloid polypeptide toxicity is membrane disruption by intermediate-sized toxic amyloid particles. Diabetes. 1999;48:491–498. doi: 10.2337/diabetes.48.3.491. [DOI] [PubMed] [Google Scholar]
- 47.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, et al. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Srodulski S, Sharma S, Bachstetter AB, Brelsfoard JM, Pascual C, Xie XS, et al. Neuroinflammation and neurologic deficits in diabetes linked to brain accumulation of amylin. Mol Neurodegener. 2014;9:30. doi: 10.1186/1750-1326-9-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Okabayashi S, Shimozawa N, Yasutomi Y, Yanagisawa K, Kimura N. Diabetes mellitus accelerates Abeta pathology in brain accompanied by enhanced GAbeta generation in nonhuman primates. PLoS One. 2015;10:e0117362. doi: 10.1371/journal.pone.0117362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gotz J, Ittner LM, Lim YA. Common features between diabetes mellitus and Alzheimer’s disease. Cell Mol Life Sci. 2009;66:1321–1325. doi: 10.1007/s00018-009-9070-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jolivalt CG, Hurford R, Lee CA, Dumaop W, Rockenstein E, Masliah E. Type 1 diabetes exaggerates features of Alzheimer’s disease in APP transgenic mice. Exp Neurol. 2010;223:422–431. doi: 10.1016/j.expneurol.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.