

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a devastating disease. Circulating tumor cells (CTC) in the blood are hypothesized as the means of systemic tumor spread. Blood obtained from healthy donors and patients with PDAC was therefore subject to size-based CTC-isolation. We additionally compared Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations in pancreatic CTC and corresponding tumors, and evaluated their significance as prognostic markers. Samples from 68 individuals (58 PDAC patients, 10 healthy donors) were analyzed; CTCs were present in patients with UICC stage IA-IV tumors and none of the controls (p < 0.001). Patients with >3 CTC/ml had a trend for worse median overall survival (OS) than patients with 0.3–3 CTC/ml (P = 0.12). Surprisingly, CTCs harbored various KRAS mutations in codon 12 and 13. Patients with a KRAS G12V mutation in their CTC (n = 14) had a trend to better median OS (24.5 months) compared to patients with other (10 months), or no detectable KRAS mutations (8 months; P = 0.04). KRAS mutations in CTC and corresponding tumor were discordant in 11 of 26 “tumor-CTC-pairs” (42%), while 15 (58%) had a matching mutation; survival was similar in both groups (P = 0.36). Genetic characterization, including mutations such as KRAS, may prove useful for prognosis and understanding of tumor biology.
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related death in the United States and Europe1,2. Incidence almost equals mortality with a 5-year survival rate of <6%2. This is mostly due to its often late diagnosis at metastatic stages, its aggressive biology and only partial response to known chemotherapies3.
To date, most treatment decisions are made based on the tumor stage evaluated by fine needle aspiration (FNA) cytology and cross-sectional imaging, and patients are “under staged” in about 20% of the cases since metastatic disease is often only visible upon operative exploration4. Conventional prognostic factors such as tumor size, nodal status and perineural invasion can be evaluated only after resection and mostly confirm the poor prognosis. Even after complete tumor resection, more than 80% of the patients develop local or distant tumor recurrence2. These figures highlight the need for a biomarker that can improve diagnosis and staging, and that contributes to our understanding of the tumor biology.
Circulating tumor cells (CTCs) in the blood stream are thought to represent disseminated tumor cells that have detached from the primary lesion and that are undetectable by clinical imaging and inaccessible to excision. These cells have been isolated and evaluated for the diagnostic workup and treatment monitoring of various cancers, including prostate, lung, colorectal, and breast cancer5–14 and are thought to undergo epithelial to mesenchymal transition (EMT) to enter the blood stream and to seed in distant organs.
While CTCs have been extensively studied in the mentioned neoplasms, their significance in PDAC at various stages is not known. There is, however, emerging evidence that CTCs may also serve as a valuable tool for outcome prediction and understanding of tumor biology in PDAC15–19. Previous studies in PDAC used techniques that depended on the CTC capture with antibodies to epithelial cell surface antigens. They have reported low rates of CTC positivity (5–50%) and CTC positivity was associated with at least a trend to worse progression-free and overall survival20–24. It is of note that if EMT does play an essential role in cancer cell spread in PDAC, CTC isolation methods that rely on epithelial surface markers alone are likely to provide an incomplete capture of the cells in the blood stream and may explain at least in part the low isolation rates and discrepant results in PDAC in the past18–21.
Point mutations in the Kirsten rat sarcoma viral oncogene homolog (KRAS) gene are present in over 90% of PDAC cases and are thought to be an early event in the development of PDAC, already occurring in PanIN 1A lesions of the pancreas25–27. The mutations typically affect the hotspot codons 12 or 13. Depending on the specific amino acid substitution, the mutations differ in their associated mRNA expression patterns, biochemical activity and transforming capacity28,29. The underlying biological processes are yet not understood.
The objective of our prospective study was to assess the impact of CTC counts on survival and the correlations of KRAS mutations in CTCs and corresponding primary tumor samples in patients with PDAC. For CTC isolation, we used a simple filtration-based technique which is independent of the CTC surface. We show that patients with >3 CTC/ml tend to have a worse overall survival (OS) than patients with 0.3–3 CTC/ml and that the KRAS mutations identified in CTCs or primary tumor may differ within the same patient.
Material and Methods
Patient selection
Patients with histologically-proven PDAC who were treated with either tumor resection or palliative bypass at our institution were included. Additionally, healthy donor blood was spiked by adding varying known quantities of PDAC cells to re-test the sensitivity of detection as partially previously described16. All experiments were performed in accordance with guidelines and regulations of good research practice. We confirm that all experimental protocols were approved by the institutional committee (independent Ethics Committee University of Freiburg). Written informed consent was obtained from each patient.
CTC isolation method
Three ml of peripheral blood samples were processed through ScreenCell® filtration devices (Paris, France) according to the manufacturer´s instructions, as previously described16,17, to capture CTCs within three hours of draw. The ScreenCell® system is an isolation-by-size method; the devices are fitted with microfilters that capture the cells on small metal-rimmed filters via low-pressure vacuum-filtration (Fig. 1a). Cell cytology was visualized with Giemsa staining, and DNA was isolated from cells captured from 6 ml blood on a parallel filter, the ScreenCell® MB device for DNA isolation. Control blood samples were obtained from healthy volunteers.
Figure 1.
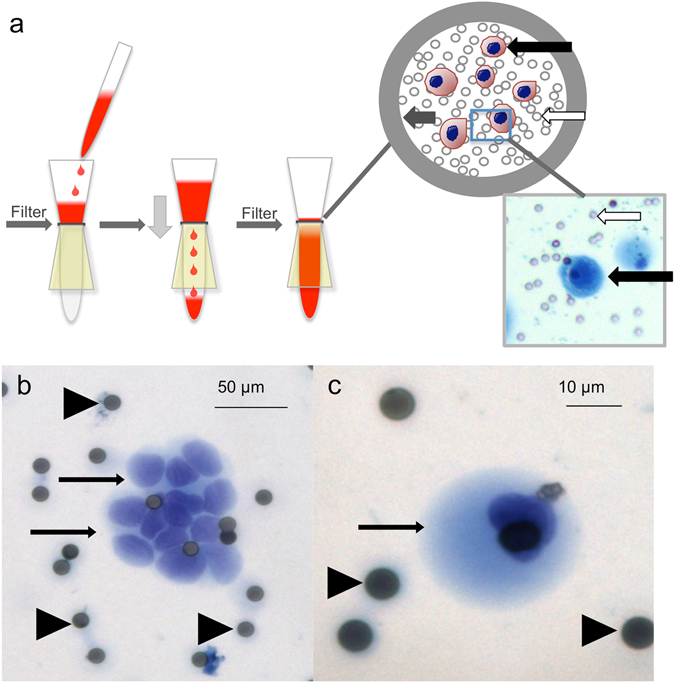
(a) Isolation method of CTC. White arrows point at filter pores, black arrows at “CTC” grey arrow points at the metal rim of the filter. Examples of CTC in patients with PDAC. (b) CTC cluster (patient 24), the scale bar represents 50 µm C single CTC (patient 23) the scale bar represents 10 µm. Arrows point at CTC, arrowheads at filter pores.
Cytological characterization of CTC
An experienced pancreatic cytopathologist (MBP) and two pathologists (PB, ST) evaluated the stained filters blinded to the histological diagnosis. The cells were categorized as positive, “suspicious”, or “negative” for malignant CTC based on established cytomorphological features for PDAC30. The results were evaluated independently. If the results showed inter-observer difference, the third observer re-evaluated the specimen and observers came to a final agreement.
Cells considered positive or diagnostic of malignant CTCs were epithelioid cells with markedly enlarged (8–20x filter pore size) irregular, hyperchromatic nuclei and scant, well-defined cytoplasm or smaller epithelioid cells (2–7x filter pore size) with round to oval nuclei, occasional nuclear groove and ill-defined but visible cytoplasm in small or large clusters (cluster and single cell Fig. 1). Cells suspicious for malignant CTCs were enlarged, clumped cells with molded nuclei but poorly-defined or absent cytoplasm. CTC were counted by three independent reviewers and the number of CTCs was divided by 3 to estimate the number of CTC/ml.
Genotyping of KRAS in CTC samples and primary tumor
For genetic analysis of CTC, six ml of blood were filtered using the ScreenCell® MB device according to the manufacturer’s instructions. DNA of cells on the filter was isolated using the Qiagen Purgene core kit (QIAGEN, Hilden, Germany). Tumor samples were randomly obtained from an unspecific part of the tumor and micro-dissected by a technician. Due to tissue availability, only one region of the tumor was analyzed. Tumor DNA was extracted from paraffin-embedded samples via the QIAamp DNA FFPE Tissue Kit (QIAGEN, Hilden, Germany). The mutational status of KRAS codons 12 or 13 was determined using a peptide nucleic acid (PNA) and polymerase chain reaction (PCR)-based assay, as previously described16,31.
Briefly, this PNA clamp anneals to the wild-type area of the KRAS gene and prevents amplification of the KRAS wild-type DNA sequences. Thus, mutations in the KRAS gene can be identified even in very low concentrations of 2 cancer cells/ml blood16. PCR products were electrophoresed on a 4% agarose gel. In the presence of a KRAS mutation (KRAS mut), PCR products were extracted using the MinElute Gel extraction kit (QIAGEN), and cloned into the pGEM-T Easy vector system (Promega, Fitchburg, WI). Five clones were subsequently selected from each CTC specimen and 5 from each tumor specimen, and all 10 clones were sequenced (GATC Biotech®, Konstanz, Germany).
Droplet digital PCR (ddPCR) was used to validate selected mutations in KRAS p.G12D, KRAS p.G12V, KRAS p.G12S, KRAS p.G12C, KRAS p.G13D, and KRAS p.G13S. Four tumor-CTC “DNA pairs”, 3 tumor DNA specimens and 7 CTC DNA specimens were analyzed with confirmatory ddPCR. Each assay was performed four times using a Bio-Rad QX100 ddPCR system as described previously32. Mutations-specific ddPCR assays for detection of the KRAS mutations mentioned above were designed in-house. For the KRAS mutations, a dual-labeled locked nucleic acid (LNA) probe strategy was used with FAM and HEX as fluorescent dyes. For details of the analysis please see the supplementary material. Briefly, the raw fluorescence amplitude was analyzed using the Quantasoft version 1.6.6 software and used to obtain the fractional abundance for a given mutation. This was reported as the allele frequency. For calculation of the allele fraction the total number of droplets (with and without DNA) was used to calculate DNA copies/ml. For this, we divided the number of mutant copies by the number of total DNA copies (mutant plus wild-type), and multiplied by 100 to give the percentage (allele fraction) of mutant DNA copies.
Statistical analysis
Statistical evaluation was performed using Graphpad Prism 6 for MAC OS X (GraphPad Software, La Jolla California USA) with p values of p < 0.05 considered significant. The association of CTCs and the different subgroups in correlation with individual clinical characteristics, including T-stage, nodal involvement, resection status, UICC classification and operation technique, were compared using one-way ANOVA, Fisher´s exact test or chi-square test. Univariate survival analysis for CTC positivity, KRAS mutation status, and different subgroups of CTC features were performed using the Kaplan-Meier method and compared using the log-rank (Mantel-Cox) test. Survival was calculated from the time of study enrollment to death of the patient. Patients who were still alive at the end of the study period (February 2016) were censored. Within each ddPCR experiment, we ran 4 wells of wild-type (WT) DNA only control (human genomic DNA digested with HaeIII). A 95% confidence interval of false-positive mutant droplets was calculated for each mutation after pooling control wells from individual plates. Using 2-tailed unpaired t-test, we compared mutant droplet counts of a given sample to assay-specific false positives. This to our best knowledge reflects common practice of ddPCR interpretation (Rare Mutation Detection Best Practices Guidelines [Internet]. Feb 2015, Available: http://www.bio-rad.com/webroot/web/pdf/lsr/literature/Bulletin_6628.pdf, accessed 24 April 2017).
Results
Patient characteristics
We analyzed the blood specimens of 58 patients with histologically-proven PDAC (median age 67 years, range 41–92 years) for CTCs between February 2012 and June 2014.
Tumor stages varied from small node negative UICC IA tumors to metastatic stage IV disease (Table 1). Thirty-seven of the 58 (63.8%) patients underwent pancreatic resection, and 21 (36.2%) were bypassed. Forty-seven patients received adjuvant or palliative chemotherapy. The median follow-up time was 24 months for living and 10 months for deceased patients. Thirteen patients were alive at the end of the study period. Tissue samples were available from 37 patients. Three of the 10 control-blood samples were from healthy donors (Table 1).
Table 1.
Demographics, tumor characteristics, circulating epithelial cell status, and median survival of patients with PDAC (n = 58) and control patients (n = 10).
| Parameter | All | CTC KRAS G12V | CTC KRAS other mutation | Cyto positive KRAS WT | CTC Negative | P |
|---|---|---|---|---|---|---|
| N | 68 | 14 | 28 | 11 | 15/5 | |
| PDAC patients, n (%) | 58 | 14 (24%) | 28 (48%) | 11 (19%) | 5 (9%) | <0.0001 |
| Control group, n (%) | 10 | 0 (0%) | 0 (0%) | 0 (0%) | 10 (100%) | |
| Median age, years | 67 | 68 | 67 | 69 | 65 | 0.88 |
| T-Stage | ||||||
| T1, n (%) | 2 (3.5%) | 0 (0%) | 1 (1.7%) | 0 (0%) | 0 (0%) | 0.409 |
| T2, n (%) | 3 (5.2%) | 1 (1.7%) | 2 (3.4%) | 0 (0%) | 0 (0%) | |
| T3, n (%) | 30 (51.8%) | 6 (10.4%) | 15 (25.9%) | 5 (8.6%) | 4 (7%) | |
| T4, n (%) | 1 (3.4%) | 0 (0%) | 1 (1.7%) | 0 (0%) | 1 (1.7%) | |
| Tx, n (%) | 19 (37.9%) | 7 (12%) | 9 (15.5%) | 6 (10.4%) | 0 (0%) | |
| N-Stage | ||||||
| N0, n (%) | 8 (13.8%) | 3 (5.2%) | 5 (6.9%) | 0 (1.7%) | 0 (0%) | 0.376 |
| N1, n (%) | 28 (48.3%) | 4 (6.9%) | 14 (24.1%) | 5 (8.6%) | 5 (8.6%) | |
| Nx, n (%) | 22 (37.9%) | 7 (12%) | 9 (15.5%) | 6 (10.4%) | 0 (0%) | |
| Resection margin | ||||||
| R0, n (%) | 28 (48.3%) | 6 (10.4%) | 14 (24.1%) | 5 (8.6%) | 3 (5.2%) | 0.275 |
| R1, n (%) | 8 | 1 (1.7%) | 4 (6.9%) | 1 (1.7%) | 2 (3.4%) | |
| Not resected, n (%) | 22 (37.9%) | 7 (12%) | 9 (15.5%) | 6 (10.4%) | 0 (0%) | |
| UICC classification | ||||||
| IA, n (%) | 1 | 0 (0%) | 1 (1.7%) | 0 (0%) | 0 (0%) | 0.549 |
| IB, n (%) | 2 | 1 (1.7%) | 0 (0%) | 1 (1.7%) | 0 (0%) | |
| IIA, n (%) | 4 | 1 (1.7%) | 3 (5.2%) | 0 (0%) | 0 (0%) | |
| IIB, n (%) | 28 | 5 (8.6%) | 14 (24.1%) | 5 (8.6%) | 4 (6.9%) | |
| III, n (%) | 11 | 4 (6.9%) | 3 (5.2%) | 1 (1.7%) | 1 (1.7%) | |
| IV, n (%) | 12 | 3 (5.2%) | 6 (10.4%) | 5 (8.6%) | 0 (0%) | |
| Operation technique | ||||||
| PPPD/Whipple, n (%) | 28 (48.3%) | 6 (10.3%) | 13 (22.4%) | 6 (10.4%) | 3 (5.2%) | 0.149 |
| Bypass/ Exploration, n (%) | 21 (36.2%) | 6 (10.4%) | 9 (15.5%) | 5 (8.6%) | 1 (1.7%) | |
| Left resection, n (%) | 9 (15.5%) | 2 (3.5%) | 5 (8.6%) | 1 (1.7%) | 1 (1.7%) | |
UICC Union international contre le cancer, n.s. not significant.
Genetic and cytologic detection of PDAC cell lines spiked in whole blood
Whole blood to which PDAC cells were added (spiked) was used to determine the sensitivity of CTC detection by cytology and KRAS mutational capture analysis. PANC-1 cells that are known to contain a KRAS G12D mutation were spiked into whole blood to final concentrations of 10, 2 and 1 cells/ml. Cytological evaluation revealed the PANC-1 cells in all samples (n = 6) spiked with 10 cell/ml and was able to detect PANC-1 cells in 1 of 2 specimens spiked with 2 cells/ml or 1cell/ml respectively (Supplementary Table 1). Using mutant KRAS as a molecular biomarker for the presence of CTC, KRAS mutations were detected in all spiked samples (n = 16), including at concentrations as low as 1 cell/ml (Supplementary Table 1).
CTC counts and survival
Thirty-nine patients (67.3%) showed CTC clusters or single CTCs based on cytomorphology (Fig. 1a,b), 2 (3.4%) had cytology suspicious for CTCs and 17 (29.3%) were cytologically negative for CTC. Inter-observer consensus was high with a κ- value of 0.82 (PB vs. ST), 0.92 (PB vs. MBP) and (ST vs. MPB) 0.89 respectively (p > 0.001). Initial disagreement was present in 5 cases, and a majority decision was performed.
The total number of visible, malignant appearing CTCs on cytology ranged from 0–13 CTC/ml. In 42 of the patients (72.4%), we found ≥1 KRAS mutation(s) in the CTC (Tables 1 and 2). Twenty-eight (48.3%) patients had both, a CTC- positive cytologic specimen and ≥1 KRAS mutation(s) in the CTC. Five (8.7%) specimens showed neither a KRAS mutation nor a positive cytologic result. None of the 10 control blood samples had CTC on cytology or a KRAS mutation.
Table 2.
Pairwise comparison of KRAS mutations and overall survival in PDAC patients.
| Parameter | CTC KRAS G12V | CTC KRAS other mutation | CTC cyto positive KRAS WT | CTC Negative |
|---|---|---|---|---|
| n = 58 | 14 (24.1%) | 28 (48.3%) | 11 (19%) | 5 (8.6%) |
| CTC KRAS G12V | P = 0.21 | P = 0.04 | P = 0.26 | |
| CTC KRAS other mutation | P = 0.21 | P = 0.23 | P = 0.90 | |
| CTC positive KRAS WT | P = 0.04 | P = 0.23 | P = 0.67 | |
| CTC Negative | P = 0.26 | P = 0.90 | P = 0.67 | |
| Median OS, months | 24.5 | 10 | 8 | 8 |
| P | 0.63 | |||
| Alive at end of study period (2/2016) | 3 | 8 | 2 | 1 |
The mere cytological presence of CTCs had no influence on overall survival (P = 0.23; median overall survival, 12 vs 8 months). However, higher numbers of CTC/ml blood were associated with a trend for shorter overall survival. Patients with more than 3 CTC/ml blood (n = 16) had a median overall survival of 11.5 months and patients with 0.3–3 CTC/ml blood (n = 23) of 20 months (P = 0.12; Fig. 2).
Figure 2.
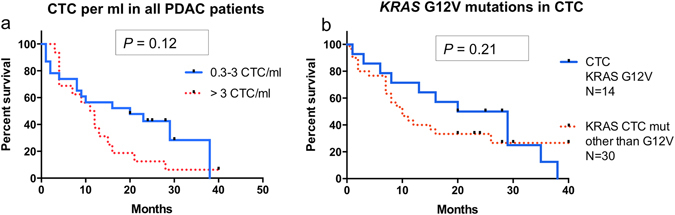
CTC and overall survival. (a) Survival analysis of PDAC patients with 0.3–3 CTC in the blood (blue, N = 23, MS 20 months) versus those with >3 CTC/ml blood (red dots, N = 16, MS 11.5 months; P = 0.12). (b) Survival analysis of PDAC patients with CTC containing a KRAS G12V mutation (MS 24.5 months) and other KRAS mutations (MS 10 months; P = 0.21).
Distribution of KRAS mutations in CTC and primary tumor
In the KRAS mutation analyses of the CTC and primary tumor specimens, we detected various types of codon 12 and 13 mutations (Table 3). The distribution of KRAS mutations in the CTC equals that of the mutations in the primary tumor, but CTC also showed rare KRAS mutations (Table 3). Mutations were present in 97.4% (n = 37) of the available 38 tissue samples. The samples mainly harbored c.35G > A (p.G12D; KRAS G12D) mutations: they were found in 57.8% of the samples (n = 22). The second most frequent mutation was c.35G > T (p.G12V; KRAS G12V) which was found in 16 of the 38 samples (42.1%). Multiple mutations were present in seven tumor specimens and eleven CTC specimens. We only observed overlap of these two groups in one patient (Table 3).
Table 3.
KRAS mutation status of the primary tumor and the CTC DNA in 58 patients; in 20 cases no primary tissue was available.
| KRAS mutation status | CTC DNA Sample (n = 58) | Primary Tumor Sample (n = 38) |
|---|---|---|
| Wild type (GGTGGC) (CTC cyto positive) | 11 | 1 |
| All KRAS mutations* | 57 | 44 |
| KRAS G12V (GTT)* | 14 | 16 |
| KRAS G12D (GAT)* | 21 | 22 |
| KRAS G13S (AGC)* | 5 | 2 |
| KRAS G13D (GAC)* | 8 | 4 |
| KRAS G12C (TGT)* | 2 | — |
| KRAS G12S (AGT)* | 6 | — |
| KRAS G12A (GCT)* | 1 | — |
| multiple KRAS mutations | 11 | 7 |
*Eleven patients had multiple mutations – all mutations are shown here; 6 patients with a KRAS G12D mutation had one additional mutation, and 3 patients with a KRAS G12V mutation had one additional mutation in their CTC. 4 patients had a combination of a KRAS G12V and a KRAS G12D mutation in the primary tumor.
The KRAS mutational analysis of CTCs revealed a c.35G > A (p.G12D; KRAS G12D) mutation in 21 specimens (Fig. 3), a c.35G > T (p.G12V; KRAS G12V) mutation in 14 specimens, and other KRAS mutations (c.35G > C (p.G12A), c.34G > T (p.G12C), c.34G > A (p.G12S), c.38G > A (p.G13D), c.37G > A (p.G13S)) in 22 specimens. Eleven patients had more than one KRAS mutation in their CTC. Two had a KRAS G12V mutation and were assigned to the KRAS G12V group, five had a KRAS G12D and one additional “other mutation” and were included into the KRAS other group, and four had multiple KRAS other mutations in their CTC (Table 3 and Fig. 4a). All patients with KRAS Non-G12V mutations are summarized as KRAS other. The frequencies of the varying mutations are in line with those previously described in PDAC (ref.33; Table 3). Five patients with available tumor samples harboring KRAS mutations did not show CTCs in the blood specimens. Among the patients with KRAS other mutations, those with KRAS G12D had a median overall survival of 9 months (n = 21). Survival analysis additionally revealed 8 months for patients with no detectable KRAS mutation in their CTC but positive CTC cytology (KRAS wt, n = 11), 10 months for patients with KRAS other mutations including KRAS G12D (n = 30) and 9 months for patients without detected CTC. Patients with KRAS G12V had the longest median overall survival with 24.5 months (n = 14); this was however not statistically significant on overall comparison (P = 0.63; Table 2). In pairwise comparison of patients with KRAS G12V and those with KRAS wt (but CTC in the parallel cytology specimen), the overall survival was significantly longer among patients with a KRAS G12V mutation (P = 0.04, Fig. 2b, Table 2).
Figure 3.
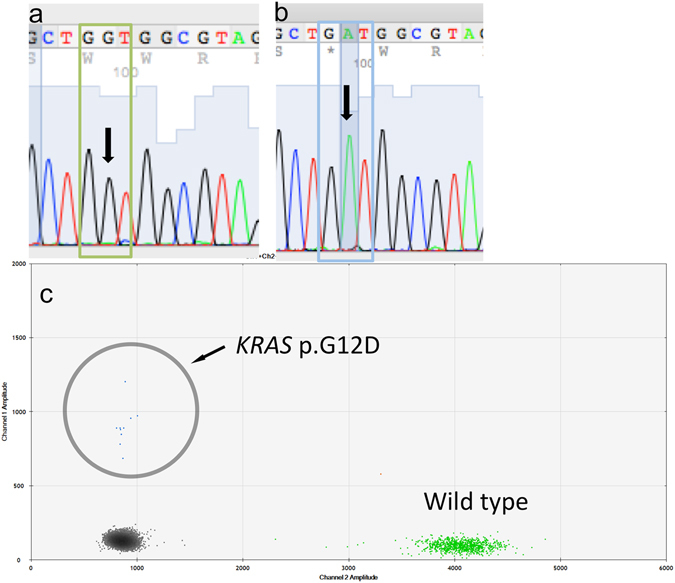
(a) Example Chromatogram (antisense strand) wild-type KRAS in a negative control sample. (b) Example Chromatogram (antisense strand) KRAS c.35G > A (pG12D) in a CTC sample. (c) Example ddPCR result (Patient 45) for KRAS G12D. The number of events is very low which represents a very low copy of mutant KRAS G12D molecules in the specimen. The circle encircles the area of interest and the approximately 6 events.
Figure 4.
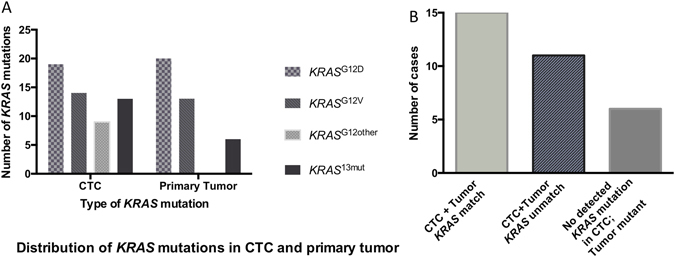
Distribution of KRAS mutations in CTC and the primary tumor. (a) KRAS mutations in CTC and the primary tumor: Several patients (n = 13) had multiple mutations in the CTC or primary tumor, in 20 cases no tissue was available. (b) Substantial discordance of KRAS mutations in CTC and the primary tumor (n = 9): those with no available tissue samples were excluded (n = 20); 5 were CTC negative. Cyto pos = cytology positive; MS = median survival (in months).
Substantial discordance in KRAS mutations in CTC and primary tumor
Of the 38 patients with KRAS information on both the CTCs and the tumor, one was KRAS wild-type in the primary tumor, and five patients had no detectable CTC, resulting in 32 tumor-CTC “pairs”: Six patients had no detected KRAS mutation in the CTCs (e.g. due to detection limitations), but in the primary tumor (18.7%). Of the 26 patients with detected mutations in both the CTC and the primary tumor, 15 (58%) patients had at least one matching mutation (CTC + Tumor KRAS match), while in 11 (42%) patients, the KRAS mutations differed between the CTCs and the tumor (CTC + Tumor KRAS unmatch; Fig. 4). Eighteen selected tumor and CTC specimens - mainly with multiple mutations - were additionally analyzed with the quantitative droplet digital PCR (ddPCR) method to confirm these heterogeneous results. In the selected cases with multiple mutations all mutations were confirmed in ddPCR, revealing extraordinary low allele-frequencies of <0.03%. It is of note, that the performed t-tests showed not significant results in about half of the cases (Table 4, Fig. 3c, Supplementary Table 2); ddPCR did however not detect all mutations in the analyzed CTC specimens (Table 4, Supplementary Table 2). Interestingly, four additional KRAS mutations were identified by the ddPCR technique (two tumor specimens, two CTC specimens).
Table 4.
Comparison of KRAS mutation results in clamping PCR and ddPCR in selected patient samples, mainly with multiple mutations in CTC or primary tumor.
| Patient Sample ID | Tumor/CTC | KRAS mutation (codon 12/13) in clamping PCR | KRAS mutation (codon 12/13) in ddPCR | Allele fraction (ddPCR): % of mutant DNA |
|---|---|---|---|---|
| 2/6T | Tumor | KRAS G12D (GAT), | KRAS G12D (GAT), | 43.6 |
| KRAS G13D (GAC) | (KRAS G12V (GTT)) | 6.7 | ||
| 2/6CTC | CTC | KRAS G12V (GTT) | (KRAS G12V (GTT)) | (0.09) |
| (KRAS G12D (GAT)) | (0.1) | |||
| 2/33T | Tumor | KRAS G13D (GAC) | KRAS G13D (GAC) | 0.13 |
| 2/33CTC | CTC | KRAS G12D (GAT) | (KRAS G12D (GAT)) | 0.18 |
| (KRAS G13D (GAC)) | 0.02 | |||
| 2/40T | Tumor | KRAS G12D (GAT) | KRAS G12D (GAT) | 20.5 |
| 2/40CTC | CTC | KRAS G12D (GAT) | KRAS G12D (GAT) | 2.5 |
| 2/45T | Tumor | KRAS G12D (GAT), | KRAS G12D (GAT), | 7.2 |
| KRAS G12V (GTT) | (KRAS G12V (GTT)) | 1.15 | ||
| (KRAS G13S (AGC)), | 4.7 | |||
| 2/45CTC | CTC | KRAS G12V (GTT), | (KRAS G12V (GTT)), | 0.7 |
| KRAS G13S (AGC) | (KRAS G13S (AGC)), | 0.5 | ||
| (KRAS G12D (GAT)) | 1.3 | |||
| 2/12T | Tumor | KRAS G12D (GAT), | KRAS G12D (GAT), | 4.1 |
| KRAS G12V (GTT) | KRAS G12V (GTT) | 8.7 | ||
| 2/18T | Tumor | KRAS G12D (GAT), | KRAS G12D (GAT), | 23.2 |
| KRAS G13D (GAC) | KRAS G13D (GAC) | 0.16 | ||
| 2/30T | Tumor | KRAS G12D (GAT), | KRAS G12D (GAT), | 36.2 |
| KRAS G12V (GTT) | KRAS G12V (GTT) | 17.7 | ||
| 2/14CTC | CTC | KRAS G13S (AGC), | KRAS G13S (AGC), | 1.1 |
| KRAS G12V (GTT) | KRAS G12V (GTT), | 2.6 | ||
| 1/26CTC | CTC | KRAS G13S (AGC) | (KRAS G13S (AGC)) | 0.12 |
| 1/28CTC | CTC | KRAS G12D (GAT), | (KRAS G12D (GAT)), | 0.03 |
| KRAS G13D (GAC) | (KRAS G13D (GAC)) | 0.03 | ||
| 1/32CTC | CTC | KRAS G12S (AGT) | (KRAS G12S (AGT)) | 0.10 |
| 2/34CTC | CTC | KRAS G13D (GAC), | KRAS G13D (GAC), | 3.3 |
| KRAS G12S (AGT) | (KRAS G12S (AGT)) | 0.5 | ||
| 2/50CTC | CTC | KRAS G12D (GAT), | KRAS G12D (GAT) | 42.7 |
| KRAS G12C (TGT) | (KRAS G12C (TGT)) | 3.9 | ||
| 2/56CTC | CTC | KRAS G12C (TGT) | (KRAS G12C (TGT)) | 0.04 |
| Spiking 2 cells/ml | “CTC” | KRAS G12D (GAT) | KRAS G12D (GAT) | 1.9 |
Statistically significant results are displayed in bold type; statistically non-significant results are displayed in parentheses. ddPCR: digital droplet PCR.
Discussion
In the present study we were able to isolate and genetically characterize CTC in all stages of PDAC from UICC stage IA to metastatic stage IV patients with a combination of cytological and genetic evaluation. Patients with a higher tumor burden in the blood (>3CTC/ml) showed a trend to poorer OS (11.5 months vs 20 months). The CTC and the primary tumors had equally-distributed genotypes of KRAS mutations, but CTC showed more diverse KRAS mutations. Surprisingly, the KRAS G12V mutation in CTC was associated with better OS (median OS 24.5 months) compared to KRAS wt (P = 0.04). Other mutations, and also CTC negative patients, had similar median overall survival times (8–9.5 months).
It is of note that of the 21 patients with KRAS mutations in the CTC and the primary tumor, only 58% had a matching mutation in CTC and tissue. The other 42% had discordant mutations. The “matching” status had no influence on patient overall survival. Additionally, 6 patients with a KRAS mutant tumor and a positive cytology specimen did not show a KRAS mutation. This may be due to a detection limit of the KRAS assay, or the cytological “positive” CTC were not tumor derived. Moreover, cytological interpretation of CTC specimens can be challenging and depends on the investigator’s experience. Furthermore, benign pancreatic lesions may also shed cells with morphological features similar to those of CTCs into the blood stream34,35. All this may lead to false positive or negative results. However, in the present study we had highly concordant scores for the patient samples from three independent and experienced investigators.
Increasing evidence suggests that characterization of CTC from cancer patients may provide important information regarding early detection, prognosis, treatment and relapse as well as improved mechanistic insight with respect to tumor invasion and metastasis. CTC are extensively investigated in breast5,10, lung11 colorectal6,12 and prostate cancer13,14. In selected studies in these entities, CTC are used for response prediction and therapy adjustment5,14. In PDAC we are far from this scenario, due to inconsistent detection, isolation and characterization of CTC in PDAC23. Several studies with the FDA-approved CellSearch® technique were rather disappointing in PDAC patients, with CTC detection rates of about 521–40%22–24 but ongoing research has developed more promising approaches16–19.
Several studies, including our own, have shown that CTC in PDAC patients are not only rare, but also a diverse population with respect to (EMT) surface markers17–19. We were now able to show that KRAS mutations are also heterogeneous in this cell population. The CTC showed KRAS G12V, KRAS G12D, KRAS G12S, KRAS G13D and other KRAS mutations. Additionally, not only the CTC but also the primary tumors showed samples with more than one KRAS mutation. A recent extensive study on distinct tumor subtypes in PDAC revealed similar results: mutations (not only KRAS) were partially diverse in the primary tumor and metastatic sites. The authors also describe a high inter-tumor diversity between patients36.
KRAS mutations in primary tumor samples and metastasis in colorectal cancer have been described as highly similar with a concordance of 93–97%37,38. Especially in colorectal cancer, however, where the KRAS mutation status is crucial for the decision of anti-EGFR treatment, there is increasing evidence for a paradigm shift. Tumors are described as heterogeneous and may harbor small subsets of cells with specific mutations not found in routine diagnostics, showing heterogeneous mutations and may thus limit - or direct eligibility for anti-EGFR treatment39–41. Additionally, KRAS mutations can be reliably found in CTC of colorectal patients with a robust concordance of mutations of 44–77% and also 27–56% discordance in small sample sizes (9–12 samples)39,40. It is possible that the discordance between primary tumor and CTC could account for the failure of anti-EGFR therapy in patients with KRAS WT tumors, who in fact harbor KRAS mutations in their CTC. The only published study on PDAC and KRAS mutations in the CTC showed a 100% concordance in the 5 tested sample pairs of CTC and primary tumor15.
There are possible biological and technical explanations for the 42% discordant KRAS mutations in the CTC and the primary tumor in this study. First, the discordant CTC could represent the cells in transit that may have departed the primary lesions before the acquisition of a fully-malignant phenotype to undergo somatic mutations or deletions at a distant site20,31,42. Second, the heterogeneity of KRAS mutation status within the primary tumor is a well-known phenomenon43–45. In fact, there could thus be more than one KRAS mutation in each PDAC of which one “matches” the tumor: Heterogeneous KRAS mutations within one tumor were found in 7 of 32 tumor samples. Third, technical limitations like cross-contaminated specimens are theoretically possible- although this is unlikely in 42% of the cases. Additionally, 18 exemplary samples were quantitatively measured by ddPCR and show results very similar to those obtained with the clamping PCR. Finally, discordance may in theory be explained by metastases from a non-detected second primary. It is of note that the used KRAS detection method with the PNA Clamp as well as the ddPCR has a detection limit and may lead to false-negative KRAS mutational analysis results, as mentioned above. The majorities of mutations that were found in ddPCR had a very low abundance and were partially not statistically significant.
In the present study, we found that patients with the specific KRAS G12V mutation in their CTC had a significantly better OS than patients without detectable KRAS mutations in the specimens (KRAS WT;(p = 0.04)). In a study on unresectable PDAC, KRAS mutations in general were negative predictors for survival, but KRAS G12V mutations were associated with better survival when found in FNA samples46. Another study also showed superior survival of the KRAS G12V subtype in resected PDAC samples. The KRAS mutation subtypes in that study had a higher impact on survival than molecular factors such as p53, p16INK4, p21WAF1, and cyclin D1, while the KRAS mutations per se were not a risk factor for poor outcome47. Other studies in colorectal cancer showed inferior survival in patients with KRAS G12V 48.
The underlying biological phenomenon remains unclear to date; the success of different chemotherapies, however, may also depend on the identification of a unique genotypic pattern. The KRAS mutation phenotype (mutant or wild-type) has been used successfully for treatment decisions for anti-EGFR treatment in colorectal cancer: patients with KRAS mutations benefited significantly less from anti-EGFR treatment than patients with KRAS wild-type tumors49. Recently, however, the specific genotype of KRAS G13D showed better response to treatment than other KRAS mutations50,51. Tumor heterogeneity may confound chemotherapeutic strategies in personalizing care for individual patients, and a more complete picture of the cancer cells inside an individual may help treatment decisions. To date, mutant KRAS is considered an undruggable target52, there is however ongoing research in this field. New approaches for blocking KRAS activity continue to be developed and further insights into the biology of the specific KRAS mutations of primary tumors and CTC may provide better therapeutic targets in the future53,54.
The present study has several limitations. The CTC isolation-by-size is independent of surface markers but may miss rather small but potentially relevant CTC; additionally, the CTC specimens have to be read by a trained cytologist to address the nuances of cell cytomorphology. The method has, however, been found to isolate more CTC than surface antigen-dependent methods such as CellSearch®19. It is of note that the used KRAS detection method with the PNA clamp as well as the ddPCR has a detection limit and may lead to false-negative KRAS mutational analysis results. High numbers of CTCs isolated on the parallel cyto-filter however suggests that this is not the case in these patients (data not shown). The used KRAS PCR with the PNA Clamp is an allele-specific blocker (ASB) PCR. Its superiority has, however, been described especially in CTC-mutation detection compared to methods that use more advanced techniques like low denaturation temperature PCR (TransgenomicTM)55. One may argue that newer methods, like digital droplet PCR (ddPCR), would lead to better results; this was however not the case in 18 exemplary specimens, where the clamping PCR found more mutations than the very sensitive ddPCR (Table 4). And although the PNA-PCR is rather “old-fashioned”, we experienced it as a very reliable and sensitive detection method. In previous studies we - and others - found a KRAS mutation when we diluted down to two tumor cells per ml blood in spiking experiments16,56, and we have now found PANC-1 cells even at final concentrations of 1cell/ml. The consecutive mini-prep additionally allows the detection of multiple mutations after direct sequencing. Furthermore, the proposed study includes only 58 patients and the results - especially with respect to survival analysis and the small group of CTC-negative patients (n = 5) in this study- should be interpreted with caution. Additionally, due to the low number of mutations, even thorough statistical analysis cannot fully exclude artefacts. On the other hand, genuine heterogeneous, partially discordant mutations in CTC and primary tumor samples should not be downscaled by rigorous statistics.
Nevertheless, this is one of the largest studies on CTC in PDAC. And despite its aforementioned limitations this is, to the best of our knowledge, the first study comparing KRAS mutation status in 26 matched primary tumors and CTC showing a substantial match but also discordant mutations.
These preliminary results in 58 PDAC patients suggest that a) higher numbers of CTCs per ml may be associated with an inferior outcome, b) the subtype of CTC with a KRAS G12V mutation may be associated with better survival, and c) KRAS mutations in the CTC (and the tumor) can be heterogeneous and can be different from the mutations in the primary. The superiority in survival of patients with the KRAS G12V mutation in CTC needs to be evaluated in larger studies. Yet, it appears that a specific mutation may be important in the biology of a cancer, but its effect on chemotherapy and resistance remains to be determined in pancreatic CTC. Finally, the divergence between the disseminated tumor cells and the primary tumor as well as intra-tumoral heterogeneity may be one hypothesis for the poor response to treatment in PDAC and should be investigated in further detail.
Electronic supplementary material
Acknowledgements
We thank Silke Hempel and Kerstin Meyer for their assistance in the KRAS PNA Assay and imaging of CTC.The article processing charge was funded by the German Research Foundation (DFG) and the Albert Ludwigs University Freiburg in the funding programme Open Access Publishing.
Author Contributions
B.K. designed the study; B.K., J.H., G.S. and H.B. drafted the manuscript; S.S., S.R., V.M., J.K., S.H. and R.F. performed the experiments and performed parts of the analysis, P.B., S.T. and M.B.P. performed the pathological analyses, T.G. and B.K. performed the statistical analysis. All authors critically reviewed the manuscript and approved the final form.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
A correction to this article is available online at https://doi.org/10.1038/s41598-017-14870-3.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04601-z
Change History: A correction to this article has been published and is linked from the HTML version of this paper. The error has been fixed in the paper.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics 2015. CA Cancer J Clin. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- 3.Conroy T, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25. doi: 10.1056/NEJMoa1011923. [DOI] [PubMed] [Google Scholar]
- 4.Diehl SJ, Lehmann KJ, Sadick M, Lachmann R, Georgi M. Pancreatic cancer: value of dual-phase helical CT in assessing resectability. Radiology. 1998;206:373–8. doi: 10.1148/radiology.206.2.9457188. [DOI] [PubMed] [Google Scholar]
- 5.Cristofanilli M, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med. 2004;351:781–91. doi: 10.1056/NEJMoa040766. [DOI] [PubMed] [Google Scholar]
- 6.Cohen SJ, et al. Prognostic significance of circulating tumor cells in patients with metastatic colorectal cancer. Ann Oncol. 2009;20:1223–9. doi: 10.1093/annonc/mdn786. [DOI] [PubMed] [Google Scholar]
- 7.Scher HI, et al. Circulating tumour cells as prognostic markers in progressive, castration-resistant prostate cancer: a reanalysis of IMMC38 trial data. Lancet Oncol. 2009;10:233–9. doi: 10.1016/S1470-2045(08)70340-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Panteleakou Z, et al. Detection of circulating tumor cells in prostate cancer patients: methodological pitfalls and clinical relevance. Mol Med. 2009;15:101–14. doi: 10.2119/molmed.2008.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaifi JT, et al. Circulating tumor cell isolation during resection of colorectal cancer lung and liver metastases: a prospective trial with different detection techniques. Cancer Biol Ther. 2015;16:699–708. doi: 10.1080/15384047.2015.1030556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ignatiadis M, et al. Liquid biopsy-based clinical research in early breast cancer: The EORTC 90091-10093 Treat CTC trial. Eur J Cancer. 2016;63:97–104. doi: 10.1016/j.ejca.2016.04.024. [DOI] [PubMed] [Google Scholar]
- 11.Pailler E, et al. Detection of circulating tumor cells harboring a unique ALK rearrangement in ALK-positive non-small-cell lung cancer. J Clin Oncol. 2013;31:2273–81. doi: 10.1200/JCO.2012.44.5932. [DOI] [PubMed] [Google Scholar]
- 12.Onstenk, W. et al. Molecular characteristics of circulating tumor cells resemble the liver metastasis more closely than the primary tumor in metastatic colorectal cancer. Oncotarget. 2016 Jun 20. doi:10.18632/oncotarget.10175 (2016). [DOI] [PMC free article] [PubMed]
- 13.Lohr JG, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol. 2014;32:479–84. doi: 10.1038/nbt.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Munzone E, et al. Changes of HER2 status in circulating tumor cells compared with the primary tumor during treatment for advanced breast cancer. Clin Breast Cancer. 2010;10:392–7. doi: 10.3816/CBC.2010.n.052. [DOI] [PubMed] [Google Scholar]
- 15.Ankeny JS, et al. Circulating tumour cells as a biomarker for diagnosis and staging in pancreatic cancer. Br J Cancer. 2016;114:1367–75. doi: 10.1038/bjc.2016.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kulemann B, et al. Circulating tumor cells found in patients with localized and advanced pancreatic cancer. Pancreas. 2015;44:547–50. doi: 10.1097/MPA.0000000000000324. [DOI] [PubMed] [Google Scholar]
- 17.Kulemann B, et al. KRAS mutations in pancreatic circulating tumor cells: a pilot study. Tumour Biol. 2016;37:7547–54. doi: 10.1007/s13277-015-4589-2. [DOI] [PubMed] [Google Scholar]
- 18.Poruk, K. E. et al. Circulating Tumor Cell Phenotype Predicts Recurrence and Survival in Pancreatic Adenocarcinoma. Ann Surg. Jan 7. [Epub ahead of print] (2016). [DOI] [PMC free article] [PubMed]
- 19.Khoja L, et al. A pilot study to explore circulating tumour cells in pancreatic cancer as a novel biomarker. Br J Cancer. 2012;106:508–16. doi: 10.1038/bjc.2011.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rhim AD, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148:349–61. doi: 10.1016/j.cell.2011.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bidard FC, et al. Circulating tumor cells in locally advanced pancreatic adenocarcinoma: the ancillary CirCe 07 study to the LAP 07 trial. Ann Oncol. 2013;24:2057–2061. doi: 10.1093/annonc/mdt176. [DOI] [PubMed] [Google Scholar]
- 22.de Albuquerque A, et al. Multimarker gene analysis of circulating tumor cells in pancreatic cancer patients: a feasibility study. Oncology. 2012;82:3–10. doi: 10.1159/000335479. [DOI] [PubMed] [Google Scholar]
- 23.Kurihara T, et al. Detection of circulating tumor cells in patients with pancreatic cancer: a preliminary result. J Hepatobiliary Pancreat Surg. 2008;15:189–195. doi: 10.1007/s00534-007-1250-5. [DOI] [PubMed] [Google Scholar]
- 24.Negin BP, et al. Characterization and prognostic significance of circulating tumor cells in the peripheral blood of patients with metastatic pancreatic cancer. ASCO Meeting Abstr. 2010;28:4127. [Google Scholar]
- 25.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol. 2008;9:517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kanda M, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730–733. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris J, Pt., Wang SC, Hebrok M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer. 2010;10:683–695. doi: 10.1038/nrc2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cully M, Downward J. SnapShot: Ras Signaling. Cell. 2008;133:1292–1292. doi: 10.1016/j.cell.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 29.Stolze B, Reinhart S, Bulllinger L, Fröhling S, Scholl C. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci Rep. 2015;5:8535. doi: 10.1038/srep08535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pitman, M. B. Pancreas in Comprehensive Cytopathology 4th edition, (eds. Bibbo and Wilbur) 759 (Elsevier 2014).
- 31.Valsangkar NP, et al. Survival in ampullary cancer: potential role of different KRAS mutations. Surgery. 2015;157:260–8. doi: 10.1016/j.surg.2014.08.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hindson BJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Analytical chemistry. 2011;83:8604–10. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Forbes SA, et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res. 2015;43:D805–11. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cauley CE, et al. Circulating Epithelial Cells in Patients with Pancreatic Lesions: Clinical and Pathologic Findings. J Am Coll Surg. 2015;221(3):699–707. doi: 10.1016/j.jamcollsurg.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rhim AD, et al. Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology. 2014;146(3):647–51. doi: 10.1053/j.gastro.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moffitt RA, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47(10):1168–78. doi: 10.1038/ng.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Albanese I, et al. Heterogeneity within and between primary colorectal carcinomas and matched metastases as revealed by analysis of Ki-ras and p53 mutations. Biochem. Biophys. Res. Commun. 2004;325:784–791. doi: 10.1016/j.bbrc.2004.10.111. [DOI] [PubMed] [Google Scholar]
- 38.Cejas P, et al. KRAS mutations in primary colorectal cancer tumors and related metastases: a potential role in prediction of lung metastasis. PLoS One. 2009;4:e8199. doi: 10.1371/journal.pone.0008199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Laurent-Puig P, et al. Clinical relevance of KRAS-mutated subclones detected with picodroplet digital PCR in advanced colorectal cancer treated with anti-EGFR therapy. Clin Cancer Res. 2015;21:1087–97. doi: 10.1158/1078-0432.CCR-14-0983. [DOI] [PubMed] [Google Scholar]
- 40.Denis, J. A. Droplet digital PCR of circulating tumor cells from colorectal cancer patients can predict KRAS mutations before surgery. Mol Oncol. Jun 7. p [Epub ahead of print] (2016). [DOI] [PMC free article] [PubMed]
- 41.Klein CA. Parallel progression of primary tumours and metastases. Nat Rev Cancer. 2009;9:302–12. doi: 10.1038/nrc2627. [DOI] [PubMed] [Google Scholar]
- 42.Rhim AD, et al. Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology. 2014;146:647–51. doi: 10.1053/j.gastro.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:1039–49. doi: 10.1056/NEJMra1404198. [DOI] [PubMed] [Google Scholar]
- 44.Murphy SJ, et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology. 2013;145:1098–1109. doi: 10.1053/j.gastro.2013.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kosmidou V, et al. Tumor heterogeneity revealed by KRAS, BRAF, and PIK3CA pyrosequencing: KRAS and PIK3CA intratumor mutation profile differences and their therapeutic implications. Hum Mutat. 2014;35(3):329–40. doi: 10.1002/humu.22496. [DOI] [PubMed] [Google Scholar]
- 46.Ogura T, et al. Prognostic value of K-ras mutation status and subtypes in endoscopic ultrasound-guided fine-needle aspiration specimens from patients with unresectable pancreatic cancer. J Gastroenterol. 2013;48:640–6. doi: 10.1007/s00535-012-0664-2. [DOI] [PubMed] [Google Scholar]
- 47.Kawesha A, et al. K-ras oncogene subtype mutations are associated with survival but not expression of p53, p16(INK4A), p21(WAF-1), cyclin D1, erbB-2 and erbB-3 in resected pancreatic ductal adenocarcinoma. Int J Cancer. 2000;89:469–74. doi: 10.1002/1097-0215(20001120)89:6<469::AID-IJC1>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 48.Andreyev NJ, et al. Kirsten ras mutations in patients with colorectal cancer: the multicenter ‘RASCAL’ study. Br J Cancer. 2001;85:692–6. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tol J, et al. Chemotherapy, bevacizumab, and cetuximab in metastatic colorectal cancer. N Engl J Med. 2009;360:563–72. doi: 10.1056/NEJMoa0808268. [DOI] [PubMed] [Google Scholar]
- 50.De Roock W, et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA. 2010;304:1812–1820. doi: 10.1001/jama.2010.1535. [DOI] [PubMed] [Google Scholar]
- 51.Tural D, et al. Association KRAS G13D tumor mutated outcome in patients with chemotherapy refractory metastatic colorectal cancer treated with cetuximab. Hepatogastroenterology. 2013;60:1035–1040. doi: 10.5754/hge12983. [DOI] [PubMed] [Google Scholar]
- 52.Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras (G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. 2013;503:548–551. doi: 10.1038/nature12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roberts PJ, Stinchcombe TE, Der CJ, Socinski MA. Personalized medicine in non-small-cell lung cancer: is KRAS a useful marker in selecting patients for epidermal growth factor receptor-targeted therapy? J Clin Oncol. 2010;28:4769–4777. doi: 10.1200/JCO.2009.27.4365. [DOI] [PubMed] [Google Scholar]
- 54.Zimmermann G, et al. Small molecule inhibition of the KRAS-PDEd interaction impairs oncogenic KRAS signalling. Nature. 2013;497:638–642. doi: 10.1038/nature12205. [DOI] [PubMed] [Google Scholar]
- 55.Mostert B, et al. KRAS and BRAF mutation status in circulating colorectal tumor cells and their correlation with primary and metastatic tumor tissue. Int J Cancer. 2013;133:130–41. doi: 10.1002/ijc.27987. [DOI] [PubMed] [Google Scholar]
- 56.Steinert G, et al. Immune escape and survival mechanisms in circulating tumor cells of colorectal cancer. Cancer Res. 2014;74:1694–704. doi: 10.1158/0008-5472.CAN-13-1885. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.