

Abstract
The RTK/ERK signaling pathway has been implicated in prostate cancer progression. However, the genetic relevance of this pathway to aggressive prostate cancer at the SNP level remains undefined. Here we performed a SNP and gene-based association analysis of the RTK/ERK pathway with aggressive prostate cancer in a cohort comprising 956 aggressive and 347 non-aggressive cases. We identified several loci including rs3217869/CCND2 within the pathway shown to be significantly associated with aggressive prostate cancer. Our functional analysis revealed a statistically significant relationship between rs3217869 risk genotype and decreased CCND2 expression levels in a collection of 119 prostate cancer patient samples. Reduced expression of CCND2 promoted cell proliferation and its overexpression inhibited cell growth of prostate cancer. Strikingly, CCND2 downregulation was consistently observed in the advanced prostate cancer in 18 available clinical data sets with a total amount of 1,095 prostate samples. Furthermore, the lower expression levels of CCND2 markedly correlated with prostate tumor progression to high Gleason score and elevated PSA levels, and served as an independent predictor of biochemical relapse and overall survival in a large cohort of prostate cancer patients. Together, we have identified an association of genetic variants and genes in the RTK/ERK pathway with prostate cancer aggressiveness, and highlighted the potential importance of CCND2 in prostate cancer susceptibility and tumor progression to metastasis.
Introduction
Prostate cancer remains the second most commonly diagnosed cancer and one of the leading causes of cancer-related deaths in men worldwide. More than 1.1 million cases of prostate cancer and 307,000 deaths were recorded around the globe in 2012, accounting for approximately 15% of new cancer cases in men and 6.6% of the total male cancer mortality1, respectively. In the United States, there are nearly 180,890 new cases diagnosed and around 26,120 deaths in 20162. While prostate cancer patients with an indolent form of the disease may be easily treated, the patients who have aggressive prostate cancer indicate worse prognosis and thus need intensive treatment. Recent genome-wide association studies (GWASs) have been applied to identify possible prostate cancer risk loci with potential in management of the disease, though the molecular pathogenesis is being unraveled3–6, especially for the aggressive form of the disease.
Many established risk factors have been implicated in prostate cancer etiology and development. These include genetics and family history, ethnic background, lifestyle, advancing age and environment. In addition, considerable evidence indicated that the signaling pathways including AR, PI3K/AKT, and RTK/ERK7–9 have been identified as the key factors for prostate cancer risk, yet the underlying genetic basis remains elusive. The association analyses in our previous work suggested that the RTK/ERK pathway might be a significant factor conferring risk of prostate cancer10–12. Moreover, recent studies demonstrated the important role of this pathway in metastasis and progression of prostate cancer13, 14. However, the potential genetic risk loci, genes and their function in this pathway associated with aggressive prostate cancer has not been defined.
With a number of genes15 such as EGFR, PDGF, GRB2, MET and cyclin D genes16 (CCND1, CCND2 and CCND3) involved, the RTK/ERK pathway regulates various biological processes including cellular signal transduction and crosstalk, cell proliferation, differentiation, survival and migration in normal and cancer development17, 18. Mutations that induce abnormalities of the structures of proteins in the pathway, may lead to cell transformation and malignancy in various types of cancer. The receptor tyrosine kinases (RTKs) lie upstream in the pathway, transduce the signal into the cells and trigger a cascade of phosphorylation for the downstream kinases such as the mitogen-activated protein kinase (MAPK) cascade, including Raf, MEK and extracellular signal-regulated kinase (ERK) kinases17, 18. The consecutive signal transduction may lead to the phosphorylation of target proteins inside the cell and control normal biological activities. In contrast, the imbalance during the cascade of signal transduction and the inappropriately response for susceptible environment stimulus may lead to various diseases, including cancer. Therefore, investigation on the associations of the RTK/ERK pathway with prostate cancer risk can provide new insight into the molecular mechanisms underlying the susceptibility, and development of prostate cancer at the pathway-level, and reveal novel links between the pathway genes and disease progression to advanced stage and metastasis.
Although the potential association of the RTK/ERK pathway with prostate cancer risk has been suggested by our previous study10–12, the genetic and functional link of the pathway to aggressive prostate cancer have not been elucidated. In this work, on the basis of the Chinese Consortium for Prostate Cancer Genetics (ChinaPCa) project, we performed SNP- and gene-based prostate cancer case-only study, as well as expression quantitative trait loci (eQTL), clinical correlation, and prognosis analyses. Whereas some loci and genes were defined, only one SNP (rs3217869 in CCND2) was reproducibly identified to be significantly associated with aggressive prostate cancer by multiple testing. The eQTL analysis revealed a significant association of the risk allele at rs3217869 with decreased mRNA levels of CCND2. Interestingly, we found that CCND2 is strikingly downregulated in advanced prostate tumors and its downregulation is associated with disease progression and poor prognosis in multiple independent cohorts of prostate cancer, suggesting that CCND2 is a new biomarker to distinguish aggressive disease and a potential metastatic tumor suppressor. Thus, our study not only investigated the effects of the RTK/ERK pathway on the progression of prostate cancer, but also revealed the genetic factors responsible for aggressive disease susceptibility, highlighted the potential role of CCND2 in disease progression.
Materials and Methods
Patients and study approval
Informed consent was obtained from all subjects. The experimental protocols for the study using the China PCa cohort was approved by the institutional review boards at Shanghai Changhai Hospital, The Second Military Medical University, Shanghai, China. The study using Finnish PCa cohort was performed with the appropriate research permissions from the Ethics Committee of the Tampere University Hospital, Finland, and the Ministry of Social Affairs and Health in Finland. All experiments and methods were performed according to these relevant guidelines and regulations.
Study population
The Chinese Consortium for Prostate Cancer Genetics (ChinaPCa) has recruited a study population comprised of Han Chinese men from southeastern China, with various areas including Shanghai, Suzhou, Guangxi and Nanjing. In current study, 1417 prostate cancer patients from Shanghai and surrounding areas genotyped in the first state of a previous GWAS19 were collected with the information of age, PSA level, Gleason score (GS) and clinical stage. The details of this cohort were presented in the study by Xu et al.19. To ensure that all of the SNPs in the RTK/ERK signaling pathway genes were covered, the region around each gene was extended by 100 kb. Finally, 50,459 single nucleotide polymorphisms (SNPs) were identified.
To investigate the association between the RTK/ERK pathway and aggressive prostate cancer, this analysis was designed to be a case-only study, in which the samples were divided into two groups: aggressive cases and non-aggressive prostate cancer cases. The aggressive cases were defined as: histologically confirmed prostate cancer, with GS ≥ 8, or clinical stage ≥T3, or with lymphatic metastasis (N+), or being metastatic (M+), or with PSA >50 ng/ml. Once the unambiguous samples met any of the above criteria, the aggressive data will be collected. Otherwise, the non-aggressive prostate cancer was defined. Finally, after the information filtering, the study included 1303 prostate cancer patients consisting of 956 aggressive and 347 non-aggressive ones20, 21. The characteristics of the collected samples are presented in Table 1.
Table 1.
The characteristics of the samples we included in our analysis.
| aggressive (n = 956) | non-aggressive (n = 347) | P | |
|---|---|---|---|
| aAge (mean ± SD, n) | 71.25 ± 8.38, 950 | 70.76 ± 7.37, 343 | 0.309 |
| bPSA (mean ± SD, n), | 152.79 ± 517.55, 855 | 15.09 ± 9.48, 347 | <0.001 |
| Classification of PSA, n (%) | |||
| ≤4.0 | 31 (3.24%) | 18 (5.19%) | |
| >4.0, <10 | 69 (7.22%) | 105 (30.26%) | |
| ≥10 | 833 (87.13%) | 224 (64.55%) | d<0.001 |
| Undefined | 23 (2.41%) | 0 (0.00%) | e<0.001 |
| cGleason score, n (%) | |||
| ≤7 | 396 (41.42%) | 347 (100%) | |
| ≥8 | 537 (56.17%) | 0 (0.00%) | f<0.001 |
| Undefined | 23 (2.41%) | 0 (0.00%) | g<0.001 |
| Clinical stage, n (%) | |||
| Stage (I, II) | 353 (36.92%) | 347 (100%) | |
| Stage (III, IV) | 491 (51.36%) | 0 (0.00%) | h<0.001 |
| Undefined | 112 (11.72%) | 0 (0.00%) | i<0.001 |
aThe information of age were missing in 6 aggressive and 4 no-aggressive samples.
bThe levels of PSA in 78 aggressive samples were too high and no measurable values; In addition, 23 aggressive samples were missing.
c23 aggressive samples were missing for Gleason score.
dThe Χ2-test for three levels of PSA (PSA ≤ 4.0, 4 < PSA < 10, PSA ≥ 10) in aggressive and non-aggressive groups.
eThe Χ2-test for three levels of PSA (PSA ≤ 4.0, 4 < PSA < 10, PSA ≥ 10 and Undefined) in aggressive and non-aggressive groups.
fThe Χ2-test for three levels of Gleason score (GS ≤ 7, GS ≥ 8) in aggressive and non-aggressive groups.
gThe Χ2-test for three levels of Gleason score (GS ≤ 7, GS ≥ 8 and Undefined) in aggressive and non-aggressive groups.
hThe Χ2-test for three levels of Clinical stage (Stage (I, II) and Stage (III, IV)) in aggressive and non-aggressive groups.
iThe Χ2-test for three levels of Clinical stage (Stage (I, II) and Stage (III, IV) and Undefined) in aggressive and non-aggressive groups.
SNP selection and association analysis
On the basis of ChinaPCa, the genotype data for the 40 genes in the RTK/ERK pathway such as EGFR, EGF, HGF, HRAS and cyclin D genes (CCND1, CCND2 and CCND3) were collected. Before conducting the association analysis, tag SNPs representing each loci in the RTK/ERK pathway relevant genes were selected using the web server SNPinfo22 (http://manticore.niehs.nih.gov/snpinfo/snptag.htm), which includes a set of web-based SNP selection tools with many functional modules involved, such as GWAS SNP Selection in Linkage Loci, Candidate Gene SNP Selection, TAG SNP Selection, SNP Function Prediction and so on22. On the basis of the powerful system, the official gene names and IDs were obtained using the HapMap database and CHB + JPT populations. Then, tag SNPs were calculated with common SNPs (minor allele frequency, MAF ≥ 0.05; r2 ≥ 0.8). Finally, 760 SNPs representing the pathway relevant genes were collected. In addition, the genotype data of the corresponding tag SNPs were extracted using PLINK tool23.
Statistical analysis
In this study, two groups of samples were defined. One is aggressive prostate cancer and the other is non-aggressive on the basis of the criteria as described above. Then, the information of ages, PSA levels, GS and Clinical stage were collected. The Student’s t test, Mann-Whitney U test, and the Χ2-test were properly applied.
To investigate the real association between the SNPs in the RTK/ERK pathway and aggressive prostate cancer, we adopted three genotype models, including dominant, recessive and additive model (http://pngu.mgh.harvard.edu/~purcell/plink/anal.shtml#model)23. The odd ratios (OR) and corresponding 95% confidence intervals (CI) were used to evaluate the power of associations. Meanwhile, Cochran–Armitage trend test was also applied (http://pngu.mgh.harvard.edu/~purcell/plink/anal.shtml#model)23. In addition, to discover the independent association between the RTK/ERK pathway relevant loci and aggressive prostate cancer, those SNPs in each gene with the lowest P values were collected as the covariates in the next conditional analysis (http://pngu.mgh.harvard.edu/~purcell/plink/anal.shtml#glm)23. All the associations were conducted by adjusting the parameter of age for every subject. In addition, the associations between the discovered loci and clinical parameters (especially for GS and clinical stage) were also conducted, which would help understand the loci more clearly on the level of possible pathogenesis. However, in our data, some of the PSA levels were a scope without pronounced values, which were only enough to define the aggressive and non-aggressive prostate cancer. Thus, we did not include these in this analysis to rule out potential bias. All the statistical analyses were performed using SPSS version 16.0 software (SPSS Inc., Chicago, IL, USA) and PLINK23 program. The statistical tests were two-tailed.
Gene-based pathway analysis
After testing the association at the SNP-level for aggressive and non-aggressive prostate cancer, the gene-based pathway analysis was conducted for further exploration using the adaptive rank truncated product (ARTP) method24. This method could potentially remedy the results discovered by single-SNP analysis and provide additional insights into the genetic mechanism underlying complex diseases. The analysis consisted of two steps: firstly, the standardized summary was acquired for the evidence of association between a gene and the outcome; secondly, these gene-level P-values were combined into a test statistic for the disease-pathway association. To use the method, three sets of information are needed: the genotypes of the SNPs, the phenotype for the samples, and the annotation of the SNPs. According to the manual of ARTP, the calculations were mainly divided into three steps: (1) Defining the phenotype and genotype data; (2) Calling the ARTP function; and (3) Calling the runPermutations and ARTP pathway functions. Then, in this study, the gene-based pathway association was conducted with 10,000-fold permutations to make the results more accurate. In all of the analyses, P-values were two-sided with R v2.13.2.
Association analysis of SNP genotype with gene expression
For the analysis of the association between the rs3217869 genotype and CCND2 expression levels, Kruskal–Wallis H test was applied to assess the statistical significance. Furthermore, we also used Matrix eQTL to test for the cis eQTL association between rs3217869 genotypes and CCND2 expression levels using data from Camcap cohort comprised of 119 prostate samples4, 25. The parameters “useModel = modelLINEAR”, “errorCovariance = numeric ()” were used with Matrix eQTL. R (version 3.2.2) was used to graphically visualize the association between rs3217869 genotypes and CCND2 expression levels.
Cell lines and transfection
The prostate cancer cell lines LNCaP, PC3 and DU145 were grown in Dulbecco’s Nodified Eagle medium containing 10% fetal calf serum (FCS) and 1.2% penicillin/streptomycin (Gibco, Thermo Fisher scientific), at 37 °C and 5% CO2. Considering the expression of CCND2 was lower in LNCaP cells than DU145 and PC3 (data not shown), LNCaP cell line was selected to perform the CCND2 overexpression experiment. The full-length of human CCND2 cDNA were cloned into pcDNA3.1(+) expression vector (Life technologies, Carlsbad, CA) and the construct was verified by sequencing. pcDNA3.1-CCND2 or parental pcDNA3.1 vector were transfected into LNCaP cells using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol.
Western blot
For detection of CCND2 overexpression in LNCaP cells were collected with a gum rubber-scraping device, lysed with RIPA Lysis Buffer (P0013C, Beyotime Institute of Biotechnology) and protein concentration was determined using BCA assay (Thermo Scientifc, Waltham, MA, USA) according to manufacturer’s information. Subsequently, 50 μg of total protein were separated by SDS-PAGE, transferred to nitrocellulose membranes and incubated with antibodies against anti-CCND2 (dilution: 1:4000, Cell Signaling, Danvers, MA, USA). Proteins levels were normalized using Actin levels as reference.
siRNA transfection and quantitative RT-PCR
To further evaluate functional role of CCND2 in prostate cancer cell growth, we performed short interfering RNA (siRNA)-mediated knockdown assays by transfecting siRNAs against CCND2 into the DU145 cells that were seeded in 96-well palates (reverse transfection, 6pmol siRNA per well) using HiPerFect Transfection Reagent (Cat No: 301705). The siRNAs used included CCND2 siRNAs: AGGAGTGTAGTTGGATCTCTA (SI00027839); AAGAAATAGACTTGCACCTTA (SI00027853); CAGGGCCGTGCGGGACCGCAA (SI03071369) and AllStars Hs Cell Death siRNA (SI04381048) and AllStars Negative Control siRNA(SI03650318) purchased from Qiagen. We used qRT-PCR (SYBR Select Master Mix, 4472920, Applied Biosystems) to evaluate knockdown effect of the siRNAs on CCND2. Primer sequences used in this experiment are TCCTGGCCTCCAAACTCAAAG (CCND2 forward), GAGGCTTGATGGAGTTGTCG (CCND2 reverse) and AGAAAATCTGGCACCACACC (ACTB forward) and AGAGGCGTACAGGGATAGCA (ACTB reverse).
Cell proliferation assay
3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide assay (MP Biomedicals, LLC) was applied to evaluate the cell viability. Transfected LNCaP cells were seeded in 96-well plates. MTT was added to each well, incubating for 4 h at 37 °C. Then, using dimethyl sulfoxide (DMSO, 100 µL), the precipitate was dissolved. The absorbance at 570 nm was measured with EPOCH2 Microplate Readers (BioTek). The measures were performed at 0 h, 24 h and 48 h. All groups were consisted of 3 wells of LNCaP cells per time point independently (four times in total). For siRNA-mediated knockdown cell proliferation experiments, cell viability and proliferation were determined by using XTT (11465015001, Roche) and values were obtained at designed time point by measuring the absorbance at 450 nm. Three independent experiments have been run, and in each experiments, each group include 3 wells of DU145 cells at given time points. Two-tailed t test was used to calculate the significance.
Differential gene expression analysis
For the differential gene expression analysis, we performed Mann-Whitney U tests or Kruskal–Wallis H test to evaluate the significance for the comparison of CCND2, PDGF-C and other gene expression levels among normal, tumor or metastasis samples from Oncomine database26. R (version 3.2.2) was used to perform these statistical analyses and box plot was used to graphically display the distribution of gene expression among different groups.
Immunohistochemistry
Aggressive prostate cancer (n = 5), non-aggressive prostate cancer (n = 4), benign prostate hyperplasia (BPH, n = 3) and one normal bladder were collected from the First Affiliated Hospital of Guangxi Medical University, Nanning, China. The study was approved by the ethics committee. All the formalin-fixed and paraffin-embedded prostate tissue sections were performed with DAB Detection Kit (Streptavidin-Biotin) according to the instructions. The anti-CCND2 (Cell Signaling, Danvers, MA, USA) was used at a 1:800 dilution. With Meyer’s hematoxylin, the sections were analyzed with light microscopy.
Analysis of association between CCND2 expression and risk of overall survival or biochemical recurrence
We assessed the association between CCND2 expression levels and prostate cancer overall survival and biochemical recurrence by using Kaplan–Meier estimator as previously reported27. For the association analysis of CCND2 expression levels and overall survival and biochemical recurrence, we used a collection of 596 prostate carcinoma samples from the Nakagawa Prostate dataset from Oncomine database26, 28. Samples were stratified into two groups based on median measure of CCND2 expression levels. For a given sample expression measurement, the expression value was subtracted by the median value of all sample expression measurements. Thus, scores above zero indicate higher expression while scores below zero indicate lower expression. We then ranked samples in each group according to CCND2 expression and defined the higher CCND2 expression group of top 50% of samples with positive scores. In a same way, we defined lower CCND2 expression group of bottom 50% of samples with negative scores.
Multivariate analysis
For analysis of the association between prostate cancer overall survival, biochemical recurrence and CCND2 expression and clinical variables including Gleason score (GS), PSA, T stage and age, we performed multivariate Cox proportional hazard analyses with the 596 samples from the Nakagawa prostate dataset from Oncomine database26, 28 in R (version 3.2.2). Samples were stratified into two groups with higher and lower expression by comparing to the median measurement of CCND2 expression. Samples were ranked based on CCND2 expression and highest 50% and lowest 50% of the samples were selected for the Cox regression analysis. The clinical relevance of overall survival and biochemical recurrence and covariates were performed in several different scenarios. We tested the association by including covariates Gleason score, PSA, age, and clinical T state.
Results
Characteristics
Prostate cancer is a common disease, affecting a large number of males worldwide. As an invasive form of the disease, aggressive prostate cancer can induce more serious damage than other forms. In our previous work11, 12, the potential link between the prostate cancer risk and the RTK/ERK pathway was observed. On the basis of these associations, we proposed to illustrate the effects of the pathway on aggressive prostate cancer, which could help understand the pathogenesis and the genetic basis underlying the disease susceptibility and progression.
SNP-based association analysis
As the iconic SNPs in a region of the human genome, the tag-SNPs were calculated with the SNPinfo22 for every gene in the RTK/ERK pathway. Finally, we obtained 760 tag SNPs. The association analysis was conducted using three common genotype models and the logistic regression after adjusting for age. For the additive model, 32 loci were discovered with P < 0.05, 31 for dominant model and 21 for recessive model (Table 2 and Supplementary Table 1), respectively. In the additive and dominant model, the lowest P value was 1.132 × 10−3 (OR = 1.393, 95% (confidence interval) CI = 1.141–1.700) and 9.800 × 10−4 (OR = 1.519, 95% CI = 1.185–1.948) for rs603781 in the PDGF-D gene (Supplementary Table 1). As for the recessive model, rs11247380 in IGF1R (P = 2.269 × 10−3, OR = 1.757 95% CI = 1.224–2.524) was suggested to be the most significant (Supplementary Table 1).
Table 2.
The association between aggressive prostate cancer and the loci of the RTK/ERK pathway assessed by the additive, dominant and recessive models, respectively.
| CHR | Gene | SNP | BP | A1 | Model | OR (95% CI) | Pa | Conditional SNP | Conditional P valueb | Pe |
|---|---|---|---|---|---|---|---|---|---|---|
| 4 | EGF | rs2255355 | 110891543 | A | Additive | 1.186 (0.991–1.421) | 6.330 × 10−2 | — | — | 6.528 × 10−2 |
| Dominant | 1.095 (0.853–1.406) | 4.770 × 10−1 | — | — | ||||||
| Recessive | 1.694 (1.147–2.504) | 8.125 × 10−3 | REFc | REF | ||||||
| rs1860129 | 110886343 | G | Additive | 1.136 (0.942–1.369) | 1.814 × 10−1 | — | — | 1.787 × 10−1 | ||
| Dominant | 1.020 (0.797–1.305) | 8.777 × 10−1 | — | — | ||||||
| Recessive | 1.841 (1.164–2.912) | 9.107 × 10−3 | rs2255355 | 3.404 × 10−1 | ||||||
| 7 | EGFR | rs1050171 | 55249063 | A | Additive | 0.707 (0.557–0.897) | 4.306 × 10−3 | REFc | REF | 4.795 × 10−3 |
| Dominant | 0.667 (0.511–0.871) | 2.890 × 10−3 | REFc | REF | ||||||
| Recessive | 0.763 (0.326–1.784) | 5.321 × 10−1 | — | — | ||||||
| rs2075101 | 55250026 | G | Additive | 0.719 (0.567–0.911) | 6.367 × 10−3 | rs1050171 | NA | 7.148 × 10−3 | ||
| Dominant | 0.681 (0.523–0.888) | 4.572 × 10−3 | rs1050171 | NA | ||||||
| Recessive | 0.764 (0.326–1.787) | 5.342 × 10−1 | — | — | ||||||
| 11 | PDGF-D | rs603781 | 103965133 | T | Additive | 1.393 (1.141–1.700) | 1.132 × 10−3 | REFc | REF | 1.415 × 10−3 |
| Dominant | 1.519 (1.185–1.948) | 9.800 × 10−4 | REFc | REF | ||||||
| Recessive | 1.477 (0.919–2.376) | 1.075 × 10−1 | — | — | ||||||
| 12 | CCND2 | rs3217892 | 4403864 | A | Additive | 1.488 (1.139–1.943) | 3.542 × 10−3 | REFc | REF | 3.438 × 10−3 |
| Dominant | 1.548 (1.143–2.097) | 4.729 × 10−3 | REFc | REF | ||||||
| Recessive | 1.970 (0.815–4.763) | 1.322 × 10−1 | — | — | ||||||
| rs3217869 | 4399970 | A | Additive | 1.511 (1.135–2.010) | 4.653 × 10 −3 | rs3217892 | 7.263 × 10−3 | 5.188 × 10−3 | ||
| Dominant | 6.907 (1.733–26.900) | 5.341 × 10 −3 | rs3217892 | 8.938 × 10−3 | ||||||
| Recessive | 1.435 (1.055–1.954) | 2.156 × 10 −2 | rs3217907 | 2.629 × 10−2 | ||||||
| rs3217907 | 4406836 | A | Additive | 1.322 (1.087–1.608) | 5.189 × 10−3 | rs3217892 | 1.631 × 10−1 | 5.589 × 10−3 | ||
| Dominant | 1.332 (1.017–1.744) | 3.754 × 10−2 | rs3217892 | 3.865 × 10−1 | ||||||
| Recessive | 1.708 (1.134–2.572) | 1.035 × 10−2 | REFc | REF | ||||||
| 12 | IGF1 | rs6214 | 102793569 | T | Additive | 1.158 (0.970–1.383) | 1.040 × 10−1 | — | — | 1.086 × 10−1 |
| Dominant | 1.474 (1.122–1.937) | 5.371 × 10−3 | rs3217892 | 2.353 × 10−2 | ||||||
| Recessive | 0.972 (0.727–1.299) | 8.471 × 10−1 | — | — | ||||||
| 15 | IGF1R | rs11247380 | 99440731 | G | Additive | 1.145 (0.955–1.372) | 1.431 × 10−1 | — | — | 1.562 × 10−1 |
| Dominant | 0.954 (0.730–1.245) | 7.274 × 10−1 | — | — | ||||||
| Recessive | 1.757 (1.224–2.524) | 2.269 × 10−3 | REFc | REF | ||||||
| rs2715417 | 99453047 | T | Additive | 1.148 (0.949–1.388) | 1.554 × 10−1 | — | — | 1.798 × 10−1 | ||
| Dominant | 0.997 (0.777–1.278) | 9.778 × 10−1 | — | — | ||||||
| Recessive | 2.077 (1.292–3.340) | 2.561 × 10−3 | rs11247380 | 2.128 × 10−1 | ||||||
| rs2684779 | 99406245 | G | Additive | 1.251 (0.991–1.580) | 6.010 × 10−2 | — | — | 6.243 × 10−2 | ||
| Dominant | 1.152 (0.877–1.512) | 3.088 × 10−1 | — | — | ||||||
| Recessive | 3.621 (1.426–9.194) | 6.789 × 10−3 | rs11247380 | 7.206 × 10−2 | ||||||
| 18 | BCL2 | rs7243091 | 60880562 | A | Additive | 1.080 (0.861–1.355) | 5.058 × 10−1 | — | — | 5.205 × 10−1 |
| Dominant | 1.255 (0.967–1.628) | 8.719 × 10−2 | ||||||||
| Recessive | 0.432 (0.228–0.815) | 9.640 × 10−3 | REFc | REF |
aThe P values are based on logistic regression analysis and adjusted for age.
bThe conditional P values are based on logistic regression analysis and adjusted for age and reference SNPs.
cThe reference SNPs were applied to be adjusted in the conditional analysis.
dOnly one SNP with the P < 0.05 was shown in additive model (for chromosome 3, 5, 6, 12, 18 and 22), dominant model (chromosome 3 and 22) and recessive model (chromosome 6 and 11).
eP for Cochran–Armitage trend test.
To confirm the association, we next performed the Cochran–Armitage trend test and conditional analysis. This analysis identified ten SNPs suggested to be associated with aggressive prostate cancer in additive and dominant models (rs603781, rs1050171, rs3217892, rs11231741, rs4705415, rs12643184, rs3217869, rs17035367, rs5757573 and rs11923427) and three loci in the additive and recessive model (rs3217869, rs9487729 and rs6828477) (Table 2 and Supplementary Table 1). Only one SNP (rs3217869 in CCND2) was shown to be significantly associated with aggressive prostate cancer after the analysis in the three models (Additive model: P = 4.653 × 10−3, OR = 1.511, 95% CI = 1.135–2.010; Dominant model: P = 5.341 × 10−3, OR = 6.907, 95% CI = 1.773–26.900; Recessive model: P = 2.156 × 10−2, OR = 1.435, 95% CI = 1.055–1.954), Cochran–Armitage trend test (P = 5.188 × 10−3) and conditional analysis (Additive model: P = 7.263 × 10−3; Dominant model: P = 8.938 × 10−3; Recessive model: P = 2.629 × 10−2) (Table 2).
We also tested the association of rs3217869 with aggressive prostate cancer in a Finnish cohort including 1729 aggressive and 693 non-aggressive cases. Consistently, rs3217869 A allele showed a tendency for risk in aggressive cases compared to non-aggressive prostate cancer though the results were not statistically significant (Dominant model: P = 0.273, OR = 1.116, 95% CI = 0.917–1.357; Recessive model: P = 0.221, OR = 1.144, 95% CI = 0.923–1.418). Together, the results from both cohorts suggest that the prostate cancer patients carrying rs3217869 A allele have a tendency to increase the risk for aggressive disease.
While only one locus was defined in the different tests, we also tried a further analysis to discover more potential loci. In this analysis, the loci identified in more than two tests were included. We tested whether these SNPs associated with the GS and clinical stages of prostate cancer. We found that the mutation of rs12643184 (PDGF-C) (Additive model: P = 3.612 × 10−2, OR = 1.374, 95% CI = 1.021–1.848; Dominant model: P = 2.094 × 10−2, OR = 1.484, 95% CI = 1.062–2.075) (Supplementary Table 1) was suggested to be the risk factor increasing the risk of aggressive prostate cancer (GS: mutation = 7.72 ± 1.45, wild type = 7.43 ± 1.71, P = 0.034) (Supplementary Table 2).
Gene-based association analysis
After conducting the analysis with the SNPs available for the genes in the RTK/ERK pathway, we focused on all genes in the pathway and performed the gene-based analysis using the ARTP method24. This analysis revealed a marginal association between the RTK/ERK pathway genes including BAD (P = 0.058), PDGF-D (P = 0.051) and CCND2 (P = 0.052) and aggressive prostate cancer.
Functional annotation of newly-identified SNPs and genes including rs3217869/CCND2
This genetic study implicates the association of the SNPs rs3217869 (CCND2) and rs12643184 (PDGF-C), and the genes BAD, PDGF-D and CCND2 in the RTK/ERK signaling pathway with aggressive prostate cancer. To gain insight into the underlying biological basis for the associated SNPs, we performed the expression quantitative trait loci (eQTL) analysis using array-based transcriptomics data derived from prostate tissues and the matched genotype information on 119 patients with prostate cancer4, 25. This analysis revealed a strong association of the risk allele A at rs3217869 with lower mRNA levels of CCND2 (Fig. 1) but not with other genes within 2 Mb of rs3217869 (Supplementary Table 3), suggesting that CCND2 is a plausible causative gene for aggressive prostate cancer. In contrast, no significant association was found for rs12643184 (PDGF-C) within this eQTL data set.
Figure 1.
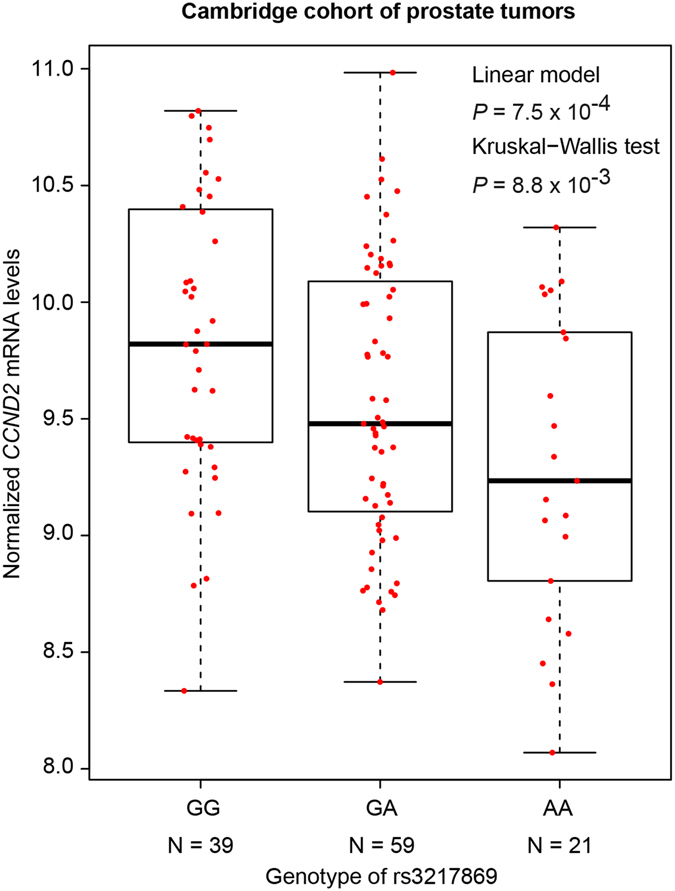
The association between rs3217869 genotype and CCND2 expression in prostate tissue samples. Note that the risk A allele of rs3217869 for aggressive prostate cancer was significantly associated with decreased mRNA expression of CCND2. The CCND2 mRNA levels were assessed by Illumina Expression BeadChip-based transcriptional profiling in a collection of 119 human prostate tissue samples4, 25. The CCND2 mRNA expression data in prostate tumor tissues are displayed as the five number distribution (minimum, first quartile, median, third quartile, and maximum) according to rs3217869 genotype The P values were examined by linear regression model and Kruskal-Wallis H test, respectively.
CCND2 was previously reported to be frequently methylated, with loss of its expression in prostate cancer29, suggesting a potential role as a tumor suppressor. Consistent with this, we found that overexpression of CCND2 in prostate cancer cell line LNCaP significantly inhibited its proliferation (Fig. 2a). Moreover, short interfering RNA (siRNA)-mediated knockdown of CCND2 in prostate cancer cells DU145 greatly promoted cell growth and viability compared to the cells with negative control siRNAs (Fig. 2b). In contrast, we observed a slight change in PC3 cells upon siRNA-mediated knockdown of CCND2 with similar cell proliferation experiments (Supplementary Fig. 1), indicating that CCND2-directed cell growth inhibition may be cell-context dependent. In addition, in multiple large cohorts of clinical prostate cancer samples30, 31, we observed significantly inverse correlations between the mRNA expression of CCND2 and MKI67 (Fig. 2c and d), the latter coding Ki67 antigen, a recognized indicator of cellular proliferation.
Figure 2.
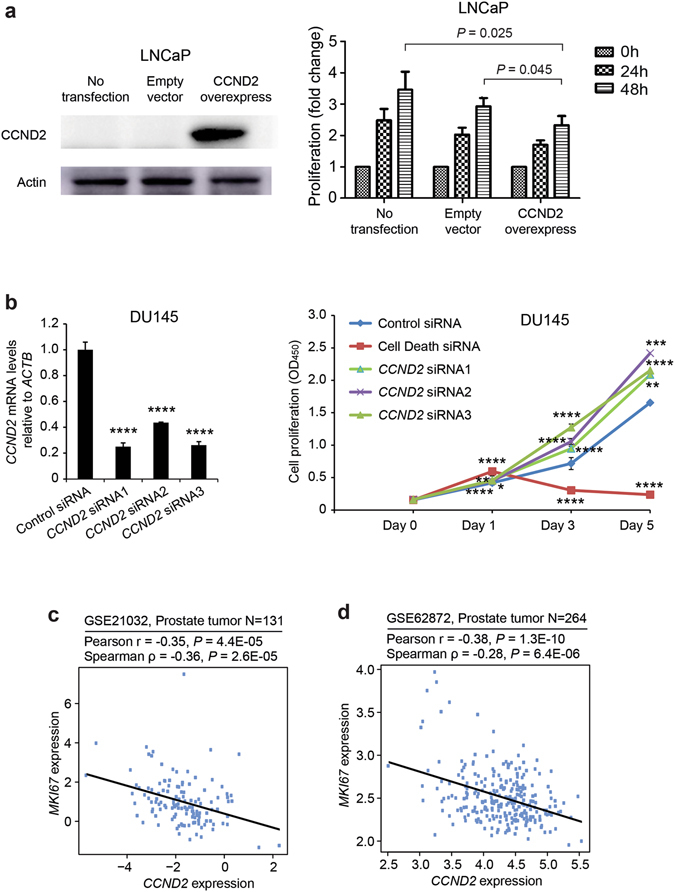
CCND2 plays a role in prostate cancer cell proliferation. (a) Ectopic expression of CCND2 in LNCaP inhibits its cellular proliferation. Left: Measurement of CCND2 overexpression by Western blot. Note that the three lanes represent LNCaP without transfection, LNCaP transfected with empty vector and LNCaP transfected with CCND2 expression plasmids, respectively. Right: The results of MTT in LNCaP cells upon CCND2 overexpression. Comparing to controls (no transfection or transfection with empty vector), the proliferations of LNCaP cells were significantly inhibited. The absorbance at 570 nm was measured with EPOCH2 Microplate Readers (BioTek), mean ± s.d. of four independent experiments. (b) Knockdown of CCND2 in DU145 promotes its cellular proliferation. Left panel: The knockdown efficiency of siRNAs against CCND2 were measured by quantitative real-time PCR. Right panel: Knockdown of CCND2 increases cell proliferation of prostate cancer DU145 cell line measured by XTT colorimetric assay (absorbance at 450 nm (OD450); mean ± s.d. of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. In (a and b), the P values were assessed by the two-tailed Student’s t test. (c,d) Expression correlation of CCND2 with MKI67 in human prostate tumor samples. Scatter plot showing the inverse correlation between CCND2 and MKI67 expression in two independent cohorts of prostate tumors (n = 131 and n = 264, respectively)30, 31.
We next investigated the status of the associated genes in human prostate tissues by analyzing the expression levels of BAD, CCND2, PDGF-C, and PDGF-D in tens of independent cohorts of clinical expression profile data sets28, 30, 32–52. The results showed that the mRNA levels of BAD, CCND2 and PDGF-D were differentially expressed in prostate tumor tissues and normal prostate gland (Figs 3a–g and Supplementary Figs 2a–j, 3a–f and 4a–d). BAD was significantly upregulated in primary and metastatic prostate tumors compared to matched normal tissues30, 34, 35, 37–39 (Supplementary Fig. 3a–f). PDGF-D showed a strong tendency to be highly expressed in metastatic prostate cancer samples34, 40, 45, 51 (Supplementary Fig. 4a–c). In particular, the transcriptional levels of CCND2 were significantly lower in prostate tumors than in normal tissues, and the results from one cohort of Chinese32 and other sixteen independent clinical data sets30, 32–47 with a total amount of 1,083 samples showed that CCND2 was markedly underexpressed upon tumor progression to advanced stage and metastasis (Fig. 3a–g and Supplementary Fig. 2a–j). We also investigated the expression of CCND2 at protein levels using immunohistochemistry staining on prostatic tissue specimens of Chinese men. This analysis revealed that loss of CCND2 expression appeared to be in aggressive prostate cancer (Fig. 4). While all the Benign Prostatic Hyperplasia (BPH, n = 3) and non-aggressive prostate cancer (n = 4) showed positive CCND2 staining, only 50% of aggressive prostate cancer (n = 5) appeared to be positive with CCND2 expression (see Methods), consistent with one previous study53 showed that CCND2 proteins were frequently lost in a cohort of prostate cancer specimens by immunohistochemistry staining analysis.
Figure 3.
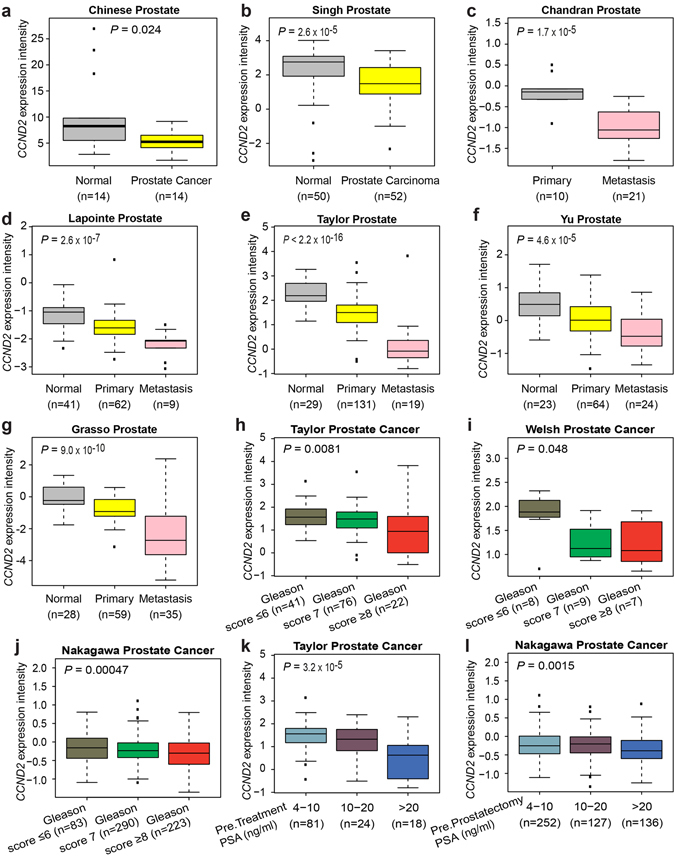
CCND2 mRNA expression is strikingly downregulated in metastatic human prostate cancer and its underexpression correlates with disease aggressiveness. (a–g) CCND2 transcript levels are greatly decreased in primary and metastatic prostate tumor tissues in several clinical data sets30, 32–37, including a cohort of prostate samples of Chinese men32. Normal, benign prostate gland. Primary, primary human prostate cancer. Metastasis, metastatic prostate samples. (h–j) Decreased CCND2 expression correlates with prostate tumor progression to high Gleason score in multiple cohorts of patients with prostate cancer28, 30, 38. (k,l) CCND2 underexpression markedly correlates with elevated serum PSA levels in the Taylor et al.30 and Nakagawa et al.28 clinical data sets. PSA is a diagnostic marker of prostate gland malignancy. Pre.Treatment or Pre.Prostatectomy PSA refer to the PSA level prior to radical prostatectomy. In (a), CCND2 expression intensity was determined by RNA sequencing32; (b–l), CCND2 expression intensity is log2 median-centered intensity as reported in Rhodes, D. R. et al.26.
Figure 4.
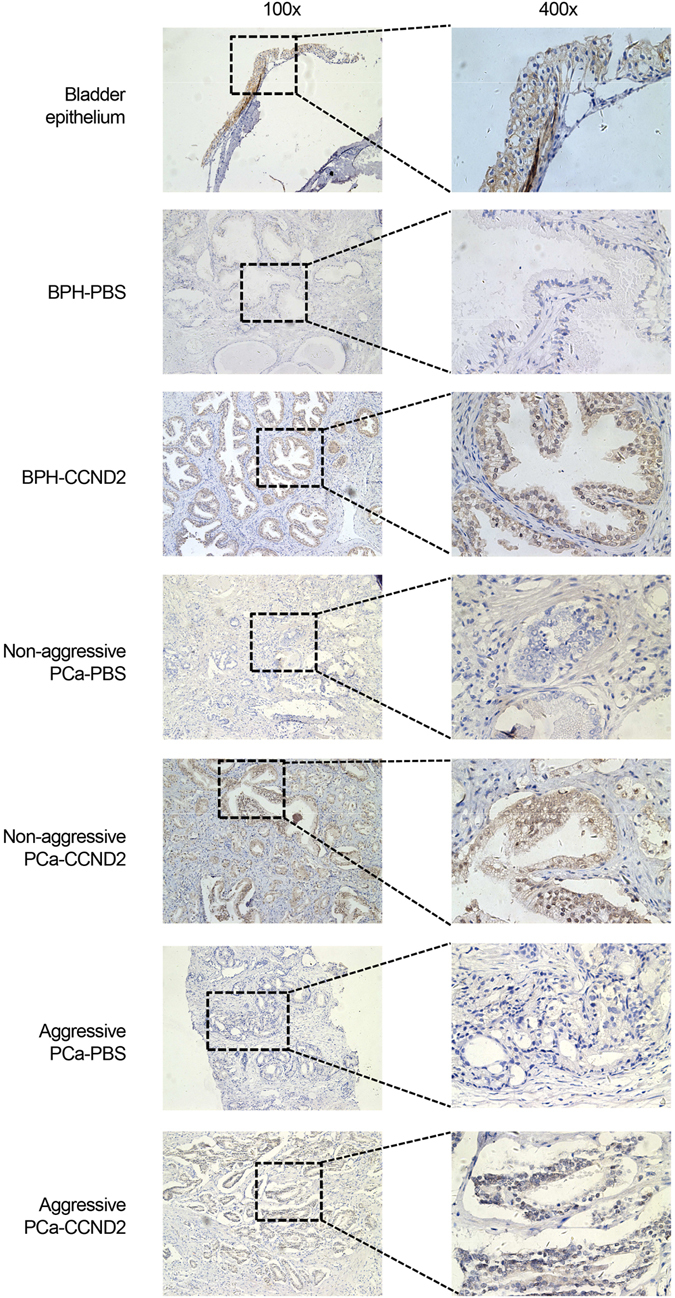
The immunohistochemistry staining analysis of CCND2 expression in the bladder epithelium, benign prostatic hyperplasia (BPH), aggressive and non-aggressive prostate cancer specimens (left: 100x and right: 400x magnification). Note that, according to BioGPS (http://biogps.org/#goto=genereport&id=12566&show_dataset=BDS_00007), the CCND2 was highly expressed in bladder epithelium cells. So, bladder epithelium was used as the positive control with strong staining signal using a CCND2-specific antibody. Staining with no antibody (PBS) as negative control. Note that, in contrast to BPH and non-aggressive prostate cancer, aggressive specimens showed less staining signal of CCND2 expression. The boxes around the area in the 100x images indicate the regions highlighted in the 400x images.
Additionally, we analyzed prostate cancer expression data from men with clinical records of Gleason score and prostate-specific antigen (PSA) levels28, 30, 38, 40, the indicators of prostate cancer aggressiveness, finding that CCND2 mRNA levels were significantly lower in tumors with high Gleason score and elevated PSA levels (Fig. 3h–l and Supplementary Fig. 2k–m). In contrast, higher transcript levels of BAD, PDGF-C and PDGF-D appeared to correlate with high Gleason score and PSA levels30, 35, 37, 40, 49–52 (Supplementary Figs 3g–l and 4e–h and 5), suggesting that these three genes may possess oncogenic functions whereas CCND2 is a potential tumor and metastasis suppressor in prostate cancer.
CCND2 indicates prognostic potential in prostate cancer
Among these analyses, CCND2 indicates the most prominent association with aggressive prostate cancer, supporting CCND2 as the most plausible causative gene through our integrated genetic association, follow-up eQTL and functional studies. We thus investigated the potential clinical relevance of CCND2 in prostate cancer in a large collection of prostate cancer cases28. Using the Kaplan-Meier analysis, we examined the association of CCND2 expression with clinical variables indicating prostate cancer aggressiveness, and found that the time to postoperative PSA recurrence and for overall survival was significantly shorter in the patient group with lower expression levels of CCND2 (P < 0.05; Fig. 5a and b), the latter association was also observed to be significant in another independent cohort of prostate cancer54 (P = 0.023; Fig. 5c). Notably, CCND2 downregulation also showed a strong tendency correlation with increased risk of biochemical relapse in an additional collection of prostate tumors30 (Supplementary Fig. 6a). In addition, to address the question of whether CCND2 levels have predictive values for low- and high-risk cases, we subdivided a large cohort of prostate cancer55, 56 by Gleason score and compared the frequency of postoperative biochemical recurrence. This analysis revealed a trend of CCND2 levels in predicting biochemical recurrence in prostate cancer patients with a Gleason score of 7 (intermediate risk, n = 246; Supplementary Fig. 6b), but not for the cases with a Gleason score of 6 (low risk, n = 44; Supplementary Fig. 6c) or the very high-risk patients (Gleason score ≥8, n = 201; Supplementary Fig. 6d), suggesting that CCND2 is a potential biomarker to distinguish the patients that are likely to recur in the patient group at intermediate risk.
Figure 5.
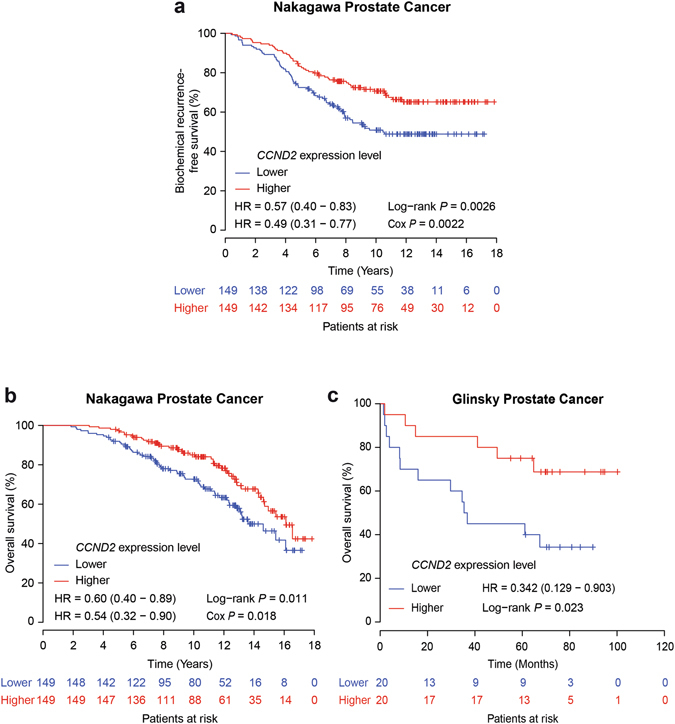
CCND2 downregulation correlates with poor prognosis of the patients with prostate cancer. Kaplan-Meier curves and estimates of the risk for biochemical recurrence (a), and overall survival (b) in a large group of prostate cancer patients28 with higher (top 50%; n = 149) or lower (bottom 50%; n = 149) expression levels of CCND2, and (c) in another independent cohort54. Overall, the patients with tumors expressing lower levels of CCND2 show elevated risk of biochemical relapse and decreased overall survival. The number of patients in each group at every 2-year (a,b) and (c) 20-month intervals was indicated. The P value was calculated by a Log-rank test (a–c) and Cox regression model (a,b), respectively.
To further assess the prognostic potential of CCND2 in prostate cancer and determine the relative dependency of PSA recurrence and overall survival, we performed multivariate analysis, including Gleason score, PSA levels, age and clinical T stage. This analysis revealed that the mRNA level of CCND2 was determined to be an independent predictor of biochemical recurrence and overall survival (P < 0.05; Fig. 5a and b). These results demonstrate a clear association of CCND2 downregulation with prostate cancer development, progression and poor prognosis in patients. Together with our genetic and eQTL evidence of the strong association between rs3217869 risk genotype and reduced CCND2 expression, this study identified CCND2 as a plausible causative gene and potential metastatic tumor suppressor underlying genetic predisposition and disease progression to aggressive prostate cancer.
Discussion
Aggressive prostate cancer indicates poor prognosis for the disease development. In our previous work10–12, a potential association was revealed between prostate cancer risk and the RTK/ERK pathway. Considering the disease development, a study on the association between aggressive prostate cancer and this pathway was needed, which could implicate the RTK/ERK pathway in the progression of prostate cancer and shed new insight into the targeted therapy for prostate cancer. Through a comprehensive analysis, we identified two SNPs rs3217869 in CCND2 and rs12643184 in PDGF-C suggested to be important. In addition, we observed a marginal association of three genes including CCND2 with aggressive prostate cancer.
The present study was conducted using the ChinaPCa data. At the SNP-level, the tag-SNPs in all 40 genes within the pathway were collected. After the association analysis, the SNPs with P < 0.05 were selected for the next analysis. As for the gene-based analysis, the ARTP method was applied. We identified three genes BAD, CCND2, and PDGF-D with a marginal association with aggressive prostate cancer. Combined with the results of SNP-level analysis, we presented two loci to be associated with prostate cancer aggressiveness. One is rs3217869 in CCND2 that was discovered by various model analyses, and suggested to be a risk factor (rs3217869 A allele) for aggressive prostate cancer. Consistently, this risk tendency of rs3217869 for aggressive prostate cancer was also observed in a large cohort of Finnish prostate cancer patients comprising 1729 aggressive and 693 non-aggressive cases. A slightly risk effect of allele A at rs3217869 was identified. This is in agreement with the eQTL analysis indicating a strong association of the allele A at rs3217869 with decreased transcript levels of CCND2, a potential tumor suppressive gene in prostate cancer and strongly implicating tumor progression. The other locus is rs12643184 (PDGF-C). rs12643184 was indicated as a risk factor related to the progression of prostate cancer with OR >1 in the additive and dominant models. Meanwhile, based on the clinical information (GS and clinical stage), it was suggested that the mutation of rs12643184 was associated with increased GS in the aggressive prostate cancer, consistent with the association result as described above.
As for the associated gene, rs3217892 is within CCND2, a member of D-type cyclins (CCND) that are known to be responsible for G1 phase progression. There are three D-type cyclins (CCND1, CCND2 and CCND3) involved in the regulation of transition from G1 to S during the cell cycle57, 58. Despite the fact that the overexpression of CCND2 has been reported in some cancers such as gastric cancer16, 59, 60, our analyses showed a striking downregulation of CCND2 and its association with disease progression and poor prognosis in multiple independent cohorts of prostate cancer, suggesting that CCND2 is a potential tumor suppressor in prostate carcinoma. Consistently, our results together with the previous reports53, 61 showed that restoration of CCND2 expression inhibited the proliferation of prostate cancer cells, and CCND2 knockdown accelerated prostate cancer cell proliferation. This is in line with our eQTL analysis that the risk allele A at rs3217869 is significantly associated with CCND2 downregulation, indicating a potential causal role of CCND2 underlying prostate cancer susceptibility and severity. In addition, a striking downregulation of CCND2 observed in this study may be partially explained by a previous report of promoter methylation of CCND2 in prostate cancer and its association with clinicopathological features of poor prognosis29.
The other SNP, rs12643184 is in PDGF-C, a member of the platelet-derived growth factor (PDGF) family. This family consists of four different polypeptide chains (PDGF-A, -B, -C and -D) that were mainly combined with two receptors (PDGFR-α and β) to perform the biological functions62. Activated PDGF-C and PDGF‑D are a high affinity ligand for PDGFR‑α and PDGFR-β homodimers, respectively63. Recent studies suggested that these two genes were associated with tumor progression and angiogenesis64, 65. PDGF-C was suggested to take part in the modulation of human melanoma cell invasiveness66. In addition, blockade of PDGF-C could inhibit pathological angiogenesis67, with the potential to be applied in cancer therapy. We thus hypothesized that the PDGF-C might be important in the metastasis and progression of prostate cancer to some extent. In line with this, we found that PDGF-C indicated a tendency to be highly expressed in advanced prostate tumors with high Gleason score and PSA levels.
PDGF-D consists of a two-domain structure with an N-terminal CUB domain (for complement C1r/C1s, Uegf, Bmp1), and a C-terminal PDGF/vascular endothelial growth factor domain68. Activated PDGF-D binds to its cognate receptor PDGFR-β inducing the removal of CUB domain and contributing to the development and progression of diseases69, 70. PDGF-D has been revealed with the potential oncogenic activity in the development of prostate cancer71. Consistently, we observed a high tendency of PDGF-D upregulation in the metastatic prostate tumors and the tumors indicating high Gleason score and PSA levels. Moreover, in metastatic prostate cancer, PDGF-D was reported to be a regulator of osteoclastic differentiation that is critical for the establishment of skeletal metastatic deposits in prostate cancer patients72.
There are limited studies to link the association between BAD gene and aggressive prostate cancer. In support of the role for BAD in prostate cancer progression, one recent study73 suggested that BAD activation through the JNK and PI3K/Akt pathways contributed to prostate tumor growth under chronic hypoxia. In addition, a study in non-small cell lung cancer74 showed that the loss of BAD independently predicted poor prognosis. In addition to this, our results showed a markedly increased expression of BAD in advanced prostate cancer samples, further supporting this genetic identification of the association of BAD with aggressive prostate cancer.
In summary, our combined SNP- and gene-based analyses revealed the association of two novel loci (rs3217869/CCND2 and rs12643184/PDGF-C), and three genes (BAD, PDGF-D and CCND2) in the RTK/ERK signaling pathway with aggressive prostate cancer. Our eQTL study together with a comprehensive analysis of tens of independent clinical data sets highlight the potential role of CCND2 as a plausible causative gene with metastatic tumor suppressive activity in aggressive prostate cancer, providing promising clues on risk stratification and targeted therapy in men with advanced prostate cancer.
Electronic supplementary material
Acknowledgements
We thank the CSC - IT Center for Science Ltd. for providing Linux High Performance Computing server. This work was supported by Guangxi Natural Science Fund for Innovation Research Team (2013GXNSFFA019002), Guangxi Collaborative Innovation Center for genomic and personalized medicine (201319), The Science and technology development plan of Guangxi (Guikegong 1355005-3-17), National Program on Key Basic Research Project (973 Program) (2012CB518303), and the Academy of Finland (284618 and 279760), University of Oulu Strategic Funds and Jane and Aatos Erkko Foundation grants awarded to G.-H.W.
Author Contributions
Y.C. (Yang Chen), Q.Z., G.-H.W. and Z.M. conceived and designed the study. Y.C. (Yang Chen) and J. L. performed genetic and statistical analysis, analysed the data. Q.Z. and G.-H.W. performed bioinformatics analysis and clinical correlation analysis. Q.W, J.X. (Jihan Xia), P.G., H.C. and Y.Y. performed the experiments and provided technical support. J.X. (Jianfeng Xu) and Y.S. provided scientific discussion and the ChinaPCa data set. C.S. and J.S. contributed to the analysis of the Finnish prostate cancer GWAS data set. Y.H provided the advices for revising paper. H.Z and X.Y participated in the epidemiological design of China PCA. Y.J, Z.Y, Y.C (Yinchun Chen), Y.G, A.T and M.L participated in the collection of samples. Y.C. (Yang Chen) and G.-H.W. wrote the manuscript. G.-H.W. and Z.M. jointly supervised the study.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Yang Chen, Qin Zhang, Qiuyan Wang and Jie Li contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04731-4
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Gong-Hong Wei, Email: gonghong.wei@oulu.fi.
Zengnan Mo, Email: zengnanmo@hotmail.com.
References
- 1.Ferlay J, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. 2015;136:E359–86. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 2.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer. J. Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 3.Chen R, Ren S, Sun Y. Genome-wide association studies on prostate cancer: the end or the beginning? Protein Cell. 2013;4:677–686. doi: 10.1007/s13238-013-3055-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitington T, et al. Gene regulatory mechanisms underpinning prostate cancer susceptibility. Nat. Genet. 2016;48:387–397. doi: 10.1038/ng.3523. [DOI] [PubMed] [Google Scholar]
- 5.Du M, et al. Chromatin interactions and candidate genes at ten prostate cancer risk loci. Sci. Rep. 2016;6:23202. doi: 10.1038/srep23202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen H, et al. Systematic enrichment analysis of potentially functional regions for 103 prostate cancer risk-associated loci. Prostate. 2015;75:1264–1276. doi: 10.1002/pros.23008. [DOI] [PubMed] [Google Scholar]
- 7.Shih MC, et al. TOPK/PBK promotes cell migration via modulation of the PI3K/PTEN/AKT pathway and is associated with poor prognosis in lung cancer. Oncogene. 2012;31:2389–2400. doi: 10.1038/onc.2011.419. [DOI] [PubMed] [Google Scholar]
- 8.Jung HY, Joo HJ, Park JK, Kim YH. The Blocking of c-Met Signaling Induces Apoptosis through the Increase of p53 Protein in Lung Cancer. Cancer. Res. Treat. 2012;44:251–261. doi: 10.4143/crt.2012.44.4.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010;24:1967–2000. doi: 10.1101/gad.1965810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ning QY, et al. Key pathways involved in prostate cancer based on gene set enrichment analysis and meta analysis. Genet. Mol. Res. 2011;10:3856–3887. doi: 10.4238/2011.December.14.10. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y, et al. RTK/ERK pathway under natural selection associated with prostate cancer. PLoS One. 2013;8:e78254. doi: 10.1371/journal.pone.0078254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen Y, et al. The RTK/ERK pathway is associated with prostate cancer risk on the SNP level: a pooled analysis of 41 sets of data from case-control studies. Gene. 2014;534:286–297. doi: 10.1016/j.gene.2013.10.042. [DOI] [PubMed] [Google Scholar]
- 13.Mulholland DJ, et al. Pten loss and RAS/MAPK activation cooperate to promote EMT and metastasis initiated from prostate cancer stem/progenitor cells. Cancer Res. 2012;72:1878–1889. doi: 10.1158/0008-5472.CAN-11-3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang J, et al. B-Raf activation cooperates with PTEN loss to drive c-Myc expression in advanced prostate cancer. Cancer Res. 2012;72:4765–4776. doi: 10.1158/0008-5472.CAN-12-0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Regad T. Targeting RTK Signaling Pathways in Cancer. Cancers (Basel) 2015;7:1758–1784. doi: 10.3390/cancers7030860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL. Cyclin D as a therapeutic target in cancer. Nat. Rev. Cancer. 2011;11:558–572. doi: 10.1038/nrc3090. [DOI] [PubMed] [Google Scholar]
- 17.Zwick E, Bange J, Ullrich A. Receptor tyrosine kinase signalling as a target for cancer intervention strategies. Endocr. Relat. Cancer. 2001;8:161–173. doi: 10.1677/erc.0.0080161. [DOI] [PubMed] [Google Scholar]
- 18.McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. 2007;26:3113–3121. doi: 10.1038/sj.onc.1210394. [DOI] [PubMed] [Google Scholar]
- 19.Xu J, et al. Genome-wide association study in Chinese men identifies two new prostate cancer risk loci at 9q31.2 and 19q13.4. Nat. Genet. 2012;44:1231–1235. doi: 10.1038/ng.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu W, et al. Association of a germ-line copy number variation at 2p24.3 and risk for aggressive prostate cancer. Cancer Res. 2009;69:2176–2179. doi: 10.1158/0008-5472.CAN-08-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Duggan D, et al. Two genome-wide association studies of aggressive prostate cancer implicate putative prostate tumor suppressor gene DAB2IP. J. Natl. Cancer Inst. 2007;99:1836–1844. doi: 10.1093/jnci/djm250. [DOI] [PubMed] [Google Scholar]
- 22.Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. 2009;37:W600–5. doi: 10.1093/nar/gkp290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Purcell S, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu K, et al. Pathway analysis by adaptive combination of P-values. Genet. Epidemiol. 2009;33:700–709. doi: 10.1002/gepi.20422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ross-Adams H, et al. Integration of copy number and transcriptomics provides risk stratification in prostate cancer: A discovery and validation cohort study. EBioMedicine. 2015;2:1133–1144. doi: 10.1016/j.ebiom.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rhodes DR, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Q, et al. A prostate cancer susceptibility allele at 6q22 increases RFX6 expression by modulating HOXB13 chromatin binding. Nat. Genet. 2014;46:126–135. doi: 10.1038/ng.2862. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa T, et al. A tissue biomarker panel predicting systemic progression after PSA recurrence post-definitive prostate cancer therapy. PLoS One. 2008;3:e2318. doi: 10.1371/journal.pone.0002318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Padar A, et al. Inactivation of cyclin D2 gene in prostate cancers by aberrant promoter methylation. Clin. Cancer Res. 2003;9:4730–4734. [PubMed] [Google Scholar]
- 30.Taylor BS, et al. Integrative genomic profiling of human prostate cancer. Cancer. Cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Penney KL, et al. Association of prostate cancer risk variants with gene expression in normal and tumor tissue. Cancer Epidemiol. Biomarkers Prev. 2015;24:255–260. doi: 10.1158/1055-9965.EPI-14-0694-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ren S, et al. RNA-seq analysis of prostate cancer in the Chinese population identifies recurrent gene fusions, cancer-associated long noncoding RNAs and aberrant alternative splicings. Cell Res. 2012;22:806–821. doi: 10.1038/cr.2012.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh D, et al. Gene expression correlates of clinical prostate cancer behavior. Cancer. Cell. 2002;1:203–209. doi: 10.1016/S1535-6108(02)00030-2. [DOI] [PubMed] [Google Scholar]
- 34.Chandran UR, et al. Gene expression profiles of prostate cancer reveal involvement of multiple molecular pathways in the metastatic process. BMC Cancer. 2007;7:64. doi: 10.1186/1471-2407-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lapointe J, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc. Natl. Acad. Sci. USA. 2004;101:811–816. doi: 10.1073/pnas.0304146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu YP, et al. Gene expression alterations in prostate cancer predicting tumor aggression and preceding development of malignancy. J. Clin. Oncol. 2004;22:2790–2799. doi: 10.1200/JCO.2004.05.158. [DOI] [PubMed] [Google Scholar]
- 37.Grasso CS, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Welsh JB, et al. Analysis of gene expression identifies candidate markers and pharmacological targets in prostate cancer. Cancer Res. 2001;61:5974–5978. [PubMed] [Google Scholar]
- 39.Varambally S, et al. Integrative genomic and proteomic analysis of prostate cancer reveals signatures of metastatic progression. Cancer. Cell. 2005;8:393–406. doi: 10.1016/j.ccr.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 40.Vanaja DK, Cheville JC, Iturria SJ, Young CY. Transcriptional silencing of zinc finger protein 185 identified by expression profiling is associated with prostate cancer progression. Cancer Res. 2003;63:3877–3882. [PubMed] [Google Scholar]
- 41.Wallace TA, et al. Tumor immunobiological differences in prostate cancer between African-American and European-American men. Cancer Res. 2008;68:927–936. doi: 10.1158/0008-5472.CAN-07-2608. [DOI] [PubMed] [Google Scholar]
- 42.Liu P, et al. Sex-determining region Y box 4 is a transforming oncogene in human prostate cancer cells. Cancer Res. 2006;66:4011–4019. doi: 10.1158/0008-5472.CAN-05-3055. [DOI] [PubMed] [Google Scholar]
- 43.Arredouani MS, et al. Identification of the transcription factor single-minded homologue 2 as a potential biomarker and immunotherapy target in prostate cancer. Clin. Cancer Res. 2009;15:5794–5802. doi: 10.1158/1078-0432.CCR-09-0911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tamura K, et al. Molecular features of hormone-refractory prostate cancer cells by genome-wide gene expression profiles. Cancer Res. 2007;67:5117–5125. doi: 10.1158/0008-5472.CAN-06-4040. [DOI] [PubMed] [Google Scholar]
- 45.Magee JA, et al. Expression profiling reveals hepsin overexpression in prostate cancer. Cancer Res. 2001;61:5692–5696. [PubMed] [Google Scholar]
- 46.Holzbeierlein J, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am. J. Pathol. 2004;164:217–227. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.LaTulippe E, et al. Comprehensive gene expression analysis of prostate cancer reveals distinct transcriptional programs associated with metastatic disease. Cancer Res. 2002;62:4499–4506. [PubMed] [Google Scholar]
- 48.Luo JH, et al. Gene expression analysis of prostate cancers. Mol. Carcinog. 2002;33:25–35. doi: 10.1002/mc.10018. [DOI] [PubMed] [Google Scholar]
- 49.Best CJ, et al. Molecular alterations in primary prostate cancer after androgen ablation therapy. Clin. Cancer Res. 2005;11:6823–6834. doi: 10.1158/1078-0432.CCR-05-0585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Barwick BG, et al. Prostate cancer genes associated with TMPRSS2-ERG gene fusion and prognostic of biochemical recurrence in multiple cohorts. Br. J. Cancer. 2010;102:570–576. doi: 10.1038/sj.bjc.6605519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tomlins SA, et al. Integrative molecular concept modeling of prostate cancer progression. Nat. Genet. 2007;39:41–51. doi: 10.1038/ng1935. [DOI] [PubMed] [Google Scholar]
- 52.Nanni S, et al. Epithelial-restricted gene profile of primary cultures from human prostate tumors: a molecular approach to predict clinical behavior of prostate cancer. Mol. Cancer. Res. 2006;4:79–92. doi: 10.1158/1541-7786.MCR-05-0098. [DOI] [PubMed] [Google Scholar]
- 53.Witt D, et al. Valproic acid inhibits the proliferation of cancer cells by re-expressing cyclin D2. Carcinogenesis. 2013;34:1115–1124. doi: 10.1093/carcin/bgt019. [DOI] [PubMed] [Google Scholar]
- 54.Glinsky GV, Glinskii AB, Stephenson AJ, Hoffman RM, Gerald WL. Gene expression profiling predicts clinical outcome of prostate cancer. J. Clin. Invest. 2004;113:913–923. doi: 10.1172/JCI20032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell163, 1011–1025 (2015). [DOI] [PMC free article] [PubMed]
- 56.Cerami E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer. Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang P. The cell cycle and development: redundant roles of cell cycle regulators. Curr. Opin. Cell Biol. 1999;11:655–662. doi: 10.1016/S0955-0674(99)00032-0. [DOI] [PubMed] [Google Scholar]
- 58.Sherr CJ. D-type cyclins. Trends Biochem. Sci. 1995;20:187–190. doi: 10.1016/S0968-0004(00)89005-2. [DOI] [PubMed] [Google Scholar]
- 59.Takano Y, Kato Y, Masuda M, Ohshima Y, Okayasu I. Cyclin D2, but not cyclin D1, overexpression closely correlates with gastric cancer progression and prognosis. J. Pathol. 1999;189:194–200. doi: 10.1002/(SICI)1096-9896(199910)189:2<194::AID-PATH426>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 60.Takano Y, et al. Cyclin D2 overexpression and lack of p27 correlate positively and cyclin E inversely with a poor prognosis in gastric cancer cases. Am. J. Pathol. 2000;156:585–594. doi: 10.1016/S0002-9440(10)64763-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kobayashi T, et al. Restoration of cyclin D2 has an inhibitory potential on the proliferation of LNCaP cells. Biochem. Biophys. Res. Commun. 2009;387:196–201. doi: 10.1016/j.bbrc.2009.06.146. [DOI] [PubMed] [Google Scholar]
- 62.Ogawa N, et al. Clinical significance of platelet derived growth factor-C and -D in gastric cancer. Oncol. Lett. 2015;10:3495–3501. doi: 10.3892/ol.2015.3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev. 2008;22:1276–1312. doi: 10.1101/gad.1653708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.LaRochelle WJ, et al. Platelet-derived growth factor D: tumorigenicity in mice and dysregulated expression in human cancer. Cancer Res. 2002;62:2468–2473. [PubMed] [Google Scholar]
- 65.Lokker NA, Sullivan CM, Hollenbach SJ, Israel MA, Giese NA. Platelet-derived growth factor (PDGF) autocrine signaling regulates survival and mitogenic pathways in glioblastoma cells: evidence that the novel PDGF-C and PDGF-D ligands may play a role in the development of brain tumors. Cancer Res. 2002;62:3729–3735. [PubMed] [Google Scholar]
- 66.Ruffini F, et al. Platelet-derived growth factor C and calpain-3 are modulators of human melanoma cell invasiveness. Oncol. Rep. 2013;30:2887–2896. doi: 10.3892/or.2013.2791. [DOI] [PubMed] [Google Scholar]
- 67.Hou X, et al. PDGF-CC blockade inhibits pathological angiogenesis by acting on multiple cellular and molecular targets. Proc. Natl. Acad. Sci. USA. 2010;107:12216–12221. doi: 10.1073/pnas.1004143107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ustach CV, Kim HR. Platelet-derived growth factor D is activated by urokinase plasminogen activator in prostate carcinoma cells. Mol. Cell. Biol. 2005;25:6279–6288. doi: 10.1128/MCB.25.14.6279-6288.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Reigstad LJ, Varhaug JE, Lillehaug JR. Structural and functional specificities of PDGF-C and PDGF-D, the novel members of the platelet-derived growth factors family. FEBS J. 2005;272:5723–5741. doi: 10.1111/j.1742-4658.2005.04989.x. [DOI] [PubMed] [Google Scholar]
- 70.Fredriksson L, Li H, Fieber C, Li X, Eriksson U. Tissue plasminogen activator is a potent activator of PDGF-CC. EMBO J. 2004;23:3793–3802. doi: 10.1038/sj.emboj.7600397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ustach CV, et al. A potential oncogenic activity of platelet-derived growth factor d in prostate cancer progression. Cancer Res. 2004;64:1722–1729. doi: 10.1158/0008-5472.CAN-03-3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang W, et al. A novel function for platelet-derived growth factor D: induction of osteoclastic differentiation for intraosseous tumor growth. Oncogene. 2012;31:4527–4535. doi: 10.1038/onc.2011.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nomura, T. et al. Targeting the Vav3 oncogene enhances docetaxel-induced apoptosis through the inhibition of androgen receptor phosphorylation in LNCaP prostate cancer cells under chronic hypoxia. Mol. Cancer. 12, 27-4598-12-27 (2013). [DOI] [PMC free article] [PubMed]
- 74.Huang Y, et al. Loss of Bad expression confers poor prognosis in non-small cell lung cancer. Med. Oncol. 2012;29:1648–1655. doi: 10.1007/s12032-011-0060-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.