

Abstract
Although multilocus sequence typing (MLST) is highly discriminatory and useful for outbreak investigations and epidemiological surveillance, it has always been controversial whether clustering and phylogeny inferred from the MLST gene loci can represent the real phylogeny of bacterial strains. In this study, we compare the phylogenetic trees constructed using three approaches, (1) concatenated blocks of homologous sequence shared between the bacterial genomes, (2) genome single-nucleotide polymorphisms (SNP) profile and (3) concatenated nucleotide sequences of gene loci in the corresponding MLST schemes, for 10 bacterial species with >30 complete genome sequences available. Major differences in strain clustering at more than one position were observed between the phylogeny inferred using genome/SNP data and MLST for all 10 bacterial species. Shimodaira-Hasegawa test revealed significant difference between the topologies of the genome and MLST trees for nine of the 10 bacterial species, and significant difference between the topologies of the SNP and MLST trees were present for all 10 bacterial species. Matching Clusters and R-F Clusters metrics showed that the distances between the genome/SNP and MLST trees were larger than those between the SNP and genome trees. Phylogeny inferred from MLST failed to represent genome phylogeny with the same bacterial species.
Introduction
Since the invention of multilocus sequence typing (MLST) in 1998, this technique has been confirmed to be highly reproducible, objective and discriminatory for molecular typing of bacteria, and can be performed easily by different laboratories for typing of strains collected in different localities1. MLST involves amplification and sequencing of multiple, usually seven, gene loci. In the past 15 years, MLST has been used widely for typing of bacteria2–7. At the moment, MLST schemes are available for more than 110 bacteria. Recently, we have developed an MLST scheme for Laribacter hongkongensis, a novel bacterium associated with fish-borne gastroenteritis and traveler’s diarrhea, which was also achieved using seven gene loci8, 9. eBURST and minimum spanning trees are commonly used to analyze MLST data for typing or cluster analysis. Despite its high discriminatory power for typing bacteria, it has been controversial whether the phylogenetic tree constructed using the sequences of the gene loci can represent the microevolution process of the bacterial strains undergoing typing, although many studies have used MLST data for further phylogenetic analysis based on the concatenation of the MLST genes10–14.
Complete genome sequencing of bacteria has not only revolutionized our understanding on multiple aspects of bacterial genetics and genomics and the phylogenetic relationships among bacteria at the species and intraspecies levels, but the availability of the genome sequences has also given us ample opportunities to solve problems that we have never been able to solve in the past. At the time of writing, more than 6,600 complete genome sequences of more than 1,900 bacterial species are available (https://www.ncbi.nlm.nih.gov/genome/browse/). Recently, we have also published the complete genome sequence of L. hongkongensis and have used genome sequencing for typing an emerging bacterium, Elizabethkingia anopheles 15, 16. During the process of constructing the MLST scheme and performing complete genome sequencing of L. hongkongensis, we were also inspired to answer the question of whether MLST phylogeny can represent the microevolution process of the genomes which can best be depicted by genome phylogeny. In this study, we used various methods to compare the phylogenetic trees constructed using three approaches, (1) concatenated blocks of homologous sequence shared between the bacterial genomes, (2) genome single-nucleotide polymorphisms (SNP) profile and (3) concatenated nucleotide sequences of the gene loci in the corresponding MLST schemes, for 10 bacterial species with more than 30 complete genome sequences available.
Materials and Methods
Bacterial genomes
All bacterial species with more than 30 complete genome sequences available by August 1 2015 were included in the analysis. The genome sequences were obtained from the National Center for Biotechnology Information database (Supplementary Table S1) and were further processed for phylogenetic tree construction using the following three methods. Only chromosomes I for Burkholderia pseudomallei were used for analysis because all MLST loci for B. pseudomallei were located on this chromosome.
Construction of genome phylogenetic tree
The downloaded genomes were aligned with the multiple genome alignment tool Mugsy by using the “-distance 1000” and “-minlength 100” options17. The Multiple Alignment Format blocks were concatenated and transformed in FASTA file format using the script available in Galaxy18–20. The resulting core alignment was filtered using Gblocks version 0.91b with the minimum length of a block set at 100 (b4 = 100) by removing poorly aligned positions and divergent regions21. An approximately maximum likelihood tree was built using FastTree 2, applying the generalized time-reversible model22. Outgroups listed in Table 1 were used for rooting the phylogenetic trees. Phylogenetic trees were visualized with MEGA623.
Table 1.
Information of outgroups and models used to construct maximum likelihood phylogenetic trees for each bacterial species in this study.
| Bacteria | Outgroup (GenBank accession no.) | Substitution models for MLST treesa |
|---|---|---|
| Burkholderia pseudomallei | Burkholderia thailandensis E254 (CP004381) | TIM1 + I + G |
| Campylobacter jejuni | Campylobacter coli 15-537360 (CP006702) | TIM1 + I + G |
| Chlamydia trachomatis | Chlamydia muridarum Nigg (AE002160) | TIM3 + I + G |
| Escherichia coli 1 | Escherichia fergusonii ATCC 35469 (CU928158) | TIM3 + I + G |
| Escherichia coli 2 | Escherichia fergusonii ATCC 35469 (CU928158) | GTR + I + G |
| Helicobacter pylori | Helicobacter acinonychis str. Sheeba (AM260522) | GTR + I + G |
| Helicobacter pylori SouthAfrica20 (CP006691) | ||
| Helicobacter pylori SouthAfrica7 (CP002336) | ||
| Klebsiella pneumoniae | Klebsiella variicola strain DSM 15968 (CP010523) | TIM1 + I + G |
| Listeria monocytogenes | Listeria innocua Clip11262 (AL592022) | TIM3 + I + G |
| Staphylococcus aureus | Staphylococcus capitis subsp. capitis strain AYP1020 (CP007601) | GTR + I + G |
| Salmonella enterica | Salmonella bongori serovar 48:z41:–str. RKS3044 (CP006692) | TrN + I + G |
| Streptococcus pyogenes | Streptococcus dysgalactiae subsp. equisimilis 167 (AP012976) | TrN + I + G |
aG, gamma distributed rate of heterogeneity; I, proportion of invariant sites.
Construction of SNP phylogenetic tree
Core genome SNPs were identified using the Parsnp program included in Harvest. A reference genome was randomly selected using the parameter ‘-r !’24. An approximately maximum likelihood tree was constructed from concatenated SNPs using FastTree 2, applying the generalized time-reversible model22. Outgroups were assigned as described above for the genome phylogenetic trees.
MLST sequence identification for construction of MLST phylogenetic tree
For each bacterial species with more than 30 complete genomes available, the nucleotide sequences of the gene loci used in its MLST scheme for one isolate were downloaded from the PubMLST database (http://pubmlst.org/). Two MLST schemes exist for Escherichia coli. The sequences obtained were used as queries in BLASTN searches against the downloaded nucleotide sequence of all the genomes of the species25. For all the strains of each bacterial species, the nucleotide sequences of the gene loci used in their MLST scheme were aligned independently with ClustalW2 using default settings26. Subsequently, Gblocks version 0.91b with the default options was used to concatenate conserved blocks into a single alignment21. Once aligned, the appropriate model of evolution was determined using corrected Akaike’s Information Criteria in jModelTest version 2.1.727. Maximum likelihood phylogenetic trees were inferred by using PhyML version 3.0, based on the concatenated alignment with the selected model listed in Table 1 28. Outgroups were assigned as described above for the genome phylogenetic trees.
Statistical analysis
Shimodaira-Hasegawa tests were performed to determine the congruence between the topologies of the phylogenetic trees derived from genome/SNP data and MLST gene fragments data29. Shimodaira-Hasegawa tests were conducted in PAUP* version 4.0b10 using 10000 RELL bootstrap replicates30. The null hypothesis in a Shimodaira-Hasegawa test is that two trees being compared are equally good explanations of the data. A P value of less than 0.05 is considered statistically significant to reject the null hypothesis and indicates that the trees are significantly different from one another.
Furthermore, differences among phylogenetic trees obtained from genome, SNP and MLST gene fragments data were evaluated with the software TreeCmp. In this analysis, two topology metrics, Matching Clusters and R-F Clusters, were utilized. A distance value of 0 indicates that the two trees under evaluation are identical and the value increases when they become more different.
Results
Bacterial genomes
A total of 639 genomes of 10 bacterial species with more than 30 complete genome sequences were analyzed (Supplementary Table S1). They included B. pseudomallei (43 genomes), Campylobacter jejuni (31 genomes), Chlamydia trachomatis (71 genomes), Escherichia coli (112 genomes), Helicobacter pylori (71 genomes), Klebsiella pneumoniae (36 genomes), Listeria monocytogenes (46 genomes), Staphylococcus aureus (70 genomes), Salmonella enterica (124 genomes) and Streptococcus pyogenes (35 genomes). The phylogenetic trees constructed using genomes (chromosome I for B. pseudomallei) and SNP data were compared to those constructed using the seven gene loci in the corresponding MLST schemes.
Phylogenies and topology comparisons
For each of the 10 bacterial species, phylogenies were inferred using genome, SNP and MLST data. Major differences in strain clustering at more than one position were observed between the phylogeny inferred using genome/SNP data and MLST for all 10 bacterial species (Figs 1–10), with the most prominent differences observed in B. pseudomallei, K. pneumoniae and S. enterica as described below.
Figure 2.
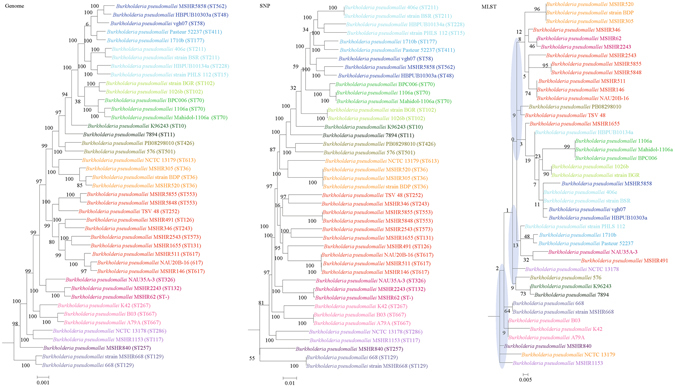
Comparison of phylogenetic trees constructed using genome data (left), SNP data (middle), and MLST data (right) for Burkholderia pseudomallei (chromosome I). Clusters that were manually selected based on the genome trees are illustrated in different colors. The unresolved polytomies are shaded in blue. A new sequence type is represented by a dash (“ST-”).
Figure 3.
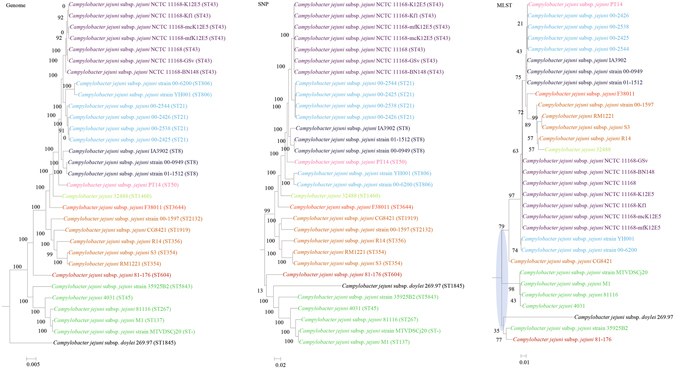
Comparison of phylogenetic trees constructed using genome data (left), SNP data (middle), and MLST data (right) for Campylobacter jejuni. Clusters that were manually selected based on the genome trees are illustrated in different colors. The unresolved polytomies are shaded in blue. A new sequence type is represented by a dash (“ST-”).
Figure 4.

Comparison of phylogenetic trees constructed using genome data (left), SNP data (middle), and MLST data (right) for Chlamydia trachomatis. Clusters that were manually selected based on the genome trees are illustrated in different colors. The unresolved polytomies are shaded in blue. A new sequence type is represented by a dash (“ST-”).
Figure 5.
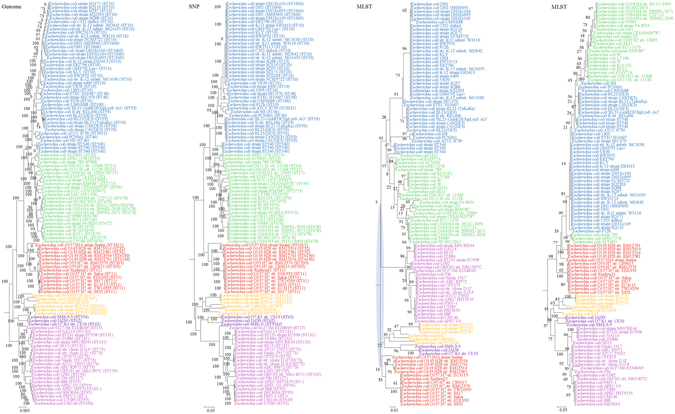
Comparison of phylogenetic trees constructed using genome data (left), SNP data (middle), and MLST data (right) for Escherichia coli (two MLST schemes). Clusters that were manually selected based on the genome trees are illustrated in different colors. The unresolved polytomies are shaded in blue. A new sequence type is represented by a dash (“ST-”).
Figure 6.
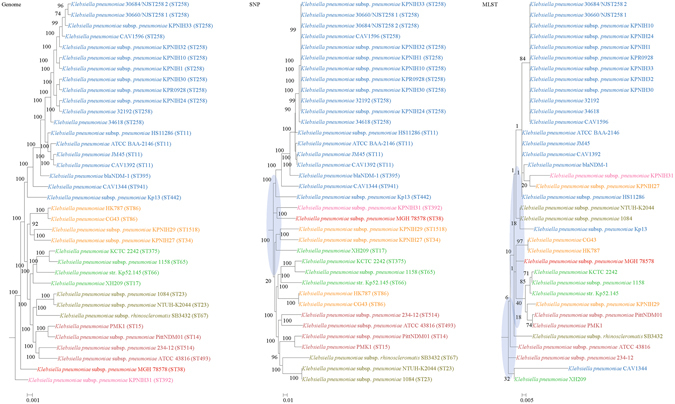
Comparison of phylogenetic trees constructed using genome data (left), SNP data (middle), and MLST data (right) for Klebsiella pneumoniae. Clusters that were manually selected based on the genome trees are illustrated in different colors. The unresolved polytomies are shaded in blue.
Figure 7.

Comparison of phylogenetic trees constructed using genome data (left), SNP data (middle), and MLST data (right) for Listeria monocytogenes. Clusters that were manually selected based on the genome trees are illustrated in different colors. The unresolved polytomies are shaded in blue. A new sequence type is represented by a dash (“ST-”).
Figure 8.
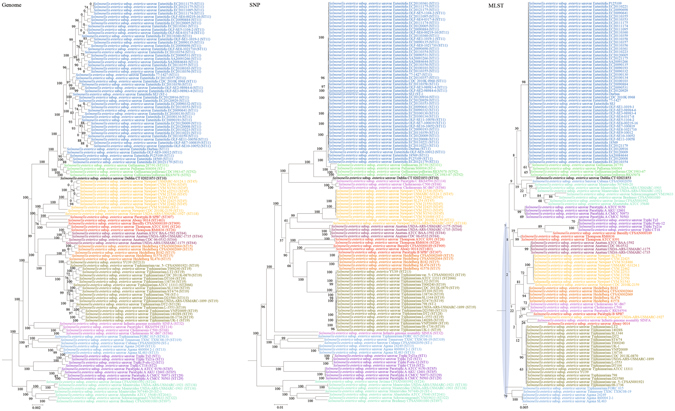
Comparison of phylogenetic trees constructed using genome data (left), SNP data (middle), and MLST data (right) for Salmonella enterica. Clusters that were manually selected based on the genome trees are illustrated in different colors. The unresolved polytomies are shaded in blue. A new sequence type is represented by a dash (“ST-”).
Figure 1.
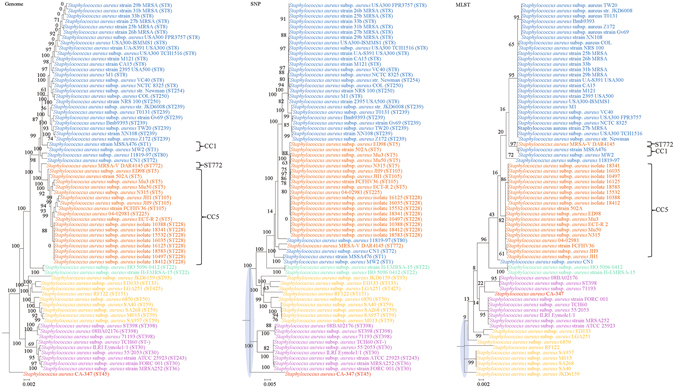
Comparison of phylogenetic trees constructed using genome data (left), SNP data (middle), and MLST data (right) for Staphylococcus aureus. Clusters that were manually selected based on the genome trees are illustrated in different colors. The unresolved polytomies are shaded in blue. A new sequence type is represented by a dash (“ST-”).
Figure 10.

Comparison of phylogenetic trees constructed using genome data (left), SNP data (middle), and MLST data (right) for Helicobacter pylori. Clusters that were manually selected based on the genome trees are illustrated in different colors. A new sequence type is represented by a dash (“ST-”).
Burkholderia pseudomallei
In both the genome and SNP phylogenetic trees, NAU35A-3 was clustered with MSHR2243 and MSHR62 with significant branch support of 100; but in the MLST tree, NAU35A-3 was clustered with MSHR491. In both the genome and SNP phylogenetic trees, PB08298010 was clustered with 576 with significant branch support of 100; but in the MLST tree, PB08298010 was clustered with TSV 48. In both the genome and SNP phylogenetic trees, NCTC 13178 was clustered with MSHR1153 with significant branch support of 100; but in the MLST tree, they were phylogenetically distinct. In both the genome and SNP phylogenetic trees, 406e, BSR HBPUB10134a and PHLS 112 were clustered together with significant branch support of 100; but in the MLST tree, they were designated in separate clades.
Klebsiella pneumonia
In both the genome and SNP phylogenetic trees, SB3432 was grouped together with 1084 and NTUH K2044 into one cluster with significant branch support of 100; but in the MLST tree, SB3432 was phylogenetically distinct. In both the genome and SNP phylogenetic trees, 234–12 was clustered with ATCC 43816 with significant branch support of 100; but in the MLST tree, 234–12 was phylogenetically distinct.
Salmonella enterica
In both the genome and SNP trees, S. enterica serovar Heidelberg was clustered with S. enterica ser. Typhimurium, with significant branch support of 100; but in the MLST tree, S. enterica ser. Heidelberg was clustered with S. enterica ser. Newport. In both the genome and SNP trees, S. enterica ser. Cubana was clustered with S. enterica ser. Tennessee with significant branch support of 100 and 99 respectively; but in the MLST tree, S. enterica ser. Cubana was clustered with serovars including S. enterica ser. Montevideo, S. enterica ser. Schwarzengrund, S. enterica ser. Bredeney, S. enterica ser. Javiana and S. enterica ser. Abaetetuba. In both the genome and SNP trees, S. enterica ser. Abony was clustered with S. enterica ser. Paratyphi B with significant branch support of 100; but in the MLST tree, S. enterica ser. Abony was clustered with S. enterica ser. Infantis.
Resolution and support for trees using different approaches
Both genome and SNP approaches yielded phylogenetic trees with strong support for most nodes. In contrast, for the trees inferred using MLST data, the bootstrap supports are generally low, with most of the nodes receiving less than 70% support (Figs 1–10). As for tree resolution, the genome phylogenetic trees are fully bifurcating; but both the SNP and MLST trees contain polytomies, with the MLST trees containing more polytomies than the SNP trees (Figs 1–9). For example, the phylogenetic trees are unresolved at the roots of the MLST trees of B. pseudomallei, C. jejuni, S. pyogenes, K. pneumoniae, S. enterica, S. aureus and E. coli.
Figure 9.
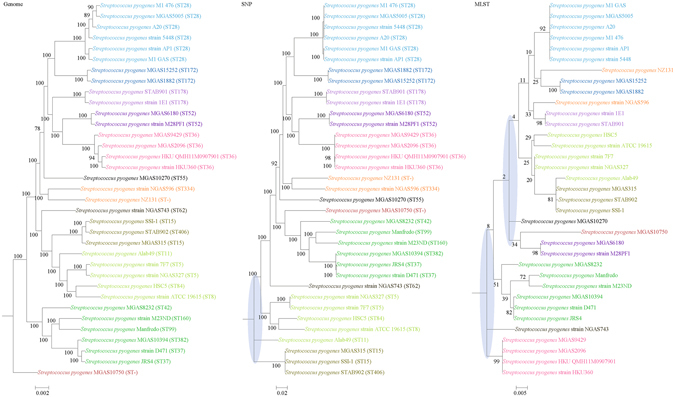
Comparison of phylogenetic trees constructed using genome data (left), SNP data (middle), and MLST data (right) for Streptococcus pyogenes. Clusters that were manually selected based on the genome trees are illustrated in different colors. The unresolved polytomies are shaded in blue. A new sequence type is represented by a dash (“ST-”).
Statistical measurement of phylogenetic incongruence
For the Shimodaira-Hasegawa test, significant difference between the topologies of the genome tree and MLST tree were present for nine (B. pseudomallei, C. jejuni, E. coli, H. pylori, K. pneumoniae, L. monocytogenes, S. aureus, S. enterica and S. pyogenes) of the 10 bacterial species, and significant difference between the topologies of the SNP tree and MLST tree were present for all the 10 bacterial species (Table 2). Matching Clusters and R-F Clusters metrics were used to quantify the difference between the phylogenetic trees constructed using the three approaches. For both Matching Clusters and R-F Clusters metrics, the distances between the genome/SNP trees and MLST trees were larger than those between the SNP trees and genome trees (Table 3).
Table 2.
Comparison by Shimodaira-Hasegawa test of log-likelihood scores between genome/SNP and MLST trees of the 10 bacterial species.
| Species | MLST Tree –ln L | Genome vs MLST | SNP vs MLST | ||||
|---|---|---|---|---|---|---|---|
| Genome Tree –ln L | Diff –ln L | P value | SNP –ln L | Diff –ln L | P value | ||
| Burkholderia pseudomallei | 5994.26511 | 6164.70585 | 170.44074 | 0.002* | 6190.62314 | 196.35803 | 0.002* |
| Campylobacter jejuni | 7762.65452 | 8088.5789 | 325.40337 | 0.000* | 8112.14513 | 349.49061 | 0.000* |
| Chlamydia trachomatis | 7810.81726 | 7844.19200 | 33.37474 | 0.065 | 7857.47013 | 46.65286 | 0.032* |
| Escherichia coli 1 | 9412.75447 | 9845.31931 | 432.56484 | 0.000* | 9836.13458 | 423.38011 | 0.000* |
| Escherichia coli 2 | 12615.10225 | 13363.74929 | 748.64704 | 0.000* | 13428.07040 | 812.96814 | 0.000* |
| Helicobacter pylori | 23840.02481 | 24459.54406 | 619.51925 | 0.000* | 24423.92980 | 583.90499 | 0.000* |
| Klebsiella pneumoniae | 5660.92269 | 5770.77356 | 109.85087 | 0.004* | 5767.36723 | 106.44455 | 0.003* |
| Listeria monocytogenes | 8418.29236 | 8563.19518 | 144.90282 | 0.002* | 8541.07333 | 122.78098 | 0.006* |
| Staphylococcus aureus | 8146.48997 | 8269.76377 | 123.27381 | 0.000* | 8288.49580 | 142.00584 | 0.000* |
| Salmonella enterica | 11447.21256 | 11769.73347 | 322.52092 | 0.000* | 11787.43639 | 340.2283 | 0.000* |
| Streptococcus pyogenes | 7481.09682 | 7682.21034 | 201.11352 | 0.000* | 7622.24838 | 141.15156 | 0.001* |
*P < 0.05.
Table 3.
Tree distances among phylogenies inferred using different approaches.
| Species | Genome vs MLST | SNP vs MLST | Genome vs SNP | |||
|---|---|---|---|---|---|---|
| Matching cluster | R-F cluster | Matching cluster | R-F cluster | Matching cluster | R-F cluster | |
| Burkholderia pseudomallei | 143 | 28 | 176 | 31 | 55 | 9 |
| Campylobacter jejuni | 156 | 25 | 176 | 24.5 | 62 | 7.5 |
| Chlamydia trachomatis | 436 | 57.5 | 466 | 58.5 | 102 | 18 |
| Escherichia coli 1 | 564 | 76.5 | 491 | 72.5 | 307 | 27 |
| Escherichia coli 2 | 531 | 74.5 | 564 | 70.5 | 307 | 27 |
| Helicobacter pylori | 257 | 45 | 212 | 42 | 171 | 32 |
| Klebsiella pneumoniae | 147 | 26 | 161 | 27 | 60 | 9 |
| Listeria monocytogenes | 112 | 28 | 124 | 28 | 48 | 8 |
| Staphylococcus aureus | 444 | 48 | 372 | 46.5 | 114 | 11.5 |
| Salmonella enterica | 1287 | 98.5 | 1242 | 97 | 214 | 31.5 |
| Streptococcus pyogenes | 95 | 17 | 127 | 17 | 72 | 7 |
Discussion
In this study, we unambiguously showed that phylogeny inferred from MLST cannot fully represent genome phylogeny. Although MLST has been shown to be highly discriminatory and hence very useful for outbreak investigations and epidemiological surveillance of infections, it has always been controversial whether clustering and phylogeny inferred from the MLST gene loci can represent the real phylogeny of the strains. Despite this controversy, numerous publications on MLST, including those published in leading infectious disease and microbiology journals, did draw conclusions on clustering of the studied strains10–14. As complete genome sequencing has become less expensive with the next generation genome sequencing technologies such as the Roche 454 sequence and Illumina systems, the number of complete bacterial genomes sequenced has been rising exponentially in recent years. As a result, we are now able to construct genome phylogenetic trees of different strains of the same bacterial species. In the present study, we employed the phylogenetic tree constructed using concatenated blocks of homologous sequence shared between bacterial genomes as the gold standard of genome phylogeny to determine if the phylogenetic tree constructed using the MLST gene loci sequences, also extracted from the same set of complete genome sequences, can represent the phylogenetic relatedness of the bacterial strains. At the moment, more than 30 complete genomes are available for 10 highly important pathogenic bacteria, B. pseudomallei, C. jejuni, C. trachomatis, E. coli, H. pylori, K. pneumoniae, L. monocytogenes, S. aureus, S. enterica and S. pyogenes. Comparison of their genome trees and MLST trees by visual inspection revealed that their topologies are different. Major differences in strain clustering at more than one position were observed in the two trees for all 10 bacterial species (Figs 1–10).
In addition to the difference in topologies observed by visual inspection, the genome tree and MLST trees were shown to be incongruent according to three independent statistical tests for determining and quantifying the incongruence between the phylogenies, which included the Shimodaira-Hasegawa test, Matching Clusters and R-F Clusters metrics. The Shimodaira-Hasegawa test determines whether two tree topologies are equally well supported by the data, while the Matching Clusters metric calculates the smallest number of moves in order to transform one tree into the other and R-F Clusters metric calculates the number of different bipartitions between two trees. In this study, for the Shimodaira-Hasegawa test, incongruence between the genome tree and MLST tree were observed for all 10 bacteria except C. trachomatis (Table 2). For the Matching Clusters and R-F Clusters metrics, large distances were observed between the genome tree and MLST tree for all 10 bacteria (Table 3). All these indicate that phylogeny and clustering of bacterial strains using MLST trees may not represent their true phylogeny and clustering and therefore must be interpreted with great caution. For example, methicillin-resistant S. aureus with Panton-Valentine leucocidin ST772, associated with severe skin and soft tissue infections, was assigned to clonal complex 131–36, as observed in the MLST tree in the present study (Fig. 1). However, the genome tree clearly showed that ST772 is more closely related to complex 5 than complex 1 with very high branch support of 100 (Fig. 1).
MLST fails to represent genome phylogeny because the seven genes used for MLST contain much less sequence information than the whole genome. In this study, the genome tree was constructed by whole genome alignment followed by extraction and concatenation of all locally collinear blocks. For example, for the genome tree for S. aureus, 618,392 bases per genome (21.8% of the genomes) were used for constructing the genome tree. On the other hand, only 3,186 bases, which belonged to seven independent genes in different parts of the genome, per genome (0.1% of the genomes) were used to construct the corresponding MLST tree. Hence, the MLST tree cannot fully represent genome phylogeny as it only contains 0.46% of the information used for genome tree construction. This is a problem in MLST, as one of these genes might even be subject of recombination, leading to conflicting results that do not correlate with the whole genome genetic information. In a typical MLST approach, MLST could be less vulnerable to recombination events by constructing trees from allelic profile data instead of total sequence similarity between strains. In recent years, the whole genome approach is vastly superior to using a single- or multiple-marker gene for examining phylogenetic relationships. The issues of recombination and horizontal gene transfer could be mitigated by using thousands or more genes in whole genome. It has also been observed that whole genome could provide much richer information than MLST and can be used to study microevolution in much finer detail in previous studies37, 38. In addition, the phylogenies inferred using MLST data are generally less resolved with low support, which is also likely due to the small number of informative sites. As genome sequencing has become much easier and cheaper than before, it should be performed for unambiguous typing of bacterial strains16. The whole genome sequencing approach provides the possibility to perform MLST on a genome-wide scale such as ribosomal MLST39, core genome MLST40 and whole genome MLST41 with increasing discriminatory power.
Interestingly, the SNP trees showed similar topologies to the genome trees by visual inspection and higher congruence to the genome trees compared to MLST trees. Genome-wide SNP trees were first used for analysis of Bacillus anthracis genomes42, 43 because of their coverage of the entire genome, relative simpler and less time consuming44–46. Although computational capacity in general doubles every 18 months, sequencing capability has been doubling every 6–9 months in recent years47. Therefore, it would be essential to look for less time consuming ways of analyzing genome phylogeny. For example, in the present study, constructing the whole genome tree for S. aureus took about 31 hours with a Xeon 5690 CPU and 48 GB memory, whereas only 3 minutes was used for constructing the SNP tree. As the conclusions drawn from SNP trees are consistent with the genome trees, the SNP trees may be considered as an alternative in whole genome epidemiology studies, particularly in situations where computational resources and time are limited.
Electronic supplementary material
Acknowledgements
This work is partly supported by the Strategic Research Theme Fund, The University of Hong Kong and Croucher Senior Medical Research Fellowships.
Author Contributions
A.K.L. Tsang, S.K.P. Lau and P.C.Y. Woo were involved in the conception and design of the study; A.K.L. Tsang and H.H. Lee were involved in acquisition of data and analysis; A.K.L. Tsang and P.C.Y. Woo were involved in interpretation of data. All authors read and approved the final manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-04707-4
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Susanna K. P. Lau, Email: skplau@hku.hk
Patrick C. Y. Woo, Email: pcywoo@hku.hk
References
- 1.Maiden MC, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:3140–3145. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Q, et al. Genotypic analysis of Klebsiella pneumoniae isolates in a Beijing Hospital reveals high genetic diversity and clonal population structure of drug-resistant isolates. PloS one. 2013;8:e57091. doi: 10.1371/journal.pone.0057091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enright MC, Day NP, Davies CE, Peacock SJ, Spratt BG. Multilocus sequence typing for characterization of methicillin-resistant and methicillin-susceptible clones of Staphylococcus aureus. Journal of clinical microbiology. 2000;38:1008–1015. doi: 10.1128/jcm.38.3.1008-1015.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Enright MC, Spratt BG. A multilocus sequence typing scheme for Streptococcus pneumoniae: identification of clones associated with serious invasive disease. Microbiology. 1998;144(Pt 11):3049–3060. doi: 10.1099/00221287-144-11-3049. [DOI] [PubMed] [Google Scholar]
- 5.Dingle KE, et al. Multilocus sequence typing system for Campylobacter jejuni. Journal of clinical microbiology. 2001;39:14–23. doi: 10.1128/JCM.39.1.14-23.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Enright MC, Spratt BG, Kalia A, Cross JH, Bessen DE. Multilocus sequence typing of Streptococcus pyogenes and the relationships between emm type and clone. Infection and immunity. 2001;69:2416–2427. doi: 10.1128/IAI.69.4.2416-2427.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Woo PC, et al. Analysis of multilocus sequence typing schemes for 35 different bacteria revealed that gene loci of 10 bacteria could be replaced to improve cost-effectiveness. Diagnostic microbiology and infectious disease. 2011;70:316–323. doi: 10.1016/j.diagmicrobio.2011.03.006. [DOI] [PubMed] [Google Scholar]
- 8.Tse CW, et al. A novel MLST sequence type discovered in the first fatal case of Laribacter hongkongensis bacteremia clusters with the sequence types of other human isolates. Emerg Microbes Infect. 2014;3:e41. doi: 10.1038/emi.2014.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woo PC, et al. Development of a multi-locus sequence typing scheme for Laribacter hongkongensis, a novel bacterium associated with freshwater fish-borne gastroenteritis and traveler’s diarrhea. BMC microbiology. 2009;9:21. doi: 10.1186/1471-2180-9-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Davies HD, et al. Multilocus sequence typing of serotype III group B streptococcus and correlation with pathogenic potential. The Journal of infectious diseases. 2004;189:1097–1102. doi: 10.1086/382087. [DOI] [PubMed] [Google Scholar]
- 11.Jones N, et al. Enhanced invasiveness of bovine-derived neonatal sequence type 17 group B streptococcus is independent of capsular serotype. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2006;42:915–924. doi: 10.1086/500324. [DOI] [PubMed] [Google Scholar]
- 12.Wertheim HF, et al. Associations between Staphylococcus aureus Genotype, Infection, and In-Hospital Mortality: A Nested Case-Control Study. The Journal of infectious diseases. 2005;192:1196–1200. doi: 10.1086/444427. [DOI] [PubMed] [Google Scholar]
- 13.Woerther PL, et al. Emergence and dissemination of extended-spectrum beta-lactamase-producing Escherichia coli in the community: lessons from the study of a remote and controlled population. The Journal of infectious diseases. 2010;202:515–523. doi: 10.1086/654883. [DOI] [PubMed] [Google Scholar]
- 14.Cassir N, et al. Clostridium butyricum Strains and Dysbiosis Linked to Necrotizing Enterocolitis in Preterm Neonates. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2015;61:1107–1115. doi: 10.1093/cid/civ468. [DOI] [PubMed] [Google Scholar]
- 15.Woo PC, et al. The complete genome and proteome of Laribacter hongkongensis reveal potential mechanisms for adaptations to different temperatures and habitats. PLoS genetics. 2009;5:e1000416. doi: 10.1371/journal.pgen.1000416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lau SK, et al. Evidence for Elizabethkingia anophelis transmission from mother to infant, Hong Kong. Emerging infectious diseases. 2015;21:232–241. doi: 10.3201/eid2102.140623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Angiuoli SV, Salzberg SL. Mugsy: fast multiple alignment of closely related whole genomes. Bioinformatics. 2011;27:334–342. doi: 10.1093/bioinformatics/btq665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giardine B, et al. Galaxy: a platform for interactive large-scale genome analysis. Genome research. 2005;15:1451–1455. doi: 10.1101/gr.4086505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Blankenberg, D. et al. Galaxy: a web-based genome analysis tool for experimentalists. Current protocols in molecular biology/edited by Frederick M. Ausube. [et al.] Chapter 19, Unit 19 10 11–21; doi:10.1002/0471142727.mb1910s89 (2010). [DOI] [PMC free article] [PubMed]
- 20.Goecks J, Nekrutenko A, Taylor J, Galaxy T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome biology. 2010;11:R86. doi: 10.1186/gb-2010-11-8-r86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- 22.Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PloS one. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 24.Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome biology. 2014;15:524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of molecular biology. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 26.Larkin MA, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 27.Keane TM, Creevey CJ, Pentony MM, Naughton TJ, McLnerney JO. Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol Biol. 2006;6:29. doi: 10.1186/1471-2148-6-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guindon S, Gascuel O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Systematic biology. 2003;52:696–704. doi: 10.1080/10635150390235520. [DOI] [PubMed] [Google Scholar]
- 29.Shimodaira H, Hasegawa M. Multiple Comparisons of Log-Likelihoods with Applications to Phylogenetic Inference. Mol Biol Evol. 1999;16:1114–1116. doi: 10.1093/oxfordjournals.molbev.a026201. [DOI] [Google Scholar]
- 30.Swofford, D. L. PAUP*: Phylogenetic analysis using parsimony (* and other methods). Version 4.0b10 (2002).
- 31.Ellington MJ, Ganner M, Warner M, Cookson BD, Kearns AM. Polyclonal multiply antibiotic-resistant methicillin-resistant Staphylococcus aureus with Panton-Valentine leucocidin in England. The Journal of antimicrobial chemotherapy. 2010;65:46–50. doi: 10.1093/jac/dkp386. [DOI] [PubMed] [Google Scholar]
- 32.Nadig S, et al. Staphylococcus aureus eye infections in two Indian hospitals: emergence of ST772 as a major clone. Clinical ophthalmology. 2012;6:165–173. doi: 10.2147/OPTH.S23878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kechrid A, et al. Molecular analysis of community-acquired methicillin-susceptible and resistant Staphylococcus aureus isolates recovered from bacteraemic and osteomyelitis infections in children from Tunisia. Clinical microbiology and infection: the official publication of the European Society of Clinical Microbiology and Infectious Diseases. 2011;17:1020–1026. doi: 10.1111/j.1469-0691.2010.03367.x. [DOI] [PubMed] [Google Scholar]
- 34.D’Souza N, Rodrigues C, Mehta A. Molecular characterization of methicillin-resistant Staphylococcus aureus with emergence of epidemic clones of sequence type (ST) 22 and ST 772 in Mumbai, India. Journal of clinical microbiology. 2010;48:1806–1811. doi: 10.1128/JCM.01867-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neela V, Ehsanollah GR, Zamberi S, Van Belkum A, Mariana NS. Prevalence of Panton-Valentine leukocidin genes among carriage and invasive Staphylococcus aureus isolates in Malaysia. International journal of infectious diseases: IJID: official publication of the International Society for Infectious Diseases. 2009;13:e131–132. doi: 10.1016/j.ijid.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 36.Goering RV, et al. Molecular epidemiology of methicillin-resistant and methicillin-susceptible Staphylococcus aureus isolates from global clinical trials. Journal of clinical microbiology. 2008;46:2842–2847. doi: 10.1128/JCM.00521-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maiden MC, et al. MLST revisited: the gene-by-gene approach to bacterial genomics. Nature reviews. Microbiology. 2013;11:728–736. doi: 10.1038/nrmicro3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sahl JW, et al. A comparative genomic analysis of diverse clonal types of enterotoxigenic Escherichia coli reveals pathovar-specific conservation. Infection and immunity. 2011;79:950–960. doi: 10.1128/IAI.00932-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jolley KA, et al. Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology. 2012;158:1005–1015. doi: 10.1099/mic.0.055459-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruppitsch W, et al. Defining and Evaluating a Core Genome Multilocus Sequence Typing Scheme for Whole-Genome Sequence-Based Typing of Listeria monocytogenes. Journal of clinical microbiology. 2015;53:2869–2876. doi: 10.1128/JCM.01193-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jackson BR, et al. Implementation of Nationwide Real-time Whole-genome Sequencing to Enhance Listeriosis Outbreak Detection and Investigation. Clinical infectious diseases: an official publication of the Infectious Diseases Society of America. 2016;63:380–386. doi: 10.1093/cid/ciw242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Keim P, et al. Anthrax molecular epidemiology and forensics: using the appropriate marker for different evolutionary scales. Infection, genetics and evolution: journal of molecular epidemiology and evolutionary genetics in infectious diseases. 2004;4:205–213. doi: 10.1016/j.meegid.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 43.Pearson T, et al. Phylogenetic discovery bias in Bacillus anthracis using single-nucleotide polymorphisms from whole-genome sequencing. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:13536–13541. doi: 10.1073/pnas.0403844101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Harris SR, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327:469–474. doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Holt KE, et al. Shigella sonnei genome sequencing and phylogenetic analysis indicate recent global dissemination from Europe. Nature genetics. 2012;44:1056–1059. doi: 10.1038/ng.2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okoro CK, et al. Intracontinental spread of human invasive Salmonella Typhimurium pathovariants in sub-Saharan Africa. Nature genetics. 2012;44:1215–1221. doi: 10.1038/ng.2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Loman NJ, et al. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nature reviews. Microbiology. 2012;10:599–606. doi: 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.