

Abstract
Outbreaks of Streptococcus pyogenes hypervirulent clones are constant public health threats. In western Switzerland, an increase of severe cases of S. pyogenes invasive infections was observed between December 2015 and March 2016. Our aim was (i) to investigate these cases by the use of Whole Genome Sequencing (WGS) and (ii) to determine the specific virulome and resistome of each isolate in order to undertake adequate public health measures. Eleven Streptococcus pyogenes strains isolated from 11 patients with severe invasive infections between December 13, 2015 and March 12, 2016 were included in our study. Practically, emm-typing, MLST and WGS were used to investigate the relatedness between the isolates. The presence of virulence and antibiotic resistance genes as well as mutations in transcriptional regulators of virulence and in genes encoding for antibiotic targets were assessed. Three and two groups of isolates shared the same emm-type and ST type, respectively. Single Nucleotide Polymorphism (SNP) analysis revealed 14 to 32 SNPs between the strains of the same emm-type group, ruling out the possibility of a clonal outbreak. Mutations found in covS and rocA could partially explain an increased virulence. As these reassuring results were obtained in less than 10 days, no specific hospital hygiene and no dedicated public health measures had to be undertaken. WGS is a powerful technique to discriminate between closely related strains, excluding an outbreak in less than 10 days. Moreover, WGS provided extensive data on the virulome and resistome of all these strains.
Electronic supplementary material
The online version of this article (doi:10.1007/s10096-017-2905-z) contains supplementary material, which is available to authorized users.
Keywords: Streptococcus pyogenes, Group a streptococcus, Whole genome sequencing, Outbreak, Emm-typing, MLST
Introduction
Streptococcus pyogenes (Group A beta-hemolytic Streptococcus, GAS) is a colonizer of the human oropharynx and skin that can cause very diverse clinical presentations ranging from common and limited diseases such as pharyngo-tonsillitis, impetigo, erysipelas or cellulitis to life-threatening invasive diseases such as necrotizing fasciitis, pneumonia or streptococcal toxic shock syndrome [1]. The burden of invasive GAS infections in western countries is significant with an average incidence of 2.45 cases per 100,000 person-year and a case fatality rate of 15% [2]. Outbreaks of hypervirulent clones have been reported [3–5]. Their dramatic spread remains a constant public health threat, which requires a quick assessment.
Typing of S. pyogenes strains was historically done using a serologic classification of the M protein described by Lancefield in 1928. Nowadays, the emm gene, encoding for the M protein, allows the typing of isolates using PCR and sequencing of the amplicons, but not to discriminate between two isolates of the same emm-type and subtype. The same limitation applies to other typing approaches such as (i) multilocus sequence typing (MLST), based on the amplification and sequencing of a few housekeeping genes [6] or (ii) pulsed-field gel electrophoresis (PFGE) of large genomic fragments, previously cut by restriction enzymes [7]. Whole genome sequencing (WGS) has recently entered diagnostic laboratories and becomes a very powerful tool that allows differentiation between isolates at the level of Single Nucleotide Polymorphism (SNP) as well as a thorough investigation of the presence of specific virulence and antibiotic resistance genes [8]. Furthermore, mutations in two-components systems, in transcriptional regulators or in regulatory proteins (e.g. covR/covS (csrR/csrS), ropB/rgg or rocA respectively) have been shown to increase GAS virulence [9, 10], and the presence of such mutations may be investigated in order to get insights into the isolate’s propensity for invasion.
From January 2016 to March 2016, an increase of severe Streptococcus pyogenes infections was reported in Valais, a Swiss alpine canton with a population of 360,000 inhabitants. Indeed, six cases of GAS bacteremia were reported from January 1, 2016 to March 20, 2016, whereas only six, ten and eight GAS bacteremia were reported in 12 months in 2013, 2014 and 2015, respectively. Thus, we investigated a potential outbreak due to a hypervirulent S. pyogenes clone using genomics and analyzed the virulome and resistome of the isolated strains.
Methods
Clinical isolates and clinical data
We investigated Streptococcus pyogenes strains isolated between December 13, 2015 and March 12, 2016 from inpatients from Valais, who were included in this study as well as all the isolates obtained from normally sterile sites or from normally non-sterile sites providing a severe clinical presentation defined as sepsis, septic shock/toxic shock syndrome, meningitis, pneumonia or necrotizing fasciitis [11, 12]. Clinical data were retrieved from the patients’ electronic charts.
DNA extraction and whole genome sequencing
Genomic DNA extraction and purification were done using the protocol for Gram-positive bacteria with the Wizard Genomic DNA Purification Kit (Promega, ref. A1120). Genomic libraries were made using Nextera XT library kit (Illumina). One hundred fifty base pairs (bp) paired-end sequencing was performed using a MiSeq sequencer (Illumina, San Diego CA).
Assembly and annotation
Reads quality was assessed using FastQC version 0.11.5 (Andrews S. (2010), available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc). Trimmomatic 0.35 was used to filter low quality reads and reads shorter than 150 bp [13]. Assemblies were done with SPAdes genome assembler version 3.6.2 using k-mer sizes ranging from 43 to 127 bases [14]. Quast version 3.1 [15] was used to select the best assembly based on lowest number of contigs and best N50. Contigs shorter than 1000 bp and low k-mer coverage contigs (<2x) were discarded from the assemblies. The remaining contigs were reordered based on the RefSeq reference genome (strain SF370). Annotation was performed using RAST version 2.0 [16].
emm typing and multilocus sequence typing
emm-types and subtypes were determined by submitting emm gene sequences to the website of the Centers for Disease Control and Prevention, Streptococcus laboratory (http://www2a.cdc.gov/ncidod/biotech/strepblast.asp). Genome assemblies were submitted online to the pubMLST (http://pubmlst.org) database in order to assess their MLST types.
Core genomes alignment and mapping — whole genome alignments
The assemblies were aligned with the 51 complete genomes available on NCBI (Table S1) using Parsnp (v1.2) [17]. FigTree version 1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/) was used to edit the phylogenic tree based on the core-genome alignment. In order to detect the genetic variants, the trimmed reads were mapped against the core-genome of a closely related reference (based on Parsnp results) using snippy version 3.0 [18].
The assemblies of closely related isolates were aligned with Mauve version 2.4.0 [19] to assess genetic gains and losses. We looked manually at the annotation with Artemis 16.0.0 [20] to identify the gained or lost coding sequences.
Virulence factors
Virulence factors were identified by BLASTing the curated Virulence Factors Database (VFDB) [21] against the assemblies (cut-offs: e-value < 10−5, amino acid identity > 90%). Variants in the two-component regulatory system covR/covS and the transcriptional regulators ropB and rocA were identified by mapping trimmed reads against a reference genome (strain SF370) using snippy version 3.0 [18].
Hyaluronic acid assay
Hyaluronic acid quantification was performed in two independent experiments. Isolates were grown in THB medium until mid-logarithmic phase, as described in Hollands et al. [22]. Five milliliters of the culture (OD600, 0,4) were centrifuged at 5000 g for 10 min. Then, supernatants were discarded and pellets of bacteria were resuspended into 500 μl of deionized water. Serial dilutions of bacterial suspension were plated on blood agar growth medium and incubated to calibrate the suspension in colony-forming unit (CFU) per ml. Four hundred microliters were mixed with 1 ml of chloroform, 1-millimeter beads and mechanically shaken with a Precellys Evolution Homogenizer (Bertin Instruments, Montigny-le-Bretonneux), three times for 30 sec at 6800 rpm. Tubes were then centrifugated at 13000 g for 10 min. Hyaluronic acid concentration in the aqueous phase was determined using Corgenix Hyaluronic Acid kit according to manufacturer’s instructions.
Resistome and antibiotic susceptibility testing
Antibiotic resistance genes were identified using Resfinder version 2.1 with a threshold of 98% identity and 60% query coverage [23]. A manual search for known mutations in parC, gyrA, folA and folP was performed. Minimal inhibitory concentration for each isolate was determined using VITEK® 2 AST Cards (Biomérieux, France).
Results
Isolates and clinical presentations
Eleven S. pyogenes isolates were included in this study. Five out of six patients with complicated pneumonia were pediatric patients, whereas skin and soft tissues as well as joint infections were seen only in adult patients (Table 1). A report could be sent after less than 10 days after the reception of the strains in our laboratory.
Table 1.
List of the isolates and the clinical presentations
| Identifier | Gender | Age (years) | Sample | Primary diagnosis | Secondary diagnosis | Outcome |
|---|---|---|---|---|---|---|
| ISR1 | F | 1 | Blood culture | Complicated pneumonia with bilateral empyemas and TSS | RSV Bronchiolitis | Good |
| ISR2 | M | 1 | Pleural fluid | Complicated pneumonia with parapneumonic effusion | RSV infection | Good |
| ISR3 | M | 4 | Blood culture | Complicated pneumonia with parapneumonic effusion and TSS | Influenza A infection | Good |
| ISR4 | M | 6 | Pharyngeal swab | Oto-mastoiditis with subsequent meningitis and TSS | Influenza B infection | Good |
| ISR5 | F | 47 | Blood culture | Pneumonia complicated by ARDS | – | Good |
| ISR6 | F | 1 | Post-mortem, pleural fluid | Complicated pneumonia with parapneumonic effusion and TSS | – | Death |
| ISR7 | M | 49 | Blood culture | Skin and soft tissues infection (cellulitis) | – | Good |
| ISR8 | M | 79 | Blood culture | Skin and soft tissues infection (cellulitis) complicated by deep venous thrombosis and acute renal failure | – | Good |
| ISR9 | F | 58 | Blood culture | Septic arthritis | – | Good |
| ISR10 | F | 76 | Blood culture | Skin and soft tissues infection complicated by TSS | – | Good |
| ISR11 | F | 15 | Blood culture | Complicated pneumonia with parapneumonic effusion | – | Good |
F female, M male, ARDS acute respiratory distress syndrome, TSS toxic shock syndrome, RSV respiratory syncytial virus
emm-type and MLST
Eight different emm-types were detected (Table 2). Three emm-types were recovered twice among the isolates: emm1, emm22 and emm28. Multilocus sequence typing was more discriminant and allowed the identification of nine different ST-types (Table 2). Eight ST-types were detected with all the loci matching exactly against the pubMLST database. ISR2 bore a new allele of murI (110), which was submitted to the database as well as its new ST-type. In summary, emm-typing and MLST did not allow discrimination between three and two pairs of strains, respectively.
Table 2.
emm-types, emm-clusters and multi-locus sequence typing (ST types and the allelic profiles corresponding to the 7 loci)
| Isolates | emm-type (.subtype) | emm-cluster | ST | gki | gtr | murI | mutS | recP | xpt | yqiL |
|---|---|---|---|---|---|---|---|---|---|---|
| ISR3 | emm1.0 | A-C3 | 28 | 4 | 3 | 4 | 4 | 4 | 2 | 4 |
| ISR4 | emm1.0 | A-C3 | 28 | 4 | 3 | 4 | 4 | 4 | 2 | 4 |
| ISR7 | emm22 | E4 | 46 | 9 | 8 | 1 | 1 | 1 | 3 | 4 |
| ISR8 | emm22 | E4 | 46 | 9 | 8 | 1 | 1 | 1 | 3 | 4 |
| ISR9 | emm28.0 | E4 | 458 | 11 | 77 | 14 | 5 | 9 | 17 | 19 |
| ISR11 | emm28.0 | E4 | 52 | 11 | 6 | 14 | 5 | 9 | 17 | 19 |
| ISR1 | emm3.1 | A-C5 | 15 | 2 | 6 | 8 | 5 | 2 | 3 | 2 |
| ISR2 | emm75.0 | E6 | 880 | 11 | 2 | 110 | 3 | 50 | 8 | 7 |
| ISR5 | emm73.0 | E4 | 331 | 43 | 2 | 2 | 47 | 1 | 3 | 4 |
| ISR6 | emm5.23 | Ya | 99 | 33 | 30 | 7 | 5 | 5 | 26 | 3 |
| ISR10 | emm89.0 | E4 | 101 | 16 | 2 | 8 | 3 | 1 | 13 | 3 |
aSingle protein emm-cluster clade Y
Core-genome alignment and mapping
Genome assembly sizes ranged from 1,689,494 to 1,868,156 bp with a mean GC content of 38.35% (assemblies statistics are provided in supplementary materials, Table S2). The core genome size was 1,445,210 bp (see Parsnp results, Fig. 1). SNPs quantification in the core genomes of the closest isolates (corresponding to the same emm-types) revealed for emm1 isolates (ISR3 and ISR4), emm22 isolates (ISR7 and ISR8) and emm28 isolates (ISR9 and ISR11), 32, 14 and 28 SNPs of difference in their core genomes, respectively.
Fig. 1.

Phylogenetic representation of the sequenced strains during our study and the complete GAS genomes publicly available on NCBI. The phylogenetic tree was made using Parsnp and is based on the core genome alignment of our 11 isolates (in red), the currently 51 complete genomes available on NCBI (Table S1) and seven other genomes of S. pyogenes recently sequenced in our institute (from CV1 to CV7, taking part in another study). The tree was rooted at midpoint. Stars indicate bootstraps below 0.9
To look for additional differences (i.e. large insertions and deletions not detected by mapping) between the genomes of the closest isolates, we aligned the complete assemblies with progressive Mauve. ISR3 and ISR4 did not show any noticeable differences. ISR8 held 2 phage regions of 13.2 Kb and 14.6 Kb, the latest bearing speK gene whereas ISR7 had a 10.0 Kb phage region not retrieved in ISR8. ISR11 bore a genomic island of 24.7 Kb with a high content in hypothetical protein CDSs that was not present in ISR9.
Virulence factors and regulation of virulence
Twelve to 24 virulence factors encoding genes were identified per isolate (Table 3). The has-operon was found in all but three isolates (ISR7, ISR8 and ISR10), which only bore the hasC gene. ISR9 and ISR11 presented a 1 bp-insertion responsible for a frame-shift and premature truncation of the hasA product at amino acid 72. All the isolates had two to five genes encoding for superantigens (mean = 4).
Table 3.
Virulence genes detected by BLASTing the Virulence Factor Database (VFDB) on the assemblies
| emm1 | emm22 | emm28 | emm3 | emm75 | emm73 | emm5 | emm89 | |||
|---|---|---|---|---|---|---|---|---|---|---|
| ISR3 | ISR4 | ISR7 | ISR8 | ISR9 | ISR11 | ISR1 | ISR2 | ISR5 | ISR6 | ISR10 |
| cpa | cpa | fbaA | fbaA | fbaA | fbaA | cpa | cpa | fbaA | fbaB | fbaA |
| emm | emm | fbp54 | fbp54 | fbp54 | fbp54 | emm | emm | fbp54 | fbp54 | fbaB |
| fbaA | fbaA | grab | grab | hasA | hasA | fbaB | fbaB | hasA | hasA | fbp54 |
| fbp54 | fbp54 | hasC | hasC | hasB | hasB | fbp54 | fbp54 | hasB | hasB | hasC |
| fctA | fctA | hylP | hylP | hasC | hasC | grab | grab | hasC | hasC | lmb |
| fctB | fctB | ideS/mac | ideS/mac | lmb | lmb | hasA | hasA | lmb | lmb | mf/spd |
| grab | grab | lmb | lmb | mf/spd | mf/spd | hasB | hasB | mf/spd | mf/spd | scpA |
| hasA | hasA | mf/spd | mf/spd | mf2 | mf2 | hasC | hasC | mf2 | mf2 | scpB |
| hasB | hasB | mf2 | mf2 | scpA | scpA | hylP | hylP | scpA | mf3 | slo |
| hasC | hasC | scpA | scpA | scpB | scpB | ideS/mac | ideS/mac | slo | scpA | smeZ |
| ideS/mac | ideS/mac | scpB | scpB | slo | slo | lmb | lmb | smeZ | scpB | speB |
| lepA | lepA | slo | slo | smeZ | smeZ | mf/spd | mf/spd | speB | slo | speG |
| lmb | lmb | smeZ | smeZ | speB | speB | scpA | scpA | speC | smeZ | |
| mf/spd | mf/spd | speB | speB | speC | speC | ska | ska | speG | speB | |
| mf3 | mf3 | speC | speC | speG | speG | slo | slo | speH | speC | |
| scpA | scpA | speG | speG | speJ | speJ | smeZ | smeZ | speI | speG | |
| ska | ska | ssa | speK | speB | speA | |||||
| slo | slo | ssa | speG | speB | ||||||
| smeZ | smeZ | speK | speG | |||||||
| speA | speA | ssa | speK | |||||||
| speB | speB | ssa | ||||||||
| speG | speG | |||||||||
| speJ | speJ | |||||||||
| srtC1 | srtC1 | |||||||||
Has operon genes are shown in bold and superantigens are underlined. Of notes, speB is not considered as a superantigen and appears in black. For gene descriptions, see online supplementary materials, Table S4
We found two mutations in covS and one in rocA that are associated with an increased virulence [24, 25] (Fig. 2a). ISR9 exhibited a non-synonymous SNP in covS responsible for an amino acid replacement; E226G and ISR11 had a single nucleotide deletion at position 528 responsible for a premature truncation of CovS at amino acid 180. These two covS mutations were already described by Ikebe et al. in an invasive emm28 (NIH35) and in an invasive emm3 isolate (NIH453), respectively [24]. ISR1 (emm3) showed a known deletion in rocA causing a premature truncation and a loss of function of the product, as described by Lynskey et al. [25]. The rest of the variants found in covR, covS, ropB and rocA (supplementary materials, Table S3) have not been linked to more invasive phenotypes yet. They seem to be emm-type specific and can also be found in strains recovered from non-invasive infections (data not shown).
Fig. 2.
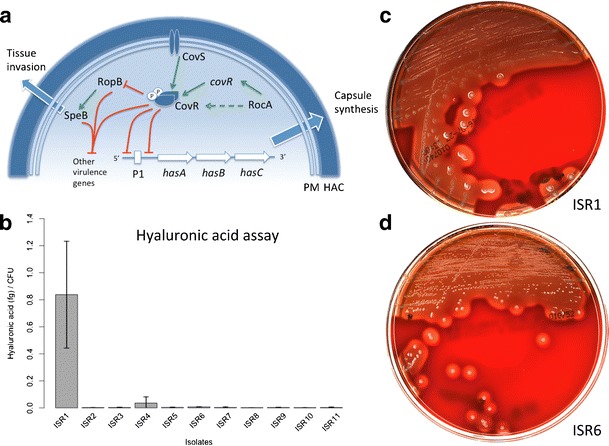
a Simplified scheme showing the best-known transcriptional regulators of virulence in Streptococcus pyogenes missense and non-sense mutations in covR and covS can increase Streptococcus pyogenes virulence by relieving CovR downregulation of many virulence genes such as the has-operon involved in hyaluronic acid synthesis [9]. RocA positively regulates covR transcription and possibly directly phosphorylates CovR [10]. Mutations resulting in premature truncation of RocA were shown to increase hyaluronic acid production and virulence. Mutations in ropB, a negative transcriptional regulator of many virulence genes, have been linked to more invasive phenotypes [24]. This regulator is also necessary for the expression of speB, a gene encoding for a cysteine protease degrading many Streptococcus pyogenes virulence factors and whose expression is inversely proportional to virulence, though speB is also known to be a major virulence factor involved in tissue invasion and in the pathogenesis of necrotizing fasciitis [9]. PM plasma membrane, HAC hyaluronic acid capsule, P1 promoter region 1 of the has operon. b Enzyme-linked binding protein assay ISR1 (emm3.1) and ISR4 (emm1.0) produced a higher amount of hyaluronic acid. c, d Blood agar plates showing a mucoid phenotype (ISR1) and a non-mucoid phenotype (ISR6)
Hyaluronic acid assay
ISR1, which was the only isolate exhibiting a frank mucoid phenotype on agar plates (Fig. 2c), showed the highest production of hyaluronic acid of the assay, which is congruent with previous findings [25]. ISR4 produced a larger amount of hyaluronic acid than the other isolates (Fig. 2b) but does not exhibit any known mutations in the transcriptional regulator genes analyzed.
Resistance genes
Two resistance genes were detected with ResFinder in the genome of the isolates. erm(B), a chromosomal gene involved in antibiotic resistance against macrolides and lincosamides, was detected in the ISR11 genome. tet(M), a tetracycline resistance gene, was found in ISR6, ISR7 and ISR8 genomes. Finally, ISR7 and ISR8 exhibited a non-synonymous mutation in parC (S79F), a gene involved in fluoroquinolone resistance [26].
Antibiotic susceptibility test
Congruent with the genomic findings, ISR11 was resistant to clindamycin (MIC > 1) and erythromycin (MIC > 8) and ISR6, ISR7 and ISR8 displayed various levels of tetracycline resistance (MIC: 4, MIC > =16 and MIC > =16, respectively). ISR7 and ISR8 were also resistant to levofloxacin (MIC: 4), as expected from their mutations located in the quinolone resistance-determining region of parC. Interestingly, ISR1 presented an intermediate sensitivity to trimethoprim-sulfamethoxazole (MIC: 40).
Discussion
An outbreak resulting from a hypervirulent clone was suspected due to the observed increased number of severe invasive GAS infections in western Switzerland over a 3-month period. In order to precisely and rapidly address this concern, we used whole genome sequencing (WGS) and we could rule out a clonal outbreak by comparing all the sequenced strains and by detecting the identification of 14 to 32 core-genome SNPs in the three pairs of closely related strains. This number of SNPs was much higher between the Swiss isolates taken less than 3 months apart compared to the 0–4 SNPs difference reported by Engelthaler et al. [4] when comparing clonal isolates drawn during a similar 3-month period. Moreover, Beres et al. [27] described a mean rate of 1.7 SNP/strain/year in the core genome, dating divergence of our closest isolates at about 8 years before sampling. We are confident that even the isolates bearing only 14 SNPs of difference in their core genome (ISR7 & ISR8) did not come from the same clone because they also had variations in their accessory genomes with different phage regions. Thus, this study showed the high discriminatory power of WGS, its possible application in clinical microbiology and emphasized the limitation of emm-typing and MLST, which did not allow differentiation of closely related strains of S. pyogenes. The use of WGS enabled rapid (<10 days) exclusion of a clonal outbreak, which reassured clinicians and public health authorities. This short time-to-result period had a positive impact on sparing hospital hygiene/public health measures, thus reinforcing its cost-effectiveness.
All strains were recovered from severe invasive GAS infections and thus, the analysis of the virulome was very interesting. However, the major limitation of the investigation of the genetic basis of the virulence of particular strains is the exhaustiveness of public databases. Here, we used only the curated and commonly used VFDB. Interestingly, all the isolates encoded between 2 to 5 genes homologous to known superantigens (mean = 4), but only five patients developed a toxic shock syndrome, thus supporting the evidence that the finding of CDS does not correlate with virulence [9]. Consistent to previous observations [28, 29], emm22 and emm89 isolates lacked the complete has-operon, although they were collected from patients with severe clinical presentation, thus emphasizing that hyaluronic acid capsule is not a prerequisite for increased virulence. Moreover, emm28 isolates (ISR9 and ISR11) had a premature truncation in the hasA product and did not produce an increased amount of hyaluronic acid despite the presence of mutations in covS, generally associated with increased hyaluronic acid production. In total, three mutations found in the regulatory genes rocA and covS (ISR1, ISR9 & ISR11) were previously correlated to more virulent phenotypes [24, 25]. While identifying mutations in the regulatory pathways of virulence is a promising way to predict the virulence of a given strain based on genomic data, no comprehensive database is currently available and the published data concern mainly emm3 and emm1 strains. Further research is thus required in this field.
Going deeper into virulence characterization, Olsen et al. [30] provided a response to a mock outbreak of GAS by performing WGS, genome-wide transcript analysis, and mouse virulence studies in a short time period. Here, we do not think that transcriptome analysis or in vivo studies were required as an outbreak of a single hypervirulent clone was already ruled out and further characterization of virulence factors would be difficult to perform and interpret due to our limited sample size with such heterogeneity of emm-types. Indeed, we only aimed to report findings from a real time investigation of a putative outbreak rather than conducting a study on virulence.
Concerning antibiotic resistances, the presence of antibiotic resistance genes tet(M) and ermB correlated with reduced antibiotic susceptibility. However, Resfinder did not detect point mutations in genes encoding for the antibiotic target such as gyrA or parC. Our study showed that a known mutation in parC conferring quinolone resistance to both emm22 isolates could only be found by additional specific analysis, emphasizing a lack of automated tools for the detection of point mutations in genes encoding for drug targets.
In conclusion, our study demonstrates the usefulness of whole genome sequencing by providing short time-to-results, by discriminating between closely related isolates when suspecting an outbreak but also by investigating the virulome, including its regulatory mechanisms as well as the antibiotic resistome. As such, WGS should be broadly implemented in clinical microbiology laboratories to improve patient care.
Accession numbers
Assemblies have been deposited on the DDBJ/EMBL/Genbank database under the study accession number PRJEB14938. The new allele and ST type were submitted to the PubMLST database.
Acknowledgements
The computations were performed at the Vital-IT (http://www.vital-it.ch) Center for high-performance computing of the SIB Swiss Institute of Bioinformatics. We would like to thank Sébastien Aeby and Maria Senra Ortiz for their significant technical help.
Electronic supplementary material
Below is the link to the electronic supplementary material.
(PDF 256 kb)
Compliance with ethical standards
Funding
The Institute of Microbiology of the Lausanne University Hospital, the Valais Hospital and the Pediatric Department of the Lausanne University Hospital funded this project.
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in this study involving human participants were in accordance with the ethical standards of the regional and national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
For this type of study, informed consent is not required.
Footnotes
S. Asner and G. Greub contributed equally to this work.
References
- 1.Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev. 2000;13:470–511. doi: 10.1128/CMR.13.3.470-511.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carapetis JR, Steer AC, Mulholland EK, Weber M. The global burden of group A streptococcal diseases. Lancet Infect Dis. 2005;5:685–694. doi: 10.1016/S1473-3099(05)70267-X. [DOI] [PubMed] [Google Scholar]
- 3.Beres SB, Sylva GL, Sturdevant DE, et al. Genome-wide molecular dissection of serotype M3 group A Streptococcus strains causing two epidemics of invasive infections. Proc Natl Acad Sci USA. 2004;101:11833–11838. doi: 10.1073/pnas.0404163101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Engelthaler DM, Valentine M, Bowers J, et al. Hypervirulent emm 59 clone in invasive group A Streptococcus outbreak, southwestern United States. Emerg Infect Dis. 2016;22:734–738. doi: 10.3201/eid2204.151582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies MR, Holden MT, Coupland P, et al. Emergence of scarlet fever Streptococcus pyogenes emm12 clones in Hong Kong is associated with toxin acquisition and multidrug resistance. Nat Genet. 2015;47:84–87. doi: 10.1038/ng.3147. [DOI] [PubMed] [Google Scholar]
- 6.McGregor KF, Spratt BG, Kalia A, et al. Multilocus sequence typing of Streptococcus pyogenes representing most known emm types and distinctions among subpopulation genetic structures. J Bacteriol. 2004;186:4285–4294. doi: 10.1128/JB.186.13.4285-4294.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goering R (2004) Pulse-field gel electrophoresis. In DH Persing, FC Tenover, J Versalovic, YW Tang, ER Unger, DA Relman, TJ White (ed) Molecular microbiology: diagnostic principles and practice. ASM Press, Washington, D.C., pp 185–196
- 8.Bertelli C, Greub G. Rapid bacterial genome sequencing: methods and applications in clinical microbiology. Clin Microbiol Infect. 2013;19:803–813. doi: 10.1111/1469-0691.12217. [DOI] [PubMed] [Google Scholar]
- 9.Cole JN, Barnett TC, Nizet V, Walker MJ. Molecular insight into invasive group A streptococcal disease. Nat Rev Microbiol. 2011;9:724–736. doi: 10.1038/nrmicro2648. [DOI] [PubMed] [Google Scholar]
- 10.Lynskey NN, Goulding D, Gierula M, et al. RocA truncation underpins hyper-encapsulation, carriage longevity and transmissibility of serotype M18 group A streptococci. PLoS Pathog. 2013;9:e1003842. doi: 10.1371/journal.ppat.1003842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goldstein B, Giroir B, Randolph A (2005) International pediatric sepsis consensus conference: Definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med 6:2–8. doi: 10.1097/01.PCC.0000149131.72248.E6 [DOI] [PubMed]
- 12.Singer M, Deutschman CS, Seymour C, et al. The third international consensus definitions for sepsis and septic shock (sepsis-3) JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bankevich A, Nurk S, Antipov D, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinforma Oxf Engl. 2013;29:1072–1075. doi: 10.1093/bioinformatics/btt086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aziz RK, Bartels D, Best AA, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Treangen TJ, Ondov BD, Koren S, Phillippy AM. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014;15:524. doi: 10.1186/s13059-014-0524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seemann T (2015) Snippy: fast bacterial variant calling from NGS reads. https://github.com/tseemann/snippy. Accessed 17 January 2017
- 19.Darling AE, Mau B, Perna NT. ProgressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One. 2010;5:e11147. doi: 10.1371/journal.pone.0011147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carver T, Harris SR, Berriman M, et al. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012;28:464–469. doi: 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L, Yang J, Yu J, et al. VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 2005;33:D325–328. doi: 10.1093/nar/gki008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hollands A, Pence MA, Timmer AM, et al. Genetic switch to hypervirulence reduces colonization phenotypes of the globally disseminated group A Streptococcus M1T1 clone. J Infect Dis. 2010;202:11–19. doi: 10.1086/653124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zankari E, Hasman H, Cosentino S, et al. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikebe T, Ato M, Matsumura T, et al. Highly frequent mutations in negative regulators of multiple virulence genes in group A streptococcal toxic shock syndrome isolates. PLoS Pathog. 2010;6:e1000832. doi: 10.1371/journal.ppat.1000832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lynskey NN, Turner CE, Heng LS, Sriskandan S. A truncation in the regulator RocA underlies heightened capsule expression in serotype M3 group A streptococci. Infect Immun. 2015;83:1732–1733. doi: 10.1128/IAI.02892-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wajima T, Morozumi M, Chiba N, et al. Associations of macrolide and fluoroquinolone resistance with molecular typing in Streptococcus pyogenes from invasive infections, 2010–2012. Int J Antimicrob Agents. 2013;42:447–449. doi: 10.1016/j.ijantimicag.2013.06.022. [DOI] [PubMed] [Google Scholar]
- 27.Beres SB, Carroll RK, Shea PR, et al. Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc Natl Acad Sci. 2010;107:4371–4376. doi: 10.1073/pnas.0911295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Flores AR, Jewell BE, Fittipaldi N, et al. Human disease isolates of serotype m4 and m22 group a streptococcus lack genes required for hyaluronic acid capsule biosynthesis. mBio. 2012;3:e00413–412. doi: 10.1128/mBio.00413-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu L, Olsen RJ, Nasser W, et al. Trading capsule for increased cytotoxin production: contribution to virulence of a newly emerged clade of emm89 Streptococcus pyogenes. mBio. 2015;6:e01378–1315. doi: 10.1128/mBio.01378-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olsen RJ, Fittipaldi N, Kachroo P, et al. Clinical laboratory response to a mock outbreak of invasive bacterial infections: a preparedness study. J Clin Microbiol. 2014;52:4210–4216. doi: 10.1128/JCM.02164-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 256 kb)