

Abstract
A thin fluid layer in alveoli is normal and results from a balance of fluid entry and fluid uptake by transepithelial salt and water reabsorption. Conventional wisdom suggests the reabsorption is via epithelial Na+ channels (ENaC), but if all Na+ reabsorption were via ENaC, then amiloride, an ENaC inhibitor, should block alveolar fluid clearance (AFC). However, amiloride blocks only half of AFC. The reason for failure to block is clear from single-channel measurements from alveolar epithelial cells: ENaC channels are observed, but another channel is present at the same frequency that is nonselective for Na+ over K+, has a larger conductance, and has shorter open and closed times. These two channel types are known as highly selective channels (HSC) and nonselective cation channels (NSC). HSC channels are made up of three ENaC subunits since knocking down any of the subunits reduces HSC number. NSC channels contain α-ENaC since knocking down α-ENaC reduces the number of NSC (knocking down β- or γ-ENaC has no effect on NSC, but the molecular composition of NSC channels remains unclear). We show that NSC channels consist of at least one α-ENaC and one or more acid-sensing ion channel 1a (ASIC1a) proteins. Knocking down either α-ENaC or ASIC1a reduces both NSC and HSC number, and no NSC channels are observable in single-channel patches on lung slices from ASIC1a knockout mice. AFC is reduced in knockout mice, and wet wt-to-dry wt ratio is increased, but the percentage increase in wet wt-to-dry wt ratio is larger than expected based on the reduction in AFC.
Keywords: nonselective cation channels, acid-sensing ion channel 1a, α-epithelial Na+ channels, alveoli, lung fluid balance, alveolar fluid clearance
acute lung injury, acute respiratory distress syndrome, and pneumonia are all pathologies characterized by lung edema and alveolar flooding. Bacterial and viral pneumonia alone affects ~450 million people globally per year (7% of the population) and results in ~4 million deaths. Pneumonia mortality is typically caused by flooding of the pulmonary alveoli preventing normal gas exchange and consequent hypoxemia. Airways normally have a critically regulated fluid layer essential for normal gas exchange and removal of foreign particulates from the airway. Maintaining this fluid layer in the alveoli depends critically on sodium reabsorption; however, the pathways and regulation of this sodium reabsorption are not completely clear. In other Na+-transporting epithelia (e.g., renal collecting duct or colon), epithelial Na+ channels (ENaC) are responsible for all Na+ transport. The situation in the lung is much less clear. If all Na+ reabsorption were via ENaC alone, then amiloride, a potent inhibitor of ENaC, should block all alveolar fluid clearance (AFC). In fact, although there is some species variability, amiloride typically blocks only about half of AFC (65). The reason for the failure to block is made clear from single-channel measurements from alveolar epithelial cells in rats and mice. There are at least two alternative Na+-permeable channels in alveolar cells with dramatically different physiological and pharmacological properties, but both appear to contribute to AFC (22, 27, 50). One channel is characterized by its low conductance, long mean open and closed times, and large permeability to Na+ compared with K+, all the characteristics of ENaC subunits expressed in heterologous expression systems. On the other hand, there is another channel present at almost the same frequency that is relatively nonselective for Na+ over K+, has a larger conductance, and has shorter mean open and closed times. These two types of channels have come to be known as highly selective cation channels (HSC) and nonselective cation channels (NSC). HSC channels are presumed to be made up of the three ENaC subunits since knocking down any of the subunits reduces the number of HSC channels (47). Although many experiments show that one of the channels is ENaC, the molecular basis for the other channel, NSC, remains unknown but appears to contain, at least, α-ENaC since knocking down this subunit reduces the number of NSC (knocking down β- or γ-ENaC has no effect on NSC number; 47). However, α-ENaC by itself does not appear capable of forming channels in heterologous expression systems (33). Thus the molecular composition of NSC channels remains unclear. The significance of the two types of channels is that the regulation of NSC channels is dramatically different from HSC channels. Thus treatments to reduce alveolar flooding based on the known properties of ENaC (HSC) may be counterproductive because NSC channels are regulated so differently (see discussion). In this paper, we now hypothesize that NSC channels are composed of, at a minimum, one α-ENaC subunit and one or more acid-sensing ion channel 1a (ASIC1a) subunits.
METHODS
Animals.
The original ASIC1 knockout (KO) mice were a generous gift of Dr. Michael Welsh at the University of Iowa (3, 71, 83), and a colony was established at Emory University. ASIC1 null mice are on a C57Bl6/N background, and we used this strain of mice obtained from Jackson Laboratories as controls. Mice were kept on a 12-h:12-h light-dark cycle and fed standard laboratory chow with tap water ad libitum. Mice used for experiments were of either sex and between 8 and 12 wk of age. Mice were euthanized by an overdose of ketamine-xylazine (8.7 mg/100 g body wt ketamine and 1.3 mg/100 g body wt xylazine) and, after reaching a surgical plane of anesthesia (assessed by toe pinch), cervical dislocation. All methods of euthanasia conform to the recommendations of the American Veterinary Medical Association Panel on Euthanasia for humane euthanasia of mice. All protocols were approved by the Emory University Animal Institutional Care and Use Committee.
Chemicals and reagents.
All chemicals and reagents used were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated.
Antibody production.
Polyclonal antibodies for the extracellular domain of the α-subunit of ENaC (Ab890) and the carboxyl terminal domains of the β (Ab60)- and γ (Ab2102)-subunits of ENaC were generated in New Zealand White rabbits by Bio-Synthesis (Lewisville, TX) using constructs made in house as described by Bao et al. (7). ASIC1 antibodies were obtained from Alomone Laboratories (Israel). Anti-ASIC1 antibody (no. ASC-014) can be used for Western blot analysis, immunoprecipitation, immunocytochemistry, and immunohistochemistry applications. It was designed to recognize ASIC1 from rat, human, and mouse samples and recognizes both ASIC1 isoforms.
SDS-PAGE and Western blot analysis.
Freshly isolated lungs, from ASIC KO and control animals, were minced, then washed once with 1× PBS, and subsequently homogenized in tissue protein extraction reagent (T-PER; Thermo Scientific) using an Omni TH homogenizer (Warrenton, VA). Tissue lysates were then centrifuged at 1,000 rpm, at 4°C, for 5 min. The supernatant was sonicated twice, on ice, for 10 s. Lysate protein concentration was determined using the bicinchoninic acid protein assay (Thermo Scientific). Twenty-five micrograms of total protein were prepared in Laemmli sample buffer (Bio-Rad, Hercules, CA) and then loaded and resolved on 7.5% Tris·HCl polyacrylamide gels using the Criterion or Protean electrophoresis systems (Bio-Rad). The separated proteins were electrically transferred onto Immobilon-P transfer membranes (Millipore, Billerica, MA). The membranes were blocked in 5% wt/vol milk in 1× Tris-buffered saline-Tween 20 (TBST; Bio-Rad) at room temperature for 1 h. The membranes were washed once with 1× TBST and then incubated with primary antibody at a dilution of 1:1,000 in 5% wt/vol milk in 1× TBST overnight at 4°C. The membranes were washed three times with 1× TBST for 5-min intervals before being incubated with horseradish peroxidase-conjugated goat-anti-rabbit secondary antibody at a dilution of 1:5,000 in 5% wt/vol milk in 1× TBST. The membranes were incubated with SuperSignal Dura Chemiluminescent Substrate for 5 min before being developed using a Kodak Gel Logic 2200 Imager and Carestream Molecular Imaging software (Carestream Health, Rochester, NY). This method was used to detect ENaC subunits (with in-house antibodies; 2, 77, 85) and ASIC1 (Alamone Laboratories).
Mammalian two-hybrid assay.
GAL4:α-ENaC, GAL4:β-ENaC, and GAL4:γ-ENaC constructs were created by inserting full-length clones of each ENaC subunit into separate DNA-binding domain (DBD) plasmid vectors, pGAL4. VP16:ASIC1a construct was created by inserting a full-length clone of ASIC1a into the transcriptional activation domain (AD) plasmid, pVP16. These constructs were transfected into alveolar type 2 cells along with the luciferase reporter plasmid. The reporter gene will only be expressed if ENaC encoded in the DBD vector binds to the ASIC1a encoded in the AD vector. Cells were incubated with transfection media to be harvested after 48 h. Harvested cells were lysed and used to assess binding of ENaC subunits to ASIC1a by measuring luciferase reporter gene expression. Levels of luciferase luminescence were measured with a plate reader (LUMIstar Omega; BMG Labtech).
Isolation of primary alveolar type 2 cells.
Mice primary alveolar type 2 (AT2) cells were isolated as previously described (42). Lungs were perfused with phosphate-buffered saline (PBS) via the pulmonary artery and lavaged with PBS via a tracheal cannula. Lungs were extracted en bloc and instilled with an elastase solution. The lobes were teased away from the heart and bronchial tissue and minced into 1- to 2-mm pieces in a DNAse solution. The solution was filtered through 100- and 40-μm filters to produce a cell suspension that was centrifuged at 1,200 rpm and resuspended in DMEM/Ham's F-12 medium containing gentamycin. The cell suspension was panned for 1 h at 37°C on plates coated with mouse IgG. Cell suspension was collected, centrifuged, and resuspended in supplemented DMEM/Ham's F-12 medium to be seeded on Snapwell inserts (Corning, Lowell, MA). Air interface was established within 12 h, and AT2 cells were used within 96 h for experiments.
Mouse lung slice preparation.
Lung slices were prepared using a previously published protocol (22, 23, 42, 43). Lungs were perfused with PBS and intratracheally instilled with warm 2% low-melting point agarose in PBS to expand air spaces. Lungs were extracted en bloc and placed in cold (4°C) PBS to solidify agarose. A small section of tissue was mounted onto a Vibratome (model VT1000s; Leica Microsystems), and slices were cut 275 μm thick. Slices were transferred to supplemented DMEM/Ham's F-12 medium and were used for experiments within 4 h of initial preparation.
Single-channel, patch-clamp analysis.
Primary AT2 cells were isolated from ASIC1a KO and C57Bl6 control mice, as previously described (20, 21). AT2 cells were prepared on a permeable support before single-channel patch clamp, as previously described for patch clamp of cells in culture (41, 48). Lung slices were used directly after washing away agar. Briefly, a microelectrode was filled with physiological buffer and applied to a single cell using suction, until a >1-GΩ seal was formed. ENaC or NSC channels were identified by characteristic channel kinetics and the current-voltage relationship for the channel.
Evans blue protein assay.
AFC was measured in mice after filling their lungs with either 0.7 ml of 5% BSA in NaCl (which was isosmotic with mouse plasma: 315 mosmol/l) containing 0.1 mg/ml Evans blue or the same solution containing 10 μM amiloride (40). The mice were euthanized with intraperitoneal injections of ketamine (8.7 mg/100 g body wt; Phenix Scientific) and xylazine (1.3 mg/100 g body wt; Vedco); a trimmed, sterile 18-gauge intravenous catheter (Surflo; Terumo Medical) was inserted caudally into the lumen of the exposed trachea; and the mouse was placed on a 37°C heating pad. The 5% BSA in NaCl was then slowly instilled into the lungs using a 1-ml syringe. The fluid was removed at the end of 15 min, and the Evans Blue concentration of the fluid was measured and compared with the concentration of the instillate. Absorbance was measured at 620 nm using a spectrophotometer (Nanodrop 2000; Thermo Scientific) to determine Evans blue concentration. AFC was calculated using the formula, AFC = [(Vi − Vf)/Vi] × 100, where Vi is the initial volume of instillate and Vf is the final alveolar fluid volume. Vf = (Vi × EBi)/EBf, where EBi is the concentration of Evans blue-labeled albumin in the instilled solution and EBf is the final concentration of Evans blue in the alveolar fluid (39).
Wet wt-to-dry wt ratios.
Whole lungs from ASIC1 KO and C57Bl6 animals were perfused, extracted, and weighed immediately following removal (wet wt). The lungs were desiccated overnight at 110°C and reweighed (dry wt). The wet wt-to-dry wt ratio was determined by dividing the wet wt by the dry wt, which represents the amount of lung fluid and is a measure of lung edema. A larger ratio indicates more fluid in the lungs initially due to attenuated AFC, whereas a smaller ratio indicates drier lungs.
Determination of bronchoalveolar lavage protein.
Mice received a lethal injection of pentobarbital sodium (100 mg/kg ip; Abbott, Chicago, IL), and a cannula was inserted into the trachea. Bronchoalveolar lavage (BAL) fluid was obtained by instilling saline into the lungs through the tracheal cannula using a volume equal to 80% of lung vital capacity [3× with 0.5 ml of Hank's balanced salt solution (pH 7.2–7.4)]. Recovered aliquots of BAL fluid were pooled together for a total of 1.5 ml. BAL fluid was immediately cooled on ice. BAL cells were pelleted by centrifugation (500 g at 4°C) for 10 min, and the volume of the pellet was measured. The supernatant was used to measure total protein concentration as an indicator of lung permeability. Protein concentrations were determined using the Bio-Rad Protein Assay (Bio-Rad, Hercules, CA).
Statistical analysis.
Data acquisition and analysis were performed as described previously (43). Data are reported as means ± SE. Statistical analysis was performed with SigmaPlot software (Systat Software). Statistical significance was evaluated using t-test, z-test, or one-way ANOVA, as appropriate, and results were considered significant if P < 0.05.
RESULTS
We have shown previously that AT2 cells in primary culture have two predominant channels in their apical membranes (Fig. 1; 15, 47, 48, 50). One HSC has all the characteristics of ENaC expressed in heterologous expression systems, but the other is a nonselective cation channel (NSC) whose molecular character is unclear. We subsequently showed in lung slices that both channels were present in both AT1 and AT2 cells in almost equal numbers (43, 50). We also showed that when we knocked down α-, β-, or γ-ENaC, the number of HSC channels decreased, but knocking down α-ENaC (not β- or γ-ENaC) reduced the number of NSC and HSC channels (Fig. 2). We concluded that HSC channels consisted of α-, β-, and γ-ENaC, and that NSC channels contained α-ENaC subunits, either by themselves or together with some other subunit protein that we did not know (47). Several investigators have embraced the idea that NSC channels are formed from α-ENaC alone, but several observations argue against this hypothesis. In particular, when α-ENaC is expressed by itself in heterologous expression systems that are known to contain no endogenous ENaC subunits, there is little, if any, current and there are no channels like NSC (87).
Fig. 1.

Electrophysiologic characteristics of NSC and HSC channels in AT2 cells in primary culture. A: representative traces for NSC channels with a conductance of γ = 21 pS. B: representative traces for HSC channels with a conductance of γ = 6 pS. “C” marks the level with all channels closed. C: current-voltage plots for HSC and NSC. Nonlinear HSC current-voltage relationship and very positive reversal potential are characteristic of ENaC in other cell types and heterologous expression systems. Each point is the mean ± SD of three or more determinations. Fits (solid lines) are to the Goldman-Hodgkin-Katz equation (7, 38, 44).
Fig. 2.

Expression of α-ENaC regulates NSC channel frequency. Knockdown of α-, β-, or γ-ENaC subunits in an AT2 cell line, L2, with shRNA silencing vectors in lentivirus. A: percentage of patches with HSC channels in knockdown or sham conditions. B: percentage of patches with NSC channels in knockdown/sham conditions. Only knocking down α-ENaC reduces the surface expression of both HSC and NSC channels (n = 4 for all conditions; HSC reduction and NSC reduction are significant, P < 0.01 by z-test).
AT2 cells express ASIC1a.
Our hypothesis that NSC channels contain ASIC1a would not be plausible if lung alveolar cells did not contain ASIC1a. Others have detected high levels of ASIC1a expression in the lower lobes of the lung, suggesting ASIC1 in the alveoli but not the airways (5, 34). Figure 3 shows PCR analysis for all ASIC isoforms in mouse lung cells. Only ASIC1a can be strongly amplified from primary cultures of AT2 cells. (A note on a mixed-up nomenclature: ACCN2 gene encodes ASIC1 protein, ACCN1 gene encodes ASIC2 protein, and ACCN3–5 genes encode ASIC3–5 protein.)
Fig. 3.

AT2 cells contain ACCN2. A: PCR analysis showing expression of ACCN isoforms in cDNA prepared from AT2 cells. Only ACCN2 (ASIC1) has a visible band. B: PCR reamplification showing expression of ACCN2 variant 2 (ASIC1a, lane 3) and lack of expression of ACCN2 variant 1 (ASIC1b, lane 2). NCBI, National Center for Biotechnology Information.
Venoms that target ASIC1 alter NSC activity.
The venom of the Texas coral snake (Micrurus tener tener) is a potent, persistent, and selective agonist for acid-sensing ion channels and is highly selective for the ASIC1 subtype at neutral pH (11). When applied to AT2 cells in primary culture, it uniformly increased NSC open probability (Fig. 4A).
Fig. 4.

NSC channels are sensitive to ASIC1-modifying toxins. A: NSC channels were activated by venom of the Texas coral snake (Micrurus tener tener; MitTx). It is a potent, persistent, and selective agonist for acid-sensing ion channels and is highly selective for ASIC1 at neutral pH (11). B: psalmotoxin-1 isolated from the venom of the spider, Psalmopoeus cambridgei (Trinidad chevron tarantula), is a potent and selective acid-sensing ion channel 1a (ASIC1a) blocker (IC50 = 0.9 nM) with no effect on ASIC1b, ASIC2a, ASIC3, and ENaC channels at concentrations up to 100 nM (13, 14, 19). When applied to AT2 cells in primary culture, it uniformly decreased NSC open probability. Neither toxin had any effect on HSC channels. *P < 0.05.
Psalmotoxin-1 is isolated from the venom of the spider, Psalmopoeus cambridgei (Trinidad chevron tarantula). It is a potent and selective acid-sensing ion channel 1a (ASIC1a) blocker (IC50 = 0.9 nM) with no effect on ASIC1b, ASIC2a, ASIC3, and ENaC channels at concentrations up to 100 nM (13, 14, 19). When applied to AT2 cells in primary culture, it uniformly decreased NSC open probability (Fig. 4B). Neither toxin had any effect on HSC channels.
NSC channel frequency strongly depends on ASIC1a and α-ENaC subunits.
We next used molecular biological methods to manipulate the amount of ASIC1a and α-ENaC in heterologous expression systems and alveolar cells and assessed the effect on HSC and NSC channels using single-channel measurements. Whereas many heterologous expression systems express one or both of our genes of interest, we found that Fisher rat thyroid cells (FRT) lack endogenous ASIC1a or α-ENaC but express low levels of γ-ENaC. We transfected the FRT cells with green fluorescent protein (GFP) alone, GFP α-ENaC, or dsRed ASIC1a + GFP α-ENaC (Fig. 5A) and found different channel properties for each condition. FRT cells after expression of α-ENaC alone produced only low-conductance channels (7.2 ± 2.0 pS) with long mean open times, whereas cells expressing α-ENaC + ASIC1a produced higher conductance channels (21 ± 5.8 pS) with shorter mean open times (Fig. 5, B and C). These channel characteristics are similar to HSC and NSC channels in alveolar cells. Cells transfected with GFP/dsRed vectors as a negative control only had small numbers of lower conductance channels (2.2 ± 1.4 pS) of unknown type. Although the channels from the α-ENaC + ASIC1a-transfected FRT cells appear similar to NSC channels in lung cells, we were hesitant to conclude that native NSC channels contain ASIC1a and α-ENaC subunits. Therefore, to further investigate the molecular composition of NSC channels, we knocked down ASIC1 and α-ENaC subunits in L2 cells, a tissue culture model of AT2 cells, with short hairpin RNA (shRNA) silencing vectors in lentivirus. The constructs contain a GFP marker to assess transduction efficiency. In Fig. 6A, we show that the reduction of ASIC1 protein positively correlates to the amount of ASIC1 shRNA used. We find little to no ASIC1 protein present at the highest concentration of 12 μg, whereas the scrambled shRNA control produces no effect on the concentration of the target protein. ASIC1 silencing in L2 cells reduces the frequency of NSC channels from 91 to 10% of patches with no effect on HSC channels, whereas α-ENaC silencing reduces NSC frequency from 86 to 12% and HSC frequency from 55 to 8% of patches (Fig. 6B). These results suggest that the presence of both α-ENaC and ASIC1a is necessary to produce functional NSC channels.
Fig. 5.

Coexpression of α-ENaC/ASIC1a produces high-conductance NSC channels. A: Western blot showing ASIC1a detection in FRT cells with and without α-ENaC. Although the blot is cropped, there are no other bands in the blot. B: conductance of channels found in patches on FRT cells transfected with GFP/dsRed vector only, GFP α-ENaC, or dsRed ASIC1a + GFP α-ENaC. C, top: representative traces from FRT cell transfected with GFP α-ENaC. Bottom: representative traces from FRT cells transfected with dsRed ASIC1a + GFP α-ENaC. Vp, voltage of the pipette.
Fig. 6.

NSC channel frequency strongly depends on ASIC1a and α-ENaC presence. A: Western blot showing ASIC1 expression in L2 cells treated with scrambled shRNA or ASIC1 silencing vectors. Although the blot is cropped, there are no other bands in the blot. Reduction of ASIC1 protein positively correlates to amount of ASIC1a shRNA used. B, left: percentage patches with NSC channels present under ASIC1 or α-ENaC knockdown conditions. Right: percentage patches with HSC channels under ASIC1 or α-ENaC knockdown conditions. Numbers above bars indicate total number of patches.
ASIC1a and α-ENaC subunits are physically associated.
If NSC channels are ASIC1a/-ENaC hybrid channels, then ASIC1a and α-ENaC subunits will directly interact to create the channel. To explicitly show this association, we used L2 cell protein lysate to immunoprecipitate (IP) for α-ENaC and immunoblot for ASIC1 and IP for ASIC1 and blot for α-ENaC (Fig. 7A). The presence of bands after coimmunoprecipitation indicates that the subunits do physically interact; however, the interaction could indicate that ASIC1a and α-ENaC might also be part of a larger complex that immunoprecipitates together. Therefore we performed a mammalian two-hybrid assay to assess the affinity of α-, β-, or γ-ENaC for ASIC1a subunits. Binding affinity of subunits to ASIC1a is proportional to normalized luciferase luminescence measurements. Quantification of assay results shows that α-ENaC affinity to ASIC1a is 6.6 ± 0.73 times higher than negative control (Fig. 7B). This strength of interaction mirrors that of α-, β-, or γ-ENaC subunits' interaction with themselves in HSC channels. On the basis of these observations, we conclude that ASIC1a and α-ENaC subunits directly interact and thus have the potential to form a channel consisting of at least one α-ENaC subunit and at least one ASIC1a subunit.
Fig. 7.

Interaction of ASIC1a and α-ENaC subunits. A, top: L2 cell protein lysate immunoprecipitated (IP) for α-ENaC and immunoblotted (IB) for ASIC1. Bottom: L2 cell protein lysate immunoprecipitated for ASIC1 and immunoblotted for α-ENaC. B: quantification of mammalian two-hybrid assay between ASIC1a and ENaC subunits. Normalized luciferase luminescence is proportional to binding affinity to ASIC1a. Negative control represents random association, and positive control represents maximum affinity. Fold increase for α-ENaC (6.6 ± 0.73) indicates a high affinity for ASIC1a. Data represent a total n of 15 (n = 3 for each condition); *P < 0.05.
NSC channels are present in native lung alveolar cells.
Results from the previous experiments support the possibility of an ASIC1a/α-ENaC hybrid channel in tissue culture and primary cell models of alveolar epithelium; however, the question remains of whether NSC channels in the native lung environment are such hybrids. To examine this question, we used lung slices that allow us to make single-channel measurements from both AT1 and AT2 cells in an environment close to their native milieu (22, 42, 43). Figure 8A shows a lung slice obtained from the lower left lobe stained for AT1 and AT2 cells. The difference in the current-voltage relationships for HSC and NSC channels from AT2 cells in lung slices (Fig. 8B) and AT2 cells in primary culture (reproduced for comparison from Fig. 1 in Fig. 8C) underscores the importance of the lung slice model for studying ion transport in alveolar cells. The main difference between the two current-voltage relationships is that in the lung slice the currents reverse near zero and HSC channels have little rectification. In tissue culture, HSC channels strongly rectify and reverse at a very positive potential (66.6 ± 9.72 mV). NSC channels reverse near +40 mV (36.0 ± 5.35). In cell-attached patches, the potential applied to the patch is the pipette potential minus the membrane potential (6). Since by their nature nonselective channels must reverse at zero patch potential, the observed reversal potential is a measure of the apical membrane potential. That is, in the tissue culture cells the membrane potential must be about −40 mV, but in the lung slice the apical membrane potential must be near zero. In addition, since ENaC is very selective for Na+ over K+, the HSC reversal potential is a measure of the ratio of external Na+ to internal Na+. In the tissue culture cells, the very depolarized reversal potential for the HSC implies an intracellular Na+ concentration 10–20-fold less than the extracellular Na+ (140 mM). This is consistent with direct measurements of intracellular Na+ of ~10 mM Na+ in other sodium-transporting epithelia (24).
Fig. 8.

Lung slice preparation. A: representative image of a lung slice used for single-channel patch clamp recording. Lung slice was obtained from lower left lobe of mouse lung. AT1 cells are labeled green with Erythrina cristagalli lectin, and AT2 cells are labeled red with LysoTracker red. B: current-voltage relationships for HSC and NSC channels from AT2 cells in lung slices. Solid line is the best fit to Goldman equation. Symbols represent means ± SE for each condition for at least three measurements. C: for comparison purposes, the current-voltage relationships of HSC and NSC channels from channels in AT2 cells in culture (reproduced from Fig. 1).
The strong rectification in cultured cells is consistent with this large difference in extracellular vs. intracellular Na+ that will produce a large difference in inwardly moving charge carriers (more current at negative potentials) vs. outwardly moving charge carriers (less current at positive potentials). On the other hand, the reversal of HSC channels near zero in lung slices implies that cells in native lung (as reflected in the lung slice) must have very high intracellular Na+. The high intracellular Na+ also implies that Na+ transport in lung cells is different from transport in other Na+-transporting epithelia like renal distal nephron. Other Na+-transporting epithelia all have large electrochemical gradients driving Na+ downhill into the cell so that the rate-limiting step for transport and the major point of control is at the Na+ entry pathway in the apical membrane. In lung, Na+ entry must be directly linked on an almost ion-for-ion basis to the Na+ extrusion step via Na,K-ATPase. Therefore control of overall transport can occur by regulating either Na+ entry or Na+ exit. This is consistent with reports of lung Na+ transport and AFC being dependent on activity of the Na,K-ATPase (1, 4, 8, 10, 17, 31).
We interpret the reversal potentials near zero as indicative of cells with many Na+-permeable channels with consequent high levels of Na+ entry at their apical membranes with the result that Na+ accumulates in the cell until the basolateral Na,K-ATPase activity increases to balance the entry. Such a situation has been described previously in other epithelia when aldosterone stimulates Na+ channel activity and apical insertion of channels (24).
Such positive apical potentials and small positive reversal potentials for HSC channels might also be viewed as indicative of cell damage associated with the slicing process. Therefore we reviewed the properties of all the patches we have made on alveolar cells in lung slices. We found that the reversal potentials of HSC channels fell in a distribution skewed toward small positive reversal potentials with the majority clustered at less than +20 mV [9.12 ± 3.99 (mean ± SD), n = 100] although there were some channels that had higher positive potentials (some as high as +60 mV; Fig. 9, left). We then examined the channel activity associated with the different HSC reversal potentials and found an inverse linear relationship between channel activity and reversal potential: the closer to zero the reversal potential, the higher the channel activity [measured as the product of the number of channels per patch and the open probability (NPo); Fig. 9, right]. This result is not consistent with damaged cells but is expected for highly transporting cells (56–58). We also examined at the distribution of reversal potentials for NSC in lung slices [−1.28 ± 7.13 mV (mean ± SD), n = 84] and the distribution in cultured AT2 cells (Fig. 10). The distributions in cultured cells suggest an apical membrane potential near −40 mV and much lower channel activity (mean number of channels/patch is <1).
Fig. 9.

Properties of HSC channels in lung slices. A: we reviewed the properties of 100 HSC channels from patches made on alveolar cells in lung slices. We found that the reversal potentials of HSC channels fell in a distribution skewed toward small positive reversal potentials with the majority clustered at less than +20 mV [9.12 ± 3.99 (mean ± SD), n = 100] although there were some channels that had higher positive potentials (some as high as +60 mV). B: we examined the channel activity associated with the different HSC reversal potentials and found an inverse linear relationship between channel activity and reversal potential: the closer to zero the reversal potential, the higher the channel activity [measured as the product of the number of channels per patch and the open probability (NPo)].
Fig. 10.

Distribution of reversal potentials for NSC in lung slices and distribution in cultured AT2 cells. Left: distribution of reversal potentials for NSC in lung slices [−1.28 ± 7.13 mV (mean ± SD), n = 84]. Middle: distribution of NSC in cultured AT2 cells. Right: distribution of HSC in cultured AT2 cells. The distributions in cultured cells suggest an apical membrane potential near −40 mV and much lower channel activity (mean number of channels/patch is <1).
These major differences in the properties of the channels and cells make it important to verify the effects of any physiological manipulation in the lung slice preparation.
Alveolar cells in lung slices from ASIC1 KO animals have no NSC channels.
As expected, we could not detect ASIC1 in lung tissue from KO animals (Fig. 11). Single-channel measurements of AT1 or AT2 cells in lung slices from wild-type mice show, as expected from our previous work, that both AT1 and AT2 cells contain HSC and NSC channels (Fig. 12A). In contrast, single-channel records from ASIC1 KO mice contain no NSC channels (Fig. 12B). These results are summarized in Fig. 13A, where the fraction of patches that contain HSC or NSC channels is given for a large number of patches; in the KO animals no NSC channels are observed. Whereas lung slices from ASIC1a KO mice lack NSC channels in both AT1 and AT2 cells, the frequency of HSC channels is slightly, but significantly, greater than HSC in wild-type mice in both types of cells (Fig. 13B). These results indicate that the presence or absence of ASIC1a subunits produces only a small, presumably compensatory, change in the number of apical HSC channels.
Fig. 11.
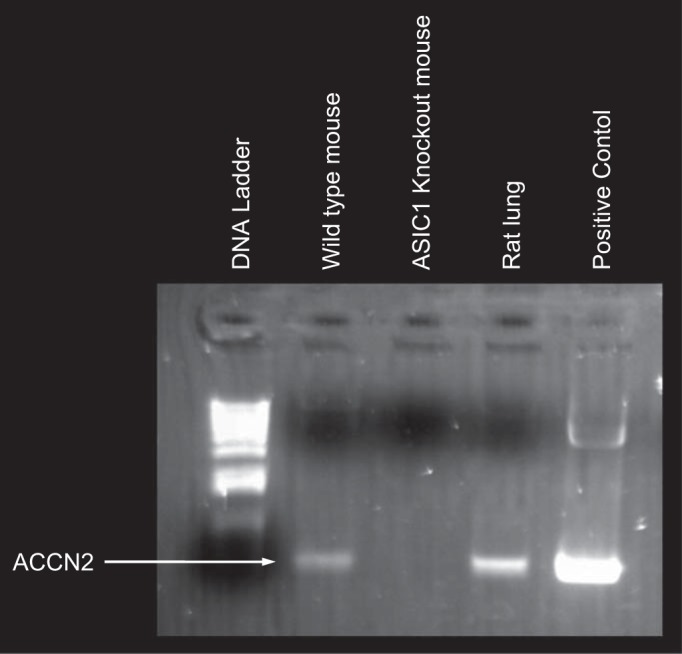
KO mice have no detectable ASIC1a. PCR analysis shows expression of ACCN2 in cDNA prepared from wild-type lungs and from rat lung as a positive control. The cDNA prepared from ASIC1 KO lung has no detectable ACCN2 (ASIC1).
Fig. 12.

NSC channels are not observable in single-channel patches on lung slices from ASIC1 KO mice. Single-channel recordings were measured from AT2 cells in lung slices. A: single-channel currents (right) and distribution of current amplitudes (left) in a patch on an AT2 cell from a wild-type lung slice, which has both NSC and HSC channels (sometimes overlapping). B: single-channel currents (right) and distribution of current amplitudes (left) in a patch on an AT2 cell from an ASIC1 KO lung slice, which has only HSC channels.
Fig. 13.

ASIC1a KO mice have no NSC channels. A: Western blot showing ASIC1a expression in lungs from wild-type mice and lack of expression in ASIC1a KO mice. Although the blot is cropped, there are no other bands in the blot. B: single-channel frequency from patches on lung slices of wild-type mice and ASIC1a KO mice. Patches on AT1 and AT2 cells from ASIC1a KO mice have similar numbers of HSC channels in AT1 and AT2 cells to wild-type mice but have no observable NSC channels in either cell. Numbers above bars in parentheses represent total number of patches for each experiment. Numbers within bars indicate the number of each type of channel observed in each type of cell. Some cell patches on both types of cells have both NSC and HSC channels (as in Fig. 8). C: number of channels per patch. Data are from the same patches as shown in B. All values for number of channels in a patch are integer values. To visualize the distribution and the density of the data, a small amount of stochastic noise was added to every data point. *P < 0.05.
Elimination of NSC increases wet wt-to-dry wt ratio and attenuates AFC.
Using single-channel patch clamp analysis, we showed that mice lacking ASIC1a subunits have no observable NSC channels. To determine whether these measurements at the individual cell level translate into changes in lung fluid balance, we assessed total lung fluid by wet wt-to-dry wt ratios and the rate of AFC in ASIC1a KO mice by Evans blue assay. Wet wt-to-dry wt ratios are ~50% larger in ASIC1 KO mice compared with age-matched wild-type mice indicating that ASIC1 KO have wetter lungs (Fig. 14A). This is a large increase in lung water but does not appear to be associated with substantial edema, since KO mice breathe normally without the labored breathing or other symptoms of hypoxia associated with edema. The Evans blue assay provides a measure of fluid uptake from the alveolar surface (Fig. 14B). This assay shows that AFC in ASIC1 KO animals is 54 ± 8.0% of the AFC in wild-type mice (from 36 ± 2.1 to 19 ± 2.6%, n = 3, P = 0.009). Instillation of 10 μM amiloride into the lungs of wild-type animals blocks 53 ± 12% of AFC, which leads to an AFC not significantly different from that of ASIC1 KO mice (17 ± 4.4 vs. 19 ± 2.6%, n = 3, P = not significant). Instilling amiloride in the lungs of ASIC1 KO mice reduces AFC to 2.5 ± 1.2%. The absolute magnitude of the reduction in AFC produced by amiloride (presumably blocking HSC channels) is not significantly different in wild-type and KO mice even though the final AFC is quite different (presumably because of the lack of NSC channels in the KO mice; Fig. 14B).
Fig. 14.

ASIC1a KO increases lung water content and reduces alveolar fluid clearance. A: lung wet wt-to-dry wt ratios. Higher wet wt-to-dry wt ratio indicates increased lung water content and decreased alveolar fluid clearance. The difference in the two groups is significant. Data represent n = 8 for each treatment group. B: Evans blue dye assay showed that alveolar fluid clearance was significantly reduced in ASIC1a KO mice compared with wild-type mice. Amiloride blocked about half of AFC in wild-type mice, but there is little residual AFC after amiloride in ASIC1 KO mice. Data represent a total n = 3 mice for each treatment group; n.s., not significant. C: bronchalveolar lavage (BAL) fluid protein from wild-type and ASIC1 KO mice. There is no significant difference in BAL protein between the two groups (n = 4 mice for each treatment group; P = 0.424).
Protein in bronchoalveolar lavage fluid is the same in wild-type and KO animals.
Because the wet wt-to-dry wt ratio was large, we thought that there might be some low-level inflammatory response in ASIC1 KO animals. However, when we measured BAL protein as a measure of the integrity of the alveolar epithelium, we found no difference between wild-type and KO animals (Fig. 14C).
ASIC1a protein expression decreases in L2 alveolar cells exposed to elevated oxygen tension.
One of the interesting differences in the regulation of NSC and HSC channels is change in expression in response to changes in oxygen tension. Several studies have shown that the expression and apical density of HSC (ENaC) subunits is reduced when oxygen tension is reduced to hypoxic levels. However, NSC expression increases in response to hypoxia (Fig. 15) in AT2 cells in primary culture. Normal oxygen tension was produced by exposing the apical surface to an air interface with 21% oxygen. Reduced oxygen tension was produced by flooding the apical surface with saline to a depth of 0.5 cm. Previous work by us has shown that this procedure reduces apical oxygen tension to ~12–15% (42). Although NSC channels do not transport Na as efficiently as HSC, they can prevent alveolar flooding in response to moderate hypoxia.
Fig. 15.

Expression of ASIC1a is dependent on oxygen tension. AT2 cells grown on permeable supports with an air interface (21% O2, high oxygen) express less ASIC1a than AT2 cells grown under liquid to reduce oxygen tension (calculated to be 12–15% O2).
DISCUSSION
Two types of cation channels in alveolar cells.
We and others have shown in several previous publications (15, 41, 43, 50, 52, 54, 55) and we show here again that there are two types of cation-selective channels in both alveolar type 1 and type 2 cells (Fig. 1). One type has characteristics that correspond to those of epithelial sodium channels (ENaC) in other sodium-transporting epithelia and that of ENaC subunits expressed in heterologous expression systems (for reviews, see 25, 26, 28, 29, 35, 36, 73, 74). Because ENaC has a selectivity for Na over K of greater than 40 to 1, these channels in the lung have come to be known as highly selective cation channels (HSC channels).
The second type of channel has a larger conductance (21–22 pS), shorter mean open and closed times (10s to 100s of ms), a lower sensitivity to amiloride (µM vs. 10s of nM), and little selectivity for Na over K (PNa/PK = 1.1). The latter property leads to the common name for these channels of “nonselective cation channels” (NSC channels). The two channels are expressed in the apical membranes of alveolar cells at approximately the same frequency in both type 1 and type 2 cells and therefore contribute to the macroscopic properties of the distal airway where approximately half of the AFC is resistant to instillation of high concentrations of amiloride (65).
The presence of the two types of channels would be of little consequence to lung fluid balance if their regulation were the same and only their selectivity and sensitivity to amiloride were different. However, their regulation is often quite different, sometimes responding to pharmacological or physiological stimuli with opposite responses. Table 1 summarizes the response of the two types of channels to different stimuli. Several of these responses are notable. Both types of channels are stimulated by β-adrenergic agents, but by different mechanisms (15). Presumably, this coactivation is why β-adrenergic agents are so effective at reducing airway fluid and edema. Activation of protein kinase C has opposite effects on the number of the two channels in the apical membrane, and purinergic stimulation decreases activity of HSC and increases activity of NSC channels. This underscores why purinergic antagonists are effective at reducing (but not eliminating) alveolar Na+ transport (45). Particularly interesting is the effect of hypoxia, which decreases HSC channels in the membrane but increases the density of NSC channels (48; see discussion of the importance of 2 channel types below).
Table 1.
Comparison of the properties of highly selective and nonselective channels
| Highly Selective | Nonselective | |
|---|---|---|
| Na/K selectivity (48, 61) | >40 | 1.1 |
| Unit conductance, pS (48, 50, 61) | 6 | 21 |
| Amiloride Ki, nM (48, 82) | 38 | 2,300 |
| Increased cellular cAMP or β-adrenergic stimulation (15, 22) |
Channel surface density increases | Po increases |
| Increased cGMP or nitric oxide (46) |
Po decreases | Po decreases |
| PKC activation (16, 23) | Po decreases and surface density decreases | Channel surface density increases |
| Increased intracellular Ca2+ (15) | No effect | Po increases |
| Purinergic stimulation (18, 59, 70, 78) | Po decreases | Po increases |
| Dopaminergic stimulation (41, 43) |
Po increases | No effect |
| Superoxide production (42) | Po increases | Channel surface density increases |
| Hypoxia (48) | Channel surface density decreases | Channel surface density increases |
Po, channel open probability; Ki, inhibitory constant, i.e., the dose that reduces open probability by 50%.
Composition of NSC channels.
Although the HSC channels are clearly the expression of ENaC in the lung, the composition of the NSC channels has been the subject of speculation for a number of years (65). Using antisense oligonucleotides directed against the three ENaC subunits, we showed that NSC channel numbers could be reduced with antisense to α-ENaC but antisense against β- or γ-ENaC did not change NSC expression (47). On the basis of our results, we speculated at the time that NSC channels were either formed from α-ENaC subunits alone or α-ENaC together with some other subunit protein. By itself, α-ENaC can form channels, but the properties of the channels are quite different from NSC channels (62). In addition, when α-ENaC is transfected into heterologous cell expression systems that contain no endogenous ENaC subunits, surface expression is quite low, which is not consistent with the frequency of observing NSC in lung cells. Therefore we favored the idea that NSC channels were formed from α-ENaC with some other membrane protein subunit. The identity of this protein was suggested to us by a report that in glioma cells, α-ENaC could form a hybrid channel with ASIC1a (51) and that these channels played a role in disease progression (72). Although these channels were in the central nervous system in neoplastic cells, we still examined lung tissue for ASIC1a and found that ASIC1a was the only ASIC family member in the lung (Fig. 3).
If ASIC1 plays a role in the formation of channels, then pharmacology might illuminate the NSC channel composition. In fact, a toxin blocker (psalmotoxin-1) strongly blocks NSC channels, and toxin agonist (Texas coral snake toxin) strongly activates the channel (Fig. 4). It is an interesting possibility that coral snake venom or a derivative might activate sodium transport through NSC channels and therefore be an effective agent in the treatment of edema.
Testing for α-ENaC/ASIC1a association and the ability to form NSC channels.
Although both ASIC1 and α-ENaC are present in lung tissue, we needed to show whether they were associated. Initially, we tried to express one or the other or both in a heterologous expression system to see whether we could produce channels with NSC properties. This idea was hampered because almost every cell line we examined had endogenous ASIC1 or endogenous α-ENaC, but finally we showed by real-time PCR that Fisher rat thyroid cells (FRT) had very limited message for α-ENaC and no detectable message for ASIC1. Expression of ASIC1a and α-ENaC in FRT cells produced channels with conductances and gating kinetics similar to NSC channels (Fig. 5).
We worried that this observation might be an artifact of overexpression and tissue culture. However, in L2 cells, a cultured type 2 cell line, we observed HSC and NSC channels. Using lentivirus to introduce ASIC-silencing shRNA, we showed that the silencing reduced ASIC1a expression and the number of NSC channels without changing the frequency of HSC channels (Fig. 6). Our previous work showed that NSC contained α-ENaC; this work implied that NSC also contained ASIC1a, at least in a tissue culture model.
We also showed that ASIC1a and α-ENaC were physically associated in lung tissue by coimmunoprecipitation and by mammalian two-hybrid assay. However, that the two are associated within the cell does not necessarily mean that the two proteins traffic to the membrane to form ion channels. An experiment that comes closer to showing this is one in which we instilled the lentiviral silencing shRNA directly into the lung and showed that the frequency of observing NSC channels in lung slices from treated lungs was much lower than the frequency of NSC channels in slices from lungs treated with scrambled shRNA (data not shown). Of course, although unlikely, the effect of the silencing RNA might be an off-target effect in which disrupting ASIC1 (or some related protein) disrupts targeting of NSC channel components to the apical membrane. Therefore it was important to show that in native lung tissue in which there was no ASIC1a, we could not observe NSC channels. Using ASIC1 KO animals, we showed that we could observe no NSC in either type 1 or type 2 cells confirming the composition of the NSC channels (Figs. 8 and 9).
Functional consequences of ASIC1 KO.
Alveolar epithelial cells and lung tissue, in general, in which there is no ASIC1 (and no NSC channels) have reduced AFC and increased wet wt-to-dry wt ratio (Fig. 14). This should not be surprising since both HSC and NSC channels in both type 1 and type 2 cells contribute to lung fluid balance (32, 50). Knocking out half of the channels that transport sodium should reduce AFC. In fluid-filled lungs, the reduction in AFC in ASIC1 KO animals is 54 ± 8.0% of the AFC in wild-type mice. This appears to be similar to the percentage change in wet wt-to-dry wt ratio. Instillation of 10 μM amiloride into the lungs of wild-type animals blocks 53 ± 12% of AFC, which leads to an AFC not significantly different from that of ASIC1 KO mice. This reduction is consistent with half of the channels in wild-type animals being NSC; removing the NSC reduces the transport capacity by about half. Instilling amiloride in the lungs of ASIC1 KO mice reduces AFC to 2.5 ± 1.2%. The percentage reduction in AFC produced by amiloride (presumably blocking HSC channels) is not significantly different in wild-type and KO mice even though the final AFC is quite different (because of the lack of NSC channels in the KO mice; Fig. 14B).
We note that the decrease in AFC does not appear to be so dramatic as the increase in wet wt-to-dry wt ratio, and therefore we hypothesize that the increase in wet wt-to-dry wt ratio is only partially due to an increase in alveolar fluid and also due to vasodilation and increased interstitial fluid. This is possible since ASIC1a is responsible for calcium entry in pulmonary vascular smooth muscle in small vessels and activation of store-operated calcium release (64). ASIC KO will cause vascular relaxation and increased vascular permeability leading to increased lung fluid (and increasing wet wt-to-dry wt ratio). The increased vascular perfusion and lack of excessive alveolar fluid will mean that gas exchange will not be compromised as much as the wet wt-to-dry wt ratio would seem to imply and could explain the relatively low level of hypoxia.
It is important to test these ideas further. Experiments examining the morphology of knockout lungs and measuring the permeability of the vasculature and alveolar epithelium, as well as examining blood chemistry, particularly oxygen and CO2 tensions, are needed. Since AFC is compromised, stressing the knockout animals by exercise or by reducing ambient oxygen tension is a possible approach. In such an experiment, we predict that the knockout mice would not tolerate exercise or reduced oxygen. It would also be helpful to examine the animals' response to β-adrenergic agents and to influenza virus. In such an experiment, we predict that knockout animals would improve AFC in response to β-adrenergic agents but would be much more likely to develop influenza pneumonia. Finally, fetal lung appears to have much higher frequency of NSC channels (60). This implies that the secretory capacity of the fetal lung might be compromised in the knockout animals or that there would be a compensatory increase in HSC in the fetal lung.
One of the interesting differences in the regulation of NSC and HSC channels is the change in expression in response to changes in oxygen tension. Several studies have shown that the expression and apical density of ENaC subunits are reduced when oxygen tension is reduced to hypoxic levels (37, 66–68, 80, 81). This would lead to dangerously reduced levels of sodium transport and a tendency for alveolar flooding. Fortunately, NSC expression increases in response to hypoxia. Although NSC channels do not transport Na as efficiently as HSC, they can prevent alveolar flooding in response to moderate hypoxia. On the other hand, severe hypoxia compromises the ability of the basolateral Na,K-ATPase to extrude Na and leads to alveolar flooding even in the face of NSC activation (17, 49, 79).
Another situation in which the two different channel types might be important is in response to viral infections. Myxoviruses like influenza and measles bind to the surface of alveolar cells and reduce the activity of HSC channels in alveolar epithelial cells through a PKC-mediated process (16, 53, 86). NSC channels are hardly affected but do not transport Na as well as HSC channels, so that there is still a risk of alveolar flooding and viral pneumonia.
Why should the lung have two types of cation channels?
Other sodium-transporting epithelial tissue like the distal nephron of the kidney and the colon do not have NSC channels (7, 9, 12, 63, 69, 75, 76, 84). In the lung, the alveolar fluid layer must be very closely controlled, so it may be important to have alternative ion transport pathways that respond differently to physiological stimuli. However, a more likely reason may have to do with providing a stable driving force for cation and anion movement across the alveolar epithelium. NSC channels contribute to the apical membrane potential causing the membrane potential to be close to zero. This will ensure that there is a driving force for the unidirectional movement of anions [through cystic fibrosis transmembrane conductance regulator (CFTR)] and for movement of sodium (through ENaC/HSC) into cells. This is necessary because of the requirement to move salt, i.e., anions plus cations. Other epithelia tend to have counterion pathways for cations that obviate the need to maintain a strong potential driving force. It is interesting that in an evolutionary context, the lung is the most recent organ to adapt to a terrestrial environment. As is typical of evolutionary processes, existing mechanisms are modified to produce a different evolutionary outcome, in this case the formation of a new channel type from preexisting building blocks. Figure 16 shows a schematic diagram for a possible structure for NSC and HSC channels.
Fig. 16.

Schematic diagram of a possible configuration of ASIC1a and ENaC subunits in NSC and HSC channels.
GRANTS
This research is supported by funds from the American Physiological Society Short-Term Research Education Program to Increase Diversity Stipend (to P. T. Trac) and National Institutes of Health Grants DK-37963 (to D. C. Eaton), K12-GM-000680 and T32-HL-076118 (to M. Greenlee), and K01-DK-099617 (to A. A. Alli).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
P.T.T., T.L.T., V.L., L.Z., M.G., Q.Y., O.K.A.-K., A.A.A., A.F.E., and D.C.E. performed experiments; P.T.T., T.L.T., V.L., Q.Y., O.K.A.-K., A.F.E., and D.C.E. analyzed data; P.T.T., T.L.T., L.Z., M.G., O.K.A.-K., and D.C.E. interpreted results of experiments; P.T.T., T.L.T., V.L., L.Z., M.G., Q.Y., O.K.A.-K., A.A.A., A.F.E., and D.C.E. approved final version of manuscript; L.Z. conceived and designed research; M.G. and D.C.E. prepared figures; D.C.E. drafted manuscript; D.C.E. edited and revised manuscript.
ACKNOWLEDGMENTS
This work has been presented in abstract form at the 2016 American Thoracic Society Conference (30).
Present address of A. F. Eaton: Dept. of Medicine, Renal and Electrolyte Division, Univ. of Pittsburgh, Pittsburgh, PA 15261.
Present address of M. Greenlee (now Mitzelfelt): American Physiological Society, 9650 Rockville Pk., Bethesda, MD 20814–3991.
REFERENCES
- 1.Adir Y, Factor P, Dumasius V, Ridge KM, Sznajder JI. Na,K-ATPase gene transfer increases liquid clearance during ventilation-induced lung injury. Am J Respir Crit Care Med 168: 1445–1448, 2003. doi: 10.1164/rccm.200207-702OC. [DOI] [PubMed] [Google Scholar]
- 2.Alli AA, Bao HF, Alli AA, Aldrugh Y, Zhou Y, Yu L, Eaton DC. Calmodulin and CaM kinase II govern MARCKS-mediated PIP2-dependent regulation of ENaC. FASEB J 26, Suppl: 867.815, 2012. [Google Scholar]
- 3.Askwith CC, Benson CJ, Welsh MJ, Snyder PM. DEG/ENaC ion channels involved in sensory transduction are modulated by cold temperature. Proc Natl Acad Sci USA 98: 6459–6463, 2001. doi: 10.1073/pnas.111155398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azzam ZS, Dumasius V, Saldias FJ, Adir Y, Sznajder JI, Factor P. Na,K-ATPase overexpression improves alveolar fluid clearance in a rat model of elevated left atrial pressure. Circulation 105: 497–501, 2002. doi: 10.1161/hc0402.102848. [DOI] [PubMed] [Google Scholar]
- 5.Babinski K, Lê KT, Séguéla P. Molecular cloning and regional distribution of a human proton receptor subunit with biphasic functional properties. J Neurochem 72: 51–57, 1999. doi: 10.1046/j.1471-4159.1999.0720051.x. [DOI] [PubMed] [Google Scholar]
- 6.Bao HF, Liu L, Self J, Duke BJ, Ueno R, Eaton DC. A synthetic prostone activates apical chloride channels in A6 epithelial cells. Am J Physiol Gastrointest Liver Physiol 295: G234–G251, 2008. doi: 10.1152/ajpgi.00366.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bao HF, Thai TL, Yue Q, Ma HP, Eaton AF, Cai H, Klein JD, Sands JM, Eaton DC. ENaC activity is increased in isolated, split-open cortical collecting ducts from protein kinase Cα knockout mice. Am J Physiol Renal Physiol 306: F309–F320, 2014. doi: 10.1152/ajprenal.00519.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barquin N, Ciccolella DE, Ridge KM, Sznajder JI. Dexamethasone upregulates the Na-K-ATPase in rat alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 273: L825–L830, 1997. [DOI] [PubMed] [Google Scholar]
- 9.Bertog M, Cuffe JE, Pradervand S, Hummler E, Hartner A, Porst M, Hilgers KF, Rossier BC, Korbmacher C. Aldosterone responsiveness of the epithelial sodium channel (ENaC) in colon is increased in a mouse model for Liddle's syndrome. J Physiol 586: 459–475, 2008. doi: 10.1113/jphysiol.2007.140459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertorello AM, Komarova Y, Smith K, Leibiger IB, Efendiev R, Pedemonte CH, Borisy G, Sznajder JI. Analysis of Na+,K+-ATPase motion and incorporation into the plasma membrane in response to G protein-coupled receptor signals in living cells. Mol Biol Cell 14: 1149–1157, 2003. doi: 10.1091/mbc.E02-06-0367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bohlen CJ, Chesler AT, Sharif-Naeini R, Medzihradszky KF, Zhou S, King D, Sánchez EE, Burlingame AL, Basbaum AI, Julius D. A heteromeric Texas coral snake toxin targets acid-sensing ion channels to produce pain. Nature 479: 410–414, 2011. doi: 10.1038/nature10607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bugaj V, Pochynyuk O, Stockand JD. Activation of the epithelial Na+ channel in the collecting duct by vasopressin contributes to water reabsorption. Am J Physiol Renal Physiol 297: F1411–F1418, 2009. doi: 10.1152/ajprenal.00371.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen X, Kalbacher H, Gründer S. Interaction of acid-sensing ion channel (ASIC) 1 with the tarantula toxin psalmotoxin 1 is state dependent. J Gen Physiol 127: 267–276, 2006. doi: 10.1085/jgp.200509409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen X, Kalbacher H, Gründer S. The tarantula toxin psalmotoxin 1 inhibits acid-sensing ion channel (ASIC) 1a by increasing its apparent H+ affinity. J Gen Physiol 126: 71–79, 2005. doi: 10.1085/jgp.200509303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen XJ, Eaton DC, Jain L. β-Adrenergic regulation of amiloride-sensitive lung sodium channels. Am J Physiol Lung Cell Mol Physiol 282: L609–L620, 2002. doi: 10.1152/ajplung.00356.2001. [DOI] [PubMed] [Google Scholar]
- 16.Chen XJ, Seth S, Yue G, Kamat P, Compans RW, Guidot D, Brown LA, Eaton DC, Jain L. Influenza virus inhibits ENaC and lung fluid clearance. Am J Physiol Lung Cell Mol Physiol 287: L366–L373, 2004. doi: 10.1152/ajplung.00011.2004. [DOI] [PubMed] [Google Scholar]
- 17.Dada LA, Sznajder JI. Mechanisms of pulmonary edema clearance during acute hypoxemic respiratory failure: role of the Na,K-ATPase. Crit Care Med 31, Suppl: S248–S252, 2003. doi: 10.1097/01.CCM.0000057895.22008.EC. [DOI] [PubMed] [Google Scholar]
- 18.Davis IC, Matalon S. Epithelial sodium channels in the adult lung: important modulators of pulmonary health and disease. Adv Exp Med Biol 618: 127–140, 2007. doi: 10.1007/978-0-387-75434-5_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dawson RJ, Benz J, Stohler P, Tetaz T, Joseph C, Huber S, Schmid G, Hügin D, Pflimlin P, Trube G, Rudolph MG, Hennig M, Ruf A. Structure of the acid-sensing ion channel 1 in complex with the gating modifier Psalmotoxin 1. Nat Commun 3: 936, 2012. doi: 10.1038/ncomms1917. [DOI] [PubMed] [Google Scholar]
- 20.Dobbs LG. Isolation and culture of alveolar type II cells. Am J Physiol Lung Cell Mol Physiol 258: L134–L147, 1990. [DOI] [PubMed] [Google Scholar]
- 21.Dobbs LG, Gonzalez R, Williams MC. An improved method for isolating type II cells in high yield and purity. Am Rev Respir Dis 134: 141–145, 1986. [DOI] [PubMed] [Google Scholar]
- 22.Downs CA, Kriener LH, Yu L, Eaton DC, Jain L, Helms MN. β-Adrenergic agonists differentially regulate highly selective and nonselective epithelial sodium channels to promote alveolar fluid clearance in vivo. Am J Physiol Lung Cell Mol Physiol 302: L1167–L1178, 2012. doi: 10.1152/ajplung.00038.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eaton AF, Yue Q, Eaton DC, Bao HF. ENaC activity and expression is decreased in the lungs of protein kinase C-α knockout mice. Am J Physiol Lung Cell Mol Physiol 307: L374–L385, 2014. doi: 10.1152/ajplung.00040.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eaton DC. Intracellular sodium ion activity and sodium transport in rabbit urinary bladder. J Physiol 316: 527–544, 1981. doi: 10.1113/jphysiol.1981.sp013804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eaton DC, Becchetti A, Ma H, Ling BN. Renal sodium channels: regulation and single channel properties. Kidney Int 48: 941–949, 1995. doi: 10.1038/ki.1995.375. [DOI] [PubMed] [Google Scholar]
- 26.Eaton DC, Chen J, Ramosevac S, Matalon S, Jain L. Regulation of Na+ channels in lung alveolar type II epithelial cells. Proc Am Thorac Soc 1: 10–16, 2004. doi: 10.1513/pats.2306008. [DOI] [PubMed] [Google Scholar]
- 27.Eaton DC, Helms MN, Koval M, Bao HF, Jain L. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol 71: 403–423, 2009. doi: 10.1146/annurev.physiol.010908.163250. [DOI] [PubMed] [Google Scholar]
- 28.Eaton DC, Marunaka Y, Ling BN. Ion channels in epithelial tissue: single-channel properties. In: Membrane Transport in Biology, edited by Schafer JA, Ussing HH, Kristensen P, and Giebisch GH. Berlin: Springer, 1992, p. 73–165. doi: 10.1007/978-3-642-76983-2_3. [DOI] [Google Scholar]
- 29.Eaton DC, Ohara A, Ling BN, Marunaka Y, Duchatelle P, Kemendy AE, Kokko KE, Matsumoto PS, Hinton CF. Cellular regulation of amiloride-blockable sodium channels. Biomed Res 12, Suppl 2: 31–35, 1991. [Google Scholar]
- 30.Eaton DC, Trac P, Al Khalili OK, Greenlee MM. ASIC1a/αEnac hybrid channels contribute to alveolar fluid clearance (Abstract). Am J Respir Crit Care Med 193, Suppl: A5782, 2016. [Google Scholar]
- 31.Factor P, Dumasius V, Saldias F, Brown LA, Sznajder JI. Adenovirus-mediated transfer of an Na+/K+-ATPase β1 subunit gene improves alveolar fluid clearance and survival in hyperoxic rats. Hum Gene Ther 11: 2231–2242, 2000. doi: 10.1089/104303400750035753. [DOI] [PubMed] [Google Scholar]
- 32.Flodby P, Kim YH, Beard LL, Gao D, Ji Y, Kage H, Liebler JM, Minoo P, Kim KJ, Borok Z, Crandall ED. Knockout mice reveal a major role for alveolar epithelial type I cells in alveolar fluid clearance. Am J Respir Cell Mol Biol 55: 395–406, 2016. doi: 10.1165/rcmb.2016-0005OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fyfe GK, Canessa CM. Subunit composition determines the single channel kinetics of the epithelial sodium channel. J Gen Physiol 112: 423–432, 1998. doi: 10.1085/jgp.112.4.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.García-Añoveros J, Derfler B, Neville-Golden J, Hyman BT, Corey DP. BNaC1 and BNaC2 constitute a new family of human neuronal sodium channels related to degenerins and epithelial sodium channels. Proc Natl Acad Sci USA 94: 1459–1464, 1997. doi: 10.1073/pnas.94.4.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev 77: 359–396, 1997. [DOI] [PubMed] [Google Scholar]
- 36.George AL Jr, Staub O, Geering K, Rossier BC, Kleyman TR, Kraehenbuhl JP. Functional expression of the amiloride-sensitive sodium channel in Xenopus oocytes. Proc Natl Acad Sci USA 86: 7295–7298, 1989. doi: 10.1073/pnas.86.18.7295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gille T, Randrianarison-Pellan N, Goolaerts A, Dard N, Uzunhan Y, Ferrary E, Hummler E, Clerici C, Planès C. Hypoxia-induced inhibition of epithelial Na(+) channels in the lung. Role of Nedd4-2 and the ubiquitin-proteasome pathway. Am J Respir Cell Mol Biol 50: 526–537, 2014. doi: 10.1165/rcmb.2012-0518OC. [DOI] [PubMed] [Google Scholar]
- 38.Goldman DE. Potential, impedance, and rectification in membranes. J Gen Physiol 27: 37–60, 1943. doi: 10.1085/jgp.27.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goodson P, Kumar A, Jain L, Kundu K, Murthy N, Koval M, Helms MN. Nadph oxidase regulates alveolar epithelial sodium channel activity and lung fluid balance in vivo via O−2 signaling. Am J Physiol Lung Cell Mol Physiol 302: L410–L419, 2012. doi: 10.1152/ajplung.00260.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hardiman KM, McNicholas-Bevensee CM, Fortenberry J, Myles CT, Malik B, Eaton DC, Matalon S. Regulation of amiloride-sensitive Na(+) transport by basal nitric oxide. Am J Respir Cell Mol Biol 30: 720–728, 2004. doi: 10.1165/rcmb.2003-0325OC. [DOI] [PubMed] [Google Scholar]
- 41.Helms MN, Chen XJ, Ramosevac S, Eaton DC, Jain L. Dopamine regulation of amiloride-sensitive sodium channels in lung cells. Am J Physiol Lung Cell Mol Physiol 290: L710–L722, 2006. doi: 10.1152/ajplung.00486.2004. [DOI] [PubMed] [Google Scholar]
- 42.Helms MN, Jain L, Self JL, Eaton DC. Redox regulation of epithelial sodium channels examined in alveolar type 1 and 2 cells patch-clamped in lung slice tissue. J Biol Chem 283: 22875–22883, 2008. doi: 10.1074/jbc.M801363200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Helms MN, Self J, Bao HF, Job LC, Jain L, Eaton DC. Dopamine activates amiloride-sensitive sodium channels in alveolar type I cells in lung slice preparations. Am J Physiol Lung Cell Mol Physiol 291: L610–L618, 2006. doi: 10.1152/ajplung.00426.2005. [DOI] [PubMed] [Google Scholar]
- 44.Hodgkin AL, Huxley AF, Katz B. Measurement of current-voltage relations in the membrane of the giant axon of Loligo. J Physiol 116: 424–448, 1952. doi: 10.1113/jphysiol.1952.sp004716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jacobson KA, Costanzi S, Joshi BV, Besada P, Shin DH, Ko H, Ivanov AA, Mamedova L. Agonists and antagonists for P2 receptors. Novartis Found Symp 276: 58–68, 2006. doi: 10.1002/9780470032244.ch6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jain L, Chen XJ, Brown LA, Eaton DC. Nitric oxide inhibits lung sodium transport through a cGMP-mediated inhibition of epithelial cation channels. Am J Physiol Lung Cell Mol Physiol 274: L475–L484, 1998. [DOI] [PubMed] [Google Scholar]
- 47.Jain L, Chen XJ, Malik B, Al-Khalili O, Eaton DC. Antisense oligonucleotides against the α-subunit of ENaC decrease lung epithelial cation-channel activity. Am J Physiol Lung Cell Mol Physiol 276: L1046–L1051, 1999. [DOI] [PubMed] [Google Scholar]
- 48.Jain L, Chen XJ, Ramosevac S, Brown LA, Eaton DC. Expression of highly selective sodium channels in alveolar type II cells is determined by culture conditions. Am J Physiol Lung Cell Mol Physiol 280: L646–L658, 2001. [DOI] [PubMed] [Google Scholar]
- 49.Jain M, Sznajder JI. Effects of hypoxia on the alveolar epithelium. Proc Am Thorac Soc 2: 202–205, 2005. doi: 10.1513/pats.200501-006AC. [DOI] [PubMed] [Google Scholar]
- 50.Johnson MD, Bao HF, Helms MN, Chen XJ, Tigue Z, Jain L, Dobbs LG, Eaton DC. Functional ion channels in pulmonary alveolar type I cells support a role for type I cells in lung ion transport. Proc Natl Acad Sci USA 103: 4964–4969, 2006. doi: 10.1073/pnas.0600855103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kapoor N, Lee W, Clark E, Bartoszewski R, McNicholas CM, Latham CB, Bebok Z, Parpura V, Fuller CM, Palmer CA, Benos DJ. Interaction of ASIC1 and ENaC subunits in human glioma cells and rat astrocytes. Am J Physiol Cell Physiol 300: C1246–C1259, 2011. doi: 10.1152/ajpcell.00199.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lazrak A, Chen L, Jurkuvenaite A, Doran SF, Liu G, Li Q, Lancaster JR Jr, Matalon S. Regulation of alveolar epithelial Na+ channels by ERK1/2 in chlorine-breathing mice. Am J Respir Cell Mol Biol 46: 342–354, 2012. doi: 10.1165/rcmb.2011-0309OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lazrak A, Iles KE, Liu G, Noah DL, Noah JW, Matalon S. Influenza virus M2 protein inhibits epithelial sodium channels by increasing reactive oxygen species. FASEB J 23: 3829–3842, 2009. doi: 10.1096/fj.09-135590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lazrak A, Samanta A, Matalon S. Biophysical properties and molecular characterization of amiloride-sensitive sodium channels in A549 cells. Am J Physiol Lung Cell Mol Physiol 278: L848–L857, 2000. [DOI] [PubMed] [Google Scholar]
- 55.Lazrak A, Samanta A, Venetsanou K, Barbry P, Matalon S. Modification of biophysical properties of lung epithelial Na+ channels by dexamethasone. Am J Physiol Cell Physiol 279: C762–C770, 2000. [DOI] [PubMed] [Google Scholar]
- 56.Lewis SA, Diamond JM. Active sodium transport by mammalian urinary bladder. Nature 253: 747–748, 1975. doi: 10.1038/253747a0. [DOI] [PubMed] [Google Scholar]
- 57.Lewis SA, Diamond JM. Na+ transport by rabbit urinary bladder, a tight epithelium. J Membr Biol 28: 1–40, 1976. doi: 10.1007/BF01869689. [DOI] [PubMed] [Google Scholar]
- 58.Lewis SA, Eaton DC, Diamond JM. The mechanism of Na+ transport by rabbit urinary bladder. J Membr Biol 28: 41–70, 1976. doi: 10.1007/BF01869690. [DOI] [PubMed] [Google Scholar]
- 59.Ma HP, Li L, Zhou ZH, Eaton DC, Warnock DG. ATP masks stretch activation of epithelial sodium channels in A6 distal nephron cells. Am J Physiol Renal Physiol 282: F501–F505, 2002. doi: 10.1152/ajprenal.00147.2001. [DOI] [PubMed] [Google Scholar]
- 60.Marunaka Y. Amiloride-blockable Ca2+-activated Na+-permeant channels in the fetal distal lung epithelium. Pflugers Arch 431: 748–756, 1996. doi: 10.1007/BF02253839. [DOI] [PubMed] [Google Scholar]
- 61.Marunaka Y, Tohda H, Hagiwara N, O'Brodovich H. Cytosolic Ca(2+)-induced modulation of ion selectivity and amiloride sensitivity of a cation channel and beta agonist action in fetal lung epithelium. Biochem Biophys Res Commun 187: 648–656, 1992. doi: 10.1016/0006-291X(92)91244-K. [DOI] [PubMed] [Google Scholar]
- 62.McNicholas CM, Canessa CM. Diversity of channels generated by different combinations of epithelial sodium channel subunits. J Gen Physiol 109: 681–692, 1997. doi: 10.1085/jgp.109.6.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mironova E, Bugay V, Pochynyuk O, Staruschenko A, Stockand JD. Recording ion channels in isolated, split-opened tubules. Methods Mol Biol 998: 341–353, 2013. doi: 10.1007/978-1-62703-351-0_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nitta CH, Osmond DA, Herbert LM, Beasley BF, Resta TC, Walker BR, Jernigan NL. Role of ASIC1 in the development of chronic hypoxia-induced pulmonary hypertension. Am J Physiol Heart Circ Physiol 306: H41–H52, 2014. doi: 10.1152/ajpheart.00269.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.O'Brodovich H, Yang P, Gandhi S, Otulakowski G. Amiloride-insensitive Na+ and fluid absorption in the mammalian distal lung. Am J Physiol Lung Cell Mol Physiol 294: L401–L408, 2008. doi: 10.1152/ajplung.00431.2007. [DOI] [PubMed] [Google Scholar]
- 66.Planès C, Blot-Chabaud M, Matthay MA, Couette S, Uchida T, Clerici C. Hypoxia and β 2-agonists regulate cell surface expression of the epithelial sodium channel in native alveolar epithelial cells. J Biol Chem 277: 47318–47324, 2002. doi: 10.1074/jbc.M209158200. [DOI] [PubMed] [Google Scholar]
- 67.Planès C, Escoubet B, Blot-Chabaud M, Friedlander G, Farman N, Clerici C. Hypoxia downregulates expression and activity of epithelial sodium channels in rat alveolar epithelial cells. Am J Respir Cell Mol Biol 17: 508–518, 1997. doi: 10.1165/ajrcmb.17.4.2680. [DOI] [PubMed] [Google Scholar]
- 68.Planès C, Randrianarison NH, Charles RP, Frateschi S, Cluzeaud F, Vuagniaux G, Soler P, Clerici C, Rossier BC, Hummler E. ENaC-mediated alveolar fluid clearance and lung fluid balance depend on the channel-activating protease 1. EMBO Mol Med 2: 26–37, 2010. doi: 10.1002/emmm.200900050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pochynyuk O, Bugaj V, Stockand JD. Physiologic regulation of the epithelial sodium channel by phosphatidylinositides. Curr Opin Nephrol Hypertens 17: 533–540, 2008. doi: 10.1097/MNH.0b013e328308fff3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pochynyuk O, Bugaj V, Vandewalle A, Stockand JD. Purinergic control of apical plasma membrane PI(4,5)P2 levels sets ENaC activity in principal cells. Am J Physiol Renal Physiol 294: F38–F46, 2008. doi: 10.1152/ajprenal.00403.2007. [DOI] [PubMed] [Google Scholar]
- 71.Price MP, Gong H, Parsons MG, Kundert JR, Reznikov LR, Bernardinelli L, Chaloner K, Buchanan GF, Wemmie JA, Richerson GB, Cassell MD, Welsh MJ. Localization and behaviors in null mice suggest that ASIC1 and ASIC2 modulate responses to aversive stimuli. Genes Brain Behav 13: 179–194, 2014. doi: 10.1111/gbb.12108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rooj AK, McNicholas CM, Bartoszewski R, Bebok Z, Benos DJ, Fuller CM. Glioma-specific cation conductance regulates migration and cell cycle progression. J Biol Chem 287: 4053–4065, 2012. doi: 10.1074/jbc.M111.311688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rossier BC, Canessa CM, Schild L, Horisberger JD. Epithelial sodium channels. Curr Opin Nephrol Hypertens 3: 487–496, 1994. doi: 10.1097/00041552-199409000-00003. [DOI] [PubMed] [Google Scholar]
- 74.Rossier BC, Pradervand S, Schild L, Hummler E. Epithelial sodium channel and the control of sodium balance: interaction between genetic and environmental factors. Annu Rev Physiol 64: 877–897, 2002. doi: 10.1146/annurev.physiol.64.082101.143243. [DOI] [PubMed] [Google Scholar]
- 75.Staruschenko A, Patel P, Tong Q, Medina JL, Stockand JD. Ras activates the epithelial Na(+) channel through phosphoinositide 3-OH kinase signaling. J Biol Chem 279: 37771–37778, 2004. doi: 10.1074/jbc.M402176200. [DOI] [PubMed] [Google Scholar]
- 76.Staruschenko A, Pochynyuk O, Vandewalle A, Bugaj V, Stockand JD. Acute regulation of the epithelial Na+ channel by phosphatidylinositide 3-OH kinase signaling in native collecting duct principal cells. J Am Soc Nephrol 18: 1652–1661, 2007. doi: 10.1681/ASN.2007010020. [DOI] [PubMed] [Google Scholar]
- 77.Stockand JD, Bao HF, Schenck J, Malik B, Middleton P, Schlanger LE, Eaton DC. Differential effects of protein kinase C on the levels of epithelial Na+ channel subunit proteins. J Biol Chem 275: 25760–25765, 2000. doi: 10.1074/jbc.M003615200. [DOI] [PubMed] [Google Scholar]
- 78.Stockand JD, Mironova E, Bugaj V, Rieg T, Insel PA, Vallon V, Peti-Peterdi J, Pochynyuk O. Purinergic inhibition of ENaC produces aldosterone escape. J Am Soc Nephrol 21: 1903–1911, 2010. doi: 10.1681/ASN.2010040377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Sznajder JI, Factor P, Ingbar DH. Invited review: lung edema clearance: role of Na+-K+-ATPase. J Appl Physiol (1985) 93: 1860–1866, 2002. doi: 10.1152/japplphysiol.00022.2002. [DOI] [PubMed] [Google Scholar]
- 80.Urner M, Herrmann IK, Booy C, Roth-Z' Graggen B, Maggiorini M, Beck-Schimmer B. Effect of hypoxia and dexamethasone on inflammation and ion transporter function in pulmonary cells. Clin Exp Immunol 169: 119–128, 2012. doi: 10.1111/j.1365-2249.2012.04595.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang S, Publicover S, Gu Y. An oxygen-sensitive mechanism in regulation of epithelial sodium channel. Proc Natl Acad Sci USA 106: 2957–2962, 2009. doi: 10.1073/pnas.0809100106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang X, Kleyman TR, Tohda H, Marunaka Y, O'Brodovich H. 5-(N-ethyl-N-isopropyl)amiloride sensitive Na+ currents in intact fetal distal lung epithelial cells. Can J Physiol Pharmacol 71: 58–62, 1993. doi: 10.1139/y93-009. [DOI] [PubMed] [Google Scholar]
- 83.Wemmie JA, Chen J, Askwith CC, Hruska-Hageman AM, Price MP, Nolan BC, Yoder PG, Lamani E, Hoshi T, Freeman JH Jr, Welsh MJ. The acid-activated ion channel ASIC contributes to synaptic plasticity, learning, and memory. Neuron 34: 463–477, 2002. doi: 10.1016/S0896-6273(02)00661-X. [DOI] [PubMed] [Google Scholar]
- 84.Wills NK, Lewis SA, Eaton DC. Active and passive properties of rabbit descending colon: a microelectrode and nystatin study. J Membr Biol 45: 81–108, 1979. doi: 10.1007/BF01869296. [DOI] [PubMed] [Google Scholar]
- 85.Yu L, Cai H, Yue Q, Alli AA, Wang D, Al-Khalili O, Bao HF, Eaton DC. WNK4 inhibition of ENaC is independent of Nedd4-2-mediated ENaC ubiquitination. Am J Physiol Renal Physiol 305: F31–F41, 2013. doi: 10.1152/ajprenal.00652.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zhao M, Liang X, Ji H. Influenza infection impairs both ENaC and CFTR function in primary mouse tracheal epithelial cells. FASEB J 25, Suppl: 1042.18, 2011. [Google Scholar]
- 87.Zhou R, Tomkovicz VR, Butler PL, Ochoa LA, Peterson ZJ, Snyder PM. Ubiquitin-specific peptidase 8 (USP8) regulates endosomal trafficking of the epithelial Na+ channel. J Biol Chem 288: 5389–5397, 2013. doi: 10.1074/jbc.M112.425272. [DOI] [PMC free article] [PubMed] [Google Scholar]