

Abstract
Several members of the SLC26A family of anion transporters associate with CFTR, forming complexes in which CFTR and SLC26A functions are reciprocally regulated. These associations are thought to be facilitated by PDZ scaffolding interactions. CFTR has been shown to be positively regulated by NHERF-1, and negatively regulated by CAL in airway epithelia. However, it is unclear which PDZ-domain protein(s) interact with SLC26A9, a SLC26A family member found in airway epithelia. We have previously shown that primary, human bronchial epithelia (HBE) from non-CF donors exhibit constitutive anion secretion attributable to SLC26A9. However, constitutive anion secretion is absent in HBE from CF donors. We examined whether changes in SLC26A9 constitutive activity could be attributed to a loss of CFTR trafficking, and what role PDZ interactions played. HEK293 coexpressing SLC26A9 with the trafficking mutant F508del CFTR exhibited a significant reduction in constitutive current compared with cells coexpressing SLC26A9 and wt CFTR. We found that SLC26A9 exhibits complex glycosylation when coexpressed with F508del CFTR, but its expression at the plasma membrane is decreased. SLC26A9 interacted with both NHERF-1 and CAL, and its interaction with both significantly increased with coexpression of wt CFTR. However, coexpression with F508del CFTR only increased SLC26A9’s interaction with CAL. Mutation of SLC26A9’s PDZ motif decreased this association with CAL, and restored its constitutive activity. Correcting aberrant F508del CFTR trafficking in CF HBE with corrector VX-809 also restored SLC26A9 activity. We conclude that when SLC26A9 is coexpressed with F508del CFTR, its trafficking defect leads to a PDZ motif-sensitive intracellular retention of SLC26A9.
Keywords: chloride channel, CAL, NHERF-1, intracellular trafficking, PDZ domain
the epithelia affected by cystic fibrosis (CF) demonstrate diverse defects in ion and fluid transport, including defective bicarbonate and/or chloride secretion across the surface airway epithelium, defective salt absorption in the airways and sweat glands, and perturbations in the balance between bicarbonate and chloride exchange in the pancreas and gut. The name given to the ion channel defective in CF, cystic fibrosis transmembrane conductance regulator (CFTR), reflected this diverse pathology, as the extent of ion transport defects suggested CFTR performed a regulatory role in addition to its ion channel function (49). Thus the study of CFTR function in the years following its identification focused both on the properties of CFTR as an anion channel as well as its role in regulating the function of other ion channels and transporters. The diversity in the pathology of CF makes the solute carrier 26 (SLC26A) family of anion transporters and ion channels ideal candidates to investigate as interacting partners with CFTR, as SLC26A exhibit diverse substrate selectivities, transport functions, and tissue distributions (21, 42).
In studies performed by Ko et al. (28), two members of the SLC26A family known to be chloride-bicarbonate exchangers, SLC26A3 and -A6 (42), were each shown to interact with CFTR in a manner that reciprocally enhanced the functions of both proteins. At a molecular level, phosphorylation of the regulatory (R) domain of CFTR induced an interaction with the sulfate transporter and anti-sigma factor antagonist (STAS) domain found in all SLC26A transporters, resulting in an increased open probability and chloride transport through CFTR, as well as a greater anion exchange rate through SLC26A3 and -A6. The interaction between CFTR and SLC26A3 or -A6 was facilitated by their class 1 postsynaptic density-95/disks large/zona occludens-1 (PDZ) motifs, which were hypothesized to tether the proteins in close association by binding to a common PDZ-domain protein. The most widespread CFTR mutation leading to CF disease is the deletion of a phenylalanine at position 508; F508del CFTR fails to fold properly and is not expressed at the plasma membrane. Based on Ko’s studies (28), a model for defective bicarbonate secretion in the CF pancreas was proposed, in which the absence of F508del CFTR at the plasma membrane results in a loss both of the electrochemical driving forces for the anion exchangers, and of the STAS domain-R domain interaction that enhanced their anion exchange rate.
Another SLC26A family member was originally identified in the lung: SLC26A9 (35). The behavior of SLC26A9 appears to be complex. It has been shown to function as a constitutively active chloride channel (6, 9, 20, 36), but may also exhibit bicarbonate sensitivity (36) and anion exchange activity (9, 61). The role of SLC26A9 in maintaining a healthy airway environment is as yet unknown, but genomewide association studies have implicated SLC26A9 as a modifier of several CF-associated pathologies, including meconium ileus (56), CF-related diabetes (7), and prenatal exocrine pancreatic damage (41). The noncoding, single-nucleotide polymorphism (SNP) in SLC26A9 predictive of pancreatic damage (41) has recently been shown to modulate the airway response to potentiator VX-770 in CF patients with the gating mutation G551D CFTR (54). Patients homozygous for the F508del CFTR mutation and this specific SLC26A9 SNP did not exhibit the same modulation; however, rescue of F508del CFTR with corrector VX-809 restored the impact of this SLC26A9 SNP. These results suggest F508del CFTR may exert a negative influence on SLC26A9 in the airway.
Smith and Welsh (53) first noted that HBE from non-CF donors exhibited both constitutive and cAMP-stimulated anion secretion, and that both components were absent in HBE from CF donors. Other studies noted that both the constitutive and cAMP-stimulated anion currents were sensitive to CFTR channel inhibitors (reviewed in 6). Thus the constitutive current has been attributed to CFTR. However, current flow through CFTR itself is preceded by phosphorylation of its R domain by the cAMP-activated protein kinase A (PKA), as well as ATP binding and dimerization of its nucleotide binding domains. Thus we hypothesized that the constitutive anion secretion could reflect CFTR regulation of current flow through SLC26A9 rather than CFTR. To test this, we examined the interaction between SLC26A9 and wt CFTR coexpressed in HEK293 cells (6), and showed that SLC26A9 mediated constitutive chloride currents, and coimmunoprecipitated wild type (wt) CFTR. In addition, we demonstrated that the anion channel inhibitor GlyH-101, originally thought to be specific for CFTR, also inhibited 67% of SLC26A9’s constitutive Cl− current, but did not evoke the same strong inward-current rectification seen with inhibition of CFTR. We used this unique pharmacological fingerprint of GlyH-101 inhibition to confirm that the constitutive current across the apical membranes of primary HBE from non-CF donors (i.e., wt CFTR monolayers; see 6) bore the properties of SLC26A9.
During these studies, we also confirmed that the magnitude of constitutive anion secretion in HBE was unaffected by intracellular ATP depletion. This further indicated that the constitutive anion current did not flow through CFTR, as we demonstrated that ATP depletion abolished the cAMP-stimulated anion secretion due to CFTR (6). The SLC26A9 fingerprint was absent in differentiated HBE homozygous for the F508del CFTR mutation, however. The loss of SLC26A9 constitutive activity when coexpressed with F508del CFTR is striking, as we and others (2, 6, 9, 20, 36, 44, 48) have shown that SLC26A9 constitutive activity is present when the protein is expressed alone. Since SLC26A9 does not require coexpression with CFTR at the plasma membrane to be constitutively active, these results (6, 54) suggest that F508del CFTR is exerting a negative influence on coexpressed SLC26A9. Because the F508del mutation prevents the normal trafficking of CFTR from the endoplasmic reticulum (ER), through the Golgi, and to the plasma membrane, accessory proteins involved in these stages of CFTR maturation may also influence the expression, trafficking, and/or function of SLC26A9.
PDZ-domain proteins have been shown to be involved in multiple steps of CFTR trafficking and expression at the plasma membrane (26, 32). In the airway, it has been proposed that cell-surface expression of CFTR is regulated by competition between the Na+/H+ exchanger-3 regulatory factor 1 (NHERF-1), and the CFTR-associated ligand (CAL) (12). NHERF-1 localizes at the plasma membrane when its ezrin recognition motif (ERM) binds ezrin, which in turn facilitates the plasma membrane colocalization and stabilization of proteins bound to either of its PDZ binding pockets (52). In contrast, CAL localizes primarily to the trans-Golgi network (TGN), where it directs the trafficking of proteins entering and within the peripheral recycling pathway; CAL has also been shown to promote the lysosomal degradation of wt CFTR (10, 13). In tests of the relative binding affinity of CFTR for these PDZ-domain proteins, a peptide comprising the last 10 amino acids of CFTR showed significantly greater affinity for NHERF-1 over CAL (16), which would favor retention of CFTR at the plasma membrane. In support of this, overexpression of CAL has been shown to reduce surface expression of wt CFTR; these experiments also showed an increase in the immature (core-glycosylated) form of CFTR, suggestive of a negative influence of CAL on ER to Golgi transport (12). Whether CAL might play a role in preventing ER to Golgi transport of misfolded F508del CFTR is unclear at this time, but CAL has been shown to have a negative effect on the surface expression of “rescued” F508del CFTR (17, 59), suggesting a role for CAL in limiting traffic to the plasma membrane.
The mechanisms underlying CFTR’s attraction to and exchange between PDZ domain proteins is an area of interest, as these may be druggable targets for improving the rescue of defective CFTR (4, 46). Guggino and colleagues have identified several proteins involved in regulating the CFTR-CAL interactome, including the N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) Syntaxin 6, which is involved in post-ER vesicle trafficking and directs CFTR to the lysosome (10); the Rho family small GTPase TC10, whose active form is associated with trafficking of CAL (and wt CFTR) from the TGN to the plasma membrane (14); and MARCH2, a ubiquitin ligase involved in targeting CFTR for lysosomal degradation (11). Naren and colleagues have found that the PDZ domain protein MAST205 competes with CAL for binding to CFTR, and they have hypothesized that MAST205 may escort CFTR from the TGN to the recycling endosome (47). Interestingly, these various interacting proteins appear to function primarily by releasing CFTR from the TGN-CAL interactome and allowing it to continue trafficking, either to the plasma membrane or lysosomes. Mutation of CFTR’s PDZ motif was shown to alleviate the impact of these proteins (10, 11, 14, 47), presumably because CFTR-ΔPDZ could traffic past the TGN-CAL interactome independently. CFTR-ΔPDZ was functional at the plasma membrane, suggesting an association with NHERF-1 at the plasma membrane was not absolutely required for channel activity. In support of this, a mutation in CFTR that abrogates its PDZ motif does not lead to CF symptoms in the lung, although it was associated with elevated sweat chloride (40).
In the present study, we demonstrate the inhibitory impact of F508del CFTR coexpression on SLC26A9 constitutive activity in HEK293 cells, and show that this inhibition results from the maturation defect in F508del CFTR. Our results indicate that SLC26A9 interacts with immature F508del CFTR, but this interaction does not alter SLC26A9 protein expression level or its glycosylation status. SLC26A9 demonstrates affinity for both NHERF-1 and CAL, and NHERF-1’s affinity is significantly weaker for SLC26A9 than for wt CFTR. We observed a significant increase in endogenous CAL coimmunoprecipitation with SLC26A9 when coexpressed with either wt or F508del CFTR vs. SLC26A9 expressed alone. For wt CFTR coexpression, this increase was accompanied by a parallel increase in NHERF-1 co-IP. Thus our data suggest that coexpression of SLC26A9 with F508del CFTR unbalances the competition between NHERF-1 and CAL binding in favor of CAL, leading to intracellular retention of SLC26A9. In support of this concept, we demonstrate that mutating the PDZ motif of SLC26A9 restores its constitutive activity when coexpressed with F508del CFTR. Similarly, correcting F508del CFTR’s trafficking defect in CF HBE restores constitutive anion secretion.
MATERIALS AND METHODS
Cell cultures and plasmid vectors.
HBE cells were cultured from excess pathological tissue following lung transplantation and organ donation under a protocol approved by the University of Pittsburgh Institutional Review Board, as previously described (19). Briefly, cells were propagated in flasks to 80–90% confluency, and then transferred to 0.33-cm2 Costar Transwell filters; apical media were removed 24 h after seeding and the filters grown at an air-liquid interface thereafter. Basolateral media were changed 3 times weekly. Twelve different patient samples were used for short-circuit current (Isc) measurements (8 non-CF, 4 CF). The non-CF HBE were obtained from patients diagnosed with idiopathic pulmonary fibrosis, or from donor lungs (no known disease), and the CF HBE used were obtained from patients with severe disease and who were at least heterozygous for the F508del CFTR mutation. For each patient sample, a minimum of three filters were tested per experimental condition. Filters were grown at air-liquid interface for at least 2 wk before testing.
HEK293 cells (ATCC CRL-1573, passages 12–50) were propagated in Falcon culture flasks (75 cm2) in a humidified atmosphere of 95% air, 5% CO2 at 37°C. The cells were passaged and fed 2 times per week with Dulbecco's Modified Eagle's Medium supplemented with 10% heat-inactivated fetal bovine serum and 2 mM l-glutamine. Cells were detached using TrypLE Express, and aliquoted into six-well plates in preparation for transfection. Transfection media was replaced with normal media after 14 h, and transfected cells were either harvested after 48 h for protein assays, or removed from the six-well plates after 24 h and plated to glass coverslips to be used for patch clamping within 18–36 h. Plates and glass coverslips were precoated with poly-l-lysine to promote cell adhesion. All cell culture medium was obtained from ThermoScientific Fisher (Invitrogen), and all chemicals from Sigma, unless explicitly noted.
HEK293 cells were transiently transfected with either SLC26A9 alone or together with CFTR. SLC26A9 expressed in a pcDNA 3.1 vector was a kind gift of Dr. Shmuel Muallem, and the entire construct was sequenced to verify that it corresponded to the open reading frame (ORF) of the published 4,815 bp isoform (35) of human SLC26A9 (NCBI Reference Sequence NM_052934). SLC26A9 with a myc epitope at the NH2 terminus (myc-SLC26A9) was prepared by excising the cDNA encoding SLC26A9 from pcDNA 3.1 and cloning it into a pcDNA 3 Myc vector (ThermoScientific Fisher). The epitope-tagged version of SLC26A9 performed similarly to untagged SLC26A9 in whole cell patch-clamp experiments (not shown). CFTR constructs in pcDNA3.1 vectors were as described (55). Variants of CFTR and SLC26A9 with mutated PDZ motifs were generated using the QuikChange II XL site-directed mutagenesis kit (Stratagene, Agilent Technologies), in accordance with manufacturer’s instructions. For SLC26A9, the −2 position T in the PDZ motif was mutated to A (T789A) using the primers 5′-GCAGAGACCCTGGCCGCCCTGTGAG (forward) and 5′-CTCACAGGGCGGCCAGGGTCTCTGC (reverse). For CFTR, the −2 position T was mutated to A (CFTR-mPDZ) with the primers 5′-GAAGAAGAGATGCAAGATGCAAGGCT (forward) and 5′-CTGCTCTCTAAAGCCTTGCATCTTGC (reverse). pMAX GFP (Lonza, Allendale, NJ) was used in transfections to balance total cDNA and identify transfected cells, except for immunofluorescence experiments, which used empty vector instead of GFP. Transfections were performed using the standard Lipofectamine 2000 protocol (ThermoScientific Fisher), with cDNA concentrations and method as previously described (6).
Electrophysiology.
HBE monolayers on filter supports were mounted in Ussing chambers, and the chambers continuously short-circuited with a VCC MC6 automatic voltage clamp (Physiologic Instruments, San Diego, CA). All experiments were performed at 37°C. Transepithelial resistance (TER) was measured by applying a 2-mV bipolar pulse every 90 s and calculated using Ohm’s law. Drug additions to each chamber were cumulative. Experiments used a serosal to mucosal chloride gradient; the serosal solution contained (in mM) 115 NaCl, 25 NaHCO3, 5 KCl, 10 HEPES, 1 MgCl2, 1.5 CaCl2, and 5 glucose, and the mucosal solution substituted 115 mM Na-gluconate for the NaCl. Chambers were constantly gassed with a mixture of 95% O2-5% CO2 to maintain pH at 7.4.
Whole cell patch clamping was performed as previously described (6). Briefly, the electronics consisted of a 200B Axopatch amplifier controlled by Clampex 8.1 software through a Digidata 1322A acquisition board (all from Axon Instruments). Solutions were applied to the imaging chamber at 2.0 ml/min and 37°C. The imaging chamber was mounted on the stage of a Nikon Diaphot microscope equipped with standard illumination and a xenon lamp with GFP filter cube (Ex 485/Em 550). Once a GFP-labeled cell was selected, the remainder of the experiment was performed under standard illumination. Seal resistances exceeded 8 GΩ and pipette capacitance was compensated; all experiments used a standard voltage-clamp protocol at a holding potential of −40 mV. NMDG-Cl was used for both the bath and pipette solutions to isolate chloride currents. The osmolarity of both solutions was measured, and NMDG-glutamate was added to the bath solution (in mM, 140 NMDG-Cl, 10 HEPES, 1 MgCl2, 1.5 CaCl2, 5 glucose, to pH 7.3 with Tris) to generate a 25 mOsm gradient that obviated cell swelling (60). The pipette solution (in mM, 140 NMDG-Cl, 10 HEPES, 1 MgCl2, 5 glucose, 1 EGTA, to pH 7.2 with Tris) included 1 mM Mg-ATP and 100 μM GTP. Pipettes using thin wall borosilicate glass were pulled to tip diameters of 1–2 μm (access resistance < 4 MΩ). All patch-clamping results reflect data pooled from a minimum of 3 separate biological samples (HEK293 with different passage numbers) and are reported as means ± SE. Student's t-test was used to determine statistical significance, with P < 0.05 considered significantly different.
Binding affinities.
Fluorescence polarization (FP) binding data were measured as previously described (16). Briefly, purified PDZ-domain proteins (CAL PDZ, NHERF-1 PDZ1 or PDZ2, or NHERF-2 PDZ1 or PDZ2) at concentrations of ~1.8 KD were incubated with 30 nM fluorescein (F*)-labeled reporter peptide (F*-iCAL36 for CAL, KD = 0.72 μM; F*-CFTR6 for the NHERF-1 domains, KD = 0.39 and 1.1 μM, respectively; F*-CFTR10 for the NHERF-2 domains, KD = 0.21 and 0.11 μM, respectively) together with increasing concentrations of unlabeled peptides corresponding to the COOH-terminal 10 residues of CFTR (CFTR-10; Tufts University Core Facility) or SLC26A9 (A9; Biomatik). Following equilibration, fluorescence anisotropy values were measured. Displacement isotherms were fit to obtain a least-squares estimate of KI for each inhibitor peptide based on comparison of experimental anisotropy values with values predicted from the competitive three-way equilibrium. For incomplete inhibitions, the FP value of free ligand was fixed based on a parallel titration with high-affinity inhibitor. All titrations were performed independently in triplicate and are reported as means ± SD.
Antibodies and densitometry.
Mouse monoclonal antibodies targeting CFTR’s second nucleotide-binding domain (NBD2, no. 596) and R domain (no. 217) were obtained via Cystic Fibrosis Foundation Therapeutics (Bethesda, MD), found at http://cftrfolding.org. These were used to detect wild-type and mutant CFTR; the F508del and G551D CFTR mutations are located in NBD1. A polyclonal antibody targeting CAL (no. A302–642A) was purchased from Bethyl Laboratories (Montgomery, TX). The manufacturer qualified the antibody for immunoblotting in HEK293, and it has previously been used to analyze CAL-CFTR interactions in the HEK293 expression system (47). A polyclonal antibody targeting NHERF-1 (aka EBP50, no. ab3452) was purchased from Abcam (Cambridge, MA). The manufacturer qualified this antibody for immunoblotting, and it has been previously used in CFTR trafficking studies (5) and specifically to detect NHERF-1 in HEK293 (31). Both rabbit polyclonal and mouse monoclonal antibodies to c-Myc were used to detect epitope-tagged SLC26A9, and we have previously demonstrated the suitability of the monoclonal 9E10 clone for immunoblotting and immunoprecipitating myc-SLC26A9 expressed in HEK293 (6). The rabbit polyclonal (no. C3956) was obtained from Sigma, and has been previously used for detecting epitope-tagged anion transporters expressed in HEK293 (34, 62). The mouse monoclonal 9E10 clone (no. MMS-150P) was obtained from Biolegend (San Diego, CA). A monoclonal antibody to β-actin (no. A1978, Sigma) was used to confirm equal protein loading (6). Secondary antibodies (goat anti-mouse and rabbit) were from Jackson Immunoresearch Laboratory (West Grove, PA). Lysis buffers included a protease inhibitor complex (PIC, Roche Diagnostics, Indianapolis, IN).
Signals were quantified from films by densitometric analysis of bands using ImageJ software (https://imagej.nih.gov/ij/). For cell-surface biotinylation experiments, background corrected values for biotinylated CFTR were normalized against background corrected values of β-actin. For PDZ-domain proteins, the background corrected value of the coimmunoprecipitated PDZ-domain protein was normalized against the background corrected value of the immunoprecipitated, dimeric plus monomeric forms of SLC26A9, immunoprecipitated under the same condition. Data are reported as the averages of normalized values from three independent experiments ± SE. Student’s t-test was used to determine statistical significance, with P < 0.05 considered significantly different.
Immunoblotting (IB) and coimmunoprecipitation (co-IP).
Forty eight hours posttransfection, cells were harvested and lysed in buffer (50 mM Tris HCl, pH 7.5, 0.15 M NaCl, 0.3% deoxycholate, 1% Igepal and 100 mM EDTA). Protein concentration was measured by the BCA method and 600 µg of protein was used for the IP of myc-SLC26A9 from the lysate. IP was done using mouse anti-c-Myc magnetic beads (no. 0088842, Pierce, Rockford, IL) applied for 2–3 h with gentle rotation at room temperature. Pellets were washed 4–5 times with the lysis buffer. Samples were then eluted by boiling with 2X Laemmli sample buffer for 5 min and subjected to SDS-PAGE on Bio-Rad gradient gels (4–15%) and immunoblotted with the indicated antibody. Five percent of the protein used for IP was subjected to SDS-PAGE and immunoblotted in parallel to show input levels of transfected and endogenous protein. To test for CFTR interactions, the protocol was modified to eliminate boiling, which can cause aggregation of CFTR. Harvested cells were lysed in RIPA buffer, and rabbit polyclonal c-Myc antibody (no. NBP1–33787, agarose immobilized, Novus Biologicals, Littleton, CO) was used for IP. Immunoprecipitated pellets were washed 4–5 times with RIPA buffer and eluted with Laemmli sample buffer at 42°C for 10 m and resolved by SDS PAGE on 6% Bio-Rad gels. Five percent of total protein was loaded in parallel as input. IB was performed with indicated antibodies. SLC26A family members have been shown to form dimers (18) that are substantially resistant to dissociation under SDS-PAGE conditions. As a result, the protein runs at two molecular weights corresponding to the values expected for a monomer and a dimer, and we therefore show both dimers and monomers for all Western blots.
Cell-surface biotinylation assay.
HEK293 cells were transfected with the desired construct(s) in replicates and grown to 70–80% confluency. Biotinylation reaction was performed 48 h posttransfection. Cells were chilled at 4°C and washed repeatedly with chilled PBS-plus (1X DPBS plus calcium and magnesium, Corning Cellgro), followed by a 60-min incubation with freshly prepared 0.5 mg/ml solution of EZ-Link Sulfo-NHS-SS-Biotin (Pierce) in PBS-plus. Reaction was then quenched by washing cells repeatedly with chilled PBS-plus containing 20 mM glycine. The entire procedure was performed in a cold room. Cells were then rinsed with PBS-plus containing PIC (Roche Diagnostics), harvested, and cell pellets lysed in Biotinylation Lysis Buffer (BLB, 10 mM Tris·HCl, pH 7.4, 1% NP40, 10 mM EGTA, 0.4% deoxycholate and PIC). Protein concentration of lysates was estimated, and equal amount of protein (500 µg) from each sample was used for affinity precipitation using 100 µl of streptavidin agarose resin (Pierce) overnight at 4°C in a rotary shaker. Beads were washed and eluted with Laemmli sample buffer. Streptavidin-bound biotinylated proteins were resolved by SDS PAGE and immunoblotted using relevant antibodies.
Glycosidase assay.
Lysates harvested as described above were subject to PNGaseF treatment in accordance with the manufacturer’s protocol (New England Biolabs). Briefly, 100 µg total protein was incubated with 10X denaturing buffer for 30 min at 37°C, then with PNGaseF in 10X G7 buffer for 1 h at 37°C. Treated lysates where then subjected to LDS-PAGE on 3–8% Tris Acetate NuPAGE gels (ThermoScientific Fisher), transferred overnight to PVDF membrane, and probed using anti-myc antibody as described above.
Immunofluorescence.
HEK293 cells plated on poly-l-lysine coated coverslips were transfected with wt or F508del GFP-CFTR (kind gift of Dr. Bruce A. Stanton) along with myc-SLC26A9 as described above. Cells were fixed in 4% PFA at room temperature for 15 min, then permeabilized and blocked in 0.3% saponin and 10% goat serum in PBS for 45 min at room temperature. Fixed and permeabilized cells were incubated with mouse monoclonal anti-C-myc and rabbit polyclonal anti-CAL primary antibodies, then labeled with goat anti-mouse IgG-Cy3 or goat anti-rabbit IgG-Cy5 conjugated secondary antibodies (Jackson ImmunoResearch). Confocal Z-stacked images were obtained on an inverted Olympus-FluoView FV1000 instrument through a PlanApomat 60 × , 1.40-NA objective using sequential line imaging. Maximum projections of stacked images were edited using Nikon element software, and cropping and minor adjustments of contrast and brightness of TIF image files were performed by Photoshop CS5.
RESULTS
SLC26A9 function in primary, human bronchial epithelia (HBE).
Electrogenic ion transport in monolayers of primary HBE can be measured using the short-circuit technique, and the identity of the transport mechanisms involved revealed through the use of channel inhibitors and manipulation of the solution composition. Using these methods, we have previously shown that primary HBE from non-CF donors (i.e., “wt CFTR monolayers”) exhibit significant, constitutive chloride secretion, and that the transport mechanism responsible for this activity bears the pharmacological fingerprint of SLC26A9 (6). Specifically, in both HBE as well as in complementary experiments using transiently transfected HEK 293 cells, the anion channel inhibitor GlyH-101 inhibited 67% of SLC26A9’s constitutive current, compared with 95% of the cAMP-stimulated current observed with CFTR expression alone. In addition, the current-voltage (I/V) relations during inhibition are strikingly different for CFTR vs. SLC26A9. In non-CF HBE, we observed good correlation between the magnitude of GlyH-101 inhibition in intact monolayers (67% of the constitutive current) and SLC26A9’s characteristic I/V relation in basolaterally permeabilized HBE (6). However, primary HBE from CF lung transplant recipients demonstrate a loss of constitutive chloride secretion, in addition to loss of cAMP-stimulated CFTR currents. This behavior is illustrated in Fig. 1, where the constitutive anion short-circuit current (Isc) is observed after addition of amiloride (to inhibit the epithelial sodium channel) and before addition of forskolin (to activate the CFTR channel). In the presence of an outward-directed chloride gradient, neither GlyH-101 nor the cAMP agonist forskolin impact Isc across HBE from a CF patient homozygous for the F508del mutation. By comparison, non-CF primary HBE display a significant GlyH-101 sensitive constitutive anion current as well as a large forskolin-stimulated current. In tests of HBE samples from 8 non-CF donors, the average constitutive current in the presence of a Cl− gradient was 33 ± 6 µA/cm2, and GlyH-101 inhibited 66 ± 5% of this current (n = 24), consistent with the identity of SLC26A9 (6). In contrast, HBE samples from 3 CF donors exhibited a constitutive current averaging 6 ± 1 µA/cm2, with 3 ± 1% inhibited by GlyH-101 (n = 9).
Fig. 1.
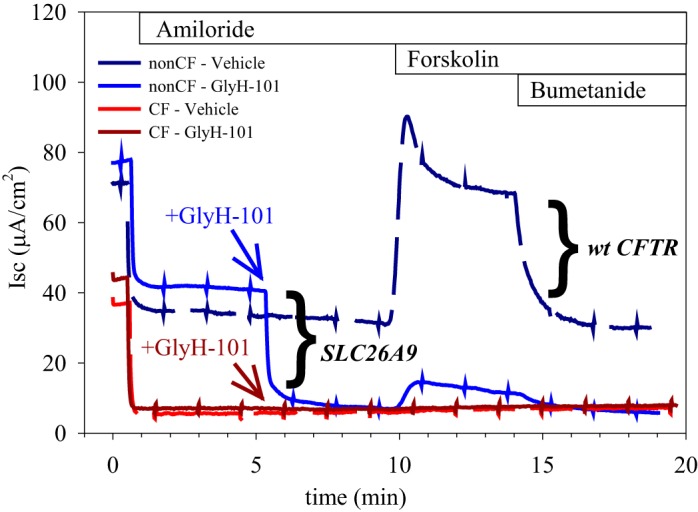
Human bronchial epithelia (HBE) from cystic fibrosis (CF) donors lack a GlyH-101-inhibitable constitutive current as well as a forskolin-stimulated current, suggesting a functional absence of both CFTR and SLC26A9 at the apical membrane. HBE from non-CF donors exhibit both constitutive and forskolin-stimulated currents. Constitutive anion secretion is measured as the short-circuit current (Isc) remaining after inhibition of ENaC with 10 µM amiloride and before addition of forskolin. GlyH-101 (50 µM) inhibits SLC26A9 activity (6), whereas 10 µM forskolin stimulates CFTR activity through activation of the cAMP/PKA cascade. Because GlyH-101 also inhibits CFTR, paired filters were treated with forskolin in the absence of GlyH-101 (Vehicle, dashed lines) to measure CFTR activity. Bumetanide (50 µM) inhibits the basolateral Na-K-2Cl cotransporter. Representative traces from a typical experiment are shown: blue traces, non-CF HBE; and red traces, CF HBE (F508del/F508del). Experiments used a chloride gradient (apical low); the small deflections at 90-s intervals reflect transepithelial resistance (TER) measurements.
F508del CFTR impacts SLC26A9 function in HEK293 cells.
Constitutive chloride secretion is observed in HEK293 transiently transfected with SLC26A9 alone or cotransfected with wt CFTR, and exhibits the same GlyH-101 fingerprint in both cases (6). To assess whether the loss of constitutive chloride secretion seen in primary HBE from CF patients was related to the failure of CFTR to properly traffic, or due to the loss of its function at the plasma membrane, we compared HEK293 coexpressing SLC26A9 with either wt, F508del, or G551D CFTR. The G551D mutation results in a CFTR channel that traffics to the plasma membrane normally, but has severe functional deficiencies (3). In whole cell patch-clamp experiments, cells coexpressing SLC26A9 with either wt or G551D CFTR exhibited similar constitutive currents even in the absence of a G551D CFTR potentiator; however, cells coexpressing SLC26A9 with F508del CFTR demonstrated a significant reduction in constitutive activity (Fig. 2A), consistent with measurements in CF HBE.
Fig. 2.
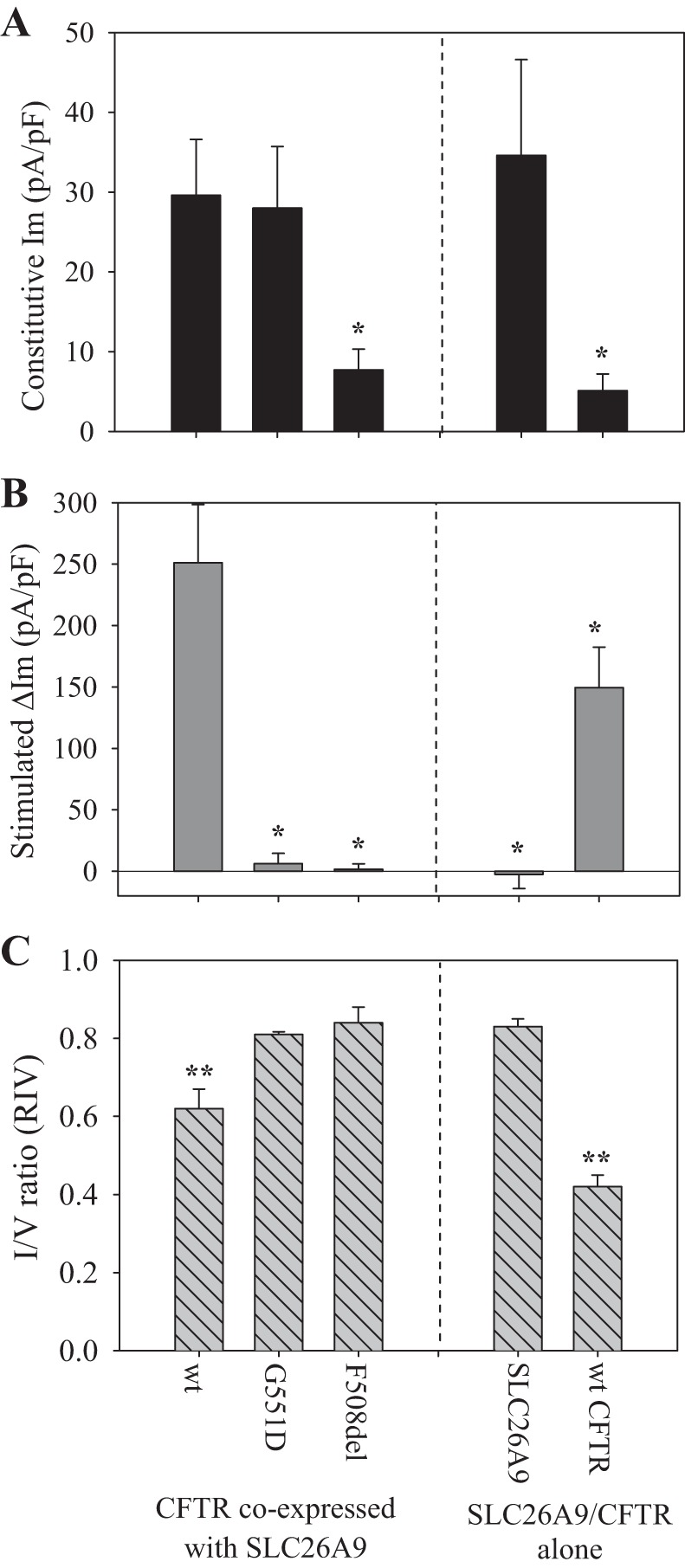
Coexpressing SLC26A9 with F508del CFTR in HEK293 cells significantly reduces constitutive current, compared with coexpression with either wt CFTR or the functionally impaired G551D CFTR. Whole cell patch-clamp measurement of membrane current (Im) or I/V ratio [RIV (6)], observed in HEK293 cells transiently transfected with myc-SLC26A9 and the indicated CFTR construct (left), or either myc-SLC26A9 or wt CFTR alone (right). A: constitutive current was measured 1 min after break-in and before any treatments. B: after measurement of constitutive Im, cells were treated with 10 µM forskolin and the change in Im (ΔIm) recorded when the response plateaued (~2 min after forskolin addition). C: after measurement of the forskolin-stimulated ΔIm, cells were treated with 50 µM GlyH-101 (in the continued presence of forskolin), and the RIV was measured to confirm the identity of the conducting channel(s). myc-SLC26A9 exhibits mild rectification (RIV ≅ 0.8) when it is the primary conducting channel, whereas wt CFTR exhibits strong rectification (RIV ≅ 0.4) when it is the primary conducting channel. Conduction through both channels results in an intermediate RIV (≅ 0.6) (6). Holding potential = −40 mV. A and B: each bar, n ≥ 5, *P < 0.05 compared with coexpression with wt CFTR. C: each bar, n = 3, **P < 0.05 compared with myc-SLC26A9 alone.
After measurement of constitutive current, cells were then stimulated with forskolin to activate CFTR via the cAMP/PKA cascade. As we have previously shown, cells coexpressing SLC26A9 and wt CFTR exhibited a significant, forskolin-stimulated current (Fig. 2B), with a magnitude reflecting synergism between the two channels (6). Cells coexpressing SLC26A9 with G551D CFTR exhibited a slight response to forskolin, consistent with published reports that the G551D mutation results in a CFTR channel gating defect exhibiting 100-fold less activity than wt CFTR (8). Of note, coexpression with SLC26A9 did not synergistically enhance G551D CFTR’s response to forskolin, as the change in current observed upon forskolin stimulation in cells coexpressing SLC26A9 with G551D CFTR was not statistically different from that observed in cells expressing G551D CFTR alone (not shown). In contrast, cells coexpressing SLC26A9 with F508del CFTR exhibited a small and physiologically insignificant response to forskolin stimulation, consistent with a failure of F508del CFTR to traffic to the plasma membrane.
Following measurement of the forskolin response, the identity of the ion channels contributing to the measured current was confirmed by assessing the degree of rectification observed in the I/V relation (RIV) during inhibition by GlyH-101 (Fig. 2C). For this protocol, 50 µM GlyH-101 was added in the continued presence of forskolin to ensure that CFTR remained active. We have previously shown (6) that GlyH-101 inhibition induces significant rectification in the I/V relation for CFTR expressed alone (RIV ≅ 0.4) compared with SLC26A9 expressed alone (RIV ≅ 0.8), and we observed the same response signature in these studies (Fig. 2C). Furthermore, when both channels contribute to the measured current, the RIV relation reflects this (RIV ≅ 0.6, Fig. 2C; Ref 6). When SLC26A9 was coexpressed with either G551D or F508del CFTR, the RIV measured during GlyH-101 inhibition in the continued presence of forskolin corresponded to that observed with SLC26A9 alone (RIV ≅ 0.8, Fig. 2C), confirming that these mutations prevented CFTR from significantly contributing to the measured current.
SLC26A9 protein expression in the presence of CFTR.
Several potential mechanisms could lead to the suppression of SLC26A9 constitutive activity in the presence of F508del CFTR. Both CFTR and SLC26A9 are known to undergo N-glycosylation (20, 25, 33); for CFTR, complex glycosylation (band C) is a hallmark of CFTR maturation, whereas the presence of only core glycosylation (band B) is associated with the F508del mutation. SLC26A9 coimmunoprecipitated both immature (band B) and mature (band C) wt CFTR in transiently transfected HEK293 cells (6). If SLC26A9 similarly interacts with F508del CFTR, it may be influenced by the latter’s failure to escape ER-associated degradation (ERAD). To confirm an interaction between SLC26A9 and CFTR, coimmunoprecipitation (co-IP) was performed on HEK293 cells coexpressing myc-SLC26A9 with either wt, G551D, or F508del CFTR. As shown in Fig. 3A, myc-SLC26A9 was able to co-IP band B CFTR in each case, suggesting that the proteins interact at an early stage in their biogenesis. In addition, SLC26A9 was also able to co-IP band C CFTR when coexpressed with either the wt or G551D forms, consistent with our previous observations for wt CFTR (6). There was no evidence for band C F508del CFTR in either the input or co-IP, confirming that this mutation inhibited forward trafficking through the Golgi irrespective of coexpression with SLC26A9.
Fig. 3.

SLC26A9 coimmunoprecipitates (co-IPs) immature (band B) CFTR from HEK293 cells coexpressing SLC26A9 with either F508del, wt, or G551D CFTR. A: myc-SLC26A9 also co-IPs mature (band C) CFTR when coexpressed with either wt or G551D CFTR, consistent with an interaction at the plasma membrane. Left panels, IP; right panels, Input. Both the monomeric (~95 kDa) and SDS-resistant dimeric (~190 kDa) forms of myc-SLC26A9 are observed in the input and are immunoprecipitated with myc, as shown. Samples were not treated with forskolin before cell lysis. B: glycosidase assay indicates that myc-SLC26A9 exhibits primarily complex glycosylation when expressed with either wt or F508del CFTR. C = control, P = PNGaseF-treated, and E = EndoH-treated. A 3–8% gradient gel was used for PAGE. C: cell-surface biotinylated myc-SLC26A9 is significantly reduced when coexpressed with F508del CFTR, as compared with wt CFTR, even though total myc-SLC26A9 is not affected. Quantification of biotinylated myc-SLC26A9 includes both monomer and dimer and is normalized by total β-actin; averages from 3 independent experiments, *P < 0.05.
Even though the co-IP results indicated that CFTR was interacting with and could thus influence SLC26A9 at an early stage of protein synthesis, immunoprecipitated myc-SLC26A9 exhibited a similar pattern whether expressed with wt, G551D, or F508del CFTR (Fig. 3A). In each case, the majority of protein runs as an SDS-resistant dimer. The migration patterns of both the mono- and dimeric forms of SLC26A9 were shown in Fig. 3 to confirm that there were no CFTR-induced differences in their molecular masses or levels of total protein expressed. We noted multiple bands at both the mono- and dimeric molecular weights for immunoprecipitated myc-SLC26A9, suggestive of core and complex glycosylation. This was confirmed by a shift in PAGE mobility following treatment with a glycosidase specific for complex glycans (Fig. 3B, lanes marked “P”), indicating that immunoprecipitated myc-SLC26A9 exhibits mature glycosylation when coexpressed with either wt or F508del CFTR, and that dimerization may precede complex glycosylation. Thus, although it is possible that F508del CFTR exerts an influence on SLC26A9 during biogenesis, the association did not result in a gross failure of SLC26A9 to be translated, exit the ER, and undergo complex glycosylation. However, the plasma membrane localization of myc-SLC26A9 was significantly reduced when coexpressed with F508del CFTR, in comparison to coexpression with wt CFTR, based on cell-surface biotinylation (Fig. 3C). Consequently, the impact of coexpression with CFTR on forward trafficking and/or recycling of mature SLC26A9 appeared to be altered when coexpressed with F508del CFTR.
SLC26A9 shares affinity for several PDZ-domain proteins with CFTR.
Because SLC26A9 exhibits complex glycosylation in the presence of F508del CFTR (Fig. 3) and thus traverses the Golgi, we considered whether PDZ-domain protein interactions were influencing its forward trafficking and function. Using a fluorescence polarization assay developed to assess CFTR:PDZ-domain binding affinities (16), a peptide comprising the last 10 amino acids of SLC26A9 was tested and showed affinity for both NHERF-1 and CAL (Table 1). Of note, the SLC26A9 peptide displayed slightly (1.8-fold) weaker affinity for CAL and substantially (6- to 40-fold) weaker affinity for NHERF-1 than was measured for CFTR. Since SLC26A9 function is suppressed by coexpression with F508del CFTR in transiently transfected HEK293 cells (Fig. 2), we tested whether SLC26A9, expressed alone or coexpressed with CFTR, interacted with endogenous NHERF-1 and CAL in HEK293 cells. As shown in Fig. 4A, myc-SLC26A9 was indeed able to co-IP endogenous NHERF-1 and CAL when expressed alone or when coexpressed with either wt or F508del CFTR. Interestingly, coexpression of either wt or F508del CFTR significantly increased the ability of myc-SLC26A9 to co-IP CAL (Fig. 4B), whereas an increase in the co-IP of NHERF-1 was only seen with coexpression of wt CFTR (Fig. 4C). Because SLC26A9 co-IPs CFTR (Fig. 3A), and CFTR itself interacts with these PDZ-domain proteins, we cannot exclude the possibility that the increases in coimmunoprecipitated CAL and NHERF-1 reflected their independent associations with CFTR. However, the increased CAL co-IP seen with F508del CFTR coexpression was unexpected, because SLC26A9 only co-IPs band B F508del CFTR (Fig. 3A), whereas CAL is thought to be primarily localized in the trans-Golgi network (12).
Table 1.
KI values for PDZ:peptide complexes
| PDZ Domain | KI for SLC26A9-10, µM | KI for CFTR-10, µM | P Value |
|---|---|---|---|
| NHERF-1 PDZ1 | 23.9 ± 6.6 | 0.6 ± 0.2 | 0.025* |
| NHERF-1 PDZ2 | 38.4 ± 4.1 | 2.4 ± 0.4 | 0.004* |
| NHERF-2 PDZ1 | 9.6 ± 3.7 | 0.53 ± 0.06 | 0.050* |
| NHERF-2 PDZ2 | 12.2 ± 1.7 | 0.17 ± 0.02 | 0.0066* |
| CAL PDZ | 670 ± 170 | 362 ± 31 | 0.037† |
Values shown are averages of three independent titrations. KI values were determined by FP displacement and fit using SOLVER to model the competitive equilibria.
t-test assuming unequal variances; equal-variance F ≤ 0.015.
t-test assuming equal variance; equal-variance F = 0.066; P value for unequal-variance t-test is 0.084.
Fig. 4.
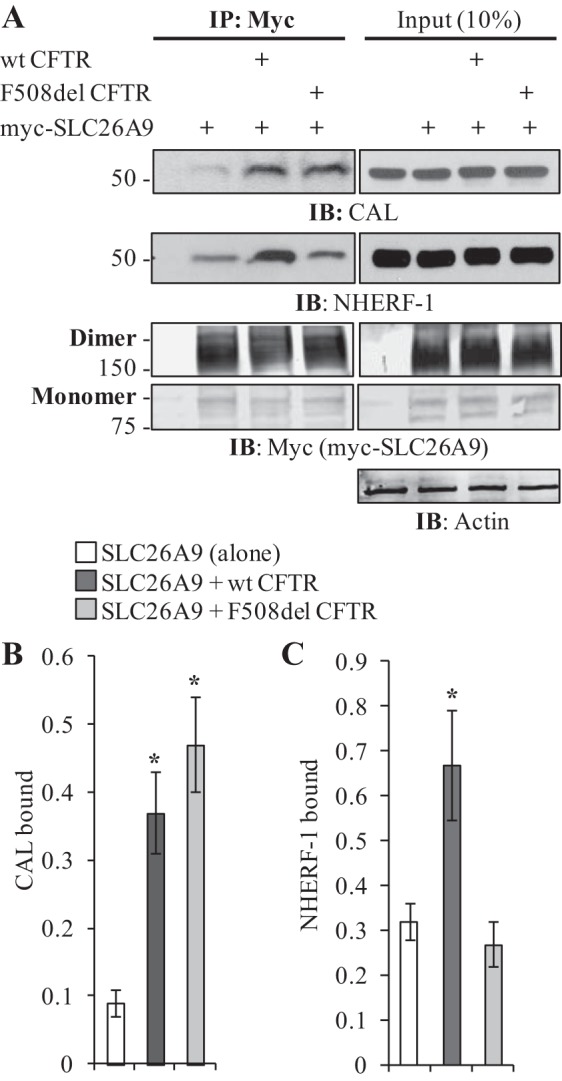
SLC26A9 coimmunoprecipitates the PDZ-domain proteins CAL and NHERF-1. A: differences in myc-SLC26A9’s ability to co-IP NHERF1 and CAL are dependent on the coexpression of CFTR. Coexpression with wt CFTR increases the co-IP of both PDZ-domain proteins, whereas coexpression with F508del CFTR only affects CAL co-IP. Left panels, IP; right panels, Input. Both monomers and dimers of myc-SLC26A9 are shown. Samples were not treated with forskolin before cell lysis. B: quantification of CAL binding indicates either form of CFTR increases myc-SLC26A9’s ability to co-IP CAL. Normalized by total immunoprecipitated myc-SLC26A9 (monomer plus dimer). Averages from 3 independent experiments, *P < 0.05. C: quantification of NHERF1 binding indicates only coexpression with wt CFTR enhances NHERF1 co-IP. Normalized by total immunoprecipitated myc-SLC26A9 (monomer plus dimer). Averages from 3 independent experiments, *P < 0.05.
To further assess the colocalization of CFTR, SLC26A9, and CAL, immunofluorescence (IF) was performed on HEK293 cells coexpressing myc-SLC26A9 with GFP-tagged versions of either wt CFTR (Fig. 5, A–D) or F508del CFTR (Fig. 5, E–H). When coexpressed with wt CFTR, SLC26A9 colocalized with CFTR both at the plasma membrane and in a perinuclear compartment; this distribution is consistent with previous studies of SLC26A9 expressed alone in HEK293 cells (33, 44). Within the perinuclear compartment, some overlap between wt CFTR, SLC26A9, and CAL was noted (Fig. 5D). In contrast, when coexpressed with F508del CFTR, SLC26A9 remained in a perinuclear compartment (Fig. 5F), where very strong colocalization with CAL was observed (Fig. 5H, arrowheads). In these experiments, F508del CFTR was absent from the plasma membrane, as expected (Fig. 5E), and showed only modest colocalization with CAL and SLC26A9 (Fig. 5H).
Fig. 5.

Immunofluorescence demonstrates CFTR-dependent differences in the intracellular localization of SLC26A9. HEK293 cells were cotransfected with myc-SLC26A9 and either wt-GFP-CFTR (A–D) or F508del-GFP-CFTR (E–H). Fixed and permeabilized cells were then immunolabeled with mouse anti-myc (to detect SLC26A9, B and F) and rabbit anti-CAL antibody (C and G). D: the overlay of wt CFTR (A), myc-SLC26A9 (B), and CAL (C) demonstrates colocalization between wt CFTR and myc-SLC26A9 in the plasma membrane region (yellow arrowheads), and in an intracellular compartment that overlaps with CAL (white arrowheads). H: the overlay of F508del CFTR (E), myc-SLC26A9 (F), and CAL (G) demonstrates that myc-SLC26A9 primarily colocalizes with CAL (white arrowheads), and that both F508del CFTR and myc-SLC26A9 are absent at the plasma membrane. Scale bar in D and H, 5 µm. Similar results were observed in 3 biological replicates.
To probe whether CAL was implicated in sequestering SLC26A9, we separately disrupted SLC26A9 and F508del CFTR PDZ interactions in coexpression assays. The PDZ motifs of CFTR and SLC26A9 conform to the Class 1 sequence X-S/T-X-L; in this motif, the S/T located 2 amino acids upstream of the COOH terminus (−2 position: S/T) play a significant role in PDZ-domain protein binding (51), and for CAL and NHERF binding, specifically (17). Mutation of the −2 position T to A in CFTR had been previously shown to disrupt its PDZ-domain protein binding (43), and we tested the effect of this mutation (mPDZ) in F508del CFTR coexpressed with SLC26A9. Similarly, we mutated the −2 position T in SLC26A9 (SLC26A9 T789A), and tested the effect of SLC26A9 T789A coexpressed with F508del CFTR. As shown and quantified in Fig. 6, A and B, mutating the PDZ motif of either myc-SLC26A9 or coexpressed F508del CFTR resulted in a substantial decrease in the co-IP of CAL, suggesting that both proteins contribute to the increase in CAL co-IP with SLC26A9. This raised the question of whether band B F508del CFTR itself directly interacted with CAL, or whether an additional, unidentified PDZ-domain protein might be involved. To address this, we tested whether F508del CFTR itself could co-IP endogenous CAL. As shown in Fig. 6C, core-glycosylated F508del CFTR was able to co-IP CAL in the absence of SLC26A9, suggesting that these PDZ-domain protein interactions commenced well before the TGN, and quite possibly in the ER.
Fig. 6.

The increase in CAL co-IP shown in Fig. 4B requires that both SLC26A9 and F508del CFTR have intact PDZ motifs. A: mutation of either myc-SLC26A9’s or F508del CFTR’s PDZ motif at the −2 position threonine abrogates the CFTR-induced increase in CAL co-IP. This mutation does not eliminate the PDZ motif from either protein, although the −2 position Thr is central to PDZ domain binding affinity (17). Left panels, IP; right panels, Input. Both monomers and dimers of myc-SLC26A9 are shown. Samples were not treated with forskolin before cell lysis. B: quantification of CAL binding indicates both proteins require intact PDZ motifs to increase myc-SLC26A9’s ability to co-IP CAL. Normalized by total immunoprecipitated myc-SLC26A9 (monomer plus dimer). Averages from 3 independent experiments, *P < 0.05. C: immature (band B) F508del CFTR co-IPs CAL when expressed alone. Left panels, IP; right panels, Input.
Disrupting PDZ-domain protein interactions modifies function and localization.
To assess whether the decrease in CAL co-IP observed with SLC26A9 T789A restored its constitutive activity, whole cell patch-clamping was performed on HEK293 cells coexpressing SLC26A9 ± T789A with either wt or F508del CFTR. As shown in Fig. 7A, a significant increase in constitutive current was observed in HEK293 expressing either form of CFTR with SLC26A9 T789A compared with SLC26A9. In particular, the constitutive current observed in cells coexpressing SLC26A9 T789A with F508del CFTR (18 ± 7 pA/pF) was indistinguishable from that in cells coexpressing SLC26A9 with wt CFTR (16 ± 2 pA/pF), indicating that disruption of PDZ-domain protein interactions essentially restored constitutive activity. Whether this also suggests that NHERF-1 is unimportant for SLC26A9 plasma membrane localization is unclear; SLC26A9 exhibits less affinity for NHERF-1 than does CFTR (Table 1), and wt CFTR is functional at the plasma membrane in the absence of its PDZ motif (40). The PDZ motif mutation did not prevent SLC26A9 T789A from interacting with wt CFTR at the plasma membrane, as the forskolin-stimulated current was statistically similar to that seen with cells coexpressing SLC26A9 with wt CFTR (Fig. 7B), and we have previously shown that this magnitude of stimulated current reflects synergy (6). However, we noted a substantial variance in the stimulated current with coexpression of SLC26A9-T789A with wt CFTR, which may reflect the loss of NHERF-1 stabilization of the complex. Thus these results suggested that PDZ-domain proteins played two roles in SLC26A9-CFTR interactions: they exerted a negative influence on forward trafficking (CAL), and a stabilizing influence on the plasma membrane association of SLC26A9 with wt CFTR (NHERF-1).
Fig. 7.
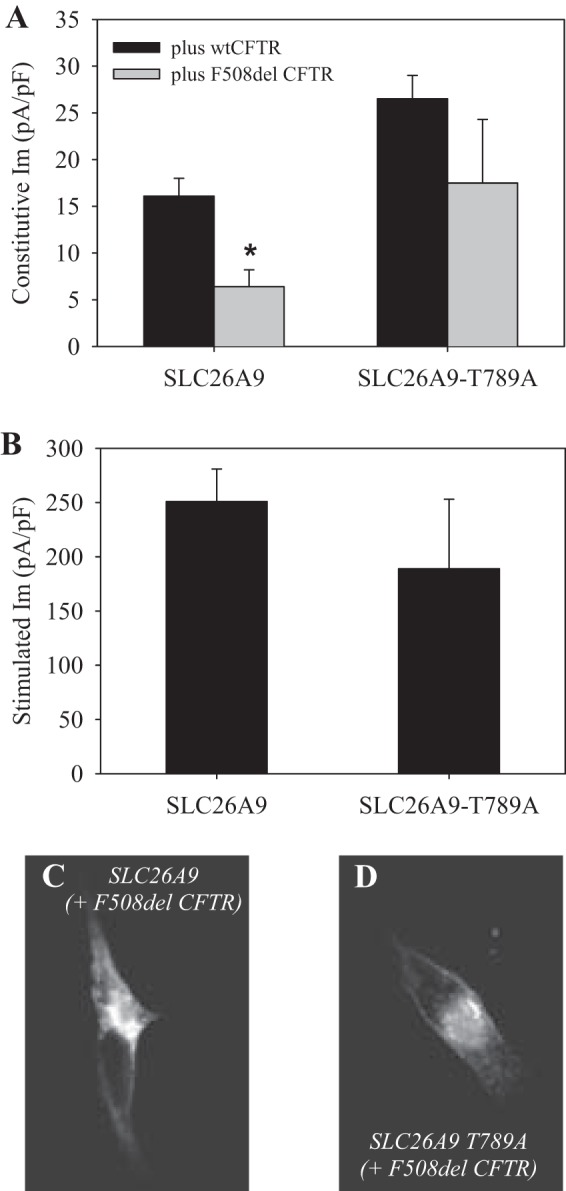
Mutating the PDZ motif of SLC26A9 restores constitutive current when coexpressed with F508del CFTR. Whole cell patch-clamp measurement of membrane current (Im) observed in HEK293 cells transiently transfected with myc-SLC26A9 ± T789A (PDZ motif mutation) and CFTR. A: constitutive current was measured 1 min after break-in and before any treatments. The indicated myc-SLC26A9 construct was coexpressed with either wt (black bars) or F508del (gray bars) CFTR. B: after measurement of constitutive Im, cells were treated with 10 µM forskolin and the change in Im recorded when the response plateaued (~2 min after forskolin addition). F508del coexpressors did not respond to forskolin (not shown). Holding potential = −40 mV; each bar, n ≥ 5, *P < 0.05 compared with coexpression with wt CFTR. C–D: IF indicates that mutating the PDZ motif of myc-SLC26A9 increases its plasma membrane expression. HEK293cells expressing myc-SLC26A9 ± T789A (PDZ motif mutation) with F508del CFTR were fixed, permeabilized, and labeled with anti-myc antibody to assess distribution of myc-SLC26A9. C: coexpression of myc-SLC26A9 + F508del CFTR. D: coexpression of myc-SLC26A9 T789A + F508del CFTR.
In addition, we examined the cellular location of SLC26A9 ± T789A coexpressed with F508del CFTR using IF. As shown in Fig. 7C, when SLC26A9 was coexpressed with F508del CFTR, the majority of the protein appeared intracellular (similar to Fig. 5F). Coexpression of the PDZ motif mutant SLC26A9 T789A with F508del CFTR resulted in a noticeable shift from intracellular to plasma membrane labeling (Fig. 7D). Thus, disrupting PDZ-domain protein interactions appeared to alleviate the intracellular retention of SLC26A9 in the presence of F508del CFTR.
Correction of the F508del CFTR trafficking mutation.
These results have indicated that in the presence of CFTR, SLC26A9 has a strong apparent affinity for the PDZ-domain protein CAL, which is known to exert a negative influence on the forward trafficking of CFTR (11, 12, 16). Furthermore, unlike coexpression with wt CFTR, coexpression of SLC26A9 with F508del CFTR did not enhance the co-IP of NHERF-1 (Fig. 4). CF HBE can be treated with correctors that allow some forward trafficking of F508del CFTR, albeit at reduced levels compared with wt CFTR (57). If loss of SLC26A9-mediated constitutive chloride secretion in CF HBE is due to the failure of F508del CFTR to exit the ER and traffic through the Golgi, then corrector treatment should restore some constitutive activity. The results of Strug et al. (54) also demonstrate that VX-809 treatment restores the impact of an SLC26A9 SNP in F508del CFTR homozygous patients. To assess this, we treated primary HBE from three separate CF patients with the corrector VX-809 for 24 h, then tested for restoration of a GlyH-101 inhibited constitutive current as well as forskolin-stimulated currents. On average, treatment with VX-809 increased both the constitutive current, as evidenced by an increase in GlyH-101 inhibition from 3 ± 1% to 13 ± 4% (n = 9, P < 0.05), and in paired experiments, the forskolin-stimulated current, from 0.8 ± 0.3 µA/cm2 to 2.2 ± 1.1 µA/cm2 (n = 9, P = 0.12). Although the increase in GlyH-101-sensitive constitutive current was statistically significant, the magnitude was still considerably smaller than observed with non-CF HBE (66%, above, Fig. 1). We traditionally rely on measurement of the I/V relation in basolaterally permeabilized HBE to confirm SLC26A9’s role when the GlyH-101-sensitive constitutive current is substantially different from our published observations (6); however, the VX-809 corrected currents were too small in this case to accurately perform this additional measurement.
DISCUSSION
In the present study, we demonstrate that F508del CFTR inhibits SLC26A9 constitutive activity in primary, human bronchial epithelia, and furthermore that this inhibition of constitutive activity is recapitulated when SLC26A9 is coexpressed with F508del CFTR in HEK293 cells (Figs. 1 and 2). Our results suggest that in the presence of F508del CFTR, SLC26A9 is absent at the plasma membrane (Figs. 2A, 3C, 5F, and 7C). PDZ-domain proteins have been shown to play a role in the trafficking and plasma membrane localization of CFTR and other SLC26A transporters (21, 26, 30, 32, 39), which led us to investigate their role. Our results identified a significant impact of PDZ-domain protein interactions, especially with CAL, on sequestration of SLC26A9. Differences in PDZ-domain protein co-IP suggest that SLC26A9’s interactions with PDZ-domain proteins are modulated by CFTR coexpression, and sensitive to the F508del mutation. In support of a PDZ-domain protein-mediated effect, we demonstrate that mutating the PDZ motifs of either SLC26A9 or F508del CFTR alleviates the CFTR-induced increase in CAL co-IP (Fig. 6), and restores SLC26A9 plasma membrane expression and constitutive activity when coexpressed with F508del CFTR (Fig. 7). These findings are summarized in Fig. 8, where we identify several key steps where CFTR appears to influence SLC26A9’s forward trafficking.
Fig. 8.
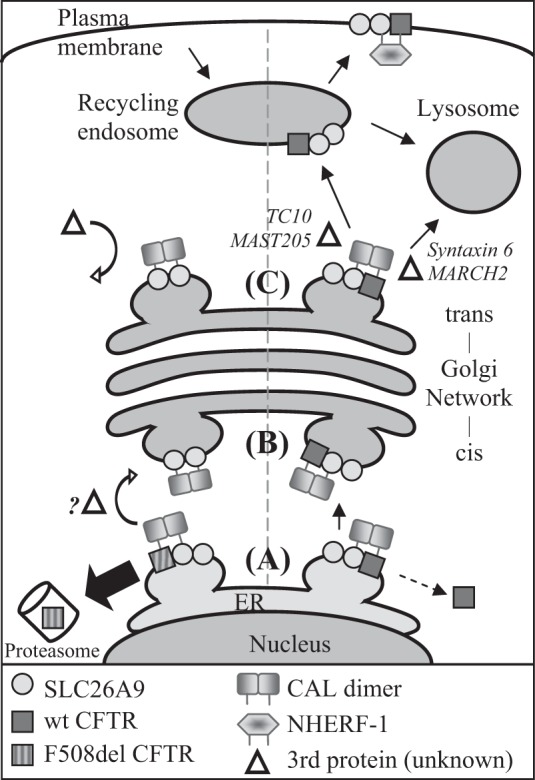
A model demonstrating potential PDZ domain protein interactions for coexpressed SLC26A9 and CFTR. Left side, F508del CFTR coexpression; right side, wt CFTR coexpression. A: the proteins commence their interaction in the ER, where the CAL dimer binds CFTR and SLC26A9. Misfolded F508del CFTR is removed from the complex and undergoes ERAD, leaving half of the CAL dimer unbound. If SLC26A9 is dimerized at this stage, removal of F508del CFTR may result in a CAL dimer-SLC26A9 dimer complex. A portion of wt CFTR may also be degraded at this stage (see text). Step B: the CAL complex traffics to and through the Golgi complex, either as a CAL-SLC26A9 dimer (F508del CFTR coexpression), or a CAL-SLC26A9-wt CFTR complex. Step C: at the TGN, the CAL-SLC26A9-wt CFTR complex may be disrupted by additional proteins which interact with CFTR, such as TC10 (14) or MAST205 (47). Since an individual SLC26A9 PDZ motif has relatively weak affinity for CAL (Table 1), the complex is released from the TGN and continues trafficking to the recycling endosome and ultimately, plasma membrane. Conversely, the lack of F508del CFTR in the CAL-SLC26A9 dimer complex impedes the recruitment of additional proteins, which, combined with a greater affinity between CAL and SLC26A9 due to avidity, halts the forward trafficking of SLC26A9.
Improperly folded F508del CFTR undergoes ERAD (38); thus the ability of SLC26A9 to co-IP band B F508del CFTR (Fig. 3A) suggests that the proteins commence their interaction in the ER. This is further supported by our evidence that F508del CFTR, expressed alone, can co-IP CAL (Fig. 6C). Since the F508del CFTR-induced increase in SLC26A9’s ability to co-IP CAL can be abrogated by mutating the PDZ motif of either SLC26A9 or F508del CFTR (Fig. 6), our data suggest that the proteins are colocalized, initially, when binding to CAL commences. Thus our data indicate that CAL is present and interacting with both proteins, at least transiently, in the ER, as shown in Fig. 8, step A. However, F508del CFTR remains core-glycosylated while the majority of co-expressed SLC26A9 exhibits complex glycosylation (Fig. 3), suggesting the proteins are not colocalized in the TGN where CAL is thought to be primarily localized (26, 32). This spatial discrepancy is also evident in the IF co-localization data (Fig. 5H). Thus our data suggest that F508del CFTR and SLC26A9 must separate before or during trafficking between the ER and Golgi complex, shown in Fig. 8, step B.
We have not identified the locus or regulatory mechanism where F508del CFTR and SLC26A9 part company, but additional regulatory mechanisms may come into play. CFTR is known to recruit and/or associate with multiple proteins in the CAL scaffolding complex that regulate its fate (10, 11, 14, 32, 47). Thus the presence of wt or F508del CFTR may differentially recruit additional proteins to the PDZ scaffolding interactome that also influence SLC26A9 (27). These additional proteins, noted in Fig. 8, have been shown to influence CFTR trafficking from the TGN, but a potential role in ER to Golgi traffic is unclear at this time. Our glycosylation data indicate that SLC26A9 traffics to the TGN whether expressed with wt or F508del CFTR (Fig. 3); thus an additional, CFTR-influenced regulatory interaction likely occurs at this stage, as shown in Fig. 8, step C.
SLC26A9 demonstrates affinity for the same PDZ domain proteins as CFTR (Table 1), and co-IPs endogenous CAL and NHERF-1 in HEK293 cells (Fig. 4A). The significant increase in the co-IP of both PDZ-domain proteins when SLC26A9 was coexpressed with wt CFTR was most intriguing (Fig. 4). Factors that could account for this increase include a CFTR-induced change in SLC26A9’s affinity for the PDZ-domain proteins, and/or the possibility that coimmunoprecipitated CFTR itself binds additional CAL and NHERF-1. However, our data are consistent with the hypothesis that binding to a common PDZ-domain protein facilitates the interaction between the STAS domain and unphosphorylated R domain (28), as discussed below.
We expect an increase in apparent binding affinity if CFTR and SLC26A9's PDZ motifs are located in close proximity, due to avidity, as both NHERF-1 and dimerized CAL (59) have two binding domains. This appears quite likely considering the relatively weak affinity CFTR and SLC26A9 individually display for CAL (Table 1). Additionally, we note that SLC26A9 forms SDS-resistant dimers (Fig. 3), which would contribute two PDZ motifs in addition to CFTR’s PDZ motif, thus potentially causing an avidity effect on their own. When and where SLC26A9 dimerizes, and how this influences the fates of F508del CFTR and SLC26A9, is currently unknown. Based on our glycosidase assay (Fig. 3), which indicates that dimers exhibiting core glycosylation are present, we suggest a model where displacement of F508del CFTR from the ER-CAL interactome (Fig. 8, step A) leads to a dimerized CAL interaction with both PDZ motifs in the SLC26A9 dimer. If this occurs, the relatively weak affinity SLC26A9 displays for CAL (Table 1) could be significantly increased due to avidity. For wt CFTR, we anticipate that dimerized CAL would continue to interact with both wt CFTR and SLC26A9 (Fig. 8, step B) during trafficking from the ER to the TGN. In this model, the continued presence of wt CFTR would attract proteins shown to disrupt the CFTR-CAL interactome to release the SLC26A9-CFTR complex, whereas the dimerized CAL-SLC26A9 interactome may lack the ability to recruit these proteins (Fig. 8, step C). This remains to be shown.
A complex interaction model has been proposed for CFTR-SLC26A family members. First, a direct interaction between the STAS domain of the SLC26A protein and the R domain of CFTR leads to their reciprocal, synergistic regulation (28); the R domain of CFTR must be phosphorylated to mediate this reciprocal activity. Second, binding of their PDZ motifs to the same PDZ-domain protein facilitates the interaction between CFTR’s R domain and the SLC26A STAS domain (21, 28, 30). Ko and colleagues (28) further showed that STAS domain, R domain binding was enhanced by PKA phosphorylation of CFTR’s R domain. The dual effect of PKA phosphorylation of CFTR on binding and activity lends some difficulty to interpreting the relevance of PDZ-domain proteins in establishing the STAS domain, R domain interaction. A better model for assessing their relevance might be found in studies of SLC26A4 (aka Pendrin), which naturally lacks a PDZ motif. SLC26A4 is found in multiple tissues that coexpress CFTR, including HBE (1), the airway serous cell line Calu-3 (22, 23), and the parotid duct (50). Despite significant evidence indicating that SLC26A4 participates in anion exchange in these cells, there is no evidence that it interacts with CFTR physically or functionally through a STAS domain, R domain interaction (1, 22, 23, 50).
When wt CFTR and SLC26A9 were coexpressed, we showed that the generation of synergistically enhanced forskolin-stimulated currents at the plasma membrane does not require that SLC26A9 have an intact PDZ motif (Fig. 7B). This result may indicate that NHERF-1 does not play a significant role in forward trafficking of SLC26A9 to the plasma membrane, although it is also possible that forskolin stimulation of CFTR compensated for loss of SLC26A9’s NHERF-1 association by enhancing the STAS domain, R domain interaction (28). Of note, we did not treat cells destined for co-IP experiments with the PKA activator forskolin, whereas cells were forskolin treated for the current measurements. Consequently, our results are consistent with the concept that PDZ-domain protein interactions are required for physical SLC26A9 STAS domain-CFTR R domain interaction when the R domain is not phosphorylated.
It is curious that the magnitude of CAL co-IP is similar whether SLC26A9 is coexpressed with F508del or wt CFTR, given that the increase in NHERF-1 co-IP seen with wt CFTR coexpression suggests that SLC26A9 (and wt CFTR) traffic out of the TGN and to the plasma membrane (Fig. 4A). There are two potential mechanisms that may account for this. First, Ward and Kopito (58) have shown that up to 75% of wt CFTR fails to mature past the ER when transiently transfected into HEK293 cells, instead undergoing premature degradation along the same trajectory as F508del CFTR. Thus a significant portion of SLC26A9 may still experience the same trafficking impediment if wt CFTR is removed from the ER-CAL complex, as we suggest occurs with F508del CFTR (Fig. 8, step A). Second, wt CFTR eventually undergoes lysosomal degradation, and this has been shown to occur via an interaction with CAL in the recycling endosome (10, 11). These additional factors may account for the similarity in CAL co-IP.
As noted, CAL, together with its interacting proteins (10, 11, 47), plays a significant role in targeting mature CFTR for lysosomal degradation (12, 13, 30). However, the association between SLC26A9 and CAL does not appear to accelerate the degradation of SLC26A9, as levels of mature SLC26A9 are similar whether coexpressed with wt or F508del CFTR (Fig. 3). CAL has been implicated in other cellular functions, such as regulation of epithelial tight junction proteins (37), inhibition of ubiquitination-dependent degradation of the mGluR5a receptor (15), and stabilization of intracellular β1-adrenergic receptor (29); thus CAL’s intracellular retention of SLC26A9 is not atypical, per se. Additional evidence for a CFTR-induced effect on CAL’s ability to bind other CF-relevant proteins is modest at this time: Gentzsch et al. (24) reported that a small portion of the Golgi-localized chloride channel ClC-3B coassociated with CFTR, and both were negatively regulated by overexpression of CAL, whereas Pelaseyed and Hansson (45) found that a CAL-induced downregulation of MUC3 could be rescued by overexpression of wt CFTR. Our study demonstrates a significant, CFTR-induced impact of CAL on SLC26A9 function that can be overcome if CFTR engages in forward trafficking through the trans-Golgi, as occurs through coexpression with wt CFTR, or by pharmacophore correction of F508del CFTR’s trafficking defect. Thus intracellular PDZ-domain protein interactions may confer a SLC26A9 regulatory mode in which wt and mutant CFTRs participate.
It is known that, at the plasma membrane, CFTR is part of a protein interactome that can include signaling molecules, receptors, and other ion channels; furthermore, PDZ-domain proteins appear to play a central role in assembling these complexes (26, 32). In the current study, we have shown that CFTR can exert a regulatory influence by modifying SLC26A9’s interactions with PDZ-domain proteins in different cellular compartments, and specifically along the protein maturation pathway. This possibility opens new avenues of approach to determining how CFTR exerts its regulatory influence. Finally, we note that SLC26A9 has the potential to restore significant chloride secretion to airway epithelia, and that it appears to be much further along the maturation pathway than is F508del CFTR, making it an alternative chloride channel to investigate in CF therapies.
GRANTS
This work was supported by Cystic Fibrosis Foundation Grant BERTRA12G0 to C. A. Bertrand, in part by a Neukom Institute CompX award and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and National Institute of General Medical Sciences Grants R01-DK-101451, P30-GM-106394, and P20-GM-113132 to D. R. Madden, and NIDDK Grants R01-DK-068196 and P30-DK-072506 and Cystic Fibrosis Foundation Therapeutics Grant FRIZZE05X0 to R. A. Frizzell.
DISCLOSURES
D. R. Madden is the coinventor of a CAL inhibitor peptide for which Intellectual Property rights are held by Dartmouth College. The peptide was not used in these studies, but CAL is a focus of the paper.
AUTHOR CONTRIBUTIONS
C.A.B., J.M.P., D.R.M., and R.A.F. conceived and designed research; C.A.B., S.M., S.K.M., X.W., and Y.Z. performed experiments; C.A.B., S.M., S.K.M., X.W., Y.Z., and D.R.M. analyzed data; C.A.B., S.M., S.K.M., X.W., Y.Z., D.R.M., and R.A.F. interpreted results of experiments; C.A.B., S.M., S.K.M., X.W., Y.Z., and D.R.M. prepared figures; C.A.B. drafted manuscript; C.A.B., D.R.M., and R.A.F. edited and revised manuscript; C.A.B., S.M., S.K.M., X.W., Y.Z., J.M.P., D.R.M., and R.A.F. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Patrick R. Cushing for performing initial PDZ:SLC26A9 binding studies, Dr. Jeyaganesh Rajamanickam for initial SLC26A9 studies, and Stefanie Brown for technical assistance with HBE cultures. The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Adams KM, Abraham V, Spielman D, Kolls JK, Rubenstein RC, Conner GE, Cohen NA, Kreindler JL. IL-17A induces Pendrin expression and chloride-bicarbonate exchange in human bronchial epithelial cells. PLoS One 9: e103263, 2014. doi: 10.1371/journal.pone.0103263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amlal H, Xu J, Barone S, Zahedi K, Soleimani M. The chloride channel/transporter Slc26a9 regulates the systemic arterial pressure and renal chloride excretion. J Mol Med (Berl) 91: 561–572, 2013. doi: 10.1007/s00109-012-0973-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anderson MP, Welsh MJ. Regulation by ATP and ADP of CFTR chloride channels that contain mutant nucleotide-binding domains. Science 257: 1701–1704, 1992. doi: 10.1126/science.1382316. [DOI] [PubMed] [Google Scholar]
- 4.Arora K, Moon C, Zhang W, Yarlagadda S, Penmatsa H, Ren A, Sinha C, Naren AP. Stabilizing rescued surface-localized δf508 CFTR by potentiation of its interaction with Na(+)/H(+) exchanger regulatory factor 1. Biochemistry 53: 4169–4179, 2014. doi: 10.1021/bi401263h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Auerbach M, Liedtke CM. Role of the scaffold protein RACK1 in apical expression of CFTR. Am J Physiol Cell Physiol 293: C294–C304, 2007. doi: 10.1152/ajpcell.00413.2006. [DOI] [PubMed] [Google Scholar]
- 6.Bertrand CA, Zhang R, Pilewski JM, Frizzell RA. SLC26A9 is a constitutively active, CFTR-regulated anion conductance in human bronchial epithelia. J Gen Physiol 133: 421–438, 2009. doi: 10.1085/jgp.200810097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blackman SM, Commander CW, Watson C, Arcara KM, Strug LJ, Stonebraker JR, Wright FA, Rommens JM, Sun L, Pace RG, Norris SA, Durie PR, Drumm ML, Knowles MR, Cutting GR. Genetic modifiers of cystic fibrosis-related diabetes. Diabetes 62: 3627–3635, 2013. doi: 10.2337/db13-0510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bompadre SG, Sohma Y, Li M, Hwang TC. G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects. J Gen Physiol 129: 285–298, 2007. doi: 10.1085/jgp.200609667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang MH, Plata C, Zandi-Nejad K, Sindić A, Sussman CR, Mercado A, Broumand V, Raghuram V, Mount DB, Romero MF. Slc26a9–anion exchanger, channel and Na+ transporter. J Membr Biol 228: 125–140, 2009. doi: 10.1007/s00232-009-9165-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng J, Cebotaru V, Cebotaru L, Guggino WB. Syntaxin 6 and CAL mediate the degradation of the cystic fibrosis transmembrane conductance regulator. Mol Biol Cell 21: 1178–1187, 2010. doi: 10.1091/mbc.E09-03-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng J, Guggino W. Ubiquitination and degradation of CFTR by the E3 ubiquitin ligase MARCH2 through its association with adaptor proteins CAL and STX6. PLoS One 8: e68001, 2013. doi: 10.1371/journal.pone.0068001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng J, Moyer BD, Milewski M, Loffing J, Ikeda M, Mickle JE, Cutting GR, Li M, Stanton BA, Guggino WB. A Golgi-associated PDZ domain protein modulates cystic fibrosis transmembrane regulator plasma membrane expression. J Biol Chem 277: 3520–3529, 2002. doi: 10.1074/jbc.M110177200. [DOI] [PubMed] [Google Scholar]
- 13.Cheng J, Wang H, Guggino WB. Modulation of mature cystic fibrosis transmembrane regulator protein by the PDZ domain protein CAL. J Biol Chem 279: 1892–1898, 2004. doi: 10.1074/jbc.M308640200. [DOI] [PubMed] [Google Scholar]
- 14.Cheng J, Wang H, Guggino WB. Regulation of cystic fibrosis transmembrane regulator trafficking and protein expression by a Rho family small GTPase TC10. J Biol Chem 280: 3731–3739, 2005. doi: 10.1074/jbc.M410026200. [DOI] [PubMed] [Google Scholar]
- 15.Cheng S, Zhang J, Zhu P, Ma Y, Xiong Y, Sun L, Xu J, Zhang H, He J. The PDZ domain protein CAL interacts with mGluR5a and modulates receptor expression. J Neurochem 112: 588–598, 2010. doi: 10.1111/j.1471-4159.2009.06454.x. [DOI] [PubMed] [Google Scholar]
- 16.Cushing PR, Fellows A, Villone D, Boisguérin P, Madden DR. The relative binding affinities of PDZ partners for CFTR: a biochemical basis for efficient endocytic recycling. Biochemistry 47: 10084–10098, 2008. doi: 10.1021/bi8003928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cushing PR, Vouilleme L, Pellegrini M, Boisguerin P, Madden DR. A stabilizing influence: CAL PDZ inhibition extends the half-life of ΔF508-CFTR. Angew Chem Int Ed Engl 49: 9907–9911, 2010. doi: 10.1002/anie.201005585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Detro-Dassen S, Schänzler M, Lauks H, Martin I, zu Berstenhorst SM, Nothmann D, Torres-Salazar D, Hidalgo P, Schmalzing G, Fahlke C. Conserved dimeric subunit stoichiometry of SLC26 multifunctional anion exchangers. J Biol Chem 283: 4177–4188, 2008. doi: 10.1074/jbc.M704924200. [DOI] [PubMed] [Google Scholar]
- 19.Devor DC, Pilewski JM. UTP inhibits Na+ absorption in wild-type and DeltaF508 CFTR-expressing human bronchial epithelia. Am J Physiol Cell Physiol 276: C827–C837, 1999. [DOI] [PubMed] [Google Scholar]
- 20.Dorwart MR, Shcheynikov N, Wang Y, Stippec S, Muallem S. SLC26A9 is a Cl(-) channel regulated by the WNK kinases. J Physiol 584: 333–345, 2007. doi: 10.1113/jphysiol.2007.135855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dorwart MR, Shcheynikov N, Yang D, Muallem S. The solute carrier 26 family of proteins in epithelial ion transport. Physiology (Bethesda) 23: 104–114, 2008. doi: 10.1152/physiol.00037.2007. [DOI] [PubMed] [Google Scholar]
- 22.Garnett JP, Hickman E, Burrows R, Hegyi P, Tiszlavicz L, Cuthbert AW, Fong P, Gray MA. Novel role for pendrin in orchestrating bicarbonate secretion in cystic fibrosis transmembrane conductance regulator (CFTR)-expressing airway serous cells. J Biol Chem 286: 41069–41082, 2011. doi: 10.1074/jbc.M111.266734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garnett JP, Hickman E, Tunkamnerdthai O, Cuthbert AW, Gray MA. Protein phosphatase 1 coordinates CFTR-dependent airway epithelial HCO3- secretion by reciprocal regulation of apical and basolateral membrane Cl(-)-HCO3- exchangers. Br J Pharmacol 168: 1946–1960, 2013. doi: 10.1111/bph.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gentzsch M, Cui L, Mengos A, Chang XB, Chen JH, Riordan JR. The PDZ-binding chloride channel ClC-3B localizes to the Golgi and associates with cystic fibrosis transmembrane conductance regulator-interacting PDZ proteins. J Biol Chem 278: 6440–6449, 2003. doi: 10.1074/jbc.M211050200. [DOI] [PubMed] [Google Scholar]
- 25.Gregory RJ, Cheng SH, Rich DP, Marshall J, Paul S, Hehir K, Ostedgaard L, Klinger KW, Welsh MJ, Smith AE. Expression and characterization of the cystic fibrosis transmembrane conductance regulator. Nature 347: 382–386, 1990. doi: 10.1038/347382a0. [DOI] [PubMed] [Google Scholar]
- 26.Guggino WB, Stanton BA. New insights into cystic fibrosis: molecular switches that regulate CFTR. Nat Rev Mol Cell Biol 7: 426–436, 2006. doi: 10.1038/nrm1949. [DOI] [PubMed] [Google Scholar]
- 27.Holcomb J, Jiang Y, Lu G, Trescott L, Brunzelle J, Sirinupong N, Li C, Naren AP, Yang Z. Structural insights into PDZ-mediated interaction of NHERF2 and LPA(2), a cellular event implicated in CFTR channel regulation. Biochem Biophys Res Commun 446: 399–403, 2014. doi: 10.1016/j.bbrc.2014.02.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ko SB, Zeng W, Dorwart MR, Luo X, Kim KH, Millen L, Goto H, Naruse S, Soyombo A, Thomas PJ, Muallem S. Gating of CFTR by the STAS domain of SLC26 transporters. Nat Cell Biol 6: 343–350, 2004. doi: 10.1038/ncb1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koliwer J, Park M, Bauch C, von Zastrow M, Kreienkamp HJ. The golgi-associated PDZ domain protein PIST/GOPC stabilizes the β1-adrenergic receptor in intracellular compartments after internalization. J Biol Chem 290: 6120–6129, 2015. doi: 10.1074/jbc.M114.605725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lamprecht G, Seidler U. The emerging role of PDZ adapter proteins for regulation of intestinal ion transport. Am J Physiol Gastrointest Liver Physiol 291: G766–G777, 2006. doi: 10.1152/ajpgi.00135.2006. [DOI] [PubMed] [Google Scholar]
- 31.Lauffer BE, Melero C, Temkin P, Lei C, Hong W, Kortemme T, von Zastrow M. SNX27 mediates PDZ-directed sorting from endosomes to the plasma membrane. J Cell Biol 190: 565–574, 2010. doi: 10.1083/jcb.201004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li C, Naren AP. Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol Ther 108: 208–223, 2005. doi: 10.1016/j.pharmthera.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Xia F, Reithmeier RA. N-glycosylation and topology of the human SLC26 family of anion transport membrane proteins. Am J Physiol Cell Physiol 306: C943–C960, 2014. doi: 10.1152/ajpcell.00030.2014. [DOI] [PubMed] [Google Scholar]
- 34.Loganathan SK, Casey JR. Corneal dystrophy-causing SLC4A11 mutants: suitability for folding-correction therapy. Hum Mutat 35: 1082–1091, 2014. doi: 10.1002/humu.22601. [DOI] [PubMed] [Google Scholar]
- 35.Lohi H, Kujala M, Makela S, Lehtonen E, Kestila M, Saarialho-Kere U, Markovich D, Kere J. Functional characterization of three novel tissue-specific anion exchangers SLC26A7, -A8, and -A9. J Biol Chem 277: 14246–14254, 2002. doi: 10.1074/jbc.M111802200. [DOI] [PubMed] [Google Scholar]
- 36.Loriol C, Dulong S, Avella M, Gabillat N, Boulukos K, Borgese F, Ehrenfeld J. Characterization of SLC26A9, facilitation of Cl(-) transport by bicarbonate. Cell Physiol Biochem 22: 15–30, 2008. doi: 10.1159/000149780. [DOI] [PubMed] [Google Scholar]
- 37.Lu R, Stewart L, Wilson JM. Scaffolding protein GOPC regulates tight junction structure. Cell Tissue Res 360: 321–332, 2015. doi: 10.1007/s00441-014-2088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lukacs GL, Mohamed A, Kartner N, Chang XB, Riordan JR, Grinstein S. Conformational maturation of CFTR but not its mutant counterpart (delta F508) occurs in the endoplasmic reticulum and requires ATP. EMBO J 13: 6076–6086, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mehta A. CFTR: more than just a chloride channel. Pediatr Pulmonol 39: 292–298, 2005. doi: 10.1002/ppul.20147. [DOI] [PubMed] [Google Scholar]
- 40.Mickle JE, Macek M Jr, Fulmer-Smentek SB, Egan MM, Schwiebert E, Guggino W, Moss R, Cutting GR. A mutation in the cystic fibrosis transmembrane conductance regulator gene associated with elevated sweat chloride concentrations in the absence of cystic fibrosis. Hum Mol Genet 7: 729–735, 1998. doi: 10.1093/hmg/7.4.729. [DOI] [PubMed] [Google Scholar]
- 41.Miller MR, Soave D, Li W, Gong J, Pace RG, Boëlle PY, Cutting GR, Drumm ML, Knowles MR, Sun L, Rommens JM, Accurso F, Durie PR, Corvol H, Levy H, Sontag MK, Strug LJ. Variants in solute carrier SLC26A9 modify prenatal exocrine pancreatic damage in cystic fibrosis. J Pediatr 166: 1152–1157.e6, 2015. doi: 10.1016/j.jpeds.2015.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mount DB, Romero MF. The SLC26 gene family of multifunctional anion exchangers. Pflugers Arch 447: 710–721, 2004. doi: 10.1007/s00424-003-1090-3. [DOI] [PubMed] [Google Scholar]
- 43.Moyer BD, Duhaime M, Shaw C, Denton J, Reynolds D, Karlson KH, Pfeiffer J, Wang S, Mickle JE, Milewski M, Cutting GR, Guggino WB, Li M, Stanton BA. The PDZ-interacting domain of cystic fibrosis transmembrane conductance regulator is required for functional expression in the apical plasma membrane. J Biol Chem 275: 27069–27074, 2000. doi: 10.1074/jbc.M004951200. [DOI] [PubMed] [Google Scholar]
- 44.Ousingsawat J, Schreiber R, Kunzelmann K. Differential contribution of SLC26A9 to Cl(-) conductance in polarized and non-polarized epithelial cells. J Cell Physiol 227: 2323–2329, 2012. doi: 10.1002/jcp.22967. [DOI] [PubMed] [Google Scholar]
- 45.Pelaseyed T, Hansson GC. CFTR anion channel modulates expression of human transmembrane mucin MUC3 through the PDZ protein GOPC. J Cell Sci 124: 3074–3083, 2011. doi: 10.1242/jcs.076943. [DOI] [PubMed] [Google Scholar]
- 46.Qian Z, Xu X, Amacher JF, Madden DR, Cormet-Boyaka E, Pei D. Intracellular delivery of peptidyl ligands by reversible cyclization: discovery of a PDZ Domain Inhibitor that rescues CFTR activity. Angew Chem Int Ed Engl 54: 5874–5878, 2015. doi: 10.1002/anie.201411594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ren A, Zhang W, Yarlagadda S, Sinha C, Arora K, Moon CS, Naren AP. MAST205 competes with cystic fibrosis transmembrane conductance regulator (CFTR)-associated ligand for binding to CFTR to regulate CFTR-mediated fluid transport. J Biol Chem 288: 12325–12334, 2013. doi: 10.1074/jbc.M112.432724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salomon JJ, Spahn S, Wang X, Füllekrug J, Bertrand CA, Mall MA. Generation and functional characterization of epithelial cells with stable expression of SLC26A9 Cl- channels. Am J Physiol Lung Cell Mol Physiol 310: L593–L602, 2016. doi: 10.1152/ajplung.00321.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwiebert EM, Benos DJ, Egan ME, Stutts MJ, Guggino WB. CFTR is a conductance regulator as well as a chloride channel. Physiol Rev 79, Suppl: S145–S166, 1999. [DOI] [PubMed] [Google Scholar]
- 50.Shcheynikov N, Yang D, Wang Y, Zeng W, Karniski LP, So I, Wall SM, Muallem S. The Slc26a4 transporter functions as an electroneutral Cl-/I-/HCO3- exchanger: role of Slc26a4 and Slc26a6 in I- and HCO3- secretion and in regulation of CFTR in the parotid duct. J Physiol 586: 3813–3824, 2008. doi: 10.1113/jphysiol.2008.154468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sheng M, Sala C. PDZ domains and the organization of supramolecular complexes. Annu Rev Neurosci 24: 1–29, 2001. doi: 10.1146/annurev.neuro.24.1.1. [DOI] [PubMed] [Google Scholar]
- 52.Shenolikar S, Voltz JW, Cunningham R, Weinman EJ. Regulation of ion transport by the NHERF family of PDZ proteins. Physiology (Bethesda) 19: 362–369, 2004. doi: 10.1152/physiol.00020.2004. [DOI] [PubMed] [Google Scholar]
- 53.Smith JJ, Welsh MJ. Fluid and electrolyte transport by cultured human airway epithelia. J Clin Invest 91: 1590–1597, 1993. doi: 10.1172/JCI116365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strug LJ, Gonska T, He G, Keenan K, Ip W, Boelle PY, Lin F, Panjwani N, Gong J, Li W, Soave D, Xiao B, Tullis E, Rabin H, Parkins MD, Price A, Zuberbuhler PC, Corvol H, Ratjen F, Sun L, Bear CE, Rommens JM. Cystic fibrosis gene modifier SLC26A9 modulates airway response to CFTR-directed therapeutics. Hum Mol Genet 25: 4590–4600, 2016. doi: 10.1093/hmg/ddw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun F, Mi Z, Condliffe SB, Bertrand CA, Gong X, Lu X, Zhang R, Latoche JD, Pilewski JM, Robbins PD, Frizzell RA. Chaperone displacement from mutant cystic fibrosis transmembrane conductance regulator restores its function in human airway epithelia. FASEB J 22: 3255–3263, 2008. doi: 10.1096/fj.07-105338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sun L, Rommens JM, Corvol H, Li W, Li X, Chiang TA, Lin F, Dorfman R, Busson PF, Parekh RV, Zelenika D, Blackman SM, Corey M, Doshi VK, Henderson L, Naughton KM, O’Neal WK, Pace RG, Stonebraker JR, Wood SD, Wright FA, Zielenski J, Clement A, Drumm ML, Boëlle PY, Cutting GR, Knowles MR, Durie PR, Strug LJ. Multiple apical plasma membrane constituents are associated with susceptibility to meconium ileus in individuals with cystic fibrosis. Nat Genet 44: 562–569, 2012. doi: 10.1038/ng.2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, Decker CJ, Miller M, McCartney J, Olson ER, Wine JJ, Frizzell RA, Ashlock M, Negulescu PA. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci USA 108: 18843–18848, 2011. doi: 10.1073/pnas.1105787108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ward CL, Kopito RR. Intracellular turnover of cystic fibrosis transmembrane conductance regulator. Inefficient processing and rapid degradation of wild-type and mutant proteins. J Biol Chem 269: 25710–25718, 1994. [PubMed] [Google Scholar]
- 59.Wolde M, Fellows A, Cheng J, Kivenson A, Coutermarsh B, Talebian L, Karlson K, Piserchio A, Mierke DF, Stanton BA, Guggino WB, Madden DR. Targeting CAL as a negative regulator of DeltaF508-CFTR cell-surface expression: an RNA interference and structure-based mutagenetic approach. J Biol Chem 282: 8099–8109, 2007. doi: 10.1074/jbc.M611049200. [DOI] [PubMed] [Google Scholar]
- 60.Worrell RT, Butt AG, Cliff WH, Frizzell RA. A volume-sensitive chloride conductance in human colonic cell line T84. Am J Physiol Cell Physiol 256: C1111–C1119, 1989. [DOI] [PubMed] [Google Scholar]
- 61.Xu J, Henriksnäs J, Barone S, Witte D, Shull GE, Forte JG, Holm L, Soleimani M. SLC26A9 is expressed in gastric surface epithelial cells, mediates Cl-/HCO3- exchange, and is inhibited by NH4+. Am J Physiol Cell Physiol 289: C493–C505, 2005. doi: 10.1152/ajpcell.00030.2005. [DOI] [PubMed] [Google Scholar]
- 62.Zheng J, Chan T, Cheung FS, Zhu L, Murray M, Zhou F. PDZK1 and NHERF1 regulate the function of human organic anion transporting polypeptide 1A2 (OATP1A2) by modulating its subcellular trafficking and stability. PLoS One 9: e94712, 2014. doi: 10.1371/journal.pone.0094712. [DOI] [PMC free article] [PubMed] [Google Scholar]