

Abstract
A new series of superarmed glycosyl donors has been investigated. It was demonstrated that the S-ethyl leaving group allows for high reactivity, which is much higher than that of equally equipped S-phenyl glycosyl donors that were previously investigated by our groups. The superarmed S-ethyl glycosyl donors equipped with a 2-O-benzoyl group gave complete β-stereoselectivity. Utility of the new glycosyl donors has been demonstrated in a one-pot one-addition oligosaccharide synthesis with all of the reaction components present from the beginning.
Mechanistic challenges in the chemical glycosylation reaction have consistently captured the attention of the synthetic community.1 Many classes of glycosyl donors have been developed2 and many strategies for oligosaccharide synthesis have emerged.3 Among the methods and strategies available, the development of the armed-disarmed strategy for chemo-selective oligosaccharide synthesis occupies an important niche.4 Reactivity tuning of various series of thioglycosides has been reported and applied to the synthesis of a variety of oligosaccharide sequences.5 Beyond the traditional scope of the armed-disarmed strategy, superarmed and superdisarmed building blocks have also been identified and studied.6 Bols and co-workers developed an approach to superarm glycosyl donors by changing the equatorial-rich 4C1 conformation to an axial-rich conformation.7 These conformational changes were induced by creating steric congestion with tert-butyldimethylsilyl (TBS) or related bulky protecting groups at the C-2, 3 and 4 positions of S-phenyl (SPh) glucosides, resulting in a skew-boat conformation. The donors showed a 20-fold increase in reactivity compared to their armed per-O-benzylated counter-parts.7c The Demchenko group also reported superarmed S-benzoxazolyl (SBox) and S-ethyl (SEt) glycosyl donors, but the superarming was based on the O2/O5-cooperative effect in glycosylation.8 Thus, it was demonstrated that donors equipped with the 2-O-benzoyl-3,4,6-tri-O-benzyl protecting group pattern are 10 times more reactive than their armed counterparts.9
Using the two different approaches to superarm glycosyl donors, our groups jointly developed a 2-O-benzoyl donor 1 with 3,4-di-O-TBS protection (Scheme 1). Over the course of that study we learned that conformational arming is a powerful tool for increasing reactivity and achieving excellent yields and the 2-O-benzoyl substituent ensured complete 1,2-trans stereoselectivity.10 The anchimeric superarming effects in the conformationally modified donor 1 are significantly weaker, to the extent that the 2-O-benzoylated SPh donor 1 is 5.8 and 4.5 times less reactive than its 2-O-TBS and 2-O-benzylated counterparts 2 and 3, respectively (Scheme 1). Although glycosylations with hybrid donor 1 were swift, high yielding and β-stereoselective,10 we feared that the reduced reactivity could translate into decreased efficacy of these building blocks in sequential chemoselective glycosylations in one-pot. This led us to a hypothesis that the use of a more reactive S-ethyl leaving group11 would help us to develop a complementary superarmed glycosyl donor with a superior reactivity profile whilst still maintaining β-stereoselectivity.
Scheme 1.

The relative reactivity of the conformationally superarmed S-phenyl glycosyl donors.10
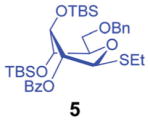
Right from the start, when donor 1 was subjected to a competition experiment with the equally protected SEt donor 5, a much higher reactivity of the latter was detected. The competition experiments for this study were conducted following essentially the same experimental conditions and ratios as in our previous study.10 Two glycosyl donors, used in equimolar amounts (1.0 equiv. each), were set to compete for excess glycosyl acceptor 4 12 (2.0 equiv.) in the presence of NIS (1.0 equiv.) and TfOH (0.1 equiv.) at −78 °C. The use of low temperature, which was allowed to gradually increase over the course of the reaction, and the use of a very limited amount of promoter helped to maintain workable reaction rates. All of the competition experiments were quenched after 1 h and the remaining glycosyl donors were isolated and quantified. Thus, as a result of the first competition experiment, SPh donor 1 remained the major monosaccharide component of the mixture and was isolated in 87% yield, whereas only 13% of the SEt donor 5 remained (Scheme 2). This translates into a 1/6.7 reactivity ratio between the two donors, or in other words, the SEt donor 5 is 6.7 times more reactive than its SPh counterpart 1.
Scheme 2.

The relative reactivity of S-phenyl versus S-ethyl glycosyl donors of the superarmed series.
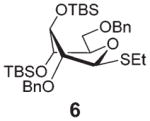
Subsequent competition experiments led to the realization that 2-O-benzoyl SEt donor 5 is nearly as reactive as 2-O-benzyl SPh donor 3 (1/1.1) and only slightly less reactive than the most reactive superarmed 2-O-TBS protected SPh donor 2 known to date (1/1.6, Scheme 2). To explore the reactivity limits of superarmed glycosyl donors of the SEt series, we obtained donor 6 equipped with the 2-O-benzyl protecting group. A competition experiment with equally protected SPh donor 3 led to the realization of the higher reactivity of donor 6 (3/6 = 1/3.8). Using a similar approach, we determined that donor 6 is 3.7 times more reactive than the 2-O-TBS SPh donor 2.10
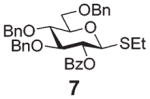
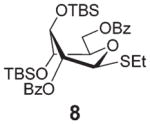
Encouraged by the first series of competition experiments, we decided to investigate the new hybrid donor 5 in the context of other SEt donors. For this study we obtained the anchimerically superarmed derivative 7 along with two conformationally superarmed donors 8 and 9, equipped with 6-O-benzoyl and 2,6-di-O-benzoyl protections, respectively. The first competition experiment that was conducted between donors 5 and 7 provided a very impressive reactivity difference: donor 5 was 95 times more reactive than donor 7 (Scheme 3). This result is more indicative of the superior reactivity of 5 than the poor reactivity of 7. The latter donor is still super-armed because it is much more reactive than its per-benzylated counterpart. In addition, donor 7 is 2.2 times more reactive than the previously developed hybrid SPh donor 1. Moreover, compound 7 is also 2.3 times more reactive than the conformationally superarmed SEt donor 8 equipped with two benzoyl groups at O-2 and O-6.
Scheme 3.

The effect of conformational and electronic superarming in a series of S-ethyl glycosyl donors.
A comparison of donors 5 and 8 showed a very significant deactivating effect of 6-O-benzoyl in comparison with 6-O-benzyl, these groups being the only structural difference between the two donors.13 Thus, the 6-O-benzyl donor 5 was 97 times more reactive than its 6-O-benzoylated counterpart 8. Donor 5 was also found to be 5.3 times more reactive than donor 9 with reverse positioning of the benzyl and benzoyl substituents: 2-O-benzyl, 6-O-benzoyl.
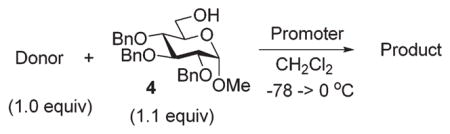
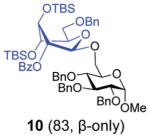
With this comprehensive set of competition experiments, we began investigating the glycosyl donor properties of compounds 5–8 with the model acceptor 4. After screening a number of promoters for the activation of thioglycosides, we chose NIS/TfOH and DMTST. These reaction conditions offered a good balance of reactivity, selectivity and yield. Other promoters, including iodine which was successfully used in our previous study of anchimerically superarmed SEt donors, led to decreased yields resulting from high rates of major side reactions: TBS cleavage and/or SEt hydrolysis. Thus, NIS/TfOH-promoted coupling between donor 5 and acceptor 4 swiftly (20 min) produced disaccharide 10 in 83% yield and complete β-stereoselectivity (entry 1, Table 1).
Table 1.
Glycosylation of acceptor 4 with different superarmed SEt donors
| |||
|---|---|---|---|
|
| |||
| Entry | Donor | Conditions,a time | Product (yield % α/β ratio) |
| 1 |
|
A, 20 min |
|
| 2 | 5 | B, 15 min | 10 (85, β-only) |
| 3 |
|
A, 15 min |
|
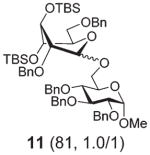
| 4 | 6 | B, 10 min | 11 (21, 0.9/1)b |
| 5 |
|
A, 30 min |
|
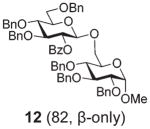
| 6 | 7 | B, 20 min | 12 (85, β-only) |
| 7 |
|
A, 30 min |
|
Conditions A: NIS/TfOH (1.3 equiv.), 3 Å mol sieves; B: DMTST (2.0 equiv.), 4 Å mol sieves.
The yield is impacted by fair stability of the TBS groups (see text).
Practically the same outcome was achieved in the DMTST-promoted reaction listed in entry 2. NIS/TfOH-promoted activation of donor 6 produced disaccharide 11 in 81% yield (entry 3). In this case, the reaction was non-stereoselective due to the absence of neighboring group participation. In this case, DMTST was less effective and the TBS protecting groups showed a high propensity to cleavage. As a result, disaccharide 11 was obtained in a poor yield of 21% (entry 4). The outcome of this reaction could be improved (44% yield) using only a slight excess of DMTST (1.3 equiv.).
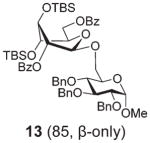
Glycosidation of the anchimerically superarmed donor 7 was successful in the case of either NIS/TfOH or DMTST-promoted activation, and disaccharide 12 was obtained with complete β-stereoselectivity in 82 or 85% yield, respectively (entries 5 and 6). In case of donor 8, only NIS/TfOH gave a practical result, whereas DMTST showed a high level of competing processes. Thus, NIS/TfOH-promoted activation of 8 produced disaccharide 13 in 85% yield and complete β-stereoselectivity (entry 7). The conformational properties of disaccharide 10 were studied using X-ray crystallography (Fig. 1). The crystals of 10 were obtained by slowly evaporating a mixture of MeOH/water. The skew-boat conformation of disaccharide 10 was deduced from the X-ray data and was consistent with the altered coupling constants obtained from its 1H NMR spectrum (Fig. 1).
Fig. 1.

The X-ray structure of disaccharide 10 (hydrogens and protecting groups have been omitted for clarity; refer to the ESI† for complete X-ray structure and data).
Using a series of glycosyl donors of differential reactivity, we began studying the applicability of this method to the one-pot oligosaccharide synthesis.14 With a number of different concepts for the one-pot synthesis, we chose the one-pot/one-addition method wherein all of the building blocks are present from the beginning. Invented by Kahne,15 and further explored by Fraser-Reid16 and Bols,7a this approach requires fine tuning of the reactivity to differentiate all of the reaction components. The general idea underpinning this approach is that the more reactive donor will react with the more reactive acceptor (hydroxyl). Subsequently, the second-step coupling will involve coupling between the less reactive donor and the less reactive acceptor.
With these considerations, we chose highly reactive donor 5 to couple with the reactive 6-OH in benzylated building block 14 equipped with the anomeric SEt group. A fast first-step reaction will permit the sequential (rather than competitive) activation of the SEt leaving group of the intermediate disaccharide to react with the less reactive acceptor 15 (Scheme 4). Building block 14 is the key reaction component in the mixture because it can react with both compounds; firstly as the more reactive acceptor and then as the less reactive donor. The role of the highly reactive superarmed donor is also essential to ensure that the first coupling step is swift. The synthesis of trisaccharide 16 was conducted by mixing together building blocks 5, 14 and 15 and adding NIS/TfOH. As a result, compound 16 was obtained via one-pot synthesis in 42% yield and high stereoselectivity (α/β = 14/1, Scheme 4). A substantial quantity of cross-coupled disaccharide resulting from the reaction between 5 and 15 indicated that the reactivity difference between primary hydroxyls in 15 and 16 is insufficient to ensure effective one-pot coupling. A simple competition experiment set up between the two acceptors and donor 5 showed that 14 is only 1.6 times more reactive than 15 (Scheme 5, see the ESI† for details).
Scheme 4.

One-pot one-addition synthesis of trisaccharides 16, 18 and 20.
Scheme 5.

The relative reactivity of glycosyl acceptors 14, 15, 17 and 19.
To improve the outcome of the one-pot synthesis we prepared secondary acceptor 17, which was deactivated by the surrounding benzoyl substituents.17 The competition experiment showed that benzylated primary acceptor 14 is 10.1 times more reactive than its benzoylated secondary counterpart 17 (Scheme 5). Theorizing that this reactivity difference would be sufficient, we set up the synthesis of trisaccharide 18 from building blocks 5, 14 and 17 that were mixed and NIS/TfOH solution that was added. As a result, trisaccharide 18 was obtained via a one-pot synthesis in 37% yield and high stereo-selectivity (α/β = 11/1, Scheme 4). No cross-coupled disaccharide was found in the reaction mixture, but attempts to push the reaction to completion promoted competitive TBS group hydrolysis. In a further search of suitable building blocks for the one-pot synthesis, we obtained acceptor 19 benzylated at C-6. This acceptor is only 2.5 times less reactive than its per-benzylated primary counterpart 14 (Scheme 5). Nevertheless, this reactivity difference was sufficient for the synthesis of trisaccharide 20, which was produced in a good yield of 65% and high stereoselectivity (α/β = 10/1, Scheme 4), showing the utility of this approach and also the necessity to fine-tune all of the reaction components.
In conclusion, we have developed a series of superarmed SEt glycosyl donors that were applied to stereoselective glycosylations and multi-step oligosaccharide synthesis in one pot. Further application of these highly reactive compounds for the glycosylation of various glycosyl acceptors in solution and on solid supports is currently underway in our laboratories.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institute of General Medical Sciences (GM111835). Funding from the National Science Foundation (MRI, CHE-0420497) for the purchase of the ApexII diffractometer is acknowledged. We thank Dr Rensheng Luo (UM – St Louis) for help with acquiring spectral data using a 600 MHz NMR spectrometer that was purchased thanks to the NSF (award CHE-0959360). Dr Winter and Mr Kramer (UM – St Louis) are thanked for the HRMS determinations.
Footnotes
Electronic supplementary information (ESI) available: Experimental details, characterization data, 1H and 13C NMR spectra for all new compounds as well as X-ray data for 10. CCDC 1509777. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ob02498j
Notes and references
- 1.(a) Capon B. Chem Rev. 1969;69:407–496. [Google Scholar]; (b) Crich D. Acc Chem Res. 2010;43:1144–1153. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]; (c) Mydock LK, Demchenko AV. Org Biomol Chem. 2010;8:497–510. doi: 10.1039/b916088d. [DOI] [PubMed] [Google Scholar]; (d) Ranade SC, Demchenko AV. J Carbohydr Chem. 2013;32:1–43. [Google Scholar]; (e) Frihed TG, Bols M, Pedersen CM. Chem Rev. 2015;115:4963–5013. doi: 10.1021/cr500434x. [DOI] [PubMed] [Google Scholar]
- 2.(a) Zhu X, Schmidt RR. Angew Chem, Int Ed. 2009;48:1900–1934. doi: 10.1002/anie.200802036. [DOI] [PubMed] [Google Scholar]; (b) Nigudkar SS, Demchenko AV. Chem Sci. 2015;6:2687–2704. doi: 10.1039/c5sc00280j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Smoot JT, Demchenko AV. Adv Carbohydr Chem Biochem. 2009;62:161–250. doi: 10.1016/S0065-2318(09)00005-5. [DOI] [PubMed] [Google Scholar]; (b) Hsu CH, Hung SC, Wu CY, Wong CH. Angew Chem, Int Ed. 2011;50:11872–11923. doi: 10.1002/anie.201100125. [DOI] [PubMed] [Google Scholar]; (c) Seeberger PH. Acc Chem Res. 2015;48:1450–1463. doi: 10.1021/ar5004362. [DOI] [PubMed] [Google Scholar]
- 4.(a) Mootoo DR, Konradsson P, Udodong U, Fraser-Reid B. J Am Chem Soc. 1988;110:5583–5584. [Google Scholar]; (b) Fraser-Reid B, Wu Z, Udodong UE, Ottosson H. J Org Chem. 1990;55:6068–6070. [Google Scholar]; (c) Fraser-Reid B, Udodong UE, Wu ZF, Ottosson H, Merritt JR, Rao CS, Roberts C, Madsen R. Synlett. 1992:927–942. references therein. [Google Scholar]
- 5.(a) Grice P, Ley SV, Pietruszka J, Priepke HWM, Walther EPE. Synlett. 1995:781–784. [Google Scholar]; (b) Douglas NL, Ley SV, Lucking U, Warriner SL. J Chem Soc, Perkin Trans. 1;1998:51–65. [Google Scholar]; (c) Zhang Z, Ollmann IR, Ye XS, Wischnat R, Baasov T, Wong CH. J Am Chem Soc. 1999;121:734–753. [Google Scholar]; (d) Ritter TK, Mong KKT, Liu H, Nakatani T, Wong CH. Angew Chem, Int Ed. 2003;42:4657–4660. doi: 10.1002/anie.200351534. [DOI] [PubMed] [Google Scholar]; (e) Hsu Y, Lu XA, Zulueta MM, Tsai CM, Lin KI, Hung SC, Wong CH. J Am Chem Soc. 2012;134:4549–4552. doi: 10.1021/ja300284x. [DOI] [PubMed] [Google Scholar]; (f) Fraser-Reid B, Lopez JC, editors. Reactivity Tuning in Oligosaccharide Assembly. Springer-Verlag; Berlin-Heidelberg: 2011. [Google Scholar]
- 6.Premathilake HD, Demchenko AV. In: Topics in Current Chemistry: Reactivity Tuning in Oligosaccharide Assembly. Fraser-Reid B, Lopez JC, editors. Vol. 301. Springer-Verlag; Berlin-Heidelberg: 2011. pp. 189–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Jensen HH, Pedersen CM, Bols M. Chem – Eur J. 2007;13:7576–7582. doi: 10.1002/chem.200700947. [DOI] [PubMed] [Google Scholar]; (b) Pedersen CM, Nordstrom LU, Bols M. J Am Chem Soc. 2007;129:9222–9235. doi: 10.1021/ja071955l. [DOI] [PubMed] [Google Scholar]; (c) Pedersen CM, Marinescu LG, Bols M. Chem Commun. 2008:2465–2467. doi: 10.1039/b801305e. [DOI] [PubMed] [Google Scholar]; (d) Heuckendorff M, Pedersen CM, Bols M. Chem – Eur J. 2010;16:13982–13994. doi: 10.1002/chem.201002313. [DOI] [PubMed] [Google Scholar]; (e) Pedersen CM, Marinescu LG, Bols M. C R Chim. 2010;14:17–43. [Google Scholar]
- 8.Kamat MN, Demchenko AV. Org Lett. 2005;7:3215–3218. doi: 10.1021/ol050969y. [DOI] [PubMed] [Google Scholar]
- 9.(a) Mydock LK, Demchenko AV. Org Lett. 2008;10:2103–2106. doi: 10.1021/ol800345j. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mydock LK, Demchenko AV. Org Lett. 2008;10:2107–2110. doi: 10.1021/ol800648d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Premathilake HD, Mydock LK, Demchenko AV. J Org Chem. 2010;75:1095–1100. doi: 10.1021/jo9021474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heuckendorff M, Premathilake HD, Pornsuriyasak P, Madsen AØ, Pedersen CM, Bols M, Demchenko AV. Org Lett. 2013;15:4904–4907. doi: 10.1021/ol402371b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.(a) Lahmann M, Oscarson S. Can J Chem. 2002;80:889–893. [Google Scholar]; (b) Yasomanee JP, Demchenko AV. Chem – Eur J. 2015;21:6572–6581. doi: 10.1002/chem.201406589. [DOI] [PubMed] [Google Scholar]
- 12.Kuester JM, Dyong I. Justus Liebigs Ann Chem. 1975:2179–2189. [Google Scholar]
- 13.Schmidt T, Madsen R. Eur J Org Chem. 2007:3935–3941. [Google Scholar]
- 14.Parameswar AR, Demchenko AV. Progress in the synthesis of complex carbohydrate chains of plant and microbial polysaccharides. In: Nifantiev NE, editor. Transworld Res Network. Kerala: 2009. pp. 463–488. [Google Scholar]
- 15.Raghavan S, Kahne D. J Am Chem Soc. 1993;115:1580–1581. [Google Scholar]
- 16.Fraser-Reid B, Lopez JC, Radhakrishnan KV, Nandakumar MV, Gomez AM, Uriel C. Chem Commun. 2002:2104–2105. doi: 10.1039/b206598c. [DOI] [PubMed] [Google Scholar]
- 17.(a) Martín-Lomas M, Cid MB, Alfonso F. Synlett. 2005:2052–2056. [Google Scholar]; (b) Kaeothip S, Akins SJ, Demchenko AV. Carbohydr Res. 2010;345:2146–2150. doi: 10.1016/j.carres.2010.08.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.