

Abstract
The development of a useful methodology for simple, scalable, and transformative automation of oligosaccharide synthesis that easily interfaces with existing methods is reported. The automated synthesis can now be performed using accessible equipment where the reactants and reagents are delivered by the pump or the autosampler and the reactions can be monitored by the UV detector. The HPLC-based platform for automation is easy to setup and adapt to different systems and targets.
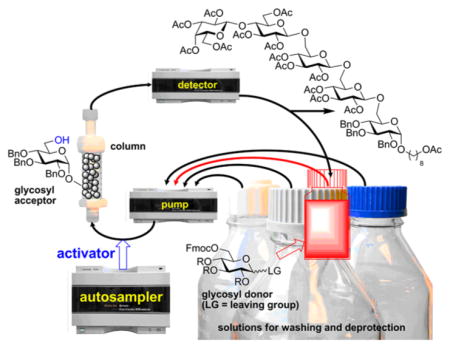
Graphical Abstract
INTRODUCTION
Glycans are oligomeric carbohydrates wherein monomers are connected via the glycosidic linkage. This linkage is obtained by a glycosylation reaction, which remains challenging to synthetic chemists due to the requirement to achieve high stereo-control1,2 and yields by suppressing side reactions.3 Beyond that, glycan synthesis may require further manipulations between each glycosylation step. Due to significant advances, the chemical synthesis of many glycans can now be streamlined by using expeditious strategies.4 Solid-phase synthesis,5,6 which eliminates the need for purifying intermediates and simplifies the removal of excess reagents, has been widely used in the preparation of peptides7,8 and oligonucleotides.9 Since 1971, solid-phase synthesis has been used for the preparation of oligosaccharides;10–14 and in 2001 Seeberger et al. reported the first automated oligosaccharide synthesis using a modified peptide synthesizer.15–18 In 2012, Seeberger et al. reported “the first fully automated solid-phase oligosaccharide synthesizer,” initially in its experimental form;19 and in 2013 it was marketed as Glyconeer 2.1. Approaches by Wong,20,21 Takahashi,22,23 Chen,24–26 Pohl,27–30 Wang,31,32 and Nokami33,34 are based on the automation of chemical, enzymatic, or chemoenzymatic syntheses in solution with or without using tags.35
In light of recent progress made in the areas of glycobiology36–39 and glycomics40 “widely applicable methods to generate both large and small quantities of glycans are needed.”41 Oligosaccharides can be obtained by isolation/release from natural sources, or prepared enzymatically and/or chemically. All three approaches are viable, each offering certain advantages, but none can significantly outperform the others. Oligosaccharide synthesis in solution requires a significant deal of know-how. The automated platform for solid-phase synthesis developed by Seeberger introduces an idea of operational simplicity and highlights that the development of accessible methods for glycan production is essential for further innovations and practical applications in all areas of glycosciences.
The development of the automated synthesizer in our laboratories began with the introduction of the surface-tethered iterative carbohydrate synthesis (STICS).43 The basis for this concept is a surface-functionalized stack of nanoporous gold plates that simplifies the transfer of the gold surface-bound molecules between reaction vessels. At the end of the synthesis, the resulting glycan can be either cleaved-off for further processing or deprotected directly on the gold surface to be used for recognition studies or immunoassay development.44 The STICS concept was developed with robotic arm automation in mind. However, we discovered that standard HPLC equipment would offer a more accessible platform for automation. This approach was discovered with nanoporous gold,45 but we have also investigated more traditional polymer supports. Using the acceptor-bound approach,46 preloaded Tentagel resin was packed in the Omnifit column and integrated into the HPLC system (Scheme 1).42 All steps were automated using a three-headed HPLC pump and the reagent consumption was monitored using a standard UV detector. Reagents were recirculated, but still 10 equiv of trichloroacetimidate donors were used for each glycosylation.42 More recently, Pentelute and co-workers investigated the HPLC-assisted synthesis of peptides.47 Other exciting developments in the area of high throughput and automated syntheses have been particularly inspiring to our own research endeavors.48,49
Scheme 1.

Original Set-up for HPLC-Assisted Synthesis.42
RESULTS AND DISCUSSION
Presented herein is the development of a broadly useful technology for simple, scalable, and transformative automation of solid-phase synthesis that does not rely on specialized equipment. Broadly available and used in most laboratories, the setup of the HPLC equipment requires no investment. This platform allows for real-time UV detector monitoring of all steps including glycosylation, which, in turn, helps reduce the reaction time and the amount of reagents and solvents needed. The use of a computer interface and standard HPLC liquid handling equipment and software will allow recording a successful automated sequence as a computer program that can then be reproduced by both specialists and nonspecialists with a “press of a button”. While this approach has a potential to revolutionize the way the automation is conducted, solid-phase synthesis suffers from many inherent limitations. Practically every aspect of solid-phase synthesis needs to be refined. Along with the introduction of the autosampler for the reagent delivery, this article is also dedicated to the refinement of some basic aspects of this methodology. Our new basic setup is using standard Agilent 1260 Infinity series HPLC system equipped with the quad pump, a UV detector, and autosampler.
Selection of Resins, Spacers, and Linkers
Our preliminary work on the HPLC-assisted synthesis was solely based on Tentagel resin.42 Previously, we compared Tentagel vs Merrifield resins using the manual approach, but saw no significant difference in efficiency and yields.46 A recent comparative study by Seeberger et al. determined that the Merrifield resin gives the best efficiency in application to their automation platform.50 To gain a better understanding of how loading, swelling, mechanical robustness, size, and other factors may affect the HPLC-assisted synthesis we performed a side-by-side comparison study of Merrifield, Wang,51 and JandaJel52 resins, all of which have been found to be excellent supports for oligosaccharide synthesis and have loading capacities up to 1.0 mmol g−1. Although identifying the best support for universal application might be simply impossible, in a series of comparative experiments we identified JandaJel as the most suitable resin for HPLC-mediated synthesis in terms of loading, reaction times, and yields.
It has become common knowledge that the type of the spacer and/or linker between the acceptor and the polymer support may be of critical importance.53–55 Factors to consider are the chemical composition, stability toward various experimental conditions, and selective (mild) conditions for its cleavage. In our preliminary study, we were using a C4 spacer in combination with succinoyl linker that worked well, and the cleavage was reliably achieved using a small amount (~2 mL) of the recirculating 0.1 M solution of NaOMe in MeOH–CH2Cl2. With the general anticipation that extension of the spacer length could move the glycosyl acceptor further out into solution and enhance the efficiency of the reaction with the solution-based glycosyl donor we performed a comparative study. In our study of glycosylations using nanoporous gold, we obtained better yields with the acceptor equipped with longer C8–O–C8 spacer than those of acceptors with shorter C4 or C8 spacers.45 With the use of polymer beads we report that while the C8–O–C8 spacer helps to enhance the yields obtained with the C4 spacer, it practically offers no advantage over the more synthetically accessible C8 spacer. Hence, all syntheses described in the article used the C8 spacer.
Loading Practices and Quantification
The resin loading capacity is important, but overcrowding of the reactive sites may prevent further elongation, particularly in case of sterically demanding and branched oligosaccharides. During our exploratory study with JandaGel and Tentagel resins, it was observed that the desired loading capacities could be achieved much faster using HPLC-based reagent delivery rather than the manual loading in a flask. Nevertheless, large-scale resin preloading (2–10 g) for this study was performed by the manual approach using the flask and the shaker as depicted in Scheme 2. Building block 1 was coupled with amine JandaJel resin in the presence of 1-ethyl-3-(3-(dimethylamino)propyl)-carbodiimide (EDC) and 4-dimethylaminopyridine (DMAP). The loading can be confirmed by weighing the unloaded versus loaded resin, as well as cleaving and quantifying of the loaded acceptor if so desired. The preloaded JandaJel resin 2 was then subjected to detritylation with 10% trifluoroacetic acid in wet CH2Cl2. The detritylation results in the formation of glycosyl acceptor 3, but it also releases triphenylcarbinol (TrOH), which could be used for the initial quantification of the loading by its isolation by evaporation and weighing. Quantification of TrOH is the key step for determining of the loading capacity of the resin.
Scheme 2.

Synthesis of the Solid-Phase-Bound Acceptor 3
Glycosylation: Reagent Delivery, Recirculation, Monitoring, and Synthetic Methods
Glycosylation is a complex multistep process, and reactions on solid supports bring additional hurdles related to the mismatch between highly reactive solution-based vs unreactive solid-phase-based reactants. This mismatch is typically addressed by using a large excess (5–10 equiv) of the solution-based reactant, most commonly the donor, and repeating the reaction 2–3 times to ensure that all solid-supported acceptor is consumed.14 Automation offers some operational simplicity to oligosaccharide synthesis, but the entire concept may suffer from the inherited drawbacks of conventional methods.
Our experience with HPLC-assisted reactions is still limited, but we already established the protocol for separate delivery of solutions of glycosyl donor and promoter using HPLC pumps.42 The primary focus of the earlier study was to determine ranges of the variables, beginning with reagent ratios, concentration, velocity, and pressure. The reaction efficiency is likely to improve with increased speed of the reagent delivery. However, this may have potential downfalls if not properly addressed. If the reagents are delivered too fast, the internal column pressure may increase to a point where the resin beads collapse or fracture.6 We have not observed this at our operating velocity of 0.5–2 mL/min (1–12 bar).
The initial reagent delivery via HPLC pump offered a notable limitation of our platform in comparison to Seeberger’s automated synthesizer that has 32 intake lines.19 In principle, essentially the same capability can be achieved with the HPLC setup by splitting of the pump intake lines with eight-way split valves. However, as further steps toward complete automation, we envisaged the use of a standard HPLC autosampler. Autosamplers are abundant, cheap, easily fit into the HPLC-automation concept, and this approach opens access to hundreds of intake/delivery lines. This approach allows us to liberate other pump intake lines for the delivery of solvent for reactions, washing, and deprotection because only one line is now used for the donor delivery and recirculation. It should be mentioned that the recirculation has already been previously optimized with the purpose of addressing the main drawback of all solid-phase syntheses: the requirement for a large excess of solution-based reagents.
The outline of the automation setup, program sequence, and the key results for basic glycosylation reactions are depicted in Scheme 3. JandaJel resin (50 mg) functionalized with glycosyl acceptor 3 (0.022 mmol) was packed in Omnifit glass chromatography column. The column was connected to the standard Agilent Infinity 1260 HPLC system and the automation sequence was programmed as follows. Pump D was programmed to deliver CH2Cl2 at a flow rate of 1.0 mL/min. After discarding the first ~5 mL of the eluate (washing, step 1, Scheme 3) the system was switched to the recirculation mode and 2 mL of CH2Cl2 was recirculated for 30 min at a flow rate of 1.0 mL/min (swelling, step 2). After that, pump C was programmed to deliver a solution of the glycosyl donor (0.10 mmol) in CH2Cl2 (2 mL) at 0.5 mL/min and the system was left recirculating for 10 min (step 3A). Beginning from this stage the synthesis was monitored using the integrated UV detector set at 254 nm. A typical trace is shown in Scheme 3.
Scheme 3.

Refinement of the Glycosylation-Cleavage Sequence for the Synthesis of Disaccharide 11
The integrated autosampler was then programmed to inject a solution of promoter (40 μL) in CH2Cl2 (3 injections of 100 μL) at 10, 12, and 14 min after the initial delivery of the donor (step 3B). The system was left recirculating for 60–90 min, and the reaction was monitored by the UV detector in real-time. When the detector trace reaches the plateau, no change in the absorbance of the recirculating solution is observed, the reaction is stopped. In principle, low-efficiency reactions can be supplemented with fresh reagents/reactants at this time. After a typical reaction time of 60–90 min, the system was switched to pump D and washed with CH2Cl2 (1.0 mL/min flow rate) to remove excess reagents (step 4). The eluate from the washing step (~10 mL) is discarded. Again, this step was monitored by the UV detector, and the washing was typically stopped after ~10 min when the detector trace reached the baseline corresponding to pure CH2Cl2.
To affect the product cleavage from the solid support, pump B was then programmed to deliver a solution of NaOMe/CH2Cl2/MeOH (0.04/1/1, v/v/v) at the flow rate of 1.0 mL/min for 10 min (step 5). This step was also monitored by the UV detector. Typically, the cleavage is completed at this stage and the use of the detector monitoring is discontinued. The resulting mixture was recirculated for an additional ~10 min. The eluate was collected, neutralized, concentrated under the reduced pressure and the residue was acetylated with Ac2O in pyridine to afford disaccharide 11. The purification of 11 was achieved by conventional column chromatography and its identity was proven by traditional spectral methods.
Our initial study of the HPLC-assisted synthesis42 was exclusively based on trichloroacetimidates56–58 as glycosyl donors. In an attempt to broaden the scope of this methodology, we performed a comparative study of other common and novel leaving groups. Thioglycosides are generally much less reactive than O-imidates and hence considered less desirable for polymer-supported synthesis. With some prior success of using thioglycosides in glycosylations using polymer46 and nanoporous supports43 we investigated S-benzoxazolyl (SBox) donor 459 and S-phenyl glycosyl donor 560 in the HPLC-automated reactions. Glycosylation of SBox donor 4 with resin-bound acceptor 3 was performed in the presence of AgOTf. Following the general programming described above, disaccharide 11 was obtained in a good yield of 50% (Scheme 3, entry 1). A similar result was achieved with SPh donor 5, wherein NIS/TfOH promoted reaction afforded disaccharide 11 in 57% yield (entry 2). While the outcome of these reactions could be improved by injecting additional quantities of reagents, we chose to explore other classes of glycosyl donors.
Recently, we developed a new class of glycosyl donors, O-benzoxazolyl (OBox) imidates, which were also tested in the HPLC-based applications, but could not outperform traditional trichloroacetimidates.61 We also introduced 3,3-difluoro-3H-indol-2-yl (OFox) imidates,62 which showed a very high reactivity and allowed us to obtain impressive results in the HPLC-based application. Thus, glycosylation of OFox donor 6 with resin-bound acceptor 3 was performed in the presence of TMSOTf. Following the general programming, disaccharide 11 was obtained in a good yield of 73% (entry 3). A very similar outcome was obtained with phosphate donor 7, a glycosylation approach frequently used in Seeberger’s automation method.63 The phosphate donor 7 also provided a very impressive result in our HPLC-based platform wherein TMSOTf-promoted activation led to disaccharide 11 in 75% yield (entry 4). Nevertheless, the most consistent result and the highest yield was obtained with trichloroacetimidate 8.64 TMSOTf-promoted activation led to disaccharide 11 in an excellent yield of 85% using only 4.4 equiv of the donor (entry 5). In order to expand this procedure to selectively protected imidates we investigated donors 9 and 1042 containing a selectively removable Fmoc protecting group at C-4 and C-6, respectively. TMSOTf promoted glycosylations afforded disaccharide 11 in 89 and 76% yields, respectively. The latter yield could be increased to 95% by using 10 equiv of donor 10.
Fmoc Deprotection and Reiteration for the Synthesis of Oligosaccharides
Having optimized conditions for glycosylation we decided to undertake the synthesis of two linear oligosaccharides 12 and 14. General programming outline is presented in Scheme 4. For the synthesis of trisaccharide 12 we selected glycosyl donor 10 equipped with the selectively removable Fmoc group at C-6. Previously, we have shown that Fmoc can be removed using mild reagents (piperidine/DMF, 2–5 min or TEA/CH2Cl2, 10–20 min using HPLC setup) and also provides a very straightforward and informative mode for monitoring the deprotection step and quantification of the glycosylation.42 To gain better yields and minimize side reactions we decided to use a larger excess of donor 10 (10 equiv). After washing and swelling of the resin containing acceptor 3 (0.022 mmol, pump D, steps 1 and 2, Scheme 4), pump C was programmed to deliver donor 10 (0.22 mmol) in CH2Cl2 (2 mL total volume) at 0.5 mL/min, which was then recirculated for 10 min. Again, all automated sequence steps have been monitored with the UV detector. The autosampler was programmed to deliver a solution of promoter in CH2Cl2 (3 injections of 100 μL each) and the resulting reaction mixture was recirculated for 60–90 min. When the UV-monitoring showed no change in absorbance of the eluate passing through the detector, the system was washed with CH2Cl2 (pump D, 1.0 mL/min rate flow for 10 min).
Scheme 4.

Automation of Glycosylation-Deprotection-Cleavage Sequences for the Synthesis of Oligosaccharides 12 and 14
A capping step in the synthetic cycle is important because it prevents the accumulation of shorter oligosaccharides due to incomplete reactions. Capping can be as simple as acetylation with Ac2O in pyridine,65 or by using benzoyl isocyanate in CH2Cl2, a procedure developed by Schmidt.66 It should be mentioned that due to high yields achieved in glycosylations of reactive primary hydroxyls with trichloroacetimidates capping was found unnecessary. To affect the deprotection of the Fmoc group, pump A was programmed to deliver a solution of triethylamine/CH2Cl2 (1/1, v/v 1.0 mL/min flow rate).
The release of the dibenzofulvene-triethylamine adduct was monitored by using the UV detector set at 312 nm. Upon reaching the baseline indicating that dibenzofulvene-triethylamine is no longer produced (20 min/20 mL total volume for step 5), the pump D was engaged for washing (step 6, 10 min). The resulting solid-phase bound disaccharide acceptor was glycosylated with donor 10 following essentially the same programming sequence as that for the first cycle. Upon completion of the glycosylation and washing (steps 7 and 8) pump B was engaged to deliver a solution of NaOMe/CH2Cl2/MeOH (0.04/1/1, v/v/v) at the flow rate of 1.0 mL/min for 10 min to remove the resulting trisaccharide (step 13). The eluate was collected, neutralized, concentrated, and the residue was acetylated with Ac2O in pyridine to afford trisaccharide 12 in 80% yield.
To investigate whether the trisaccharide sequence achieved during the synthesis of 12 could be extended further we explored a possibility for the chain elongation. For this purpose, we repeated the same steps 1–8 as those described for the synthesis of 12. It should be noted that in this case a completely automated sequence was reproduced simply by using the same program as previously. The solid phase bound trisaccharide intermediated was subjected to Fmoc deprotection (step 9) and washing (step 10). The subsequent glycosylation step was performed using lactosyl donor 1367 with the main aim of determining the scope of using larger building blocks (step 11). The glycosylation with disaccharide donor 13 was performed following essentially the same programming sequence as that for other glycosylations described in this article. Upon completion of the glycosylation and washing (steps 11 and 12) pump B was engaged to deliver a solution of NaOMe/CH2Cl2/MeOH (0.04/1/1, v/v/v) at the flow rate of 1.0 mL/min for 10 min followed by recirculation for additional 10 min to remove the resulting pentasaccharide (step 13). The eluate was collected, neutralized, concentrated, and the residue was acetylated with Ac2O in pyridine to afford pentasaccharide 14 in 67% yield overall.
CONCLUSIONS
In conclusion, we optimized the synthetic and operational strategies for HPLC-based automation, and have created a generally useful tool for accelerating glycan synthesis. This automated technology offers a transformative, semiautomatic approach to synthesis. Automated HPLC-based synthesis introduces rather sophisticated yet affordable in situ monitoring and reagent recirculation concepts. This basic approach provided a solid basis for the implementation of a standard autosampler system for the fully automated delivery of reagents. Further optimization of HPLC technology and its application using different resin, spacers, linkers is currently underway. Our efforts are also focusing on developing efficient protocols for the synthesis of branched hetero-oligosaccharides as well as using the autosampler for delivering all sugar building blocks and deprotecting reagents necessary for the synthesis.
EXPERIMENTAL SECTION
General Methods
The reactions were performed using commercial reagents and the ACS grade solvents were purified and dried according to standard procedures. Column chromatography was performed on silica gel 60 (70–230 mesh), reactions were monitored by TLC on Kieselgel 60 F254. The compounds were detected by examination under UV light and by charring with 10% sulfuric acid in methanol. Solvents were removed under reduced pressure at <40 °C. CH2Cl2 was distilled from CaH2 directly prior to application. Pyridine and acetonitrile were dried by refluxing with CaH2 and then distilled and stored over molecular sieves (3 Å). Molecular sieves (4 Å), used for reactions, were crushed and activated in vacuo at 390 °C during 8 h in the first instance and then for 2–3 h at 390 °C directly prior to application. Dowex Monosphere 650C (H+) was washed three times with MeOH and stored under MeOH. Optical rotations were measured using a polarimeter. 1H NMR spectra were recorded at 300 or 600 MHz, 13C NMR spectra were recorded at 75 or 150 MHz. The 1H chemical shifts are referenced to the signal of the residual CHCl3 (δH = 7.24 ppm). The 13C chemical shifts are referenced to the central signal of CDCl3 (δC = 77.23 ppm). HRMS determinations were made with the use of a mass spectrometer with FAB ionization and ion-trap detection. Agilent 1260 infinity HPLC System and Agilent 1260 Variable Wavelength UV–vis Detector were used to assemble the automated synthesizer.
Synthesis of Glycosyl Acceptor 3
8-(tert-Butyldiphenylsilyloxy)-oct-1-yl 2,3,4-Tri-O-benzyl-6-O-triphenylmethyl-α-D-glucopyranoside (17)
A mixture of ethyl 2,3,4-tri-O-benzyl-1-thio-6-O-triphenyl-methyl-α-D-glucopyranoside (15,68 3.0 g, 4 mmol), 8-(tert-butyldiphenylsilyloxy)octan-1-ol (16,69 1.3 g, 3.3 mmol), and freshly activated molecular sieves (4 Å, 3.0 g) in diethyl ether (100 mL) was stirred under argon for 1 h at rt. N-Iodosuccinimide (NIS, 1.8 g, 8.0 mmol) and TfOH (71 μL, 0.8 mmol) were added, and the resulting mixture was stirred for 20 min at rt. After that, the solids were filtered off and washed successively with CH2Cl2. The combined filtrate (~200 mL) was washed with sat. aq. Na2SO4 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford compound 17 (2.27 g, 65%) as a colorless foam. Analytical data for 17: Rf = 0.62 (ethyl acetate/hexanes, 1/4, v/v); [α]D25 + 25.3 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 0.89 (s, 9H, C(CH3)3), 1.12–1.20 (m, 8H, 4 × CH2), 1.38, 1.50 (2 m, 4H, 2 × CH2), 3.03 (dd, 1H, J5,6b = 4.8 Hz, J6a,6b = 9.9 Hz, H-6a), 3.31–3.32 (m, 2H, H-6b, OCH2a), 3.42–3.50 (m, 4H, H-2, 4, CH2), 3.56 (m, 1H, OCH2b), 3.68 (m, 1H, H-5), 3.82 (dd, 1H, J3,4 = 9.2 Hz, H-3), 4.13 (d, 1H, 2J = 10.4 Hz, 1/2 CH2Ph), 4.52–4.72 (m, 5H, H-1, 2 x CH2Ph), 4.81 (d, 1H, 2J = 10.6 Hz, 1/2 CH2Ph), 6.69–7.52 (m, 40H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 19.4, 25.9 (×3), 26.4, 29.5 (×2), 29.6 (×4), 30.2, 32.7 (×2), 63.8, 64.2, 70.5, 73.3, 75.2, 76.1, 78.5, 80.6, 82.5, 86.4, 96.7, 126.9 (×2), 127.1, 127.7 (×6), 127.8 (×4), 127.9 (×3), 128.1, 128.3 (×2), 128.4, 128.6 (×3), 128.9 (×4), 129.0 (×3), 129.6 (×3), 134.3, 135.7 (×6), 138.1, 138.7, 139.0, 144.1, 144.7 ppm; HR-FAB MS [M+Na]+ calcd for C70H78O7SiNa 1081.5415, found 1081.5435.
8-Hydroxyoct-1-yl 2,3,4-Tri-O-benzyl-6-O-triphenylmethyl-α-D-glucopyranoside (18)
Tetrabutylammonium fluoride (TBAF, 1.72 mL, 1.995 mmol) was added to a solution of 17 (2.0 g, 1.995 mmol) in THF (14 mL) and the resulting mixture was stirred under argon for 3 h at rt. After that, the reaction mixture was diluted with CH2Cl2 (~150 mL), washed with water (20 mL), sat. aq. NaHCO3 (20 mL), and water (20 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–toluene gradient elution) to afford compound 18 (1.68 g, 95%) as a colorless foam. Analytical data for 18: Rf = 0.45 (ethyl acetate/hexanes, 1/2, v/v); [α]D26 + 19.7 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.21–1.29 (m, 8H, 4 × CH2), 1.43, 1.61 (2 m, 4H, 2 × CH2), 3.17 (dd, 1H, J5,6a = 3.0 Hz, J6a,6b = 9.8 Hz, H-6a), 3.45–3.61 (m, 6H, H-2, 4, 6b, OCH2a, OCH2), 3.66 (m, 1H, OCH2b), 3.83 (m, 1H, H-5), 3.96 (dd, 1H, J3,4 = 10.2 Hz, H-3), 4.27 (d, 1H, 2J = 10.4 Hz, 1/2 CH2Ph), 4.67 (d, 1H, 2J = 10.3 Hz, 1/2 CH2Ph), 4.69 (d, 1H, 2J = 12.1 Hz, 1/2 CH2Ph), 4.76–4.86 (m, 3H, H-1, CH2Ph), 4.95 (d, 1H, 2J = 10.6 Hz, 1/2 CH2Ph), 6.83–7.46 (m, 30H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 25.8, 26.3, 29.4, 29.5, 29.6, 32.9, 62.8, 63.1, 68.1, 70.5, 73.2, 75.2, 76.0, 78.4, 80.6, 82.4, 86.4, 96.7, 127.0 (×3) 127.7, 127.8, 127.9 (×6), 128.0 (×3), 128.2 (×2) 128.3 (×5), 128.4 (×4), 128.9 (×6), 138.0, 138.6, 138.9, 144.1 (×2) ppm; HR-FAB MS [M+Na]+ calcd for C54H60O7Na 843.4237, found 843.4257.
8-(3-Carboxypropanoyloxy)oct-1-yl 2,3,4-Tri-O-benzyl-6-O-triphenylmethyl-α-D-glucopyranoside (1)
Succinic anhydride (0.440 g, 4.39 mmol) and 4-dimethylaminopyridine (DMAP, 0.053 g, 0.438 mmol) were added to a solution of compound 18 (1.3 g, 1.464 mmol) in pyridine (5.0 mL) and the resulting mixture was stirred under argon for 16 h at 65 °C. After that, the volatiles were removed under the reduced pressure and the residue was coevaporated with toluene (3 × 10 mL) and purified by column chromatography on silica gel (ethyl acetate–toluene gradient elution) to afford the title compound (1.30 g, 97%) as a colorless foam. Analytical data for 1: Rf = 0.25 (ethyl acetate/hexanes, 1/1, v/v); [α]D27 + 21.8 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.28–1.37 (m, 8H, CH2), 1.54–1.66 (m, 4H, 2 × CH2), 2.55–2.63 (m, 4H, 2 × CH2), 3.17 (dd, 1H, J5,6a = 4.9 Hz, J6a,6b = 9.8 Hz, H-6a), 3.44–3.47 (m, 2H, H-6b, OCH2a), 3.53–3.61 (m, 2H, H-2, 4), 3.69–3.72 (m, 1H, OCH2b), 3.82 (m, 1H, H-5), 3.95 (dd, 1H, J3,4 = 9.2 Hz, H-3), 4.03 (t, 2H, J = 6.6 Hz, CH2), 4.25 (d, 1H, 2J = 10.3 Hz, 1/2 CH2Ph), 4.66 (d, 1H, 2J = 10.4 Hz, 1/2 CH2Ph), 4.68 (d, 1H, 2J = 12.1 Hz, 1/2 CH2Ph), 4.75–4.84 (m, 4H, H-1, 1 1/2 CH2Ph) 4.93 (d, 1H, 2J = 10.7 Hz, 1/2 CH2Ph), 6.83–7.44 (m, 30H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 25.1, 26.1, 28.5, 28.7, 28.9, 29.1, 29.3, 20.4, 62.7, 65.0, 67.9, 70.3, 73.1, 75.1, 75.9, 78.3, 80.4, 82.2, 86.3, 95.5, 126.9 (×3), 127.7 (×2), 127.8 (×5), 127.9 (×2), 128.1 (×3), 128.2 (×3), 128.3 (×5), 128.4, 128.8 (×5), 137.8, 138.4, 138.6, 138.8, 144.0 (×3), 144.5, 172.2 ppm; HR-FAB [M+Na]+ calcd for C58H64NaO10 943.4397, found 943.4371.
Resin-Bound Acceptor 3
JandaJel amine resin (1% cross-linked polystyrene, 500 mg, 0.25 mmol) was added to a solution of 1 (253 mg, 0.275 mmol), 1-ethyl-3-(3-(dimethylamino)propyl)carbodiimide hydrochloride (EDC, 105.4 mg, 0.55 mmol), and DMAP (30 mg, 0.25 mmol) in CH2Cl2 (5.0 mL) and the resulting suspension was agitated under argon for 18 h at rt. When the Kaiser test70 showed the negative result, the resin was filtered off; washed with CH2Cl2 (3 × 20 mL), methanol (3 × 20 mL), and CH2Cl2 (3 × 20 mL); and dried in vacuo for 4 h. The resulting resin 2 was swelled in CH2Cl2 (10 mL) for 60 min at rt. A 10% solution of TFA in wet CH2Cl2 (5.0 mL) was added dropwise and the resulting suspension was agitated for 3 h at rt. The resin was filtered off; washed with CH2Cl2 (3 × 20 mL), methanol (3 × 20 mL), and CH2Cl2 (3 × 20 mL); and dried in vacuo for 6 h to afford the title compound. The loading (0.44 mmol/g) was determined by the quantification of TrOH formed as a result of the treatment with TFA.
Synthesis of Glycosyl Donors
Benzoxazolyl 2,3,4,6-Tetra-O-benzoyl-1-thio-β-D-glucopyranoside (4)
The synthesis of the title compound was performed in accordance with the reported procedure and its analytical data was in accordance with that previously described.59
Phenyl 2,3,4,6-Tetra-O-benzoyl-1-thio-β-D-glucopyranoside (5)
The synthesis of the title compound was performed in accordance with the reported procedure and its analytical data was in accordance with that previously described.71
3,3-Difluoro-3H-indol-2-yl 2,3,4,6-Tetra-O-benzoyl-α-D-glucopyranoside (6)
The synthesis of the title compound was performed in accordance with the reported procedure and its analytical data was in accordance with that previously described.62
Bis(buthyl)phosphoryl 2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranoside (7)
A mixture of 5 (700 mg, 1.0 mmol), dibutyl phosphate (0.58 mL, 3.0 mmol), and freshly activated molecular sieves (4 Å, 1.5 g) in CH2Cl2 (15 mL) was stirred under argon for 1 h at rt. After that, NIS (265 mg, 1.2 mmol) and TfOH (10 μL, 0.12 mmol) were added and the resulting mixture was stirred for 18 h at rt. The solid was then filtered off and rinsed successively with CH2Cl2. The combined filtrate (~40 mL) was washed with sat. aq. Na2SO4 (10 mL) and water (3 × 10 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–hexane gradient elution) to afford the title compound (1.30 g, 97%) as a clear syrup. Analytical data for 7: Rf = 0.24 (ethyl acetate/hexanes, 1/1, v/v); [α]D26 + 33.9 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 0.64, 0.79 (2 t, 6H, 2 × CH3), 0.90 (2 m, 4H, 2 × CH2), 1.18–1.69 (m, 4H, 2 x CH2), 3.73 (m, 2H, OCH2a), 3.99 (m, 2H, OCH2b), 4.28 (m, 1H, H-5), 4.46 (dd, 1H, J5,6a = 5.0 Hz, J6a,6b = 12.3 Hz, H-6a), 4.64 (dd, 1H, J5,6b = 2.5 Hz, H-6b), 5.60–5.75 (m, 3H, H-1, 2, 4), 5.91 (dd, 1H, J3,4 = 10.5 Hz, H-3), 7.15–7.80 (m, 20H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 13.4, 13.5, 18.2, 18.5, 31.7, 31.8, 32.0, 62.6, 67.0, 68.0, 68.1, 68.2, 69.0, 71.7, 71.8, 72.5, 73.0, 96.6, 128.3 (×4), 128.5 (×4), 128.6, 128.7, 129.4, 129.8 (×5), 129.9 (×4), 133.3, 133.4, 133.5, 133.6 ppm. HR-FAB [M+Na]+ calcd for C42H45O13PNa 811.2495, found 811.2505.
2,3,4,6-Tetra-O-benzoyl-β-D-glucopyranosyl Trichloroacetimidate (8)
The synthesis of the title compound was performed in accordance with the reported procedure and its analytical data was in accordance with that previously described.64,72
2,3,6-Tri-O-benzoyl-4-O-(9-fluorenylmethoxycarbonyl)-α/β-D-glucopyranosyl Trichloroacetimidate (9)
The synthesis of the title compound was performed in accordance with the reported procedure and its analytical data was in accordance with that previously described.42
2,3,4-Tri-O-benzoyl-6-O-(9-fluorenylmethoxycarbonyl)-α/β-D-glucopyranosyl Trichloroacetimidate (10)
The synthesis of the title compound was performed in accordance with the reported procedure and its analytical data was in accordance with that previously described.42
O-(2,3,4,6-Tetra-O-benzoyl-β-D-galactopyranosyl)-(1→4)-2,3,6-tri-O-benzoyl-β-D-glucopyranosyl Trichloroacetimidate (13)
The synthesis of the title compound was performed in accordance with the reported procedure and its analytical data was in accordance with that previously described.67
HPLC-Mediated Synthesis of Oligosaccharides. General Procedure for Glycosylation and Cleavage
Functionalized JandaJel resin 3 (50 mg, 0.022 mmol) was packed in an Omnifit glass chromatography column and the latter was integrated into the HPLC system. Pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 5 min (5 mL, step 1). The system was then switched to the recirculation mode and the delivery of CH2Cl2 continued for 30 min at 1.0 mL/min (swelling, step 2). After that, pump D was stopped and pump C was programmed to deliver a solution of glycosyl donor (4–10, 0.10 mmol) in CH2Cl2 (2 mL) at a flow rate of 0.5 mL/min (step 3). This step was monitored by the integrated UV detector (λmax = 254 nm). The integrated autosampler was programmed to inject a solution of the promoter in CH2Cl2 (3 × 100 μL) at 10, 12, and 14 min and the resulting mixture (~2.3 mL) was recirculated for 60–90 min until the UV detector recorded no change in absorbance of the eluate. After that, pump C was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 4). After that, pump D was stopped and pump B was programmed to deliver a 0.1 M solution of NaOMe in CH3OH/CH2Cl2 (10 mL, 0.04/1/1, v/v/v) that was recirculated at 1.0 mL/min for 20 min (step 5). Pump B was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min for 10 min, and the combined eluate was neutralized with Dowex (H+) resin. The resin was filtered off, washed successively with CH2Cl2 and CH3OH, and the combined filtrate was concentrated in vacuo to afford the crude residue that was subjected to subsequent acetylation.
General Procedure for Acetylation of Released Disaccharide
A crude residue was redissolved in pyridine (2.0 mL), Ac2O (73 μL, 0.771 mmol) was added dropwise, and the resulting mixture was stirred for 16 h at rt. The reaction mixture was quenched with CH3OH (~1.0 mL) and the volatiles were removed under the reduced pressure. The residue was diluted with CH2Cl2 (20 mL) and washed with 1 N HCl (2 × 10 mL), water (20 mL), sat. aq. NaHCO3 (20 mL), and water (2 × 20 mL). The organic phase was separated, dried with MgSO4, and concentrated in vacuo. The residue was purified by column chromatography on silica gel (ethyl acetate–toluene gradient elution) to afford disaccharide 11.
8-Acetyloxyoctan-1-yl O-(2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (11)
The title compound was synthesized from glycosyl donors 4–10 and glycosyl acceptor 3 in 50–89% yield. Analytical data for 11: Rf = 0.57 (ethyl acetate/hexanes, 1/1, v/v); [α]D27 + 8.90 (c = 1.0, CHCl3); 1H NMR (300 MHz, CDCl3): δ, 1.22–1.29 (m, 8H, 4 × CH2), 1.50–1.58 (m, 4H, 2 × CH2), 1.91–2.07 (5 s, 15H, 5 × COCH3), 3.32–3.50 (m, 3H, H-2, 4, OCH2a), 3.55–3.75 (m, 4H, 1/2 OCH2a, H-5, H-6b, H-5′), 3.95 (dd, 1H, J3,4 = 9.3 Hz, H-3), 3.98–4.23 (m, 5H, H-6a, 6′a, 6′b, OCH2b), 4.47 (d, 1H, J1′,2′ = 7.5 Hz, H-1′), 4.50 (dd, 1H, 2J = 9.8 Hz, 1/2 CH2Ph), 4.60 (d, 1H, 2J = 12.0 Hz, 1/2 CH2Ph), 4.68 (d, 1H, J1,2 = 3.5 Hz, H-1), 4.74 (d, 1H, 2J = 12.0 Hz, 1/2 CH2Ph), 4.76 (d, 1H, 2J = 10.9 Hz, 1/2 CH2Ph), 4.83 (d, 1H, 2J = 10.7 Hz, 1/2 CH2Ph), 4.96 (d, 1H, 2J = 11.0 Hz, 1/2 CH2Ph), 5.02 (dd, 1H, J2′,3′ = 9.2 Hz, H-2′), 5.05 (dd, 1H, J4′,5′ = 9.6 Hz, H-4′), 5.15 (dd, 1H, J3′,4′ = 9.4 Hz, H-3′), 7.22–7.31 (m, 15H, aromatic) ppm; 13C NMR (75 MHz, CDCl3): δ, 20.8 (×2), 20.9 (×2), 21.3, 26.1, 26.3, 28.7, 29.4, 29.5, 29.6, 57.2, 62.1, 64.8, 68.2, 68.3, 68.5, 68.6, 69.6, 71.4, 71.9, 73.0, 73.2, 73.3, 75.1, 75.8, 80.2, 96.8, 100.8, 101.8, 127.7, 128.0 (×2), 128.1, 128.2, 128.3 (×2), 128.5 (×2), 128.6 (×2), 128.7 (×2), 138.3, 138.4, 139.0, 169.2, 169.5, 170.5, 170.8, 171.4 ppm; HR-FAB [M+Na]+ calcd for C51H66O17 Na 973.4198, found 973.4175.
8-Acetyloxyoct-1-yl O-(2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4-tri-O-acetyl-β-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-acetyl-α-D-glucopyranoside (12)
Functionalized JandaJel resin 3 (50 mg, 0.022 mmol) was packed in an Omnifit glass chromatography column and the latter was integrated into the HPLC system. Pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 5 min (5 mL, step 1). The system was then switched to the recirculation mode and the delivery of CH2Cl2 continued for 30 min at 1.0 mL/min (swelling, step 2). After that, pump D was stopped and pump C was programmed to deliver a solution of donor 10 (188 mg, 0.22 mmol) in CH2Cl2 (2 mL) at a flow rate of 0.5 mL/min (step 3). This step was monitored by the integrated UV detector (λmax = 254 nm). The integrated autosampler was programmed to inject a solution of TMSOTf (81 μL, 0.44 mmol) in CH2Cl2 (3 × 100 μL) at 10, 12, and 14 min and the resulting mixture (~2.3 mL) was recirculated for 60–90 min until the UV detector recorded no change in absorbance of the eluate. After that, pump C was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 4). After that, pump D was stopped and pump A was programmed to deliver a solution of TEA/CH2Cl2 (1/1, v/v) for 20 min at 1.0 mL/min (step 5). This step was monitored by the integrated UV detector (λmax = 312 nm). After that, pump A was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 6). After that, pump D was stopped and pump C was programmed to deliver a solution of donor 10 (188 mg, 0.22 mmol) in CH2Cl2 (2 mL) at a flow rate of 0.5 mL/min (step 7). This step was monitored by the integrated UV detector (λmax = 254 nm). The integrated autosampler was programmed to inject a solution of TMSOTf (81 μL, 0.44 mmol) in CH2Cl2 (3 × 100 μL) at 10, 12, and 14 min and the resulting mixture (~2.3 mL) was recirculated for 60–90 min until the UV detector recorded no change in absorbance of the eluate. After that, pump C was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 8). After that, pump D was stopped and pump B was programmed to deliver a 0.1 M solution of NaOMe in CH3OH/CH2Cl2 (10 mL, 0.04/1/1, v/v/v) that was recirculated at 1.0 mL/min for 20 min (step 9). Pump B was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min for 10 min, and the combined eluate was neutralized with Dowex (H+) resin. The resin was filtered off, washed successively with CH2Cl2 and CH3OH, and the combined filtrate was concentrated in vacuo to afford the crude residue that was subjected to subsequent acetylation in accordance with the general procedure, as described for the synthesis of compound 11. The crude residue was purified by column chromatography on silica gel (ethyl acetate–toluene gradient elution) to afford trisaccharide 12 in 80% yield. Analytical data for 12: Rf = 0.44 (ethyl acetate/hexanes, 1/1, v/v); [α]D27 + 6.34 (c = 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ, 1.22–1.28 (m, 8H, 4 × CH2), 1.58–159 (m, 4H, 2 × CH2), 1.95–2.06 (8 s, 24H, 8 × COCH3), 3.31 (m, 1H, OCH2a), 3.45–3.49 (m, 2H, H-2, 4), 3.56–3.63 (m, 4H, H-5′, 5″, 6′a, OCH2b), 3.74–3.70 (m, 2H, H-5, 6a), 3.81 (d, 1H, J6′a,6′b = 10.5 Hz, H-6′b), 3.96 (dd, 1H, J3,4 = 9.1 Hz, H-3), 3.93–4.08 (m, 4H, H-6″a, 6b, CH2), 4.22 (dd, 1H, J5″,6″b = 4.4 Hz, J6″a,6″b = 12.3 Hz, H-6″b), 4.46–4.49 (m, 3H, H-1′, 1″, 1/2 CH2Ph), 4.60 (d, 1H, 2J = 12.1 Hz, 1/2 CH2Ph), 4.68 (d, 1H, J1,2 = 2.8 Hz, H-1), 4.75 (dd, 2H, 2J = 13.7 Hz, CH2Ph), 4.86–4.82 (m, 2H, H-4′, 1/2 CH2Ph), 4.90–5.01 (m, 4H, H-2′, 2″, 4″, 1/2 CH2Ph), 5.08–5.12 (m, 2H, H-3′, 3″), 7.23–7.32 (m, 15H, aromatic) ppm; 13C NMR (150 MHz, CDCl3): δ, 20.7, 20.8, 20.9 (×2), 20.92, 21.2, 25.9, 26.4, 28.7, 29.4, 29.5, 29.6, 61.9, 64.7, 68.0, 68.1, 63.3, 68.4, 69.2, 69.7, 71.2, 71.5, 72.1, 72.8, 73.1, 73.2, 73.4, 75.1, 75.8, 77.6, 80.2, 82.0, 96.9, 100.5, 100.9, 127.7, 128.0 (×3), 128.1 (×3), 128.2 (×2), 128.4, 128.5 (×2), 128.6 (×2), 128.7, 128.8 (×2), 138.3, 138.4, 139.0, 169.2, 169.3, 169.5, 169.7, 170.3, 170.4, 170.8, 171.4 ppm; HR-FAB [M+Na]+ calcd for C63H82O25Na 1261.5025, found 1261.5071.
8-Acetyloxyoct-1-yl O-(2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl)-(1→4)-O-(2,3,6-tri-O-acetyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4-tri-O-acetyl-β-D-glucopyranosyl)-(1→6)-O-(2,3,4-tri-O-acetyl-β-D-glucopyranosyl)-(1→6)-2,3,4-tri-O-benzyl-α-D-glucopyranoside (14)
Functionalized JandaJel resin 3 (50 mg, 0.022 mmol) was packed in an Omnifit glass chromatography column and the latter was integrated into the HPLC system. Pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 5 min (5 mL, step 1). The system was then switched to the recirculation mode and the delivery of CH2Cl2 continued for 30 min at 1.0 mL/min (swelling, step 2). After that, pump D was stopped and pump C was programmed to deliver a solution of donor 10 (188 mg, 0.22 mmol) in CH2Cl2 (2 mL) at a flow rate of 0.5 mL/min (step 3). This step was monitored by the integrated UV detector (λmax = 254 nm). The integrated autosampler was programmed to inject a solution of TMSOTf (81 μL, 0.44 mmol) in CH2Cl2 (3 × 100 μL) at 10, 12, and 14 min and the resulting mixture (~2.3 mL) was recirculated for 60–90 min until the UV detector recorded no change in absorbance of the eluate. After that, pump C was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 4). After that, pump D was stopped and pump A was programmed to deliver a solution of TEA/CH2Cl2 (1/1, v/v) for 20 min at 1.0 mL/min (step 5). This step was monitored by the integrated UV detector (λmax = 312 nm). After that, pump A was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 6). After that, pump D was stopped and pump C was programmed to deliver a solution of donor 10 (188 mg, 0.22 mmol) in CH2Cl2 (2 mL) at a flow rate of 0.5 mL/min (step 7). This step was monitored by the integrated UV detector (λmax = 254 nm). The integrated autosampler was programmed to inject a solution of TMSOTf (81 μL, 0.44 mmol) in CH2Cl2 (3 × 100 μL) at 10, 12, and 14 min and the resulting mixture (~2.3 mL) was recirculated for 60–90 min until the UV detector recorded no change in absorbance of the eluate. After that, pump C was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 8). After that, pump D was stopped and pump A was programmed to deliver a solution of TEA/CH2Cl2 (1/1, v/v) for 20 min at 1.0 mL/min (step 9). This step was monitored by the integrated UV detector (λmax = 312 nm). After that, pump A was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 10). After that, pump D was stopped and pump C was programmed to deliver a solution of donor 13 (266 mg, 0.22 mmol) in CH2Cl2 (2 mL) at a flow rate of 0.5 mL/min (step 11). This step was monitored by the integrated UV detector (λmax = 254 nm). The integrated autosampler was programmed to inject a solution of TMSOTf (81 μL, 0.44 mmol) in CH2Cl2 (3 × 100 μL) at 10, 12, and 14 min and the resulting mixture (~2.3 mL) was recirculated for 60–90 min until the UV detector recorded no change in absorbance of the eluate. After that, pump C was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min, and the eluate was discarded after washing for 10 min (10 mL, step 12). After that, pump D was stopped and pump B was programmed to deliver a 0.1 M solution of NaOMe in CH3OH/CH2Cl2 (10 mL, 0.04/1/1, v/v/v) that was recirculated at 1.0 mL/min for 20 min (step 13). Pump B was stopped and pump D was programmed to deliver CH2Cl2 at 1.0 mL/min for 10 min, and the combined eluate was neutralized with Dowex (H+) resin. The resin was flltered off, washed successively with CH2Cl2 and CH3OH, and the combined filtrate was concentrated in vacuo to afford the crude residue that was subjected to subsequent acetylation in accordance with the general procedure, as described for the synthesis of compound 11. The crude residue was purified by column chromatography on silica gel (ethyl acetate–toluene gradient elution) to afford pentasaccharide 14 in 67% yield. Analytical data for 14: Rf = 0.26 (ethyl acetate/hexanes, 1/1, v/v); [α]D27 −1.94 (c = 1.0, CHCl3); 1H NMR (600 MHz, CDCl3): δ, 1.26–1.30 (m, 8H, 4 x CH2), 1.55–159 (m, 4H, 2 × CH2), 1.90–2.12 (m, 42H, 14 × COCH3), 3.30 (m, 1H, OCH2a), 3.44–3.46 (m, 2H, H-2, 4), 3.53–3.63 (m, 6H, H-5′, 5″, 5‴, 6′a, 6″a, OCH2b), 3.68–3.73 (m, 2H, H-6a, 6″b), 3.77–3.86 (m, 4H, H-5, 5⁗, 6⁗ a, 6′b), 3.94 (dd, 1H, J3,4 = 9.2 Hz, H-3), 4.03–4.10 (m, 7H, H-2″, 2⁗, 6b, 6″b, 6⁗b, CH2), 4.42–4.52 (m, 6H, H-1′, 1″, 1‴, 1⁗, 6‴b, 1/2 CH2Ph), 4.59 (d, 1H, 2J = 12.0 Hz, 1/2 CH2Ph), 4.69 (d, 1H, J1,2 = 3.1 Hz, H-1), 4.73 (dd, 2H, 2J = 11.6 Hz, CH2Ph), 4.82–7.97 (m, 6H, H-2′, 2‴, 4′, 4″, 4‴, 1/2 CH2Ph), 4.99 (dd, 1H, J3⁗,4⁗ = 8.4 Hz, H-3⁗), 5.06–5.16 (m, 4H, H-3′, 3″, 3‴, 1/2 CH2Ph), 5.31 (d, 1H, J4⁗,5⁗ = 2.1 Hz, H-4⁗), 7.24–7.32 (m, 15H, aromatic) ppm; 13C NMR (150 MHz, CDCl3): δ, 20.6 (×2), 20.7 (×3), 20.8, 20.9 (×2), 21.1, 26.0, 26.2, 28.7, 29.4, 29.5, 29.8 (×2), 62.0, 64.4 (×2), 66.6, 68.0, 68.1, 69.0, 69.1, 69.2 (×2), 69.7, 70.7 (×2), 71.0, 71.2, 71.4 (×2), 71.5, 72.8, 72.9, 73.0, 73.1 (×2), 73.2, 75.0, 75.7, 76.2, 77.5, 80.1, 81.9, 96.7, 100.5, 100.7, 100.9, 101.1, 127.6, 127.9 (×7), 128.0, 128.1 (×3), 128.4 (×3), 128.5 (×3), 128.6 (×3), 138.3, 138.4, 138.9, 169.1, 169.2, 169.3, 169.5, 169.6, 169.8, 170.2, 170.3, 170.4 (×3), 171.3 ppm; HR-FAB [M+Na]+ calcd for C87H114O41 Na 1837.6716, found 1837.6703.
Supplementary Material
Acknowledgments
This work was supported by a grant from the National Institute of General Medical Sciences (GM111835). S.G.P is indebted to — the UM St. Louis Graduate School for awarding him with the Dissertation Fellowship. We thank Dr. Rensheng Luo (UM— St. Louis) for help with acquiring spectral data using 600 MHz NMR spectrometer that was purchased thanks to the NSF — (award CHE-0959360). Dr. Winter and Mr. Kramer (UM St. Louis) are thanked for HRMS determinations.
Footnotes
Notes
The authors declare no competing financial interest.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.6b01439.
Spectra for all new compounds (PDF)
References
- 1.Mydock LK, Demchenko AV. Org Biomol Chem. 2010;8:497. doi: 10.1039/b916088d. [DOI] [PubMed] [Google Scholar]
- 2.Crich D. Acc Chem Res. 2010;43:1144. doi: 10.1021/ar100035r. [DOI] [PubMed] [Google Scholar]
- 3.Christensen HM, Oscarson S, Jensen HH. Carbohydr Res. 2015;408:51. doi: 10.1016/j.carres.2015.02.007. [DOI] [PubMed] [Google Scholar]
- 4.Smoot JT, Demchenko AV. Adv Carbohydr Chem Biochem. 2009;62:161. doi: 10.1016/S0065-2318(09)00005-5. [DOI] [PubMed] [Google Scholar]
- 5.Fruchtel JS, Jung G. Angew Chem, Int Ed Engl. 1996;35:17. [Google Scholar]
- 6.Winter M. In: Combinatorial peptide and nonpeptide libraries: a handbook. Jung G, editor. VCH; Wienheim, New York, Basel, Cambridge, Tokyo: 1996. p. 465. [Google Scholar]
- 7.Merrifield B. Br Polym J. 1984;16:173. [Google Scholar]
- 8.Krishnamurthy VR, Dougherty A, Kamat M, Song X, Cummings RD, Chaikof EL. Carbohydr Res. 2010;345:1541. doi: 10.1016/j.carres.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Toy PH, Lam Y, editors. Solid-Phase Organic Synthesis. John Wiley & Sons, Inc; Hoboken: 2012. [Google Scholar]
- 10.Schuerch C, Frechet JM. J Am Chem Soc. 1971;93:492. [Google Scholar]
- 11.Schmidt RR, Jonke S, Liu K. In: ACS Symp Ser (Frontiers in Modern Carbohydrate Chemistry) Demchenko AV, editor. Vol. 960. Oxford Univ. Press; 2007. p. 209. [Google Scholar]
- 12.Seeberger PH. J Carbohydr Chem. 2002;21:613. [Google Scholar]
- 13.Seeberger PH, Haase WC. Chem Rev. 2000;100:4349. doi: 10.1021/cr9903104. [DOI] [PubMed] [Google Scholar]
- 14.Tanaka K, Fukase K. In: Solid-Phase Organic Synthesis. Toy PH, Lam Y, editors. John Wiley & Sons, Inc; Hoboken: 2012. p. 489. [Google Scholar]
- 15.Plante OJ, Palmacci ER, Seeberger PH. Science. 2001;291:1523. doi: 10.1126/science.1057324. [DOI] [PubMed] [Google Scholar]
- 16.Seeberger PH. Chem Soc Rev. 2008;37:19. doi: 10.1039/b511197h. [DOI] [PubMed] [Google Scholar]
- 17.Plante OJ, Palmacci ER, Seeberger PH. Adv Carbohydr Chem Biochem. 2003;58:35. doi: 10.1016/s0065-2318(03)58002-7. [DOI] [PubMed] [Google Scholar]
- 18.Seeberger PH. Acc Chem Res. 2015;48:1450. doi: 10.1021/ar5004362. [DOI] [PubMed] [Google Scholar]
- 19.Krock L, Esposito D, Castagner B, Wang CC, Bindschadler P, Seeberger PH. Chem Sci. 2012;3:1617. [Google Scholar]
- 20.Sears P, Wong CH. Science. 2001;291:2344. doi: 10.1126/science.1058899. [DOI] [PubMed] [Google Scholar]
- 21.Hsu CH, Hung SC, Wu CY, Wong CH. Angew Chem, Int Ed. 2011;50:11872. doi: 10.1002/anie.201100125. [DOI] [PubMed] [Google Scholar]
- 22.Tanaka H, Matoba N, Tsukamoto H, Takimoto H, Yamada H, Takahashi T. Synlett. 2005:0824. [Google Scholar]
- 23.Machida K, Hirose Y, Fuse S, Sugawara T, Takahashi T. Chem Pharm Bull. 2010;58:87. doi: 10.1248/cpb.58.87. [DOI] [PubMed] [Google Scholar]
- 24.Sugiarto G, Lau K, Qu J, Li Y, Lim S, Mu S, Ames JB, Fisher AJ, Chen X. ACS Chem Biol. 2012;7:1232. doi: 10.1021/cb300125k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, Thon V, Li Y, Yu H, Ding L, Lau K, Qu J, Hie L, Chen X. Chem Commun. 2011;47:10815. doi: 10.1039/c1cc14034e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muthana MM, Qu J, Li Y, Zhang L, Yu H, Ding L, Malekan H, Chen X. Chem Commun. 2012;48:2728. doi: 10.1039/c2cc17577k. [DOI] [PubMed] [Google Scholar]
- 27.Jaipuri FA, Pohl NL. Org Biomol Chem. 2008;6:2686. doi: 10.1039/b803451f. [DOI] [PubMed] [Google Scholar]
- 28.Song EH, Osanya AO, Petersen CA, Pohl NLB. J Am Chem Soc. 2010;132:11428. doi: 10.1021/ja103351m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L, Pohl NLB. Org Lett. 2011;13:1824. doi: 10.1021/ol2003435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang SL, Pohl NL. Org Lett. 2015;17:2642. doi: 10.1021/acs.orglett.5b01013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen C, Zhang Y, Xue M, Liu XW, Li Y, Chen X, Wang PG, Wang F, Cao H. Chem Commun. 2015;51:7689. doi: 10.1039/c5cc01330e. [DOI] [PubMed] [Google Scholar]
- 32.Li L, Liu Y, Ma C, Qu J, Calderon AD, Wu B, Wei N, Wang X, Guo Y, Xiao Z, Song J, Sugiarto G, Li Y, Yu H, Chen X, Wang PG. Chem Sci. 2015;6:5652. doi: 10.1039/c5sc02025e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nokami T, Hayashi R, Saigusa Y, Shimizu A, Liu C-Y, Mong K-KT, Yoshida J-i. Org Lett. 2013;15:4520. doi: 10.1021/ol402034g. [DOI] [PubMed] [Google Scholar]
- 34.Nokami T, Isoda Y, Sasaki N, Takaiso A, Hayase S, Itoh T, Hayashi R, Shimizu A, Yoshida J. Org Lett. 2015;17:1525. doi: 10.1021/acs.orglett.5b00406. [DOI] [PubMed] [Google Scholar]
- 35.Pistorio SG, Stine KJ, Demchenko AV. In: Carbohydrate Chemistry: State-of-the-art and challenges for drug development. Cipolla L, editor. Imperial College Press; London: 2015. p. 247. [Google Scholar]
- 36.Varki A, Cummings RD, Esko JD, Freeze HH, Bertozzi CR, Stanley P, Hart GW, Etzler ME. Essentials of Glycobiology. 2. CSH Laboratory Press; New York: 2009. [PubMed] [Google Scholar]
- 37.DeMarco ML, Woods RJ. Glycobiology. 2008;18:426. doi: 10.1093/glycob/cwn026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bertozzi CR, Kiessling LL. Science. 2001;291:2357. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- 39.Dwek RA. Chem Rev. 1996;96:683. doi: 10.1021/cr940283b. [DOI] [PubMed] [Google Scholar]
- 40.Cummings RD, Pierce JM. Chem Biol. 2014;21:1. doi: 10.1016/j.chembiol.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Transforming Glycoscience: A Roadmap for the Future. 2012 http://dels.nas.edu/Report/Transforming-Glycoscience-Roadmap/13446. [PubMed]
- 42.Ganesh NV, Fujikawa K, Tan YH, Stine KJ, Demchenko AV. Org Lett. 2012;14:3036. doi: 10.1021/ol301105y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pornsuriyasak P, Ranade SC, Li A, Parlato MC, Sims CR, Shulga OV, Stine KJ, Demchenko AV. Chem Commun. 2009:1834. doi: 10.1039/b817684a. [DOI] [PubMed] [Google Scholar]
- 44.Stine KJ. In: Carbohydrate Nanotechnology. Stine KJ, editor. Wiley; Hoboken, NJ: 2016. [Google Scholar]
- 45.Ganesh NV, Fujikawa K, Tan YH, Nigudkar SS, Stine KJ, Demchenko AV. J Org Chem. 2013;78:6849. doi: 10.1021/jo400095u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Parlato MC, Kamat MN, Wang H, Stine KJ, Demchenko AV. J Org Chem. 2008;73:1716. doi: 10.1021/jo701902f. [DOI] [PubMed] [Google Scholar]
- 47.Simon MD, Heider PL, Adamo A, Vinogradov AA, Mong SK, Li X, Berger T, Policarpo RL, Zhang C, Zou Y, Liao X, Spokoyny AM, Jensen KF, Pentelute BL. ChemBioChem. 2014;15:713. doi: 10.1002/cbic.201300796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li J, Ballmer SG, Gillis EP, Fujii S, Schmidt MJ, Palazzolo AME, Lehmann JW, Morehouse GF, Burke MD. Science. 2015;347:1221. doi: 10.1126/science.aaa5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santanilla AB, Regalado EL, Pereira T, Shevlin M, Bateman K, Campeau LC, Schneeweis J, Berritt S, Shi ZC, Nantermet P, Liu Y, Helmy R, Welch CJ, Vachal P, Davies IW, Cernak T, Dreher SD. Science. 2015;347:49. doi: 10.1126/science.1259203. [DOI] [PubMed] [Google Scholar]
- 50.Collot M, Eller S, Weishaupt M, Seeberger PH. Beilstein J Org Chem. 2013;9:97. doi: 10.3762/bjoc.9.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang SS. J Am Chem Soc. 1973;95:1328. doi: 10.1021/ja00785a602. [DOI] [PubMed] [Google Scholar]
- 52.Nguyen SH, Trotta AH, Cao J, Straub TJ, Bennett CS. Org Biomol Chem. 2012;10:2373. doi: 10.1039/c2ob06883d. [DOI] [PubMed] [Google Scholar]
- 53.James IW. Tetrahedron. 1999;55:4855. [Google Scholar]
- 54.Guillier F, Orain D, Bradley M. Chem Rev. 2000;100:2091. doi: 10.1021/cr000014n. [DOI] [PubMed] [Google Scholar]
- 55.Brase S, Dahmen S. Handbook of Combinatorial Chemistry. 2004;1:59. [Google Scholar]
- 56.Schmidt RR, Jung KH. In: Carbohydrates in Chemistry and Biology. Ernst B, Hart GW, Sinay P, editors. Vol. 1. Wiley-VCH; Weinheim, NY: 2000. p. 5. [Google Scholar]
- 57.Schmidt RR, Kinzy W. Adv Carbohydr Chem Biochem. 1994;50:21. doi: 10.1016/s0065-2318(08)60150-x. [DOI] [PubMed] [Google Scholar]
- 58.Schmidt RR, Michel J. Angew Chem, Int Ed Engl. 1980;19:731. [Google Scholar]
- 59.Kamat MN, Rath NP, Demchenko AV. J Org Chem. 2007;72:6938. doi: 10.1021/jo0711844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ferrier RJ, Furneaux RH. In: Methods in Carbohydrate Chemistry. Whistler RL, BeMiller JN, editors. Vol. 8. Academic Press; New York - London: 1980. p. 251. [Google Scholar]
- 61.Nigudkar SS, Parameswar AR, Pornsuriyasak P, Stine KJ, Demchenko AV. Org Biomol Chem. 2013;11:4068. doi: 10.1039/c3ob40667a. [DOI] [PubMed] [Google Scholar]
- 62.Nigudkar SS, Stine KJ, Demchenko AV. J Am Chem Soc. 2014;136:921. doi: 10.1021/ja411746a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Plante OJ, Andrade RB, Seeberger PH. Org Lett. 1999;1:211. doi: 10.1021/ol9905452. [DOI] [PubMed] [Google Scholar]
- 64.Colonna B, Harding VD, Nepogodiev SA, Raymo FM, Spencer N, Stoddart JF. Chem - Eur J. 1998;4:1244. [Google Scholar]
- 65.Carrel FR, Seeberger PH. J Org Chem. 2008;73:2058. doi: 10.1021/jo701349c. [DOI] [PubMed] [Google Scholar]
- 66.Wu X, Schmidt RR. J Org Chem. 2004;69:1853. doi: 10.1021/jo0354239. [DOI] [PubMed] [Google Scholar]
- 67.Sandbhor MS, Soya N, Albohy A, Zheng RB, Cartmell J, Bundle DR, Klassen JS, Cairo CW. Biochemistry. 2011;50:6753. doi: 10.1021/bi200449j. [DOI] [PubMed] [Google Scholar]
- 68.Ottosson H. Carbohydr Res. 1990;197:101. [Google Scholar]
- 69.Clausen MH, Madsen R. Carbohydr Res. 2004;339:2159. doi: 10.1016/j.carres.2004.06.012. [DOI] [PubMed] [Google Scholar]
- 70.Kaiser E, Colescott RL, Bossinger CD, Cook PI. Anal Biochem. 1970;34:595. doi: 10.1016/0003-2697(70)90146-6. [DOI] [PubMed] [Google Scholar]
- 71.Dinkelaar J, de Jong AR, van Meer R, Somers M, Lodder G, Overkleeft HS, Codee JDC, van der Marel GA. J Org Chem. 2009;74:4982. doi: 10.1021/jo900662v. [DOI] [PubMed] [Google Scholar]
- 72.Verduyn R, Douwes M, van der Klein PAM, Mösinger EM, van der Marel GA, van Boom JH. Tetrahedron. 1993;49:7301. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.