

Abstract
A combined structural, functional, and genetic approach was used to investigate inhibition of bacterial RNA polymerase (RNAP) by sorangicin (Sor), a macrolide polyether antibiotic. Sor lacks chemical and structural similarity to the ansamycin rifampicin (Rif), an RNAP inhibitor widely used to treat tuberculosis. Nevertheless, structural analysis revealed Sor binds in the same RNAP β subunit pocket as Rif, with almost complete overlap of RNAP binding determinants, and functional analysis revealed that both antibiotics inhibit transcription by directly blocking the path of the elongating transcript at a length of 2–3 nucleotides. Genetic analysis indicates that Rif binding is extremely sensitive to mutations expected to change the shape of the antibiotic binding pocket, while Sor is not. We suggest that conformational flexibility of Sor, in contrast to the rigid conformation of Rif, allows Sor to adapt to changes in the binding pocket. This has important implications for drug design against rapidly mutating targets.
Keywords: rifampicin, RNA polymerase, sorangicin, transcription
Introduction
Eubacterial RNA polymerase (RNAP), the enzyme responsible for transcription of DNA into RNA, is the target of several low-molecular-weight inhibitors (Darst, 2004). The best-known RNAP inhibitor, rifampicin (Rif; Sensi et al, 1960; Sensi, 1983), is widely used in combination therapy to treat tuberculosis. Bacterial strains resistant to Rif arise with appreciable frequency and compromise treatment of the disease (Ramaswamy and Musser, 1998; Heep et al, 2000). Development of antibiotics that can act on resistant bacteria is thus of considerable significance.
The structure of Thermus aquaticus (Taq) core RNAP bound to Rif (Campbell et al, 2001) corroborated and explained previous genetic and biochemical results obtained with Escherichia coli (Ec) RNAP (Ezekiel and Hutchins, 1968; Wehrli et al, 1968b; McClure and Cech, 1978; Ovchinnikov et al, 1983; Lisitsyn et al, 1984; Jin and Gross, 1988; Severinov et al, 1993, 1994). Rif binds in a pocket of the RNAP β subunit deep within the DNA/RNA channel and blocks the RNA exit pathway. As a result, RNAP bound to Rif is able to initiate RNA chain synthesis, but is unable to elongate the RNA product beyond a length of 2–3 nucleotides (nt).
The vast majority of Rif-resistant (RifR) mutants harbor substitutions in RNAP β subunit residues that either make direct contacts with Rif or are located near the binding pocket (Campbell et al, 2001). The substitutions decrease Rif binding affinity to the enzyme, making the mutant enzyme fully or partially insensitive to the drug (Ovchinnikov et al, 1983; Lisitsyn et al, 1984; Jin and Gross, 1988; Severinov et al, 1993, 1994). In principle, structural analysis of the Taq RNAP-Rif complex should help the rational design of more potent Rif-based inhibitors. However, Taq RNAP is much less sensitive to Rif than Ec RNAP (Campbell et al, 2001).
Rif is a member of the ansamycin class of antibiotics. Several other ansamycins, such as streptovaricin, are chemically similar to Rif and likely share the same RNAP binding site and mechanism of transcription inhibition (Wehrli, 1977). An unrelated antibiotic, streptolydigin, has a binding site that partially overlaps with the Rif binding site, but functions in an entirely different way by inhibiting RNAP catalytic activity (Cassani et al, 1971). In this work, we perform structural, functional, and genetic analyses of RNAP interactions with a macrolide polyether antibiotic, sorangicin A (Sor). Sor was isolated from the myxobacterium Sorangium cellulosum (Jansen et al, 1985) and shown to be a potent inhibitor of bacterial, but not eukaryotic, RNAPs (Irschik et al, 1985). Despite the lack of apparent chemical similarity between Sor and Rif (Figure 1), Sor was observed to inhibit transcription initiation, but not elongation, similar to Rif (Irschik et al, 1985). A previous study found that all isolated mutants of Ec RNAP exhibiting Sor resistance showed partial to strong Rif resistance (Römmele et al, 1990). In contrast, only 50% of mutants isolated for Rif resistance showed strong or partial Sor resistance, while 50% retained sensitivity to Sor. This led to the conclusion that the binding sites for each antibiotic largely overlap. However, the finding that some highly RifR mutants were Sor sensitive (SorS) suggested that there are determinants of RNAP that are necessary for interacting with Rif but not Sor, leading to the conclusion that there are subtle differences in the way the two antibiotics interact with RNAP (Römmele et al, 1990). Another study of antibiotic resistance in Staphylococcus aureus RNAP came to the same conclusion (O'Niell et al, 2000).
Figure 1.
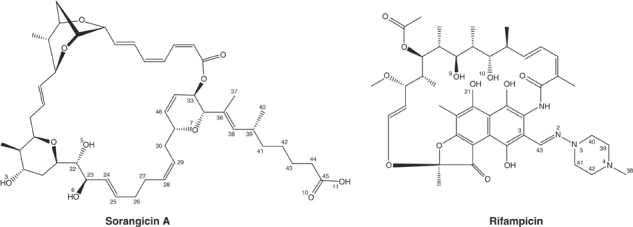
Chemical formulas for RNAP inhibitors Sor (top) and Rif (bottom). For clarity, only selected atoms discussed in the text are numbered.
In this study, we determined the X-ray crystal structure of Taq RNAP in complex with Sor in order to compare it to the previously determined RNAP-Rif structure. In addition, we performed a detailed functional analysis of Sor and Rif inhibition of Ec and Taq RNAPs, as well as a systematic analysis of crossresistance in Ec RNAP. The results show that Sor occupies the same RNAP β subunit pocket as Rif, with an almost complete overlap of RNAP binding determinants, and that Sor inhibits transcription by the same mechanism as Rif. On the other hand, while Rif binding and inhibition are very sensitive to amino-acid substitutions that would be expected to alter the shape of the antibiotic binding pocket, Sor is able to bind and inhibit these RifR RNAPs effectively. We propose that intrinsic conformational flexibility of Sor allows it to adapt to changes in the shape of the antibiotic binding pocket. This may be an important general principle for the design of inhibitors against rapidly mutating targets (Das et al, 2004).
Results
Sor inhibits transcription by Ec and Taq RNAP
The ability of Sor and Rif to inhibit transcription by Ec and Taq RNAP holoenzymes initiating at the T7 A1 promoter was investigated. Both Rif and Sor effectively inhibited transcription by the Ec enzyme (Figure 2, lanes 1 and 9). In the absence of antibiotics, RNAP produced the full-sized, 127 nt run-off transcript (RO), a 105 nt terminated transcript (T), which arose due to the presence of the tR2 terminator between the promoter and the end of the template, as well as two abortive transcripts. The abortive transcripts were likely to be the trimer CpApU, initiated from the CpA primer, as well as a dimer, pppApU, initiated from the ATP present in the reaction. The production of RO and T was essentially completely inhibited when the concentration of either antibiotic exceeded 1 μM (lanes 5–8 and 13–16). However, the amount of abortive products increased dramatically with increasing amounts of each antibiotic. At the highest Sor concentration of 1 mM (lane 16), Sor decreased the amounts of abortive products, which is likely due to nonspecific inhibition of transcription.
Figure 2.
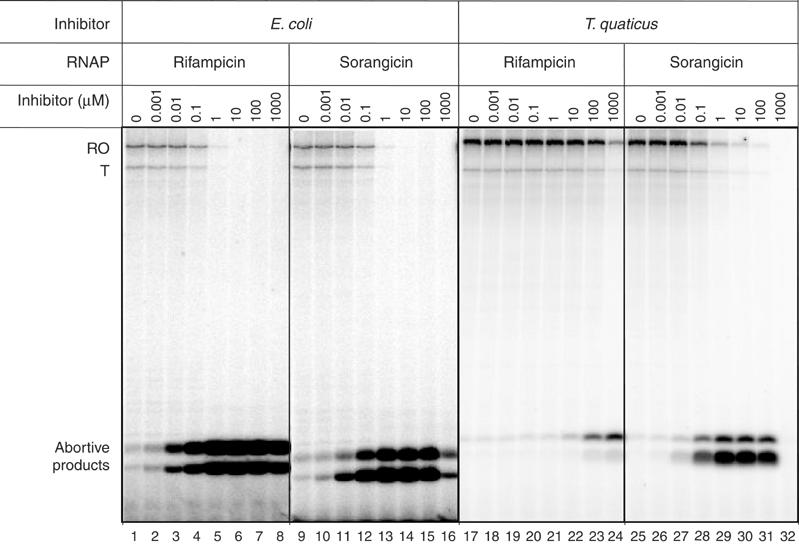
Rif and Sor inhibition of Ec and Taq RNAPs. Autoradiographs showing the radioactive RNA produced by Ec (lanes 1–16) and Taq (lanes 17–32) RNAP holoenzymes transcribing a template containing the T7 A1 promoter and the tr2 terminator, analyzed on a 20% polyacrylamide gel and quantitated by phosphorimagery. Lanes 2–8 and 18–24 show the effect of increasing concentrations of Rif. Lanes 10–16 and 26–32 show the effect of increasing concentrations of Sor.
The behavior of Taq RNAP in response to the drugs was different. As with Ec RNAP, increasing amounts of both Rif and Sor inhibited synthesis of the long transcripts (RO and T) while causing a dramatic increase in abortive products. As observed previously, the Taq enzyme was resistant to the effect of Rif—only at the highest concentrations of Rif (0.1–1 mM) was there a significant effect (lanes 23 and 24). Even at the highest concentration of Rif (1 mM), the production of long transcripts was only partially inhibited (lane 24). In contrast, Taq RNAP was as sensitive to Sor as Ec RNAP; very few full-sized transcripts were produced when the Sor concentration exceeded 1 μM, and the expected abortive transcripts were dramatically overproduced (lanes 28–31). From these experiments, we conclude that (i) Rif and Sor appear to inhibit transcription in a similar way, and (ii) Sor is an equally effective inhibitor for Ec and Taq RNAPs, while Rif is relatively ineffective against Taq RNAP, supporting the hypothesis that there are differences between the interaction of RNAP and each antibiotic.
Rif-resistant mutations and crossresistance to Sor
In order to determine whether known RifR mutations (Ovchinnikov et al, 1983; Severinov et al, 1993) in the Ec rpoB gene (coding for the RNAP β subunit) also lead to Sor resistance, we performed systematic crossresistance comparisons (Table I). Since these mutations were studied with the Ec enzyme but analyzed in the context of the Taq RNAP structure, throughout this manuscript we will refer to the mutations in Ec rpoB numbering, followed by the Taq numbering (Ec/Taq). Mutations causing Rif resistance occur in four distinct clusters in rpoB. The clusters are far from each other in the β primary sequence, but come together to line the Rif binding pocket in the three-dimensional structure (Campbell et al, 2001). One cluster of RifR mutants occurs in conserved segment B of the β subunit (the N-terminal cluster); substitutions at position Val146/137 lead to strong Rif resistance. Two additional clusters, Rif clusters I and II, harbor the majority of RifR mutants and occur between amino-acid positions 507–534/387–414 and 559–574/439–454, respectively. Finally, a single site in conserved segment E, at amino-acid position EcArg687/TaqThr566, marks Rif cluster III.
Table 1.
Effects of RNAP β subunit substitutions on Rif and Sor resistance
| Mutant (Ec/Taq)a | RifR | SorR | Classb |
|---|---|---|---|
| Single amino-acid substitutions | |||
| WT | − | − | |
| V(146/137)W | ++ | + | II |
| S(512/392)P | ++ | ++ | I |
| Q(513/393)R | −c | ++ | III |
| D(516/396)N | ++ | ++ | I |
| S(522/402)F | ++ | ++ | I |
| H(526/406)Y | ++ | ++ | I |
| H(526/406)P | ++ | ++ | I |
| H(526/406)Q | + | + | I |
| R(529/409)H | ++ | + | II |
| R(529/409)L | + | + | I |
| R(529/409)C | −c | − | |
| S(531/411)F | ++ | + | II |
| S(531/411)Y | ++ | − | II |
| A(532/412)V | ++ | + | II |
| A(532/412)E | + | + | I |
| L(533/413)P | ++ | − | II |
| G(534/414)D | + | − | II |
| I(572/452)F | ++ | − | II |
| S(574/454)F | + | ++ | III |
| Multiple substitutions or deletions | |||
| R(687/T566)H | − | − | |
| V(137/128)/I(138/129)R | − | − | |
| N(139/130)G/R(143/134)W | − | − | |
| N(139/130)K/R(143/134)W | − | − | |
| V(144/135)L/I(145/136)R | + | + | I |
| Δ540–544/420–424 | − | − | |
| Δ540–545/420–425(L) | − | + | III |
| Δ540–543/420–423(P) | − | + | III |
| Δ538–540/D418–420 | − | − | |
| Δ535–542/415–422 |
+ |
+ |
I |
| aMutations with differential effects on Rif versus Sor resistance are denoted in bold. Levels of resistance are denoted as follows: −, sensitive; +, mild resistance; ++, strong resistance. | |||
| bClass I (RifR/SorR); Class II (RifR/SorS); Class III (RifS/SorR). | |||
| cSome mutants originally isolated as RifR did not score as RifR in this assay because of the 24 h incubation time—longer incubation times (48 h) revealed weak Rif resistance. | |||
Ec DH5α cells (RifS/SorS) were transformed with expression plasmids overproducing 29 different β subunit mutants that conferred either Rif or stretpolydigin resistance (Severinov et al, 1993, 1994, 1995). The transformed cells were streaked on plates containing linear gradients of Rif or Sor (from 0 to 50 μg/ml) and allowed to grow for 24 h. In this semiquantitative in vivo assay, the extent of cell growth along the antibiotic gradient (relative to wild type (wt)) indicated the level of resistance. The results were grouped as follows: no resistance (−), mild resistance (+), and strong resistance (++), relative to the wt enzyme (−). Table I compares the crossresistance between Rif and Sor for each substitution tested. Substitutions that cause more resistance to one antibiotic relative to the other are highlighted in bold. The results can be summarized as follows:
Cells overproducing wt β subunit cease to grow when the concentration of Rif in the medium exceeded ∼7.5 μg/ml. The same cells stopped growing when the concentration of Sor reached ∼15 μg/ml. Since Rif and Sor inhibit Ec RNAP with a similar concentration profile in vitro (Figure 2), these results suggest that the uptake of Sor by Ec is less efficient than that of Rif.
Many of the RifR mutants (17 out of 29 tested) were also SorR, and the level of Rif resistance sometimes predicted Sor resistance levels. We call these mutants Class I (RifR/SorR). For example, Ser(522/402)Phe and His(526/406)Tyr were highly resistant to both Rif and Sor, while Ala(532/412)Glu was mildly resistant to both antibiotics. For RifR mutations of this class, at positions where multiple RifR substitutions were tested, different substitutions affected resistance levels to both antibiotics similarly. For example, His(526/406)Tyr and His(526/406)Pro resulted in high levels of resistance for both antibiotics, while His(526/406)Gln produced medium levels or resistance.
Some strong RifR mutations resulted in mild (Arg(529/409)His, Ser(531/411)Phe, Ala(532/412)Val, and Val (146/137)Trp) or very low, if any, Sor resistance (Ser(531/411)Tyr and Leu(533/413)Pro). We call these mutants Class II (RifR/SorS).
Two substitutions, Ser(574/454)Phe and Gln(513/393)Arg, resulted in medium to low levels of Rif resistance but were more strongly resistant to Sor. We call these mutants Class III (RifS/SorR).
These results confirm that the Rif and Sor binding sites of RNAP largely overlap, but the lack of crossresistance at all of the tested positions suggests differences in the mode of interaction.
Only five positions (Ser(512/392), Asp(516/396), Ser(522/402), His(526/406), and Ser(574/454)) out of 11 that resulted in RifR mutations when substituted also resulted in strong Sor resistance. Because our collection of mutants was originally selected for Rif resistance, some possible substitutions that may result in Sor resistance may not have been detected. We attempted to isolate SorR mutations directly, by plating Ec cells on plates containing Sor. However, we found that SorR colonies arose with a frequency that was at least 100 times higher than the frequency of spontaneous RifR mutants. The vast majority of SorR mutants obtained this way had the wt rpoB gene, and RNAP purified from these mutants was SorS in vitro. Thus, we conclude that these SorR mutants were probably uptake mutants.
RNAP-Sor crystallization and overall structure
Crystals of Taq core RNAP were grown by a modification of the previously described conditions (Zhang et al, 1999; see Materials and methods). Crystals were then incubated overnight with 1 mM Sor, followed by cryopreservation as described in Materials and methods.
The Sor-RNAP crystals were isomorphous to the native crystals and difference Fourier maps revealed strong electron density located in the Rif pocket (Figure 3A). The previously determined X-ray structure of Sor (Jansen et al, 1989) was easily placed in the density. Small adjustments of Sor were made to better fit the density. In addition, some side chains of amino acids surrounding Sor displayed slightly different positions as well as lower B-factors relative to the apoenzyme, indicating that Sor stabilized the position of these residues. Adjustments of these residues into the density were made and the structure refined to 3.2 Å (Table II).
Figure 3.
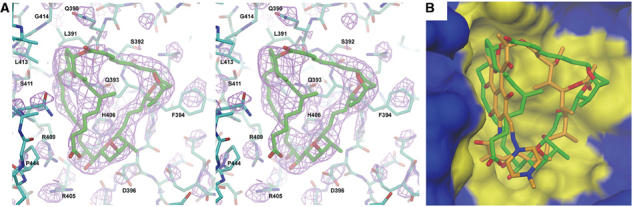
Sor-RNAP cocrystal structure and comparison with Rif. (A) Stereo view of Sor in its binding pocket of Taq core RNAP. Atoms are color-coded as follows: carbon atoms of the RNAP β subunit, cyan; carbon atoms of Sor, green; oxygen, red; nitrogen, blue; sulfur, yellow. Electron density, calculated using (∣FoSor−Fonat∣) coefficients (Sor denotes the Sor-RNAP cocrystal, nat denotes the native core RNAP crystal), is shown in magenta (contoured at 3σ), and was computed using phases from the native RNAP model. Selected amino-acid residues discussed in the text are labeled. (B) View of the antibiotic binding pocket of the RNAP β subunit (same view as A). The RNAP is shown as a surface view, with the β subunit colored blue, but with residues within 4 Å of Sor colored yellow (to define the antibiotic binding pocket). Superimposed in the binding pocket are the structures of Sor (green carbon atoms) and Rif (orange carbon atoms).
Table 2.
Crystallographic data
| RNAP-Rifa | RNAP-Sor | |
|---|---|---|
| Diffraction data | ||
| Crystal type (precipitant/cryoprotectant) | (NH4)2SO4/saturated sucrose | NH4COOH/NH4COOH |
| Space group | P41212 | P41212 |
| Unit cell | a=b=199.45 Å | a=b=199.39 Å |
| c=289.13 Å | c=290.97 Å | |
| Parameter | Total/last shell | Total/last shell |
| Resolution range (Å) | 30–3.3/3.42–3.3 | 40–3.2/3.31–3.2 |
| Rmerge | 0.077/0.344 | 0.084/0.466 |
| Completeness (%) | 86.1/71.1 | 94.1/94.2 |
| I/σ(I) | 10.7/1.7 | 13.0/2.2 |
| No. of reflections | 75 420/6173 | 91 202/8999 |
| No. of unique obs. | 214 453/11 549 | 433 405/34 210 |
| Refinement | ||
| Rcryst | 0.27 | 0.28 |
| Rfree |
0.33 |
0.34 |
| aAs described by Campbell et al (2001). | ||
Sor occupies the Rif pocket
The structure revealed that Sor exactly occupies the Rif binding pocket (Figure 3B). Comparison of RNAP residues interacting with Rif or Sor (defined as RNAP residues within 4 Å of each antibiotic) revealed essentially a one-to-one correspondence (Figure 4A and B), such that all residues that interact with Rif also interact with Sor. Superimposition of the two antibiotics within the RNAP binding pocket indicates that the overlapping binding determinants are possible because of the remarkable correspondence in the overall shape of each antibiotic (Figure 3B).
Figure 4.
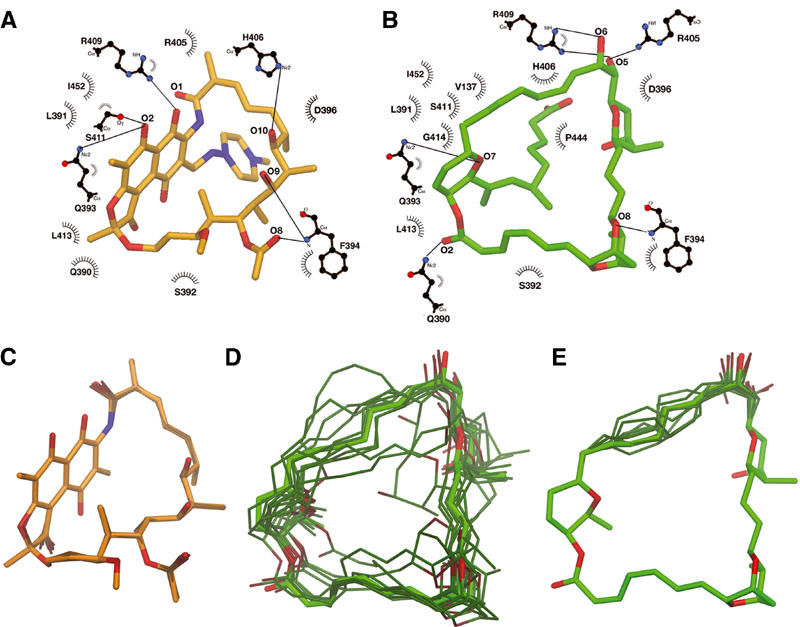
Rif and Sor interactions with RNAP, and conformational flexibility. (A, B) Schematic drawing of RNAP β subunit interactions with Rif (A) and Sor (B), modified from LIGPLOT (Wallace et al, 1995). Residues forming van der Waal's interactions are indicated; those participating in hydrogen bonds are shown in a ball-and-stick representation, with hydrogen bonds depicted as lines. (C–E) Results of molecular dynamics simulations. Starting conformations of Rif (C) or Sor (D, E) are shown as thick bonds and colored light orange (Rif) or light green (Sor). The final conformations after 10 independent molecular dynamics simulations are shown as thinner bonds and darker color. In (E), only atoms C22–C30/O5–O6 were allowed to move during the simulation. In all of the images, flexible, branched segments of each antibiotic that do not interact with RNAP (C38–C43/N2–N4 for Rif, C36–C45/O10–O11 for Sor) were included in the simulations, but were not included in the alignment and are not shown.
Determinants for Rif and Sor activity
Derivatives of Rif and their effects on antimicrobial activity have been analyzed extensively (Brufani et al, 1964; Lancini and Zanichelli, 1977; Arora, 1981, 1983, 1985; Arora and Main, 1984). Modifications of the ansa bridge, O1 or O2 of the napthol ring, or the hydroxyls O9 or O10 greatly reduce its inhibition activity in vitro. Modifications are only tolerated off C3 of the napthol ring (Figure 1). This is supported by the RNAP-Rif structure, where most of the interactions occur with the ansa bridge and its oxygens, and the napthol ring and its oxygens (Campbell et al, 2001).
Similar structure–activity studies have been carried out on Sor (Jansen et al, 1990; Schummer et al, 1992). SorA contains three hydroxyls (O3, O5, and O6), of which two (O5 and O6) are critical for transcription inhibition. These two oxygens participate in hydrogen bonds with Arg(525/405) and Arg(529/409) (Figure 4B), and our genetic studies confirm that these interactions are critical for Sor activity (Table I).
Breaking the ring structure of Sor, along with other gross chemical or stereochemical changes, eliminated all antibiotic activity, suggesting that the overall structure of Sor is critical for activity (Jansen et al, 1990). This is in agreement with our structure, since the overall shape of Sor fits very well into the Rif binding pocket (Figure 3B).
The inhibition mechanism for Sor and Rif is identical
Previously, analysis of the RNAP-Rif complex in the context of a model for the ternary elongation complex (Korzheva et al, 2000) revealed a severe steric clash between Rif, bound in its β subunit pocket, and the path of the elongating RNA transcript within the RNA/DNA hybrid (Campbell et al, 2001). The structure indicated that the presence of Rif would allow only a 2 or 3 nt RNA transcript (depending on the 5′-phosphorylation state of the 5′ nucleotide) without a steric clash, consistent with biochemical data showing that Rif inhibits production of RNA transcripts longer than 3 nt, and causes a dramatic build-up of 2–3 nt abortive products (Figure 2). Our biochemical studies of Sor indicate a very similar inhibition mechanism (Figure 2), and structural modeling indicates that Sor would sterically clash with RNA transcripts longer than 2 or 3 nt (again depending on the 5′-phosphorylation state of the 5′ nucleotide). We therefore conclude that Sor inhibits RNAP transcription by the same mechanism as Rif, by blocking the path of the elongating RNA transcript at a length of 2–3 nt.
Structural analysis of resistance mutants
Class I mutants. As mentioned above, Rif and Sor share the same binding pocket in the RNAP β subunit. In fact, all 12 residues that contact Rif share an interaction with Sor (Figure 4A and B). Three additional residues that line the binding pocket are within the 4 Å cutoff for interaction with Sor, Val(146/137), Gly(534/414), and Pro(564/444). Our genetic results support the structural results; many substitutions that gave rise to Rif resistance also gave rise to similar levels of Sor resistance (Class I; Table I). An important example is His(526/406); substitution of this residue comprises 36% of RifR Mycobacterium tuberculosis (Mtb) clinical isolates (Ramaswamy and Musser, 1998). His(526/406) participates in a critical hydrogen bond with Rif-O10, and makes extensive van der Waal's interactions with Sor (Figure 4A and B). Substitutions of His(526/406) with Tyr, Pro, or Gln led to strong resistance against both antibiotics (Table I).
Class II mutants. A second important residue, Ser(531/411), is substituted in 41% of Mtb RifR clinical isolates (Ramaswamy and Musser, 1998). This residue typifies the Class II mutants; substitutions of Ser(531/411) with bulky hydrophobic residues (Phe or Tyr) led to strong Rif resistance but mild or no Sor resistance (Table I). Substitution of this residue would remove a potentially important hydrogen bond with Rif-O2 (Brufani et al, 1964; Lancini and Zanichelli, 1977; Arora, 1981, 1983, 1985; Arora and Main, 1984). Modeling indicates that substitution of Ser(531/411) with Phe or Tyr would also introduce a severe steric clash with the napthol rings of Rif.
Ser(531/411) does not participate in hydrogen-bonding with Sor, but is sufficiently close to make van der Waal's interactions with Sor-C29 and C30 (Figure 4B). Interestingly, molecular modeling indicates that substitution of Ser(531/411) with the bulky hydrophobic residues Phe or Tyr would also introduce a severe steric clash with Sor. Nevertheless, Ser(531/411)Phe and Ser(531/411)Tyr are relatively sensitive to Sor (Table I).
Another Class II mutant illustrates a similar case. Ile(572/452) makes favorable van der Waal's interactions with the Rif napthol rings, and with Sor-C27 and C28 (Figure 4A and B). Modeling indicates that substitution of this residue with Phe would introduce steric clashes with both antibiotics, yet the substitution Ile(572/452)Phe is RifR but SorS.
Two additional Class II mutants involve residues that line the antibiotic binding pocket, Val(146/137) and Gly(534/414). Val(146/137) makes van der Waal's contact with Sor-C27 and C29, while Gly(534/414) makes van der Waal's contact with Sor-C46. Both Val(146/137) and Gly(534/414) are close to the Rif napthol ring system. Modeling indicates that the Class II substitutions Val(146/137)Trp and Gly(534/414)Asp would introduce severe steric clashes with both Rif and Sor, yet these mutants are RifR but weakly, or not at all SorR (Table I).
All of these mutants described above, Val(146/137)Trp, Ser(531/411)Phe, Ser(531/411)Tyr, Gly(534/414)Asp, and Ile(572/452)Phe, involve substitutions of bulky residues that would be expected to introduce steric clashes with both Rif and Sor. As expected, these substitutions cause strong Rif resistance. Unexpectedly, these substitutions are weakly or not at all SorR, indicating that Sor is somehow able to bind in the pocket while accommodating these bulky substitutions.
Leu(533/413) makes van der Waal's contacts with Sor-C33 and C46, and makes extensive van der Waal's interactions with the face of the Rif napthol ring system. The Class II substitution Leu(533/413)Pro may remove favorable van der Waal's contacts to Rif and Sor, but this substitution would also be expected to alter the path of the polypeptide backbone, which would introduce distortions in the antibiotic binding pocket since several important residues in the vicinity of Leu(533/413) line the pocket and make interactions with Rif and/or Sor (Ser(531/411), Leu(533/413), Gly(534/414)). The mutant Leu(533/413)Pro is strongly RifR but is SorS, suggesting that Sor is also capable of accommodating distortions to the shape of the binding pocket.
In the same region of the binding pocket as 531–534/411–414, the side chain of Ala(532/412) faces away from the antibiotic pocket, but the bulky substitution Ala(532/412)Val would also be expected to perturb the polypeptide backbone in this region, thereby distorting the shape of the binding pocket. Consistent with this, Ala(532/412)Val is strongly RifR. Nevertheless, Ala(532/412)Val is completely SorS, supporting the suggestion that Sor is able to accommodate distortions in the shape of the binding pocket.
Collectively, analysis of the Class II mutants indicates that Rif binding is extremely sensitive to changes in the shape of the antibiotic binding pocket, while Sor is not. These Class II mutants are in the vicinity of the Rif napthol ring system, a particularly rigid, inflexible structure. This raises the interesting possibility that the different response of these two antibiotics may be due to differences in their conformational (torsional) flexibility. We explore this idea further below.
Class III mutants. The final category of mutants, Class III (RifS/SorR), comprises two substitutions, Gln(513/393)Arg and Ser(574/454)Phe. Gln(513/393) forms a critical hydrogen bond with Rif-O2 and a hydrogen bond with Sor-O7, as well as contributing van der Waal's contacts to both antibiotics. Modeling suggests that, because of the details of the binding interface, an Arg at this position might be accommodated by the Rif, and the side-chain guanidinium group could donate a hydrogen bond to Rif-O2. On the other hand, Arg at this position would introduce severe steric clashes with Sor (along the alkyl chain from C25 to C28) and would also introduce a hydrophilic, positively charged group in a very hydrophobic environment of the antibiotic.
The second substitution that causes the RifS/SorR phenotype, Ser(574/454)Phe, probably asserts its effect by rearranging residues located spatially close by. Among these residues are Arg(525/405) and Arg(529/409), whose side chains would have to adopt different conformations to accommodate the bulky substitution. In the absence of a structure of the mutant, we cannot predict the response of the two Arg residues to the Ser(574/454)Phe substitution, but apparently the new arrangement is compatible with binding of Rif but not Sor.
Conformational flexibility of Rif and Sor
The analysis of Class II (RifR/SorS) mutants indicated that Rif was very sensitive to substitutions predicted to introduce steric clashes or alter the shape of the binding pocket in the vicinity of the napthol ring system. On the other hand, these mutants remained SorS, leading us to suggest that Sor is able to adapt somewhat to changes in the binding pocket due to conformational flexibility of the antibiotic. Clearly, the planar, napthol ring system of Rif is a very rigid structure with no torsional degrees of freedom. Moreover, physical molecular models (Harvard Apparatus, CPK Atomic Models) indicate that almost the entire Rif molecule is a very static structure. The ansa bridge of Rif is tightly packed against the napthol ring system, and there are essentially no degrees of torsional freedom for bonds along the backbone of the ring structure. On the other hand, Sor has far more degrees of torsional freedom, even when considering only the closed, 31-membered ring that forms the backbone of the structure.
We compared the conformational flexibility of the two antibiotics more objectively using molecular dynamics simulations (see Materials and methods). Conformations of each antibiotic obtained in this way were aligned and compared with the starting conformations (Figure 4C–E). The alignments and comparisons excluded torsionally flexible, branched segments of each molecule that are not involved in RNAP interactions (the N-amino-N-methylpiperazine moiety, C38–C43 plus N2–N4 of Rif, or the carboxylic acid side chain, C37–C45/O10–O11 of Sor; Figure 1). Alignment of 10 independent Rif conformations yielded an average root mean square deviation (r.m.s.d.) of 0.10±0.02 Å. All of the conformations are very close to the starting conformation (Figure 4C), with no torsional flips of any bonds within the ansa-bridge backbone, consistent with the molecular modeling. In contrast, alignment of 10 independent Sor conformations (Figure 4D) yielded an r.m.s.d. of 1.21±0.73 Å. The alignments reveal a few conformations close to the starting conformation (the smallest r.m.s.d. was 0.52 Å), but many are clearly far from the starting conformation (the largest r.m.s.d. was 2.45 Å).
We performed an additional test in which most of the Sor structure was fixed and not allowed to move, while the carbon backbone atoms from C22 to C30, along with the associated hydroxyls (O5 and O6) were free to move during the simulation, perhaps mimicking a situation where most of the Sor molecule was bound to RNAP, but one region needed to adjust or adapt to a change in the shape of the binding pocket (the region allowed to move is the segment of the structure that would clash with many of the Class II mutants). Even in this constrained situation, the segment shows a great deal of torsional flexibility (Figure 4E).
Discussion
In this work, we have used a combined biochemical, genetic, and structural approach to compare bacterial RNAP inhibitors Rif and Sor. The results led us to the following major conclusions:
Rif and Sor bind RNAP in the same β subunit pocket (Figure 3B), with an almost complete overlap of RNAP binding determinants (Figure 4A and B). This is supported by the Class I resistance mutants (Table I), which cause resistance to both antibiotics. The overlapping binding determinants are possible because the two antibiotics share a very similar overall shape and hydrophobic nature, despite being chemically unrelated (Figure 3B).
The two antibiotics inhibit RNAP transcription in the same way, by blocking the synthesis of transcripts longer than 2–3 nt in length. This is clearly by virtue of occupying the same site, which directly blocks the path of the elongating RNA product within the growing RNA/DNA hybrid.
The two antibiotics respond to a set of amino-acid substitutions differently (Class II, RifR/SorS). The Class II substitutions all have a common feature in that they are expected to distort the shape of the antibiotic binding pocket, either directly through bulky substitutions that would be expected to clash with the antibiotics, or indirectly by altering the path of the polypeptide backbone near the binding pocket.
Rif and Sor bind in the same site on RNAP (Figure 3B), and inhibit Ec RNAP by the same mechanism and with similar potency in vitro (Figure 2), yet Rif is extremely sensitive to amino-acid substitutions that cause changes in the shape of the antibiotic binding pocket, while Sor is not. We believe that the intrinsic conformational flexibility of Sor, in contrast to the very rigid conformation of Rif (Figure 4C–E), allows Sor to adapt to these changes in the binding pocket. Conformational flexibility of Sor might also explain why Sor is as effective against Taq RNAP as it is against Ec RNAP, while the concentration of Rif required to inhibit Taq RNAP is 2–3 orders of magnitude higher than for Ec (Figure 2; Campbell et al, 2001). This is an important point that has broad implications for drug design against rapidly mutating targets.
In general, the optimal situation for a high-affinity protein–ligand complex is thought to involve a conformationally rigid ligand interacting with the protein in a preorganized binding site with minimal conformational strain (Babine and Bender, 1997). While conformational rigidity before and after binding to a preorganized, complementary protein site would be expected to favor high-affinity binding (due to minimizing adverse entropic effects), in the milieu of a rapidly mutating target, small changes in the shape or chemical environment of the binding pocket due to single amino-acid substitutions can easily render a rigid, high-affinity inhibitor ineffective. This is exactly the situation with the use of Rif in clinical situations, where resistance to the drug arises with high frequency. With tuberculosis near epidemic proportions in some parts of the world, the rapid increase in drug-resistant strains of Mtb prompted the World Health Organization to declare tuberculosis a global public health emergency (Raviglioni et al, 1995).
In a study of reverse transcriptase inhibitors against wt and drug-resistant HIV-1 variants, Das et al (2004) concluded that conformational flexibility (‘wiggling') and/or repositioning and reorientation (‘jiggling') of an inhibitor within its binding pocket were critical for potency against a wide range of drug-resistant HIV-1 reverse transcriptase mutants. We propose here that the conformational flexibility of Sor (Figure 4D and E) allows it to adapt to shape changes of its binding pocket by a combination of ‘wiggling' and ‘jiggling'.
One might expect that conformational flexibility of a ligand would have an unfavorable effect on binding affinity due to adverse entropic effects, but this is not necessarily an insurmountable effect, since the conformationally rigid Rif and the flexible Sor bind and inhibit Ec RNAP with the same potency in vitro. Further structural studies of Sor bound to Class II mutants (Table I) may provide details of Sor conformational changes within the binding pocket that would aid the design of more effective antibiotics directed against the bacterial RNAP-Rif binding pocket.
Materials and methods
Transcription assays
Recombinant RNAP from Ec and Taq were prepared as previously described (Borukhov and Goldfarb, 1993; Kuznedelov et al, 2003). Multiple-round in vitro transcription reactions using a T7 A1 promoter-containing DNA fragment as a template (Figure 2) were performed as previously described (Minakhin et al, 2001).
Plating assays
Plasmids pMKSe2 with the rpoB mutations leading to Rif resistance were previously described (Severinov et al, 1993, 1994). Plasmids were transformed into Ec DH5α (RifS/SorS) cells, and ampicillin-resistant colonies were selected at 30°C on LB agar plates containing 200 μg/ml ampicillin. Levels of Rif and Sor resistance were tested on LB plates prepared with a linear concentration gradient of antibiotic (from 0 to 50 μg/ml for both Rif and Sor; Bryson and Szybalski, 1952; Severinov et al, 1994). Approximately 105 cells grown on ampicillin were resuspended in 1 ml of LB, and 15 μl was streaked along the drug gradient on the plates and allowed to grow for 24 h at 30°C.
Purification and crystallization
Native Taq core RNAP was purified and crystallized as previously described (Zhang et al, 1999) except that the crystallization solution was modified to 100 mM Tris–HCl, pH 8.0, 10 mM MgCl2, and 4.0 M ammonium formate. Crystals were stored in stabilization solution (crystallization solution with 4.2 M ammonium formate), and cryoprotected by soaking in cryoprotection solution (crystallization solution with 6.0 M ammonium formate) for 30 min before flash freezing in liquid ethane. Crystals prepared in this way (using ammonium formate as precipitant and cryoprotectant) diffract slightly better than crystals prepared as previously described (using ammonium sulfate as precipitant and saturated sucrose as cryoprotectant; Zhang et al, 1999), and are also more radiation resistant, allowing the collection of data sets with better statistics and completeness (details to be described elsewhere).
Crystals of RNAP with Sor were obtained by soaking native crystals in stabilization solution with 1 mM Sor for at least 12 h. Data were collected at the National Synchrotron Light Source Beamline X25 using 0.3° oscillations, and processed using the HKL suite (Otwinowski and Minor, 1997).
Structure determination
An improved native Taq core RNAP model, obtained by refinement against the ammonium formate crystals described above (to be described elsewhere), was used as a starting model for rigid body and positional refinement against the observed amplitudes from the Sor-RNAP crystal (FoSor) using REFMAC (Murshodov et al, 1997) with TLS restraints (Winn et al, 2001). An initial Fourier difference map, calculated using (∣FoSor−Fonat∣) amplitude coefficients with phases calculated from the native core RNAP model (φnat), clearly revealed density for the Sor molecule, as well as changes in nearby residues (Figure 3A). After placing the Sor X-ray structure (Jansen et al, 1985) in the difference density, the Sor structure and side chains surrounding the antibiotic were manually adjusted to fit the difference density using the program O (Jones et al, 1991), and the structure was refined using REFMAC (Murshodov et al, 1997) with TLS restraints (Winn et al, 2001), to a final R/Rfree of 0.28/0.34 at 3.2 Å.
The Rif-RNAP was also refined incorporating the updated native core model. Difference Fourier maps ((FoRif−FcRif) and (2FoRif−FcRif)) were calculated using phases calculated from the new native core RNAP structure with Rif was placed as in the previous structure. Residues surrounding Rif were adjusted to fit the density, and REFMAC (Murshodov et al, 1997) with TLS restraints (Winn et al, 2001) was used to refine the updated Rif-RNAP yielding a final R/Rfree of 0.27/0.33 at 3.3 Å.
Molecular dynamics
Crystal structures of Rif (Brufani et al, 1964) and Sor (Jansen et al, 1985) were used as initial conformations for MM2 molecular dynamics within the environment of the program Chem3D Ultra (CambridgeSoft) running on an Apple Powerbook G4. Default parameters for the MM2 molecular dynamics were used except that the step interval was 1.0 fs.
Accession codes
The coordinates for the refined RNAP-Sor and RNAP-Rif structures have been deposited in the Protein Data Bank (accession codes 1YNJ and 1YNN, respectively).
Acknowledgments
This work was initiated as a collaboration with Drs G Höfle, and H Reichenbach from the GBF Natural Product Chemistry and Biology groups at Braunschweig, Germany. We thank T Kapoor for invaluable discussions, and H Dormann for help in translating papers into English. We are indebted to M Becker and L Berman at the National Synchrotron Light Source Beamline X25, and A Joachimiak, SL Ginell, and FJ Rotella at the Advanced Photon Source Beamline SBC19ID, for support. OP was a recipient of a Waksman Summer Undergraduate Fellowship. This work was supported by NIH grants GM64530 to KS and GM61898 to SAD.
References
- Arora SK (1981) Structural investigations of mode of action of drugs III. Acta Crystallogr B 37: 152–157 [Google Scholar]
- Arora SK (1983) Correlation of structure and activity in ansamycins: molecular structure of sodium rifamycin. Mol Pharmacol 23: 133–140 [PubMed] [Google Scholar]
- Arora SK (1985) Correlation of structure and activity in ansamycins: structure, conformation, and interactions of antibiotics rifamycin SV. J Antibiot 37: 178–181 [DOI] [PubMed] [Google Scholar]
- Arora SK, Main P (1984) Correlation of structure and activity in ansamycins: molecular structure of cyclized rifamycin SV. J Antibiot 37: 1099–1102 [DOI] [PubMed] [Google Scholar]
- Babine RE, Bender SL (1997) Molecular recognition of protein–ligand complexes: applications to drug design. Chem Rev 97: 1359–1472 [DOI] [PubMed] [Google Scholar]
- Borukhov S, Goldfarb A (1993) Recombinant Escherichia coli RNA polymerase: purification of individually overexpressed subunits and in vitro assembly. Protein Expres Purif 4: 503–511 [DOI] [PubMed] [Google Scholar]
- Brufani M, Fedeli W, Ciacomello G, Vaciago A (1964) The X-ray analysis of the structure of rifamycin B. Experientia 20: 339–342 [DOI] [PubMed] [Google Scholar]
- Bryson V, Szybalski W (1952) Microbial selection. Science 115: 45–51 [PubMed] [Google Scholar]
- Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA (2001) Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 104: 901–912 [DOI] [PubMed] [Google Scholar]
- Cassani G, Burgess RR, Goodman HM, Gold L (1971) Inhibition of RNA polymerase by streptolydigin. Nat New Biol 230: 197–200 [DOI] [PubMed] [Google Scholar]
- Darst SA (2004) New inhibitors targeting bacterial RNA polymerase. Trends Biochem Sci 29: 159–162 [DOI] [PubMed] [Google Scholar]
- Das K, Clark AD Jr, Lewi PJ, Heeres J, De Jonge MR, Koymans LM, Vinkers HM, Daeyaert F, Ludovici DW, Kukla MJ, De Corte B, Kavash RW, Ho CY, Ye H, Lichtenstein MA, Andries K, Pauwels R, De Bethune MP, Boyer PL, Clark P, Hughes SH, Janssen PA, Arnold E (2004) Roles of conformational and positional adaptability in structure-based design of TMC125-R165335 (etravirine) and related non-nucleoside reverse transcriptase inhibitors that are highly potent and effective against wild-type and drug-resistant HIV-1 variants. J Med Chem 47: 2550–2560 [DOI] [PubMed] [Google Scholar]
- Ezekiel DH, Hutchins JE (1968) Mutations affecting RNAP associated with rifampicin resistance in Escherichia coli. Nature 220: 276–277 [DOI] [PubMed] [Google Scholar]
- Heep M, Rieger U, Beck D, Lehn N (2000) Mutations in the beginning of the rpoB gene can induce resistance to rifamycins in both Helicobacter pylori and Mycobacterium tuberculosis. Antimicrob Agents Chemother 44: 1075–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irschik H, Jansen R, Gerth K, Höfle G, Reichenbach H (1985) The sorangicins, novel and powerful inhibitors of eubacterial RNA polymerase isolated from myxobacteria. J Antibiot 40: 7–13 [DOI] [PubMed] [Google Scholar]
- Jansen R, Irschik H, Reichenbach H, Schomburg D, Wray V, Hofle G (1989) Antibiotics from gliding bacteria. XXXVII. Sorangicin A, a highly active antibiotic with novel macrolide-polyether structure from Sorangium cellulosum, So cel2: spectroscopic structure elucidation, crystal and solution structure. Leibigs Ann Chem 1989: 111–119 [Google Scholar]
- Jansen R, Schummer D, Irschik H, Hoefle G (1990) Antibiotics from gliding bacteria, XLII. Chemical modification of Sorangicin A and structure–activity relationship I: carboxyl and hydroxyl group derivatives. Liebigs Ann Chem 1990: 975–988 [Google Scholar]
- Jansen R, Wray H, Irschik H, Reichenbach H, Höfle G (1985) Isolation and spectroscopic structure elucidation of sorangicin A, a new type of macrolide-polyether antibiotic from gliding bacteria-XXX. Tetrahedron Lett 26: 6031–6034 [Google Scholar]
- Jin DJ, Gross CA (1988) Mapping and sequencing of mutations in the Escherichia coli rpoB gene that lead to rifampicin resistance. J Mol Biol 202: 45–58 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan S, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Korzheva N, Mustaev A, Kozlov M, Malhotra A, Nikiforov A, Goldfarb A, Darst SA (2000) A structural model of transcription elongation. Science 289: 619–625 [DOI] [PubMed] [Google Scholar]
- Kuznedelov K, Minakhin L, Severinov K (2003) Preparation and characterization of recombinant Thermus aquaticus RNA polymerase. Methods Enzymol 370: 94–108 [DOI] [PubMed] [Google Scholar]
- Lancini G, Zanichelli W (1977) Structure–activity relationships in rifamycins. In Structure–activity Relationship in Semisynthetic Antibiotics, Perlman D (ed) pp 531–600ar. New York: Academic Press [Google Scholar]
- Lisitsyn NA, Sverdlov ED, Moiseyeva EP, Danilevskaya ON, Nikiforov V (1984) Mutation to rifampicin resistance at the beginning of the RNA polymerase beta subunit gene in Escherichia coli. Mol Gen Genet 196: 173–174 [DOI] [PubMed] [Google Scholar]
- McClure WR, Cech CL (1978) On the mechanism of rifampicin inhibition of RNA synthesis. J Biol Chem 253: 8949–8956 [PubMed] [Google Scholar]
- Minakhin L, Nechaev S, Campbell EA, Severinov K (2001) Recombinant Thermus aquaticus RNA polymerase—a new tool for structure-based analysis of transcription. J Bacteriol 183: 71–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshodov GN, Vagin AA, Dodson EJ (1997) Refinement of macromolecular structures by maximum-likelihood method. Acta Crystallogr D 53: 240–255 [DOI] [PubMed] [Google Scholar]
- O'Niell A, Oliva B, Storey C, Hoyle A, Fishwick C, Chopra I (2000) RNA polymerase inhibitors with activity against rifampin-resistant mutants of Staphylococcus aureus. Antimicrobial Agents Chemother 44: 3163–3166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Ovchinnikov YA, Monastyrskaya GS, Guriev SO, Kalinina NF, Sverdlov ED, Gragerov AI, Bass IA, Kiver IF, Moiseyeva EP, Igumnov VN, Mindlin SZ, Nikiforov VG, Khesin RB (1983) RNA polymerase rifampicin resistance mutations in Escherichia coli: sequence changes and dominance. Mol Gen Genet 190: 344–348 [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Musser JM (1998) Molecular genetic basis of antimicrobial agent resistance in Mycobacterium tuberculosis: 1998 update. Tuberc Lung Dis 79: 3–29 [DOI] [PubMed] [Google Scholar]
- Raviglioni M, DE Snider J, Kochi A (1995) Global epidemiology of tuberculosis. JAMA 273: 220–226 [PubMed] [Google Scholar]
- Römmele G, Gabriela W, Renate S, Klaus V, Gruner J, Wehrli W (1990) Resistance of Escherichia coli to rifampicin and sorangicin A—a comparison. J Antibiot 43: 88–91 [DOI] [PubMed] [Google Scholar]
- Schummer D, Irschik H, Hoefle G (1992) Antibiotics from gliding bacteria, L. Chemical modification of Sorangicin A and structure–activity relationship II: derivatives obtained by reduction, oxidation and rearrangement reactions. Liebigs Ann Chem 1992: 293–304 [Google Scholar]
- Sensi P (1983) History of the development of rifampin. Rev Infect Dis 5 (Supp 3): 402–406 [DOI] [PubMed] [Google Scholar]
- Sensi P, Greco AN, Ballotta R (1960) Isolation and properties of rifamycin B and rifamycin complex. Antibiot Ann 1959–1960: 7: 262–270 [PubMed] [Google Scholar]
- Severinov K, Markov D, Severinova E, Nikiforov V, Landick R, Darst SA, Goldfarb A (1995) Streptolydigin-resistant mutants in an evolutionarily conserved region of the β' subunit of Escherichia coli RNA polymerase. J Biol Chem 270: 23926–23929 [DOI] [PubMed] [Google Scholar]
- Severinov K, Soushko M, Goldfarb A, Nikiforov A (1993) Rifampicin region revisited. New rifampicin-resistant and streptolydigin-resistant mutants in the beta subunit of Escherichia coli RNA polymerase. J Biol Chem 268: 14820–14825 [PubMed] [Google Scholar]
- Severinov K, Soushko M, Goldfarb A, Nikiforov V (1994) RifR mutations in the beginning of the Escherichia coli rpoB gene. Mol Gen Genet 244: 120–126 [DOI] [PubMed] [Google Scholar]
- Wallace AC, Laskowski RA, Thornton JM (1995) LIGPLOT: a program to generate schematic diagrams of protein–ligand interactions. Protein Eng 8: 127–134 [DOI] [PubMed] [Google Scholar]
- Wehrli W (1977) Ansamycins. Chemistry, biosynthesis and biological activity. Top Curr Chem 72: 21–49 [DOI] [PubMed] [Google Scholar]
- Wehrli W, Neusch J, Knüsel F, Staehelin M (1968b) Action of rifamycin on RNA polymerase from sensitive and resistant bacteria. Biochem Biophys Res Commun 32: 284–288 [DOI] [PubMed] [Google Scholar]
- Winn MD, Isupov MN, Murshudov GN (2001) Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr D 57: 122–123 [DOI] [PubMed] [Google Scholar]
- Zhang G, Campbell E, Minakhin L, Richter C, Severinov K, Darst SA (1999) Crystal structure of Thermus aquaticus core RNA polymerase at 3.3 Å resolution. Cell 98: 811–824 [DOI] [PubMed] [Google Scholar]