

Abstract
Flap endonuclease-1 (FEN1) is a key enzyme for maintaining genomic stability and replication. Proliferating cell nuclear antigen (PCNA) binds FEN1 and stimulates its endonuclease activity. The structural basis of the FEN1–PCNA interaction was revealed by the crystal structure of the complex between human FEN1 and PCNA. The main interface involves the C-terminal tail of FEN1, which forms two β-strands connected by a short helix, the βA–αA–βB motif, participating in β–β and hydrophobic interactions with PCNA. These interactions are similar to those previously observed for the p21CIP1/WAF1 peptide. However, this structure involving the full-length enzyme has revealed additional interfaces that are involved in the core domain. The interactions at the interfaces maintain the enzyme in an inactive ‘locked-down' orientation and might be utilized in rapid DNA-tracking by preserving the central hole of PCNA for sliding along the DNA. A hinge region present between the core domain and the C-terminal tail of FEN1 would play a role in switching the FEN1 orientation from an inactive to an active orientation.
Keywords: DNA clamp, flap endonuclease, repair, replication, X-ray
Introduction
DNA replication in eukaryotes is a highly coordinated process involving many proteins that work cooperatively to ensure the accurate and efficient replication of DNA (Waga and Stillman, 1998). In this process, flap endonuclease-1 (FEN1) plays a crucial role in the removal of RNA primers during Okazaki fragment maturation in lagging strand DNA synthesis (Liu et al, 2004). FEN1 belongs to the XPG-like family of structure-specific nucleases that include bacteriophage and bacterial 5′-nucleases. Flap DNA removal by FEN1 is also essential during long-patch base excision repair (Klungland and Lindahl, 1997). In yeast, FEN1 mutants display severely impaired phenotypes such as UV sensitivity, deficient chromosome segregation, conditional lethality and accumulation in S phase. In mice, haplo-insufficiency of FEN1 leads to rapid tumor progression (Kucherlapati et al, 2002; Henneke et al, 2003). Moreover, FEN1 has been shown to participate in physical and functional interactions with the Werner Syndrome protein WRN, which is a member of the RecQ helicase family (Brosh et al, 2001; Hickson, 2003). Werner Syndrome is a human premature aging disorder well characterized by chromosomal instability. Thus, FEN1 appears to be a key player in maintaining genomic stability by participating in the DNA replication and repair processes, which are the early events that modulate cancer susceptibility and tumorigenesis (Henneke et al, 2003). Despite its pronounced importance in biology and medicine, no three-dimensional structure of eukaryotic FEN1 has been elucidated.
In vitro experiments have shown that the FEN1 activity is markedly stimulated by proliferating cell nuclear antigen (PCNA), which is well known as the ‘DNA sliding clamp' (Kelman, 1997; Tsurimoto, 1998). This stimulation is induced by direct binding of FEN1 to PCNA, leading to a 10- to 50-fold increase in its nuclease activity (Li et al, 1995; Wu et al, 1996; Tom et al, 2000). The interaction between FEN1 and PCNA is an essential prerequisite that provides for the subsequent FEN1 functionality found in cells. Mutations in FEN1 that disrupt the interaction with PCNA decrease the cleavage efficiency of flap DNA at the replication fork (Stucki et al, 2001), thus leading to the generation of unfavorably long flap DNA strands (Gary et al, 1999).
PCNA forms a trimeric ring with three-fold symmetry perpendicular to the ring plane, and possesses a central hole that can be used to clamp DNA (Krishna et al, 1994). Importantly, PCNA binds several DNA-editing enzymes that function in DNA metabolic processes ranging from DNA methylation, base excision repair, nucleotide excision repair, mismatch repair, DNA replication and translesion DNA synthesis (Maga and Hubscher, 2003). It appears that PCNA acts as a platform for these enzymes, including FEN1, and facilitates the efficient functioning of these proteins. To date, crystal structures of complexes between PCNA and peptides from several PCNA-interacting proteins have been reported since the first structure of human PCNA bound to a peptide from the cyclin-dependent protein kinase inhibitor p21CIP1/WAF1 (hereafter referred to as p21) had been determined (Gulbis et al, 1996). However, the precise molecular mechanism by which these entire proteins cooperate with PCNA on the DNA remains unknown. Studying the entire complex would add to previous knowledge of peptide-based interactions.
PCNA-binding proteins including p21 possess a short PCNA-binding motif, QXX(I/L/M)XXF(F/Y), located either at the N- or C-terminal region (reviewed in Warbrick, 1998; Tsurimoto, 1999; Matsumoto, 2001), while the crystal structure of human PCNA complexed with the C-terminal peptide of p21 (the PCNA–p21 complex) revealed the presence of extensive interactions with over 18 residues of p21 containing the motif (Gulbis et al, 1996). Interestingly, human FEN1 possesses a C-terminal tail consisting of ∼45 residues containing the short PCNA-binding motif. This long C-terminal tail represents one of the features common to eukaryotic FEN1s. Flanking residues of the motif may be utilized in interactions with DNA (Stucki et al, 2001).
We now report on the crystal structure of human FEN1 complexed with PCNA. This is the first structure of eukaryotic FEN1 and the structure of the entire complex providing the first evidence pertaining to the presence of protein–protein interactions within the FEN1–PCNA complex. The structure revealed the FEN1–PCNA interfaces consisting of protein–peptide and protein–protein interactions. The main interaction involves the N-terminal half of the C-terminal tail of each FEN1 molecule, forming the βA–αA–βB motif anchored to one PCNA subunit. The second interaction involves the enzyme core domain of FEN1. This protein–protein interaction maintains the enzyme in an inactive ‘locked-down' complex, as implied in the complex between the little finger (LF) domain of the Y-family DNA polymerase Pol IV and the Escherichia coli β-clamp processivity factor (Bunting et al, 2003), and preserves the central hole of PCNA for the purposes of DNA tracking. FEN1 possesses a short linker region containing small residues between the core domain and the C-terminal tail. This linker region acts as a hinge, which endows FEN1 with a degree of freedom to swing the core domain, and may play a role in switching the enzyme activity.
Results
Overall structure of the FEN1–PCNA complex
The crystal structure of human FEN1 complexed with human PCNA was determined by the molecular replacement method (see Materials and methods and Table I). The structure revealed three FEN1 molecules (Figure 1, molecules X, Y and Z) bound to one PCNA trimer. Human FEN1 and PCNA consist of 380 (42 kDa) and 261 (29 kDa) amino-acid residues, respectively. The total mass (213 kDa) of the complex calculated from the structure agreed with the results from gel-filtration chromatography and dynamic light scattering, both of which yielded a monodisperse peak in solution (Sakurai et al, 2003).
Table 1.
Crystallographic statistics of the human FEN1–PCNA complex
| X-ray data | |
| Space group | P212121 |
| Cell parameters, a, b, c (Å) | 82.2, 143.4, 246.7 |
| Resolution (Å)a | 50–2.9 (3.0–2.9) |
| Completeness (%)b | 85.1 (58.3) |
| I/σI | 13.3 (2.9) |
| Rmerge (%) | 8.0 (28.0) |
| Refinement | |
| Number of residues included | |
| FEN1 (X, Y, Z) | 313, 312, 349 (of 380) |
| PCNA (A, B, C) | 256, 258, 255 (of 261) |
| Number of atoms | 13,162 |
| Number of reflections (total/unique) | 234,329/55,775 |
| R/Rfree (%)c | 22.0/28.4 |
| Average B-factor (Å2) | |
| Total | 59.2 |
| FEN1 (X, Y, Z) | 55.7, 78.8, 85.2 |
| PCNA (A, B, C) | 36.1, 40.1, 46.9 |
| R.m.s. bond length (Å), angles (deg) | 0.007, 1.4 |
| aStatistics for the outer resolution shell are given in parentheses. | |
| bIntensities (I/σI>1.0) were merged and used in the refinement. | |
| cR=∑∣∣Fobs∣−∣Fcalc∣∣/∑∣Fobs∣. Rfree is the same as R, but for a 5% subset of all reflections that were never used in the crystallographic refinement. | |
Figure 1.
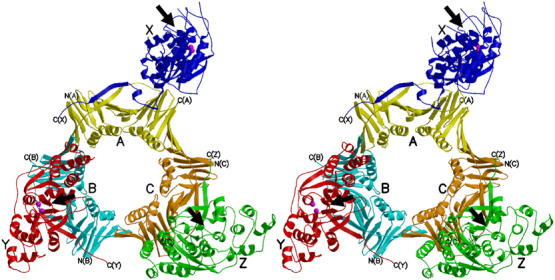
A stereo view of the human FEN1–PCNA complex. Three FEN1 molecules are colored in blue (X), red (Y) and green (Z), and the three subunits of the PCNA trimer in yellow (A), cyan (B) and orange (C). The C-termini of FEN1 and PCNA are labeled. Metal ions bound to the active sites of FEN1 (X and Y) are shown in magenta. Proposed catalytic faces of FEN1 are indicated by arrows.
Three PCNA subunits (subunits A, B and C in Figure 1) are tightly associated to form a closed ring. Each subunit contains two topologically identical domains, which are connected by the interdomain connector (IDC) loop. Each domain consists of two β–α–β–β–β motifs that are related by a pseudo two-fold symmetry. In the trimer, six β sheets form a circular outer layer that supports 12 α helices forming the inner surface. This architecture is essentially the same as those previously reported for PCNA (Krishna et al, 1994; Gulbis et al, 1996) and all secondary structural elements are preserved in our PCNA complex. The structures of human PCNAs bound to FEN1 and a p21 peptide are closely superimposable over the core secondary structural elements, with a small root-mean-square (r.m.s.) deviation (0.6 Å) for 247 Cα carbon atom positions in the trimer, while local structural differences are found in the IDC loops, caused by differences in the interactions with FEN1 as described later. Other differences are found in the prominent loop (βD2–βE2) residues 186–193, which are visible in our complex but were invisible in the p21 complex. Each PCNA subunit binds one FEN1 molecule (molecules A, B and C bind X, Y and Z, respectively).
Structure of human FEN1
The structure of human FEN1 consists of the nuclease core domain (residues 1–332) and the C-terminal tail (333–380) (Figure 2A). The N-terminal half (333–359) of the C-terminal tail is visible, whereas the C-terminal half (∼20 residues) was not observed in the density map. It has been suggested that the C-terminal half, which is rich in lysine residues, is involved in DNA binding (Stucki et al, 2001) and might be flexible without DNA. The visible C-terminal tail region forms two β-strands and one helical conformation. This βA–αA–βB motif represents the main interacting interface with PCNA. In fact, strand βA and the flanking helix αA are formed by the conserved short PCNA-binding motif QXXLXXFF.
Figure 2.
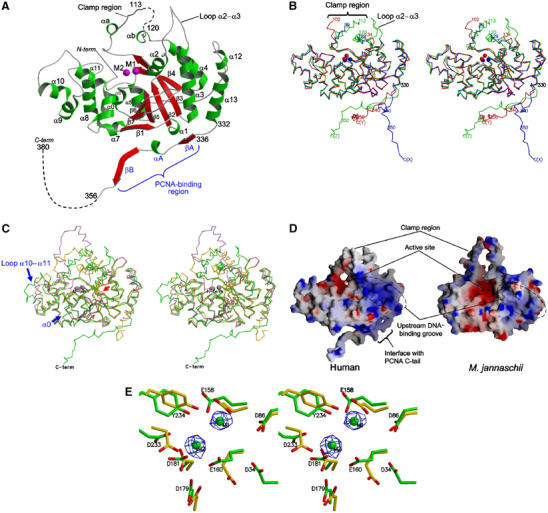
Structure of human FEN1. All diagrams of FEN1 molecules are viewed from the same viewpoint as in (A). (A) Ribbon model of the molecular structure of human FEN1 (molecule Z). Two metal ions (M1 and M2) in the active site of molecule X are superimposed and depicted in magenta. (B) A stereo diagram showing superimposition of three human FEN1 molecules (X, Y and Z) including metal ions. Each molecule is shown in the same color as in Figure 1. (C) A stereo diagram showing superimposition of human FEN1 (green), M. jannachii FEN1 (Hwang et al, 1998) (violet) and P. furiosus FEN1 (Hosfield et al, 1998) (gold). The insertion sites at helix α0 and loop α10–α11 in human FEN1 are indicated by blue arrows and the deletion site at loop β6–β7 is shown as a red arrow. (D) Electrostatic molecular surfaces of human and M. jannachii FEN1s. Each surface was colored in the range <−10 to >+10kBT, where kB is the Boltzman constant and T is the temperature. The positive potential is shown in blue and the negative potential in red. (E) A closed-up stereo view of the active sites of human (green, molecule Y) and P. furiosus (gold) FEN1s. The orientation viewed is approximately that of a 90° rotation from (A) along the horizontal axis. The omit Fo–Fc electron density maps for the two metals (M1 and M2) are shown in blue (contoured at 5σ). The metal ions and side chain of Asp34 in P. furiosus FEN1 have not been deposited in PDB.
The core domain is folded into an α/β structure with a groove formed by a twisted seven-stranded mixed β-sheet in the topology β1–β7–β6–β5–β2–β3–β4 and two helical regions: one formed by helices α0 and α5–α11, and the other by helices α1–α4, α12 and α13 (Figure 2A). These two helical regions are located on both sides of the β-sheet, which forms a central groove. The region between strand β3 and helix α4 contains 46 residues that project from the main body of the domain. In the electron density map, 29 (residues 100–128), 31 (103–133) and six (114–119) residues of this large looping-out region were not observed for molecules X, Y and Z, respectively. This region, containing many charged and hydrophobic residues, is thought to thread along the single strand of the DNA substrate (Tom et al, 2000; Storici et al, 2002) and hereafter is referred to as the ‘clamp region'. A superimposition of the three FEN1 molecules in the crystal (Figure 2B) shows the diverse conformations of the clamp regions. The loop between helices α2 and α3 (loop α2–α3) also seems to be flexible. Indeed, those for molecules X and Y are disordered. Finally, the C-terminal tails of the three FEN1 molecules project from the core domains in different directions. Excluding these flexible regions, the core domains of the three molecules are superimposed with an r.m.s. deviation of 0.8 Å (for 265 Cα).
Comparison with archaeal FEN1s
The structure of human FEN1 (molecule Z) was compared with those of two FEN1s from the hyperthermophilic archaea, Methanococcus jannaschii (Hwang et al, 1998) and Pyrococcus furiosus (Hosfield et al, 1998) (Figure 2C). These archaeal FEN1s exhibit 34 and 38% sequence identity to human FEN1, respectively (Figure 3). From archaea to humans, the primary structures of the clamp regions are mostly conserved without any deletion or insertion. In archaeal FEN1 and related bacteriophage enzymes, the clamp regions have been referred to as a ‘helical arch' in T5 5′-exonuclease (Ceska et al, 1996), or a ‘L1 loop' or ‘helical clamp' in M. jannaschii (Hwang et al, 1998) and P. furiosus (Hosfield et al, 1998) FEN1s, respectively. In our crystal, this region contains only two short helices, αa and αb. Even single amino-acid substitutions in this clamp region of human FEN1 have been reported to significantly decrease the nuclease activity (Storici et al, 2002). Deletion of the region in M. jannaschii FEN1 completely abolished the nuclease activity (Hwang et al, 1998). However, no common structure was observed in the FEN1 structures (Figure 2C). We believe that the clamp region of FEN1s would be intrinsically flexible, which could allow for effective tracking of flap DNA.
Figure 3.
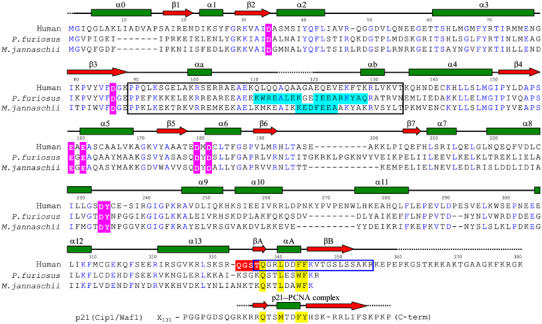
Aligned amino-acid sequences of human and two archaeal FEN1s. The alignment was adjusted according to the three-dimensional structures. Secondary structure elements of human FEN1 (as in Figure 2A) are shown at the top, and residues whose electron densities were not observed in any of the three molecules are shown as dashed lines. Identical residues between human and archaeal FEN1s are shown in blue letters. The active-site residues which bind metal ions (pink) and the hinge region (red) are highlighted. The clamp region is boxed with black lines, and helices observed in the structures of two archaeal FEN1s (Hosfield et al, 1998; Hwang et al, 1998) are highlighted in light-blue. The PCNA-binding region of human FEN1 is boxed with blue lines, and those of human p21 have been aligned at the bottom. The X subscript indicates the number of followed core-domain residues.
Compared with the archaeal FEN1s, human FEN1 possesses two major and one minor insertion sites. One major insertion is located at the N-terminus (residues 9–15) and the other (residues 267–274) at the loop region between helices α10 and α11 (Figure 3). The former insertion forms an additional helix, α0, which packs against helices α5–α8 and a strand β7. The latter insertion induces re-orientation of helix α10 (Figure 2C). Helix α0 induces significant changes in the shape of the helical region on the left side of the central β-sheet, narrowing the central groove (Figure 2D). A minor insertion (two residues) is located at loop α11–α12. Human FEN1 has three one-residue deletion sites at loop α2–α3, loop α8–α9 and loop α13–βA. In P. furiosus FEN1, insertion residues between β6 and β7 form an antiparallel β ribbon that restricts the active site groove, whereas no insertion is present in human or M. jannaschii FEN1s.
The electrostatic potential surface of human FEN1 is comparable to that of M. jannaschii FEN1 (Figure 2D). The bottom of the clamp region forming the central cleft is dominated by negatively charged residues for the active site containing metal ions. Both sides of the central cleft contain basic residues, which provide complementarity to the negative charges of the DNA substrate. In human FEN1, these basic residues are located at helix α0, loop α8–α9 and loop α10–α11 on one side and helix α3, loop β6–β7 and helix α13 on the other side. Compared with M. jannaschii FEN1, the negatively charged cleft of human FEN1 is more closed, partly due to the additional helix α0. On the other hand, human FEN1 possesses a more positively charged groove at the helical region on the right side of the cleft. This region corresponds to the binding site for the upstream DNA strand containing 3′-flap in the crystal structure of Archaeoglobus fulgidus FEN1 bound to a 3′-flap DNA fragment (Chapados et al, 2004).
Active site of human FEN1
The active site of human FEN1 is located at the central cleft with two possible metal ions (Figure 2A) and formed by two clusters of conserved acidic residues (Figure 2E). Four residues (Asp34, Asp86, Glu158 and Glu160) form the first metal ion-binding site (M1). Three residues (Asp179, Asp181 and Asp233) form the second metal ion-binding site (M2). These acidic residues are conserved in all known FEN1 enzymes, and the prevailing catalytic mechanism is thought to be universal. In the case of FEN1 molecules X and Y in our crystal, the electron densities for two metal ions were clearly observed in the difference Fourier map (Figure 2E). The density height for M1 is over 5σ in both X and Y, and that for M2 is 3σ and 5σ in X and Y, respectively. In the case of archaeal FEN1s, like our human FEN1, M2 appeared only at a lower electron density level (Hosfield et al, 1998; Hwang et al, 1998). Since no bivalent cations were added during the purification or crystallization procedures, two metal ions would have been tightly bound to FEN1 during these steps. In the present structure, these metal ions were assigned to Mg2+. M1 is known to play an important role in catalysis, probably in the nucleophilic attack of the phosphodiester bonds of DNA, while it has been suggested that M2 is involved in DNA binding (Shen et al, 1997). In P. furiosus FEN1, the conserved Tyr234 (numbered for human) was also involved in the active site via a hydrogen bond to Glu158 and a water-mediated contact with Asp181 (Hosfield et al, 1998). In the present structure, this Tyr residue was observed at the same position; however, the density for the water molecule was not identified, probably due to insufficient resolution.
FEN1–PCNA interactions
The interactions between FEN1 and PCNA are schematically summarized in Figure 4A. Overall, the binding of each FEN1 to PCNA buries large molecular surface area (∼3580 Å2 for molecules X and A). The interface between FEN1 and PCNA consists of a peptide-protein interaction involving the extended C-terminal tail of FEN1 (residues 336–356) and a channel on the surface of PCNA, and also a protein–protein interaction involving the FEN1 core domain. The latter interaction is mainly mediated by the PCNA-bound FEN1 tail and the edge of each PCNA monomer. This bipartite interaction is reminiscent of that observed in the crystal structure of the complex between the LF domain of E. coli Pol IV and the β-clamp (Bunting et al, 2003), while the protein–protein interaction in the Pol IV–LF–β-clamp complex involves two β-clamp monomers.
Figure 4.
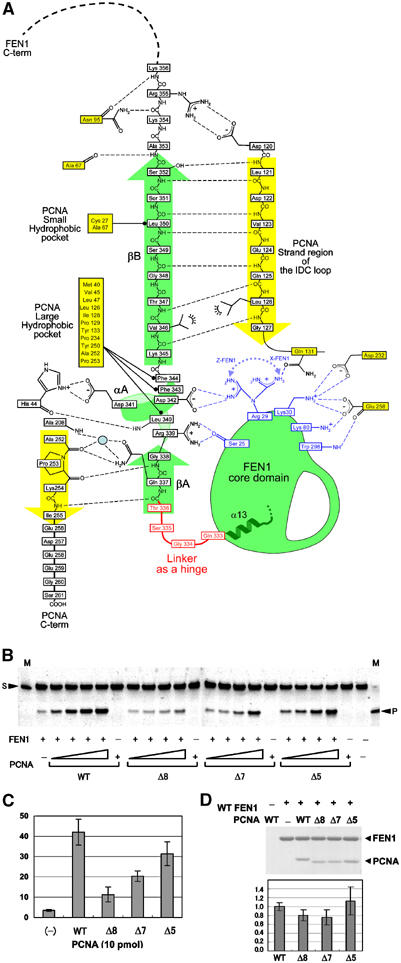
Interactions between FEN1 and PCNA. (A) Schematic depiction of interactions between FEN1 (green) and PCNA (yellow). The FEN1 hinge region is colored in red. Residues shown in blue participate in protein–protein interactions in one or two FEN1 molecules out of three: Ser25 in Y and Z, Arg29 in Z and X, and Lys30, Lys80 and Trp298 in X (see text). A tentative water molecule observed between molecules Z and C is indicated by a blue circle. (B) Human FEN1 endonuclease activity enhanced by human wild type (WT) and deletion mutant, Δ8 (254–261), Δ7 (255–261) and Δ5 (257–261), PCNAs. The mobility of the top band is 33 nucleotides (for the downstream primer) and that of the cleaved product is 20 nucleotides (for the flap DNA from the downstream primer). The size markers show 33 nucleotides, as indicated by an arrowhead with S and 20 nucleotides with P. The SF substrate DNA (0.5 pmol) was added into a solution containing FEN1 (0.3 pmol) and PCNA (0, 1, 2, 5, 10 pmol). Control experiments showed little detectable nuclease activity in the absence of PCNA. (C) The bar graph documents the cleavage (%) quantified from the gel with 10 pmol wild-type or mutant PCNA. Each cleavage was calculated from the results of three independent experiments. (D) Pull-down assays of wild-type and mutant recombinant PCNAs by FEN1(CHis). The presence of both FEN1(CHis) and PCNA bands in a single lane indicates the formation of an in vitro complex of the two proteins. The bar graph documents the relative binding by quantifying the protein amounts and normalizing with the beads-bound FEN1 amount. Each binding was calculated from the results of three independent experiments.
Peptide-binding interface
The interactions that are mediated by the βA–αA–βB motif located at the C-terminal tail of FEN1 bury ∼2500 Å2 of total surface area at the interface and can be divided into three parts. While the interaction mode resembles that found in the PCNA–p21 complex (Gulbis et al, 1996), many differences are found. The short FEN1 strand βA (residues 336TQG338) binds the PCNA C-terminal residues (Ala252–Ile255) in the form of antiparallel β–β-like interactions (βA–C-term interactions). The interactions involve the side chain of FEN1 Gln337, which is the key residue in the consensus PCNA-binding motif and is conserved in other PCNA-binding proteins including p21 (Figure 3). The hydrogen bond from the main chain of PCNA Pro253 to the main chain of FEN1 Gly338 in our complex is replaced by a hydrogen bond to the side chain of p21 Thr145 in the PCNA–p21 complex. Three PCNA residues (Leu251, Ala252 and Pro253) are strictly conserved from archaea to humans. Among these residues, Pro253 might be involved in coordinating two oxygen atoms in the backbone of Ala252 and Pro253 toward βA of FEN1. In fact, a double mutation of P252A and K253A (P253 and K254 in human) in yeast PCNA affected the stimulation of FEN1 activity (Gomes and Burgers, 2000).
Mutational studies showed that the βA–C-term interactions, especially the main-chain interactions, are important for enhancing the nuclease activity of human FEN1 (Figures 4B and C). Deletion of PCNA C-terminal residues Δ8 (254–261) resulted in a substantial reduction in the stimulation of FEN1 nuclease activity without a significant reduction in the binding affinity to FEN1 (Figure 4D). The β-like structure of the PCNA C-terminal residues, 252APKI255, could act as a rigid joint that directs the FEN1 core domain toward the DNA substrate. In contrast, deletion of Δ5 (257–261) caused no significant changes in the binding affinity or nuclease activity of FEN1. The extreme C-terminal residues (two, three and six residues for molecules A, B and C, respectively) of this region were disordered in our crystal.
Nonpolar contacts are found between FEN1 helix αA (339RLDDFF344) and a large hydrophobic pocket within PCNA. The six FEN1 residues adopt a mixture of 310 and α helical conformations at the present resolution. In the Y FEN1 molecule, the helix is stabilized by intramolecular interactions, where the side chain of Asp342 accepts hydrogen bonds from both the main chain and side chain of Arg339. Three nonpolar FEN1 residues (Leu340, Phe343 and Phe344) located on one side of the helix dock into the hydrophobic pocket of the PCNA molecular surface. These interactions are similar to those in the PCNA–p21 complex. Interestingly, on the opposite side of the helix, FEN1 has an acidic residue, Asp341, which forms a salt bridge with PCNA His44 in our complex. The position corresponding to Asp341 is replaced by threonine in p21, but is restricted to an acidic residue (Asp or Glu) in all FEN1s. The position corresponding to His44 in human PCNA is restricted to a basic residue (His or Arg) in PCNAs, suggesting that salt bridge formation would represent a characteristic feature of FEN1–PCNA interactions.
Antiparallel β–β association (βB–IDC loop interactions) is formed between the FEN1 strand βB (residues 345–352) and the PCNA IDC loop (121–127). Our observation of this β–β interaction that has been found in the PCNA–p21 complex is somewhat surprising, given the lack of sequence homology between FEN1 and p21 strand βBs. Consequently, side chain–side chain interactions between the associated β strands in our complex are different from those in the PCNA–p21 complex. In our complex, two FEN1 strand βB nonpolar residues (Val346 and Leu350) make contact with hydrophobic residues from PCNA: Val346 makes contact with Leu126 of the PCNA IDC loop and Leu350 is packed against PCNA nonpolar residues Cys27 and Ala67, located at the small hydrophobic pocket formed at the junction between the IDC loop and the underlying β sheet of the PCNA N-terminal domain. In the PCNA–p21 complex, however, electrostatic complementarity seems to be dominant. Two basic residues located at strand βB of the p21 peptide form salt bridges with negatively charged PCNA residues, while the small hydrophobic pocket of PCNA accommodates a hydrophobic residue of the p21 peptide, which corresponds to Leu350 of FEN1. As previously pointed out (Gulbis et al, 1996), the molecular surface of the human PCNA trimer involved in peptide binding is negatively charged. In our complex, a FEN1 basic residue (Arg355) located at the N-terminal flanking region of strand βB forms a salt bridge with a PCNA acidic residue (Asp120) at the N-terminal end of the IDC loop. Moreover, a notable difference is found in the main chain–main chain interactions. In the PCNA–p21 complex, strand βB of p21 forms a well-aligned β–β interaction with the IDC loop residues, whereas in the FEN1–PCNA complex a one-residue insertion of Gly348 in strand βB interrupts a regular hydrogen bond network between the main chains in the β–β interaction (Figure 4A). This distortion results in a slight decrease in the number of interactions with PCNA, which might be one of the reasons why the binding affinity of FEN1 to PCNA (Kd=60 nM) is lower than that of p21 (Kd=10–15 nM) (Chen et al, 1996). The archaeal FEN1s completely lack the residues to form these interactions (Figure 3).
The aforementioned FEN1–PCNA interactions are essentially conserved in all three FEN1 molecules in our complex. Figure 6A shows the superimposition of the βA–αA–βB motifs derived from the three FEN1 molecules, resulting in a small r.m.s. deviation (0.6 Å), together with the p21 peptide at the PCNA-binding region: the peptide is superimposed on FEN1 with an r.m.s. deviation of 1.3 Å. The relatively large r.m.s. deviation between FEN1 and p21 is due to the aforementioned one-residue insertion in FEN1 βB. Overlap of the binding sites on the PCNA molecular surface could account for the observed inhibition by p21 of the binding of FEN1 to PCNA by replacing and occupying the entire binding site on PCNA.
Figure 6.
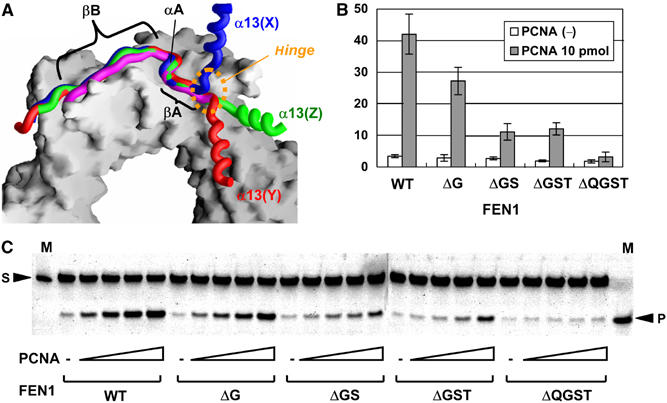
The hinge region of human FEN1. (A) Superposition of the C-terminal tails from three human FEN1 molecules. The C-terminal tail plus helix α13 from three FEN1 molecules (in the same colors as in Figure 1) and the p21 peptide (magenta) illustrated as backbone-worm models are superimposed on part of the PCNA ring illustrated as a surface model. The orientation viewed is the same as in Figure 1 (PCNA-subunit A at the top). (B) Human FEN1 endonuclease activity enhanced by human PCNA. The deletion mutants of FEN1 are ΔG for deletion of G334, ΔGS for deletion of G334 and S335, ΔGST for deletion of residues 334–336, and ΔQGST for deletion of residues 333–336. The bar graph documents the cleavage (%) quantified from the gel in C with 10 pmol PCNA from the results of three independent experiments. Control experiments showed no significant changes in the activities without PCNA. (C) Reactions (10 μl) containing the SF substrate DNA (0.5 pmol), FEN1 (0.3 pmol) and PCNA (0, 1, 2, 5, 10 pmol). The mobility of the top band is 33 nucleotides (for the downstream primer) and that of the cleaved product is 20 nucleotides (for the flap DNA from the downstream primer).
Protein–protein interface
The FEN1 core domains are involved in intermolecular contacts with its C-terminal tail bound to the PCNA groove, in addition to intermolecular contacts with PCNA. The interface involving the core domain of FEN1 molecule Y resembles that of molecule Z, although the two FEN1 molecules within the complex make different lattice contacts in the crystal. This strongly suggests that their similar orientation with respect to PCNA is determined by the stability of that particular interaction with PCNA and is not an artifact of crystal packing. At the heart of the interface, helix α1 and strand β2 from the FEN1 core domain make contact with strand βA and helix αA from the PCNA-bound FEN1 tail (Figures 2A and D left): the main chain of Ser25 of the core domain forms hydrogen bonds with the conserved Arg339 of the C-terminal tail. In Z, an additional salt bridge is formed between Arg29 of the core domain and Asp342 of the C-terminal tail helix αA.
Compared with molecules Y and Z (∼700 Å2), the interface involving the core domain of FEN1 molecule X is large and buries 1490 Å2 of accessible surface area. Thus, the X-position may be more stable than the other two, Y and Z. Strands β2 and β3 and loop α11–α12 from the FEN1 core domain make contact with PCNA residues at the C-terminal flanking region of the IDC loop and the C-terminal tail: four FEN1 core-domain residues (Arg29, Lys30, Lys80 and Trp298) interact directly with three PCNA residues (Gln131, Asp232 and Glu258). The FEN1 molecule X displays a different orientation of its core domain compared to molecules Y and Z, although the PCNA trimer preserves the central hole that is utilized in the interaction with DNA.
The FEN1 hinge region
The FEN1 core domain is linked to the PCNA-binding tail by a short linker (Figure 4A). We found that this linker, 333QGST336, functions as a hinge (Figure 6A). The torsion angles of these residues are markedly different among the three FEN1 molecules. This hinge region connects helix α13 of the core domain and strand βA of the C-terminal tail, thus providing a degree of freedom that allows for a possible swing motion of the core domain. The QGST motif of the hinge region is conserved in higher eukaryotic FEN1s (Figure 5).
Figure 5.

Aligned amino-acid sequences of the putative hinge regions and the consensus motifs for PCNA binding derived from eukaryotic FEN1s. Residues within putative hinge regions are boxed and highlighted in red. Residues within the consensus motif for PCNA binding are in bold and highlighted in yellow.
We investigated the importance of the short linker in terms of the nuclease activity of FEN1 bound to PCNA by assaying the ability of PCNA to stimulate the nuclease activity of FEN1 and FEN1 deletion mutants that possess a shorter linker (or no linker) between the core domain and the PCNA-binding tail (Figures 6B and C). Deletion of more than two linker residues resulted in seriously decreased nuclease activity, while complete deletion of the linker, ΔQGST, exhibited no stimulation by PCNA. These results clearly indicate the importance of the linker, probably acting as a hinge, to direct the FEN1 core domain toward the DNA substrate.
In our complexed structure, the nuclease core domains of the Z and Y FEN1 molecules swing about ∼90° and ∼100°, respectively, from that of molecule X around Arg332 toward the center of the PCNA ring (Figure 7A). During this swing displacement, the active site tracks on a large spherical surface with a radius of 40–50 Å. FEN1 locates its core domain in front of the PCNA C-terminal face, while the active site of molecules Y and Z faces toward the conceivable position occupied by the DNA substrate. The swing-in motion might be utilized in threading the flap DNA through the clamp region. Suppose that dsDNA is passing through the center of PCNA ring as in Figure 7 and that the DNA occupies a linear orientation; approximately 50° more swing-in displacement of the FEN1 core domain would be needed to place its active site on the cleavage site (the junction of dsDNA and flap-ssDNA). A similar displacement would also be needed for a proposed kinked DNA substrate (Chapados et al, 2004), since the upstream duplex of the kinked DNA would run through PCNA just as a linear DNA duplex.
Figure 7.
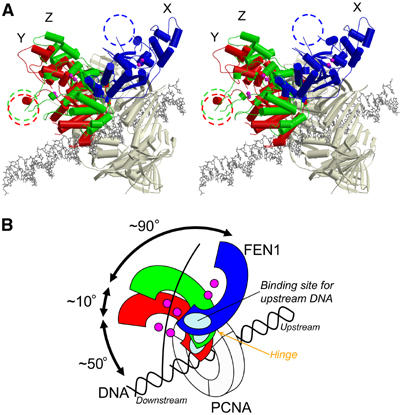
The hinge flexibility of human FEN1. (A) A stereo diagram showing three FEN1 molecules superimposed on PCNA (a gray model) as in Figure 6A, with a linear dsDNA in a wire model. The looping-out clamp regions are marked as dashed circles. The conceivable DNA model is superimposed at the center of PCNA. (B) Schematic model showing the orientation of the FEN1 core domains in (A).
Discussion
Our structure of the FEN1–PCNA complex has several implications concerning the molecular mechanism of action of FEN1 on PCNA. This represents the first reported structure involving a full-length FEN1 bound to the DNA clamp and has revealed the interfaces involving the core domain of FEN1. The interactions at the interfaces maintain the enzyme core domain in an inactive orientation, whereby the FEN1 molecule would have no access to a DNA duplex running perpendicularly through the center of PCNA. Our FEN1–PCNA complex possessing protein–protein interfaces involving the FEN1 core domain with no access to DNA may correspond to the inactive ‘locked-down' complex seen in the complex between Pol IV-LF and β-clamp (Bunting et al, 2003). As suggested for Pol IV, FEN1 could exist in an equilibrium between two states: a ‘locked-down' and an active ‘tethered' complex capable of productive interaction with DNA. In our complex structure, the FEN1 molecule X displays the maximum protein–protein interactions. Thus, the X-position may be the more populated ‘inactive' form and the other two forms may be ‘intermediate' on the way to a cleavage-competent position.
In solution, DNA-clamped PCNA is known to freely slide on double-stranded (ds) DNA (Kelman, 1997). Our structure suggests that FEN1 can bind PCNA with the core domains that do not critically disturb the sliding-clamp function of PCNA. The binding models relating to the FEN1 core domains in our crystal might be utilized in rapid DNA tracking by preserving the central PCNA hole for the purposes of sliding along the DNA. The FEN1–PCNA complex held onto the DNA by PCNA results in an effective increase in the local concentration of FEN1 on the DNA. Both the tracking and binding to the cleavage site could then be enhanced. This mechanism is consistent with in vitro kinetic investigations of FEN1 catalysis showing a large decrease (11- to 12-fold) in the Michaelis constant, Km, following the addition of PCNA (Tom et al, 2000).
A productive orientation allowing access to DNA would require disruption of the protein–protein interface. In a proposed active complex, FEN1 could swing-in the PCNA ring and induce a kink in the substrate DNA (Chapados et al, 2004). This can be achieved by hinge-like movements at the short linker that connects the core domain and the C-terminal PCNA-binding tail, while still maintaining the interaction between PCNA and the C-terminal tail. The flexible nature of the FEN1 core domains of molecules Y and Z against the bound PCNA trimer is suggested by their higher B-factors (80.1 and 85.7 Å2, respectively) compared to those of the PCNA subunits (Table I). The most likely trigger to allow FEN1 to swing into position for cleavage would be interactions between the single-stranded flap DNA and FEN1. The 5′-flap DNA would be flexible enough to interact with the clamp region of FEN1 in an inactive ‘locked-down' complex. The tracking of 5′-flap DNA through the clamp region would disrupt the protein–protein interface between FEN1 and PCNA and allow FEN1 to swing into the position for cleavage. The 3′-flap DNA also strongly interacts with FEN1 and would also help to position the FEN1 active site on the cleavage site.
We believed that the FEN1 binding to PCNA through the conserved PCNA-binding motif is essential for the proposed role of the hinge region in the structural switch from ‘locked-down' to ‘tethered' complexes. In fact, the importance of the PCNA-binding motif has been shown by several deletion and mutation studies of the motif (Gary et al, 1999; Gomes and Burgers, 2000; Stucki et al, 2001). The βA–C-term interactions are important in providing the basis for the hinge flexibility of FEN1. Our mutation experiments consistently suggest the importance of this interaction in stimulating FEN1 activity. In the present structure, Ile255 forms a main chain–main chain hydrogen bond with Thr336 of FEN-1, which provides a boundary to the hinge region (Figure 4A). Thus, the βA–C-term interactions might play a role in the form of a rigid joint that can provide effective swing displacement of the core domain toward the DNA. The controlled swing of the core domain may contribute to the observed moderate increase in VMAX (Tom et al, 2000) and also to a proposed tracking of flap DNA through the clamp region (Murante et al, 1995; Hosfield et al, 1998; Hwang et al, 1998; Tom et al, 2000).
The βA–C-term interactions in our crystal do not involve residues 256–261 at the extreme C-terminus of PCNA. Consistent with this finding, we showed that deletion of Δ5 (257–261) did not affect the binding affinity or nuclease activity of FEN1. The experimental result is in sharp contrast with the proposal that β-zipper formation between an FEN1 peptide and the PCNA C-terminus may assist in the positioning of FEN1 on the PCNA surface to facilitate flap endonuclease action (Chapados et al, 2004). The proposal has been based on crystal structures of Archaeoglobus fulgidus PCNA in the short peptide-bound forms. β-Zipper formation might be an artifact, probably due to using very short peptides in the crystallization procedure. Yet, in apparent conflict with our experimental data, the archaeal system may be somewhat different from the eukaryotic system because of the poor conservation of the hinge sequences and lack of the interaction with the IDC loop of PCNA.
It should be noted that location of the hinge region is directly adjacent to the 3′-flap-binding site. Mechanically, the hinge motion would initially position the binding site for the upstream duplex onto the substrate DNA. This initial binding to the upstream duplex and the 3′-flap may assist in positioning the FEN1 active site precisely on the cleavage site of the DNA. Recently, analysis of mutations in the upstream duplex and 3′-flap-binding site of human FEN1 demonstrated a less stringent cleavage pattern (Friedrich-Heineken and Hubscher, 2004).
DNA replication and base excision repair are the sequential processes involving other PCNA-binding enzymes such as DNA polymerase δ/ɛ, FEN1 and DNA ligase I. PCNA is thought to equally stimulate these enzymes on the DNA by binding to their consensus PCNA-binding motif. In both processes, once the excision of flap DNA has been completed by FEN1, the enzyme needs to release its core domain away from the excision point to provide access for incoming proteins, such as ligase I. The hinge region of FEN1 might prevent steric conflicts of the core domains in the exchange process when ligase I replaces FEN1 on PCNA. Otherwise, the FEN1–PCNA complex may slide away, followed by the arrival of another PCNA with ligase I. Should only one FEN1 molecule bind PCNA as in Figure 7, it might be possible that ligase I binds one of the two unoccupied binding sites on PCNA, thus forming a FEN1–ligase I–PCNA (and DNA) complex. Our structure demonstrated that the hinge region existing between the core domain and the tail can switch FEN1 and other enzymes bound to the same PCNA ring: FEN1 swings-out its core domain (typically as in molecule X) to allow for a space probably large enough for ligase I (and also polymerase δ/ɛ) to interact with the same PCNA and DNA. In fact, the simultaneous binding of FEN1, ligase and/or polymerase to the same PCNA has been reported for the archaeal proteins from Sulfolobus solfataricus (Dionne et al, 2003). Considering the structure of bacteriophage RB69 polymerase, which binds the PCNA-homologous sliding clamp (gp45), a similar hinge region should be found between its core domain and the RB69-binding region (Shamoo and Steitz, 1999). Flexibility in the tail region might represent a common feature of some PCNA-binding enzymes (Hingorani and O'Donnell, 2000). The swift high-performance characteristic of such a multi-protein complex has been noted both in terms of replication and repair pathways (Hosfield et al, 1998; Hingorani and O'Donnell, 2000; Matsumoto, 2001).
Materials and methods
X-ray data and structure determination
Detailed procedures of purification, crystallization and native data collection of the recombinant FEN1–PCNA complex were performed as previously described (Sakurai et al, 2003). The crystal contained one complex (three FEN1 monomers and one PCNA trimer) in the asymmetric unit. The intensity data were collected at 100 K at SPring-8 in Japan and merged to a resolution of 2.9 Å (Table I). Molecular replacement (MR) was performed by MOLREP (CCP4, 1994) using a PCNA monomer (from human p21–PCNA complex, PDB code: 1AXC (Gulbis et al, 1996)) as a search model. Three distinct solutions corresponding to each monomer of the PCNA trimer were obtained. On the other hand, MR trials using FEN1 monomers (PDB code: 1B43 (Hosfield et al, 1998) and 1A76 (Hwang et al, 1998), respectively) failed. Therefore, the initial electron density map was calculated using only the phases of the trimeric PCNA structure. This map was of good quality for PCNA but not interpretable for FEN1, except for the C-terminal PCNA-binding regions. Using MOLREP or FFFEAR (CCP4, 1994), three FEN1 monomer models based on the P. furiosus FEN1 structure were successfully fitted into residual electron densities in the Fo–Fc map. The density map was markedly improved by noncrystallographic symmetry (NCS) averaging using DM (CCP4, 1994) for the three FEN1 core domains and for the three PCNA monomers. The structure was modeled by O (Jones et al, 1991) and finally refined by CNS (Brünger et al, 1998) in the absence of NCS restraints. The refinement statistics are summarized in Table I.
The quality of the final electron density map was good enough for most part of the model including side chains, while those of some loop and terminal regions are poor. These contained loop α2–α3 and part of the clamp region of FEN1 and the C-terminal residues of both PCNA and FEN1. Disordered residues in these regions are not included in the current model (Table I and see text). The stereochemical quality of the model was monitored using the program PROCKECK (Laskowski et al., 1993) and 97.6% of residues were in the favorable/allowed region in the Ramachandran plot: poorly defined residues are S157 (molecule Y), Q122 (Z), K135 (Z), and E304 (Z) in the FEN1 model, and V188 (B), A241 (B), Q108 (C) and E191 (C) in the PCNA model. Superimpositions of human and archaeal FEN1s were performed by LSQMAN (Kleywegt and Jones, 1997) and figures were prepared by MOLSCRIPT (Kraulis, 1991), CONSCRIPT (Lawrence and Bourke, 2000) and GRASP (Nicholls et al, 1991). The atomic coordinates have been deposited in the PDB (accession code 1UL1).
FEN1–PCNA-binding assay
Human FEN1 with a six-histidine tag at the C-terminal end, FEN1(CHis), was overexpressed in E. coli strain BL21(DE3) and purified to homogeneity via Ni-NTA Agarose column (Qiagen) and Mono S chromatography (Amersham Bioscience). For in vitro binding, 10 μg of FEN1(CHis) was incubated with 5 μg of mutant PCNA in 100 μl of buffer A (50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 100 mM NaCl, 10% glycerol, 0.5 mg/ml bovine serum albumin and 5 mM β-mercaptoethanol) at 4°C for 1 h. TALON Metal Affinity Resin (Clontech) was then added and the mixture was further incubated at 4°C for 2 h with continuous gentle shaking. The resins were washed three times with 100 μl of buffer A and bound FEN1–PCNA complexes were eluted with buffer B (50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 100 mM NaCl, 10% glycerol, 5 mM β-mercaptoethanol and 100 mM imidazole). Collected samples were then analyzed by separation in 12.5% SDS–polyacrylamide gels and quantified using Coomassie Blue staining.
Flap endonuclease assay
Flap endonuclease assays were performed according to a previously described procedure (Harrington and Lieber, 1994). Briefly, the FEN1 endonuclease activity was measured in a 10 μl reaction volume containing 50 mM Tris–HCl (pH 8.0), 10 mM MgCl2, 62.5 mM NaCl, 0.5 mM β-mercaptoethanol, 100 μg/ml bovine serum albumin, 0.5 pmol of end-labeled DNA substrate and 0.3 pmol of human FEN1 with titration of 0, 1, 2, 5 and 10 pmol as trimer of wild-type or mutant PCNA (Fukuda et al, 1995; Oku et al, 1998). Single- or double-flap (SF or DF) DNA substrates were prepared by annealing three oligonucleotides: Upstream primer (5′-CAGCAACG CAAGCTTG-3′ or 5′-CAGCAACGCAAGCTTGG-3′), Template (5′-GTCGACCTGCAGCCCAAGCTTGCGTTGCTG-3′) and Downstream primer (5′-(FITC)-ATGTGGAAAATCTCTAGCAGGCTGCAGGTCGAC-3′). Reactions were carried out at 30°C for 15 min and terminated by adding 10 μl of loading buffer (95% formamide, 10 mM EDTA, 1 mg/ml bromophenol blue). Samples were heated to 95°C for 5 min and then electrophoresed through a 15% (w/v) polyacrylamide gel containing 7 M urea and 1 × TBE (89 mM Tris, 89 mM boric acid and 2 mM EDTA (pH 8.0)). Percentages of the cleaved products were estimated using a fluorimaging analyzer (Fuji FLA-2000). Results of mutation effects obtained for the SF substrate were basically similar to those for the DF substrate.
Acknowledgments
This work was supported by a Grant-in-Aid for Scientific Research on Priority Area (B) Metal sensor and a Protein 3000 project on Intracellular Signal Transduction to TH from the Ministry of Education, Culture, Sports, Science and Technology (MEXT). SS was supported by a Grant-in-Aid for the 21st Century COE Research from MEXT and a research fellowship for Young Scientist from the Japanese Society for the Promotion of Science. KK was supported by a grant from the NAIST foundation.
References
- Brosh RM Jr, von Kobbe C, Sommers JA, Karmakar P, Opresko PL, Piotrowski J, Dianova I, Dianov GL, Bohr VA (2001) Werner syndrome protein interacts with human flap endonuclease 1 and stimulates its cleavage activity. EMBO J 20: 5791–5801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, Warren GL (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D 54: 905–921 [DOI] [PubMed] [Google Scholar]
- Bunting KA, Roe SM, Pearl LH (2003) Structural basis for recruitment of translesion DNA polymerase Pol IV/DinB to the beta-clamp. EMBO J 22: 5883–5892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceska TA, Sayers JR, Stier G, Suck D (1996) A helical arch allowing single-stranded DNA to thread through T5 5′-exonuclease. Nature 382: 90–93 [DOI] [PubMed] [Google Scholar]
- Chapados BR, Hosfield DJ, Han S, Qiu J, Yelent B, Shen B, Tainer JA (2004) Structural basis for FEN-1 substrate specificity and PCNA-mediated activation in DNA replication and repair. Cell 116: 39–50 [DOI] [PubMed] [Google Scholar]
- Chen J, Chen S, Saha P, Dutta A (1996) p21Cip1/Waf1 disrupts the recruitment of human Fen1 by proliferating-cell nuclear antigen into the DNA replication complex. Proc Natl Acad Sci USA 93: 11597–11602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D 50: 760–763 [DOI] [PubMed] [Google Scholar]
- Dionne I, Nookala RK, Jackson SP, Doherty AJ, Bell SD (2003) A heterotrimeric PCNA in the hyperthermophilic archaeon Sulfolobus solfataricus. Mol Cell 11: 275–282 [DOI] [PubMed] [Google Scholar]
- Friedrich-Heineken E, Hubscher U (2004) The Fen1 extrahelical 3′-flap pocket is conserved from archaea to human and regulates DNA substrate specificity. Nucleic Acids Res 32: 2520–2528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda K, Morioka H, Imajou S, Ikeda S, Ohtsuka E, Tsurimoto T (1995) Structure–function relationship of the eukaryotic DNA replication factor, proliferating cell nuclear antigen. J Biol Chem 270: 22527–22534 [DOI] [PubMed] [Google Scholar]
- Gary R, Park MS, Nolan JP, Cornelius HL, Kozyreva OG, Tran HT, Lobachev KS, Resnick MA, Gordenin DA (1999) A novel role in DNA metabolism for the binding of Fen1/Rad27 to PCNA and implications for genetic risk. Mol Cell Biol 19: 5373–5382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes XV, Burgers PM (2000) Two modes of FEN1 binding to PCNA regulated by DNA. EMBO J 19: 3811–3821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulbis JM, Kelman Z, Hurwitz J, O'Donnell M, Kuriyan J (1996) Structure of the C-terminal region of p21WAF1/CIP1 complexed with human PCNA. Cell 87: 297–306 [DOI] [PubMed] [Google Scholar]
- Harrington JJ, Lieber MR (1994) The characterization of a mammalian DNA structure-specific endonuclease. EMBO J 13: 1235–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henneke G, Friedrich-Heineken E, Hubscher U (2003) Flap endonuclease 1: a novel tumour suppresser protein. Trends Biochem Sci 28: 384–390 [DOI] [PubMed] [Google Scholar]
- Hickson ID (2003) RecQ helicases: caretakers of the genome. Nat Rev Cancer 3: 169–178 [DOI] [PubMed] [Google Scholar]
- Hingorani MM, O'Donnell M (2000) Sliding clamps: a (tail)ored fit. Curr Biol 10: R25–R29 [DOI] [PubMed] [Google Scholar]
- Hosfield DJ, Mol CD, Shen B, Tainer JA (1998) Structure of the DNA repair and replication endonuclease and exonuclease FEN-1: coupling DNA and PCNA binding to FEN-1 activity. Cell 95: 135–146 [DOI] [PubMed] [Google Scholar]
- Hwang KY, Baek K, Kim HY, Cho Y (1998) The crystal structure of flap endonuclease-1 from Methanococcus jannaschii. Nat Struct Biol 5: 707–713 [DOI] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M (1991) Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A 47: 110–119 [DOI] [PubMed] [Google Scholar]
- Kelman Z (1997) PCNA: structure, functions and interactions. Oncogene 14: 629–640 [DOI] [PubMed] [Google Scholar]
- Kleywegt GJ, Jones TA (1997) Detecting folding motifs and similarities in protein structures. Methods Enzymol 277: 525–545 [DOI] [PubMed] [Google Scholar]
- Klungland A, Lindahl T (1997) Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J 16: 3341–3348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis PJ (1991) MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr 24: 946–950 [Google Scholar]
- Krishna TS, Kong XP, Gary S, Burgers PM, Kuriyan J (1994) Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 79: 1233–1243 [DOI] [PubMed] [Google Scholar]
- Kucherlapati M, Yang K, Kuraguchi M, Zhao J, Lia M, Heyer J, Kane MF, Fan K, Russell R, Brown AM, Kneitz B, Edelmann W, Kolodner RD, Lipkin M, Kucherlapati R. (2002) Haploinsufficiency of Flap endonuclease (Fen1) leads to rapid tumor progression. Proc Natl Acad Sci USA 99: 9924–9929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowski RA, MacArthur MW, Moss DS, Thornton JM (1993) PROCHECK: a program to check the stereochemical quality of protein structure. J Appl Crystallogr 26: 283–291 [Google Scholar]
- Lawrence MC, Bourke P (2000) CONSCRIPT: a program for generating electron density isosurfaces for presentation in protein crystallography. J Appl Crystallogr 33: 990–991 [Google Scholar]
- Li X, Li J, Harrington J, Lieber MR, Burgers PM (1995) Lagging strand DNA synthesis at the eukaryotic replication fork involves binding and stimulation of FEN-1 by proliferating cell nuclear antigen. J Biol Chem 270: 22109–22112 [DOI] [PubMed] [Google Scholar]
- Liu Y, Kao HI, Bambara RA (2004) FLAP ENDONUCLEASE 1: a central component of DNA metabolism. Annu Rev Biochem 73: 589–615 [DOI] [PubMed] [Google Scholar]
- Maga G, Hubscher U (2003) Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci 116: 3051–3060 [DOI] [PubMed] [Google Scholar]
- Matsumoto Y (2001) Molecular mechanism of PCNA-dependent base excision repair. Prog Nucleic Acid Res Mol Biol 68: 129–138 [DOI] [PubMed] [Google Scholar]
- Murante RS, Rust L, Bambara RA (1995) Calf 5′ to 3′ exo/endonuclease must slide from a 5′ end of the substrate to perform structure-specific cleavage. J Biol Chem 270: 30377–30383 [DOI] [PubMed] [Google Scholar]
- Nicholls A, Sharp KA, Honig B (1991) Protein folding and association: insights from the interfacial and thermodynamic properties of hydrocarbons. Proteins 11: 281–296 [DOI] [PubMed] [Google Scholar]
- Oku T, Ikeda S, Sasaki H, Fukuda K, Morioka H, Ohtsuka E, Yoshikawa H, Tsurimoto T (1998) Functional sites of human PCNA which interact with p21 (Cip1/Waf1), DNA polymerase δ and replication factor C. Genes Cells 3: 357–369 [DOI] [PubMed] [Google Scholar]
- Sakurai S, Kitano K, Okada K, Hamada K, Morioka H, Hakoshima T (2003) Preparation and crystallization of human flap endonuclease FEN-1 in complex with proliferating-cell nuclear antigen, PCNA. Acta Crystallogr D 59: 933–935 [DOI] [PubMed] [Google Scholar]
- Shamoo Y, Steitz TA (1999) Building a replisome from interacting pieces: sliding clamp complexed to a peptide from DNA polymerase and a polymerase editing complex. Cell 99: 155–166 [DOI] [PubMed] [Google Scholar]
- Shen B, Nolan JP, Sklar LA, Park MS (1997) Functional analysis of point mutations in human flap endonuclease-1 active site. Nucleic Acids Res 25: 3332–3338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storici F, Henneke G, Ferrari E, Gordenin DA, Hubscher U, Resnick MA (2002) The flexible loop of human FEN1 endonuclease is required for flap cleavage during DNA replication and repair. EMBO J 21: 5930–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stucki M, Jónsson ZO, Hübscher U (2001) In eukaryotic flap endonuclease 1, the C terminus is essential for substrate binding. J Biol Chem 276: 7843–7849 [DOI] [PubMed] [Google Scholar]
- Tom S, Henricksen LA, Bambara RA (2000) Mechanism whereby proliferating cell nuclear antigen stimulates flap endonuclease 1. J Biol Chem 275: 10498–10505 [DOI] [PubMed] [Google Scholar]
- Tsurimoto T (1998) PCNA, a multifunctional ring on DNA. Biochim Biophys Acta 1443: 23–39 [DOI] [PubMed] [Google Scholar]
- Tsurimoto T (1999) PCNA binding proteins. Front Biosci 4: D849–D858 [DOI] [PubMed] [Google Scholar]
- Waga S, Stillman B (1998) The DNA replication fork in eukaryotic cells. Annu Rev Biochem 67: 721–751 [DOI] [PubMed] [Google Scholar]
- Warbrick E (1998) PCNA binding through a conserved motif. BioEssays 20: 195–199 [DOI] [PubMed] [Google Scholar]
- Wu X, Li J, Li X, Hsieh CL, Burgers PM, Lieber MR (1996) Processing of branched DNA intermediates by a complex of human FEN-1 and PCNA. Nucleic Acids Res 24: 2036–2043 [DOI] [PMC free article] [PubMed] [Google Scholar]