

Abstract
The ERCC1-XPF heterodimer is a structure-specific endonuclease involved in both nucleotide excision repair and interstrand crosslink repair. Mice carrying a genetic defect in Ercc1 display symptoms suggestive of a progressive, segmental progeria, indicating that disruption of one or both of these DNA damage repair pathways accelerates aging. In the hematopoietic system, there are defined age-associated changes for which the cause is unknown. To determine if DNA repair is critical to prolonged hematopoietic function, hematopoiesis in Ercc1−/− mice was compared to that in young and old wild-type mice. Ercc1−/− mice (3-week-old) exhibited multilineage cytopenia and fatty replacement of bone marrow, similar to old wild-type mice. In addition, the proliferative reserves of hematopoietic progenitors and stress erythropoiesis were significantly reduced in Ercc1−/− mice compared to age-matched controls. These features were not seen in nucleotide excision repair-deficient Xpa−/− mice, but are characteristic of Fanconi anemia, a human cancer syndrome caused by defects in interstrand crosslink repair. These data support the hypothesis that spontaneous interstrand crosslink damage contributes to the functional decline of the hematopoietic system associated with aging.
Keywords: Fanconi anemia, hematopoietic progenitors, nucleotide excision repair, progeria, senescence
Introduction
The hematopoietic system possesses a robust proliferative capacity that provides 1012 blood cells per day in an adult human (Effros and Globerson, 2002), as well as a large reserve that can be mobilized following acute challenges including blood loss or systemic infection. However, the hematopoietic stem cells that give rise to all blood cell lineages are limited in their replicative and repopulating abilities (Van Zant and Liang, 2003) and the hematopoietic system functionally declines with age. This is evidenced by the mild cytopenia, particularly anemia, found in 13% of otherwise healthy individuals over 70 years of age (Salive et al, 1992) and by a reduced capacity for bursts of red blood cell (RBC) production following hypoxia (Udupa and Lipschitz, 1984b) and neutrophil production after endotoxin challenge (Timaffy, 1962). Age-associated hematopoietic deficits result in decreased physical performance (Penninx et al, 2003) and mortality (Wilkinson and Warren, 2003). The cause(s) of this functional decline is unknown but may include decreased hematopoietic stem cell reserves, imbalance in the cytokines required for effective blood cell production, alterations in the hematopoietic microenvironment that reduce output and/or cell mobilization to the periphery. Recently, the reported increase in genomic instability in the peripheral blood of aged individuals has suggested that an accumulation of genetic defects in hematopoietic progenitors may be involved (Ben Yehuda et al, 2000).
ERCC1-XPF is a protein heterodimer that is essential for the multistep, ‘cut and patch' nucleotide excision repair (NER) pathway that eliminates a wide range of helix-distorting DNA lesions induced by UV irradiation and numerous chemicals (Biggerstaff et al, 1993; Sijbers et al, 1996). Together the proteins act as a structure-specific endonuclease, which makes an incision 5′ of DNA lesions to allow removal of the damaged patch of DNA (Mu et al, 1995; de Laat et al, 1998). The ERCC1-XPF protein complex has functions outside of NER, for instance it is essential for DNA interstrand crosslink (ICL) repair (Niedernhofer et al, 2004). Mutations in Ercc1 have not been described in man, but Ercc1−/− mice are viable. However, their phenotype is severe and quite distinct from other NER-deficient mice (McWhir et al, 1993; Weeda et al, 1997). Whereas most NER-deficient mice are phenotypically normal, albeit prone to UV- and chemical-induced cancer (de Vries et al, 1995), Ercc1−/− mice develop normally but quickly display severe wasting that culminates in death in the fourth week of life. Many of the features of the Ercc1−/− mice are reminiscent of mammalian aging, including ataxia, kyphosis, osteopenia, weight loss, skin atrophy, sarcopenia and hepatocellular polyploidization (Hasty et al, 2003). This led to the designation of the Ercc1−/− phenotype as a highly accelerated and segmental progeria (Weeda et al, 1997).
We utilized Ercc1−/− mice to examine the effects of loss of DNA repair pathways on degeneration of the hematopoietic system. We demonstrate here for the first time that both basal hematopoiesis and reserve capacity under stress are severely reduced in Ercc1−/− mice, in a manner consistent with that found in normal aging. This supports the validity of the Ercc1−/− mouse as a progeroid model of hematopoietic aging. Furthermore, we show that progenitor activity is only mildly affected in the absence of NER alone, and Ercc1−/− progenitors are hypersensitive to exogenous ICL damage, suggesting that the loss of ICL repair may play an important role in spontaneous tissue exhaustion in these mice. In support of this, the results with the Ercc1−/− mice create a striking parallel with the deficits exhibited by the human ICL-sensitive inherited syndrome Fanconi anemia. These data support a significant role for ICL repair in the maintenance of hematopoietic function.
Results
Ercc1−/− mice have multilineage peripheral blood cytopenia
To determine if the hematopoietic system was affected in the DNA repair-deficient Ercc1−/− mice, we analyzed the peripheral blood of 3-week-old, prematurely aged Ercc1−/− mice (n=34) and their wild-type (wt) littermates (Table I). The mutant mice had significantly decreased white blood cell and platelet counts compared to wt mice. White blood cell counts were not affected in 13- to 15-day (d)-old Ercc1−/− mice compared to littermates, demonstrating that this hematopoietic deficit was not due to a developmental defect, but rather declined over the lifetime of the mice. Platelet levels were suppressed in the Ercc1−/− mice as early as 15 d of age (Table I). Peripheral RBCs and hemoglobin levels were not significantly affected in the Ercc1−/− mice, although there was a trend toward decreased erythropoiesis. Reticulocyte counts were also within normal ranges in knockout mice, suggesting that RBC lifespan in these mice was unaffected (data not shown).
Table 1.
Peripheral blood parameters of wt and Ercc1−/− mice at 2 and 3 weeks of age
| Cohort (n) | Age (weeks) | WBC | Plt | RBC | HCT | Hb | MCHC |
|---|---|---|---|---|---|---|---|
| wt (10) | 2 | 5.7±1.5 | 1051±112 | 6.9±0.3 | 0.34±0.02 | 6.7±0.4 | 20.0±0.4 |
| Ercc1−/− (9) | 2 | 4.4±3.3 | 517±211** | 6.1±1.3 | 0.33±0.02 | 6.9±1.3 | 21.0±1.0 |
| wt (27) | 3 | 5.4±0.5 | 795±44 | 6.6±0.2 | 0.32±0.01 | 6.8±0.2 | 20.9±0.1 |
|
Ercc1−/− (34) |
3 |
3.9±0.5* |
614±28** |
6.4±0.2 |
0.32±0.01 |
6.6±0.2 |
21.3±0.2 |
| WBC, white blood cells (× 109/l); Plt, platelets (× 109/l); RBC, red blood cells (× 1012/l); HCT, hematocrit; Hb, hemoglobin (arbitrary units); MCHC, mean corpuscular hemoglobin concentration (arbitrary units). | |||||||
| Significant difference between Ercc1−/− mice and wt mice as determined by Student's t-test, *P<0.05, **P<0.005. | |||||||
Systemic, progressive hematopoietic deficits in Ercc1−/− mice
Histological examination of the femoral bone marrow (BM) space of 3-week-old Ercc1−/− mice revealed striking hypocellularity compared to age-matched wt mice (n=3; Figure 1A). The BM space exhibited substantial adipose transformation and closely resembled bone sections from aged wt mice. Bone architecture in the Ercc1−/− mice was normal, including osteoclast-mediated lacunae formation and the endosteal osteoblast lining of these spaces that normally accompanies hematopoietic colonization, suggesting a hematopoietic-specific defect. Sections from 15-d-old Ercc1−/− revealed slightly reduced cellularity and early fatty changes compared to aged-matched animals. The BM of 1-d-old mutant and that of wt mice were indistinguishable. This demonstrates progressive BM degeneration in Ercc1−/− mice.
Figure 1.
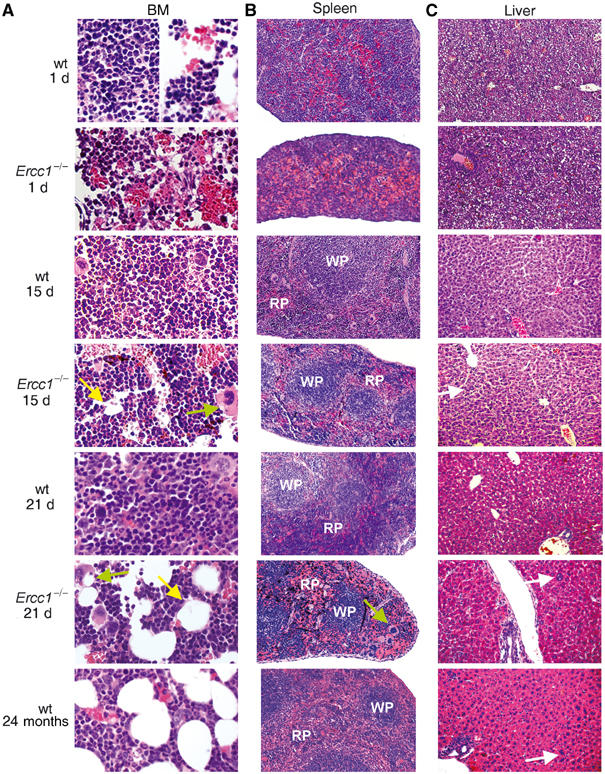
Progressive systemic hematopoietic deficits in Ercc1−/− mice. (A) Hematoxylin- and eosin-stained histologic sections of the femoral BM space from Ercc1−/− mice and wt littermates at 1, 15 and 21 d of age, as well as from an aged, 2-year-old wt mouse. At 1 d of age, the BM of Ercc1−/− and that of wt mice show equivalent cellularity and areas of erythropoiesis. By 15 d of age, the BM of Ercc1−/− mice shows early signs of fatty replacement (yellow arrow) and accumulation of large debris-laden macrophages (green arrow), which progresses by 21 d of age. Adipose deposition in the BM is a hallmark of aged mice. (B) Splenic sections of the same Ercc1−/− and wt mice. Early postnatal splenic development is unchanged in the Ercc1−/− background. By 15 d of age, there is evidence of replacement of the hematopoietic red pulp (RP; white pulp, WP) by mature erythrocytes in the mutant mice. This becomes exaggerated by 21 d of age in the Ercc1−/− mice and is also seen in the aged wt mouse spleen. Hemosiderin deposition is apparent in the spleens of 15- and 21-d-old Ercc1−/− mice (black deposits), indicating increased erythrocyte death. (C) Liver sections of the same mice. Sections from 1- and 15-d-old wt and Ercc1−/− mice are indistinguishable, showing an equivalent amount of residual hematopoiesis. Thus, Ercc1−/− mice do not have a developmental delay of hematopoiesis. Polyploid nuclei in hepatocytes (white arrows) and perivascular lymphocytic infiltrates are evident in the 21 d Ercc1−/− mouse and aged mouse livers.
In mice, the primary site of hematopoiesis migrates during development, transitioning perinatally from the fetal liver to the spleen and then to the BM by 2 weeks of age. To examine the possibility that hematopoietic colonization of the BM is merely delayed in Ercc1−/− mice, spleen and liver were also analyzed. The ratio of splenic red pulp to lymphocytic white pulp was lower in both Ercc1−/− and aged wt mice compared to young wt mice (Figure 1B). While significant hematopoiesis was observed in the splenic red pulp of young wt mice, the red pulp of young Ercc1−/− and aged wt mice was predominantly filled with mature RBCs. Furthermore, there was evidence of accumulation of splenic hemosiderin with increasing age of the Ercc1−/− mice, suggesting high turnover of erythrocytes. In line with the severely decreased platelet levels observed in the peripheral blood of Ercc1−/− mice, both BM and splenic sections from these mice showed reduced megakaryopoiesis, even compared to the normal, aged animals. No significant liver hematopoiesis was observed in any mice, regardless of age or genotype (Figure 1C). The decreased BM hematopoiesis observed in the 3-week-old Ercc1−/− mice does not therefore reflect delayed hematopoietic migration to the BM, but rather recapitulates many of the physiological features found in 2-year-old wt mice.
Livers from both the Ercc1−/− and the 2-year-old wt mice (n=3 of each genotype) contained increased numbers of polyploid cells with dramatically enlarged nuclei and prominent nuclear inclusions (Figure 1C) as previously reported (Weeda et al, 1997). Both features are hallmarks of liver aging (Epstein, 1967). There was also a pronounced infiltration of mature lymphocytes, monocytes and granulocytes in the livers of all Ercc1−/− and aged wt mice and this was not observed in young wt mice. This infiltrate is also seen in another DNA repair-deficient, progeroid mouse model expressing a mutant form of the double-strand break (DSB) end-joining repair protein Ku86 (Vogel et al, 1999). Together these observations suggest profoundly accelerated tissue damage in Ercc1−/− mice and emphasize the parallels between ERCC1-deficient progeria and natural aging.
Loss of Ercc1 decreases immature BM progenitor proliferative potential
Studies of aged humans and mice demonstrate age-related hematopoietic changes affecting multiple cell lineages, suggesting failure in a multipotent hematopoietic progenitor compartment, although this remains controversial (reviewed in Van Zant and Liang, 2003). In addition to the overall hypocellularity observed in Ercc1−/− BM, the number of low-density progenitor cells was specifically decreased two-fold compared to wt controls (Figure 2A). This cellular fraction is enriched for both lineage-committed and multipotent progenitor activity. To determine if the multipotent early progenitors were specifically affected, we plated low-density progenitors in a cytokine cocktail that stimulates the specific expansion of immature cells (Engelhardt et al, 1997; Ladd et al, 1997). Over 8 d in culture, wt progenitors expanded an average of 65-fold (±8), while Ercc1−/− cells achieved only a 12-fold (±1.6) expansion during the same time period (Figure 2B).
Figure 2.
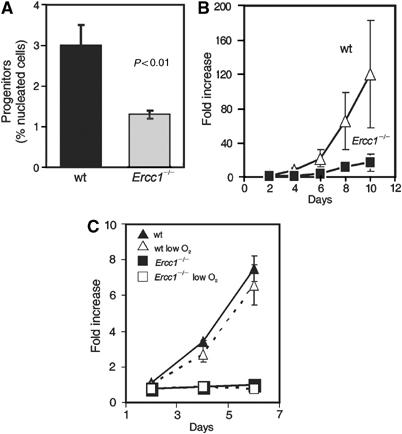
Decreased hematopoietic progenitors in Ercc1−/− mice. (A) Progenitors were isolated from 3-week-old Ercc1−/− mice and wt littermates and directly counted. The mean (±s.e.m.) for five pools of mice (2–4 mice per pool) is plotted. (B) Progenitor cells were cultured in a cytokine-rich environment to stimulate maximal proliferation. Every 2 d, cells were counted and the results plotted as fold increase from day 0. Each point represents the mean (±s.e.m.) of at least three samples. (C) Total BM was isolated from 3-week-old mice and plated with lineage-specific cytokines under 20 or 3% (low) oxygen. Progenitor numbers are expressed as fold increase from day 1. Each point represents the mean (±s.e.m.) of three mice.
The high level of oxygen present in normal tissue culture conditions causes oxidative damage, which may account for both the decreased proliferation rate and early senescence of murine fibroblasts compared to human fibroblasts (Parrinello et al, 2003). However, when total BM was plated in the cytokine conditions utilized above, the proliferation rate of progenitors grown in low-oxygen conditions (3% versus the normal 20%) was not altered during the first 6 d of culture, regardless of genotype (Figure 2C). These data support a cell-intrinsic proliferative defect in the early progenitor compartment of Ercc1−/− mice that is independent of in vitro culture conditions.
Decreased lineage-committed progenitors in Ercc1−/− mice
To determine if the hematopoietic defect extended to lineage-committed progenitors, we assayed the ability of progenitor cells to proliferate and produce differentiated cells in response to various cytokines in vitro. The number of lineage-committed progenitors capable of forming colonies was dramatically decreased across all lineages examined in Ercc1−/− BM (Figure 3A). The severity of this decrease varied by lineage, with both the erythroid lineage (BFU-E) and granulocytic lineage progenitors (CFU-G) being decreased approximately seven-fold, while the bipotent myeloid lineage progenitors (CFU-GM) demonstrated a two-fold decline in the absence of ERCC1. The proliferative capacity of those Ercc1−/− progenitors that did form colonies was also decreased, again most severely in the BFU-E and CFU-G compartments, producing far fewer terminally differentiated cells during 6 d of in vitro culture (Figure 3B). All appropriate cell types were present in each cytokine condition, demonstrating that terminal differentiation of particular lineages was not blocked (data not shown). Together, these data suggest a severe decrease in the proliferative potential of both Ercc1−/− early and lineage-committed progenitors that could account for the cytopenia observed in these mice at 3 weeks of age.
Figure 3.
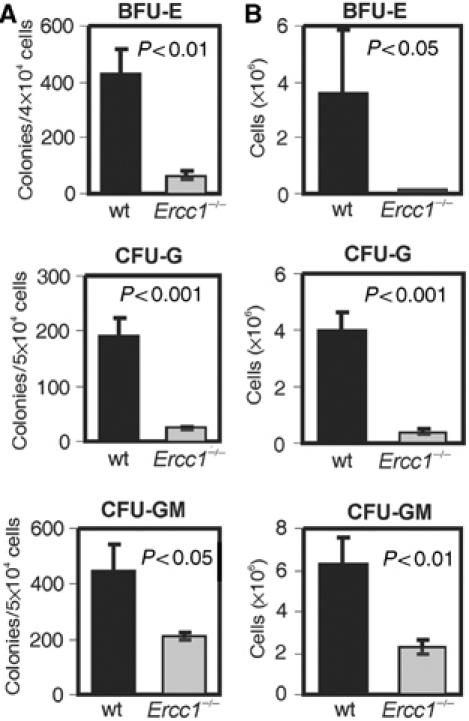
Lineage-committed progenitor numbers are decreased in Ercc1−/− BM. (A) Hematopoietic progenitors were isolated from 3-week-old mice. The indicated numbers of cells (y-axis) were seeded in methylcellulose with lineage-specific cytokines and grown for 6 d prior to colony counting. Bar graphs represent the mean (±s.e.m.) of at least four independent cell pools (2–4 mice per pool). (B) Outgrowth of cells from lineage-committed progenitors. Following colony counting, cultures were harvested and total cell numbers determined.
Suboptimal stress erythropoiesis is associated with Ercc1−/− erythroid progenitor senescence
The liver is the primary site of hematopoiesis during late embryogenesis in both mice and man. RBC production during this period is 3–5 times higher than that found during basal adult erythropoiesis (reviewed in Palis et al, 1999) and therefore represents a period of greatly increased hematopoietic demand. Deficient fetal erythropoiesis translates to a lack of optimal adult stress erythropoiesis capacity in mice (Masuoka and Townes, 2002). Although basal hematocrit and RBC levels were not significantly affected in 3-week-old Ercc1−/− mice (Table I), the proliferative defect observed in erythroid lineage progenitors (Figure 3) suggested that the hyperproliferation required under stress conditions could be more severely affected.
The number of fetal liver hematopoietic progenitors was decreased by two-fold in the Ercc1−/− mice (Figure 4A). BFU-E progenitor numbers were also specifically decreased (data not shown). Fetal liver erythroid progenitors can be cultured with differentiation-promoting cytokines in vitro, and under these conditions cells synchronously arrest, accumulate hemoglobin, reduce in size and extrude their nuclei to become mature erythrocytes (von Lindern et al, 2001). Ercc1−/− fetal liver erythroid progenitors differentiated normally (Figure 4B) and quantification of hemoglobin accumulation showed neither a delay nor a decrease (data not shown). The observed defects in fetal liver hematopoiesis do not stem, therefore, from a block in terminal differentiation capacity, in agreement with the normal differentiation observed in BM BFU-E colonies from 3-week-old Ercc1−/− mice.
Figure 4.
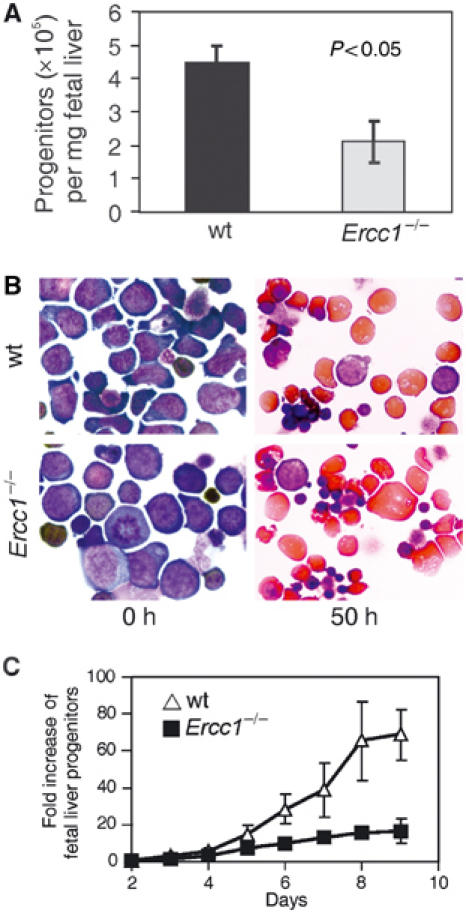
Fetal liver progenitor number, but not differentiation capacity, is decreased in Ercc1−/− mice. (A) Fetal liver hematopoietic progenitors were isolated from E12.5 embryos, counted, corrected for liver weight and plotted (±s.e.m.; n⩾4 mice). (B) Erythroid progenitors were plated with cytokines promoting differentiation. Neutral benzidine staining of cytospins demonstrates normal hemoglobin accumulation and cell size reduction in the Ercc1−/− samples. (C) Progenitors isolated from fetal livers of Ercc1−/− (▪) and wt (Δ) mice were plated in a cytokine-rich environment promoting maximal expansion of erythroid progenitors. Cells were counted daily and the results plotted as the fold increase from day 0.
Fetal liver erythroid progenitors can also be induced to expand in vitro in response to a proliferative cytokine cocktail. Maximal proliferation in this system requires glucocorticoid stimulation, as does stress erythropoiesis in vivo. Therefore, growth under these conditions is considered an in vitro model of stress erythropoiesis (Bauer et al, 1999; von Lindern et al, 2001). In the presence of glucocorticoids, Ercc1−/− progenitor cells expanded four-fold less than did wt cells (Figure 4C) with an average doubling time that was 16 h longer than that calculated for wt cells under identical conditions (36 h±1.4 versus 20.6 h±1.3 respectively, P<0.005). To determine if this decreased proliferation was the result of accelerated or increased differentiation, we analyzed proliferating progenitor cultures for the presence of terminally differentiated erythrocytes. There was a low level of spontaneous differentiation under proliferative conditions, as previously reported (von Lindern et al, 2001), which remained constant during the exponential growth period. This was not altered in Ercc1−/− cells (data not shown).
To determine the cause of the diminished proliferative potential in Ercc1−/− progenitors, we measured the frequency of apoptosis and cellular senescence. The viability of Ercc1−/− progenitors, determined by Trypan blue exclusion, remained high throughout the early culture period of fetal liver progenitors (Figure 5A). This was supported by the observation that there was no significant difference in the cell cycle profile of Ercc1−/− and wt BM progenitor cells (data not shown), including the fraction of sub-G1 cells representing apoptotic/necrotic cells (Figure 5B). This corroborated the low level of cell death found previously in Ercc1mutant mice (Weeda et al, 1997). Direct detection of apoptotic cells using the TUNEL assay also did not reveal a difference in the number of apoptotic cells between Ercc1−/− and wt BM progenitors for the length of the culture (Figure 5C). We conclude that cell death is not the predominant factor affecting proliferation in Ercc1−/− progenitors.
Figure 5.
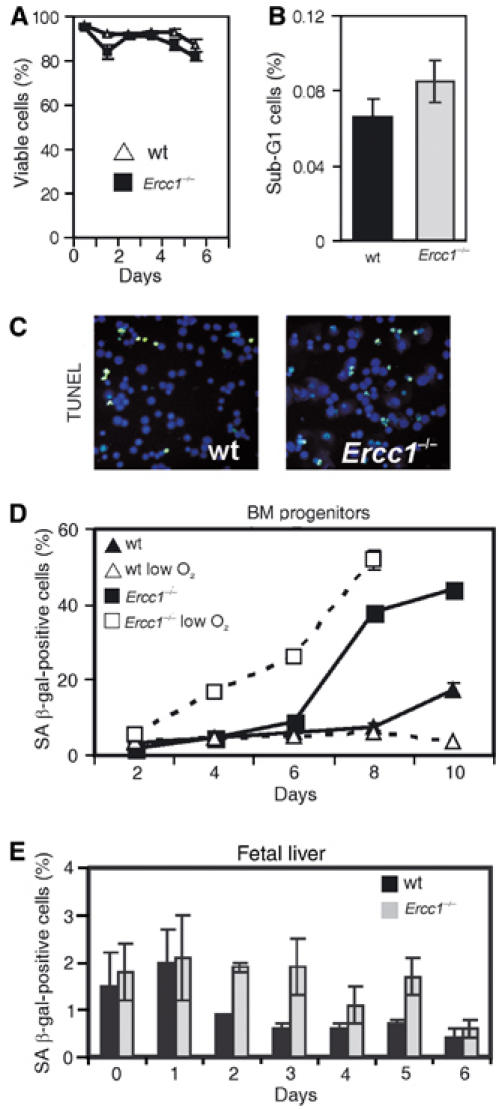
Decreased reserve capacity in the Ercc1−/− fetal liver erythroid progenitor pool is associated with progenitor senescence. (A) Fetal liver progenitor cultures were stained with the vital dye Trypan blue to determine viability at multiple time points after the initiation of cultures. A minimum of 200 cells were analyzed at each time point and the fraction of viable cells plotted. (B) BM progenitors were isolated from 21-d-old Ercc1−/− mice and wt littermates (three pools of two animals each), stained with propidium iodide to determine DNA content and analyzed by flow cytometry. Cells with less than 2N DNA content were gated and the percent calculated from the total number of viable cells. (C) TUNEL assay on fetal liver progenitors. Cells were isolated and cultured under growth-promoting conditions. Replica platings were harvested and analyzed for apoptosis every 24 h after isolation for a total of 8 d. No difference in the fraction of apoptotic cells between Ercc1−/− and wt cultures was detected. An example image from day 5 is depicted. (D) BM progenitors were isolated from femurs of 3-week-old Ercc1−/− mice and wt littermates (n=3, each genotype) and cultured for 10 d under growth-promoting conditions at either 20 or 3% (low) O2. A replicate of the culture was fixed every 2 d and stained for SA β-gal. Positively staining cells were counted and plotted as a percentage (mean±s.e.m.). (E) Fetal liver progenitors were isolated from three mice of each genotype and cultured as described in (A), then washed, fixed and stained for SA β-gal. The percentage of positively staining, senescent cells is plotted (mean±s.e.m.).
It was previously reported that both Ercc1−/− mouse embryo fibroblasts and liver cells appear to undergo a premature senescence, withdrawing from the cell cycle and exhibiting a flattened morphology (McWhir et al, 1993; Weeda et al, 1997). Senescing cells of multiple types (Dimri et al, 1995), including BM hematopoietic progenitors (Meng et al, 2003), accumulate senescence-associated β-galactosidase (SA β-gal) in their cytoplasm and this activity is widely accepted as a marker of cellular senescence. The fraction of SA β-gal-positive cells increased in both wt and Ercc1−/− BM progenitor cultures with increasing time ex vivo (Figure 5D). However, the fraction of senescence cells rose much more sharply in the Ercc1−/− cells, ranging from two- to –five-fold above the levels in wt cultures.
Cell culture conditions including oxidative stress can induce senescence in vitro (reviewed in Sherr and DePinho, 2000; Parrinello et al, 2003). However, incubation of Ercc1−/− progenitors at physiological oxygen tension (3%), rather than 20% typical of tissue culture conditions, had no effect (Figure 5D). To determine if cellular senescence was a late event or intrinsic to Ercc1 deficiency, we also examined progenitors isolated from fetal livers (n=7 mice of each genotype). While SA β-gal-positive cells were rare in fetal material, a significantly larger fraction of cells were SA β-gal positive in Ercc1−/− livers, reaching approximately 2% of the total progenitor population (Figure 5E). Thus, even progenitors isolated from young, asymptomatic Ercc1−/− animals are prone to senesce. In summary, these data suggest that hematopoietic progenitor senescence occurs with greater frequency in the absence of ERCC1. This increased senescence causes a drop in hematopoietic proliferative reserve capacity and a suboptimal response to stress conditions in vivo and in vitro.
Progenitor number and function are unaffected by NER loss alone, but are dramatically decreased following ICL induction
ERCC1 is involved in two distinct DNA repair pathways: NER, which removes bulky lesions restricted to one DNA strand, and ICL repair, which processes highly cytotoxic double-strand lesions. Having demonstrated the sensitivity of hematopoietic progenitor cells to premature senescence in the absence of ERCC1, we now sought to determine the contribution of each of these types of DNA lesions to this progeroid phenotype in Ercc1−/− mice.
To determine if loss of NER alone causes hematopoietic progenitor failure similar to that observed in Ercc1−/− mice, we isolated BM cells from the femurs of young mice in which Xpa (xeroderma pigmentosum complementation group A) was genetically deleted (Xpa−/− mice; de Vries et al, 1995). Cells from these mice are completely deficient in NER activity but retain ICL repair capacity. There were no differences in CFU-G, CFU-GM or BFU-E numbers in 4-week-old Xpa−/− mice compared to littermate controls (Figure 6) and colony size remained within the normal range (data not shown). Both the previous characterization of these mice (reviewed in Wijnhoven and van Steeg, 2003) and our own observations revealed no differences in peripheral blood values or BM cell counts between newly weaned Xpa−/− mice and wt littermate controls. However, CFU-GM (but not CFU-G) colony number was significantly reduced in 1-year-old Xpa−/− mice, indicating that an absence of NER does affect hematopoietic progenitors, but only later in life and in a lineage-restricted manner. These data demonstrate that loss of NER alone is not sufficient to cause the decrease in BM progenitor activity observed in Ercc1−/− mice.
Figure 6.
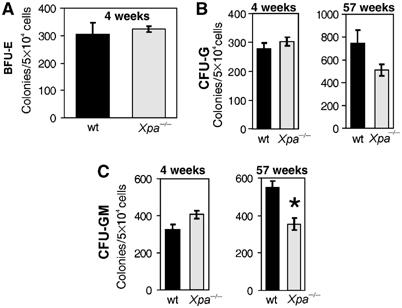
Normal progenitor pools in NER-deficient Xpa−/− mice. Hematopoietic progenitor cells were isolated from young (4 weeks) and old (56–58 weeks) wt (black bars) and Xpa−/− (gray bars) mice and plated in CFU-G or CFU-GM (or in the case of young mice BFU-E) stimulating cytokine conditions. The colony numbers were counted on the 6th d of culture. The mean (±s.e.m.) of three cell pools (two mice per pool) is shown for the young mice and three individual animals for the aged mice. The only significant difference between wt and Xpa−/− hematopoietic progenitor activity was detected in the aged mice and was restricted to the granulocyte–macrophage lineage (*P<0.05).
To determine if ICL damage induces hematopoietic progenitor failure, we treated BM progenitors from 3-week-old Ercc1−/− or wt littermates with the ICL-inducing agent mitomycin C (MMC). Figure 7 demonstrates a decrease in progenitor capacity with increasing concentrations of MMC in both wt and Ercc1−/− progenitors. Ercc1−/− BM progenitor cells were dramatically more sensitive to MMC than wt cells, regardless of the age of the animal from which the cells were derived. In contrast, the sensitivity of NER-deficient Xpa−/− progenitors was indistinguishable from wt cells. Thus, the exquisite sensitivity of the Ercc1−/− cells to ICL-inducing drugs is a unique property of the progeroid animal-derived cells. We were unable to detect a decrease in ICL repair in BM progenitors isolated from 18 month-old wt mice, indicating that if DNA repair capacity would decline with age, it is a late event.
Figure 7.
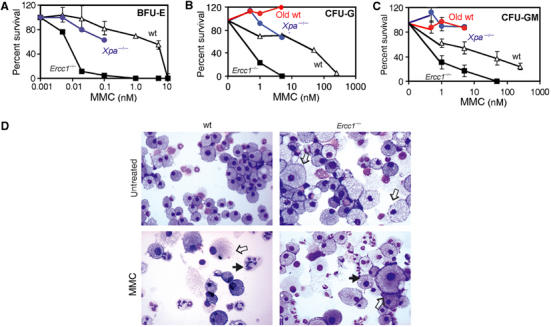
Hypersensitivity of Ercc1−/− progenitors to ICL damage. Progenitor cells from 3-week-old wt (Δ), Ercc1−/− (▪) or Xpa−/− (blue •) mice and 78-week-old wt (red •) mice were assayed for BFU-E (A), CFU-G (B) and CFU-GM (C) activity in the presence of increasing concentrations of the crosslinking agent MMC. Colonies were counted after 6 d in culture. Each data point represents the mean colony number (±s.e.m. from at least three cell pools) as a percentage of the colony number detected in untreated samples. (D) Cytology of wt and Ercc1−/− CFU-GM cultures before and after exposure to 50 or 1 nM MMC, respectively. Filled arrows indicate nuclear fragmentation; open arrows indicate enlarged macrophages.
Analysis of CFU-GM colonies revealed striking similarities between wt cells treated with high concentrations of MMC and untreated Ercc1−/− cells (Figure 7D). Untreated Ercc1−/− cultures exhibited abundant, dramatically enlarged macrophages, as well as cells with fragmented nuclei. These abnormalities, while rare in untreated wt cultures, became prevalent upon MMC treatment, suggesting that these changes may be cellular responses to and/or consequences of ICL damage. Treatment of wt cells with an ICL-inducing agent therefore recapitulates the cytological profile and progenitor capacity loss found in Ercc1−/− mice, supporting the hypothesis that the phenotype of the Ercc1−/− mice is due to a failure to repair spontaneous DNA ICLs.
Discussion
Hematopoietic changes in progeroid Ercc1−/− mice mimic normal human aging
Age-associated changes in the human hematopoietic system include mild cytopenia, in particular anemia (Salive et al, 1992), as well as a reduced capacity for bursts of RBC production following hypoxia (Udupa and Lipschitz, 1984b) and neutrophil production after endotoxin challenge (Timaffy, 1962). These deficits impact physical performance and survival (Penninx et al, 2003; Wilkinson and Warren, 2003). We investigated the hematopoietic system of DNA repair-deficient Ercc1−/− mice and discovered striking parallels with human aging.
Ercc1−/− mice (3-week-old) are mildly cytopenic, and time-course analysis indicates that this cytopenia is progressive over their lifespan (Table I). The prognostic value of mild peripheral cytopenia in elderly patients is not well established. However, we demonstrate here that the mild cytopenia in Ercc1−/− mice is a direct reflection of dramatically diminished numbers of BM hematopoietic progenitors (see below), which leads to suboptimal stress hematopoiesis. Thus, our results support recent data indicating that aged individuals with low–normal peripheral blood counts are at elevated risk of morbidity and mortality during periods of acute hematopoietic stress including blood loss or systemic infection (Wilkinson and Warren, 2003).
The prevalence of anemia increases with age and at least 20% of age-associated anemia have no identifiable cause beyond aging itself (Rothstein, 2003). Surprisingly, the RBC counts of progeroid Ercc1−/− mice were not significantly reduced (Table I). This may be attributable to the fact that the lifespan of Ercc1−/− mice is shorter than the 45 d circulatory lifespan of mouse RBCs. It is possible that if the mice survived longer, anemia would become critical. In support of this, a patient with dramatically reduced levels of ERCC1-XPF as a consequence of a mutation in XPF had a severe progeria that included anemia (LJ Niedernhofer and JHJ Hoeijmakers, unpublished data).
Despite the lack of peripheral anemia, there are several lines of evidence suggesting a severe, underlying erythropoietic deficit in Ercc1−/− mice. First, BM erythroid progenitor activity was the most severely impaired of all hematopoietic lineages in colony forming assays (Figure 3). Second, erythropoiesis was reduced in Ercc1−/− mice during fetal development and under in vitro conditions mimicking stress erythropoiesis (Figure 4; Masuoka and Townes, 2002). This mirrors the reduced progenitor expansion and RBC production seen in aged mice under hypoxic stress conditions (Udupa and Lipschitz, 1984a). In both aged and Ercc1−/− mice, erythroid differentiation, measured by hemoglobin accumulation per cell, was normal. Rather, erythropoietic deficits were due to decreased progenitor proliferation (Figure 4C; Udupa and Lipschitz, 1984b). Thus, the reduced stress erythropoiesis found in Ercc1−/− mice recapitulates that seen in aged wt mice and the decline of BM hematopoietic reserves that cause inadequate acute response in humans (Globerson, 2001). These data extend the range of tissues for which premature signs of aging are detected as a result of ERCC1-XPF deficiency (McWhir et al, 1993; Weeda et al, 1997). Furthermore, these data promote the Ercc1−/− mice as a valid model for studying degeneration of the hematopoietic system in aging.
The decreased proliferative potential in Ercc1−/− mice is caused by progenitor senescence
The mechanism(s) underlying the age-associated functional decline in hematopoietic progenitors remains controversial but may include increases in both cellular senescence and apoptosis (reviewed in Van Zant and Liang, 2003). The reduced capacity of Ercc1−/− hematopoietic progenitor cells to proliferate is not due to increased cell death (Figure 5). Similarly, no increased apoptosis is detected in the liver, kidney or spleen of Ercc1−/− mice compared to wt littermates (Weeda et al, 1997). Rather, in the absence of ERCC1, a significant fraction of hematopoietic progenitors senesce, accumulating SA β-gal (Figures 2 and 5). These data suggest that premature senescence is the primary mechanism underlying progenitor proliferative defects. Importantly, DNA damage promotes senescence of mouse fibroblasts, supporting the significance of senescence in DNA repair-deficient Ercc1−/− cells (Parrinello et al, 2003).
Hematopoietic deficits in Ercc1−/− mice are linked to the loss of ICL repair and mimic Fanconi anemia
The ERCC1-XPF endonuclease is required for NER of large bulky DNA lesions and a distinct DNA repair pathway for ICLs (Niedernhofer et al, 2004). Deficiency in NER alone is not sufficient to explain the phenotype of the Ercc1−/− mice, including their hematopoietic deficits (Figure 6; de Vries et al, 1995; Weeda et al, 1997). Genetic disruption of Xpa in the mouse results in a complete loss of NER. However, unlike the Ercc1−/− mice, the hematopoietic system of young Xpa−/− mice is normal as determined by peripheral blood cell counts and lineage-committed progenitor numbers in the BM. However, 1-year-old Xpa−/− mice display a reduction in granulocyte–macrophage-committed progenitors. This relatively mild phenotype associated with NER deficiency is further supported by normal peripheral blood counts observed in human xeroderma pigmentosum patients (Polani, 1979). Thus, an inability to excise bulky DNA adducts via NER is detrimental to the hematopoietic system, but is insufficient to explain the dramatic deficits observed in Ercc1−/− mice.
Several lines of evidence support a link between the loss of ICL repair in Ercc1−/− mice and the hematopoietic phenotype. First, Ercc1−/− primary hematopoietic progenitor cells are very sensitive to the ICL-inducing agent MMC (Figure 7; Murray et al, 2002). Second, ICL damage leads to the formation of DSBs (Bredberg et al, 1982), and in the absence of ERCC1-XPF, these ICL-induced DSBs are not repaired (Niedernhofer et al, 2004). Senescent cells and fragmented nuclei are common in untreated Ercc1−/− hematopoietic progenitor cultures and induced in wt cells by the crosslinking agent MMC (Figure 7). Thus, ICLs are capable of inducing the cellular changes observed in Ercc1−/− hematopoietic cells. Importantly, DSBs are known to accumulate in senescent human cells and in the nuclei of aged mouse tissues (Sedelnikova et al, 2004), indicating that ICL repair intermediates could contribute to the pathophysiology of aging. Third, numerous other proteins implicated in ICL repair, when mutated in humans or mice, impact the hematopoietic system including FancC, FancG, Rev3 and Rad50 (Hadjur et al, 2001; Bender et al, 2002; Koomen et al, 2002). Interestingly, peripheral blood mononuclear cells from elderly patients exhibit decreased ICL repair following cisplatin treatment (McHugh et al, 2001), suggesting that a decrease in ICL repair mechanisms may also be involved in natural aging.
The progeroid hematopoietic phenotype of the Ercc1−/− mice is spontaneous. Thus, for ICLs to play a causative role, there must be an endogenous source of crosslink damage. Spontaneous ICLs have not yet been detected in vivo, most likely because the number of lesions required to elicit a cytotoxic response is well below the limit of detection (Dronkert and Kanaar, 2001). However, endogenous compounds, which are chemically capable of forming DNA ICLs under physiological conditions, are known (Kasai et al, 1998; Niedernhofer et al, 2003). The majority of these compounds are by-products of lipid peroxidation, which is triggered when oxygen free radicals attack polyunsaturated membrane fatty acids. Importantly, elevated levels of lipid peroxidation and reactive oxygen species promote DNA damage and accelerate aging (Finkel and Holbrook, 2000; Barja, 2002; Pamplona et al, 2002).
The strongest evidence for a causal role of ICLs in the loss of hematopoietic progenitors is the striking parallel between the Ercc1−/− phenotype and that of Fanconi anemia, which too is linked with defective ICL repair. Fanconi anemia patients have pancytopenia, progressive BM failure and increased risk of myeloid leukemia (Wong and Buchwald, 2002). Diagnosis is based on hypersensitivity of the patient cells specifically to drugs that induce DNA ICLs. Like the Ercc1−/− mice, hematopoietic progenitors isolated from Fanconi anemia patients proliferate poorly (Martinez-Jaramillo et al, 2000). Also similar to Ercc1−/− mice, this defect is cell intrinsic, not caused by progenitor apoptosis and unchanged by low-oxygen culture conditions (Alter et al, 1991; Bagnara et al, 1992). Curiously, Fanconi anemia mouse models, established for three of the eight disease complementation groups, do not display hematopoietic abnormalities until challenged with MMC or increased reactive oxygen species. Under these stress conditions, the progenitors lose substantiative proliferative capacity (Wong and Buchwald, 2002). The parallels between the phenotypes of Fanconi anemia patients, Ercc1−/− mice and Fanconi mouse models challenged with ICL-inducing agents indicate that repair of ICL damage is important to the maintenance of hematopoietic reserves.
The spontaneous hematopoietic phenotype of Fanconi anemia patients is more severe than that caused by genetic disruption of this pathway in mice. This may indicate an increased burden of spontaneous DNA ICLs in human hematopoietic tissue compared to mice. Ercc1−/− murine progenitors are hypersensitive to crosslink damage (50–1000 ×; Figure 7) compared to progenitors isolated from FancA−/− mice (Rio et al, 2002). Accordingly, Ercc1−/− mice have a more severe phenotype than Fanc−/− mice, and ERCC1 deficiency is apparently lethal in humans. Because of their hypersensitivity to crosslink damage, the Ercc1 mutant mice therefore represent a good animal model of the human disease Fanconi anemia.
Interestingly, a recent study showed that hematopoietic stem cells in mice lacking the ‘ataxia telangiectasia mutated' (Atm) gene are hampered in their self-renewal abilities due to activation of the p16INK4a-Rb pathway. This manifests as a reduced reconstitutive capacity and progressive BM failure reminiscent of Ercc1−/− mice (Ito et al, 2004). Like ERCC1, ATM is involved in maintaining genomic stability, and inactivation of Atm results in a clinical syndrome with features of premature aging. Although the hematopoietic phenotype of Atm−/− mice is not as acute as in animals lacking Ercc1 (the hematopoietic defects in Atm−/− mice become apparent only 24 weeks after birth), it is tempting to speculate that failure to respond to the same spontaneous DNA damage causes the reduced hematopoietic activity in both mouse strains, although this needs further study.
In summary, this report demonstrates that DNA repair-deficient Ercc1−/− mice have decreased responses to hematopoietic stress and show exhaustion of hematopoietic progenitor activity, features that reflect symptoms of human aging. We propose that this phenotype is a consequence of progenitor cell depletion due to premature senescence of the hematopoietic stem cell and progenitor cell compartment, rather than apoptotic cell death and cell cycle changes in the progeny of these cells, and likely results from an accumulation of unrepaired endogenous DNA damage, specifically DNA ICLs.
The world's population is increasingly aged and therefore increasingly at risk for development of diseases of aging such as cancer. Elderly cancer patients tolerate current chemotherapeutic regimens poorly, and are often excluded from clinical trials due to comorbidities (reviewed in Rosti et al, 2003). The BM is a major target of most genotoxic chemotherapeutic agents and prolonged myelosuppression is one of the most common side effects of chemotherapy in the elderly (Balducci and Extermann, 1997). Therefore, there is a great need for animal models in which to test chemotherapeutic modalities in the context of an aged hematopoietic environment. Recapitulation of aged hematopoietic phenotypes in Ercc1−/− mice makes these mice not only a model of Fanconi anemia, but also a model in which to examine tissue sensitivity and rebound capacity of aged BM in response to current and novel cancer chemotherapy regimens.
Materials and methods
Mice
Ercc1−/− and wt littermates were bred from heterozygous crossings in a mixed (C57Bl/6:FVB/n) genetic background. Mice were killed at postnatal days 13–23 when the Ercc1−/− mice displayed symptoms of premature aging but were not moribund. Xpa−/− mice and old wt mice were C57Bl/6. Blood was obtained by post-mortem heart puncture and analyzed on an Animal Blood Counter (ABX Diagnostics). Femurs and tibiae were collected for BM isolation, or sectioning after fixation in 2% paraformaldehyde in sodium phosphate buffer, pH 7.4, 16 h, 4°C, then decalcification with 14% EDTA. Tissues were embedded in paraffin, sectioned (5 μm) and stained with hematoxylin and eosin.
BM proliferation and colony forming unit assays
Bones were cleaned and pulverized by mortar and pestle. BM cells were collected, washed, depleted of macrophages and fractionated by equilibrium density centrifugation on a Percoll gradient (Amersham Biosciences). Progenitor cells were enriched in the lightest density fraction as previously described (Sugiura et al, 1992). For proliferation studies, cells were plated in CellGro media (Sigma) with mIL-3 (10 ng/ml), mSCF (100 ng/ml; R&D Systems), dexamethasone (10−9 U/ml; Sigma), flt-3 ligand (0.052 μg/ml) and hTPO (0.01 μg/ml), and then cultivated at 5% CO2 and 20% O2 (or 3% where indicated) at a density of 0.8 × 106/ml. BM progenitors were harvested and then incubated in the presence of bromodeoxyuridine for 30 min prior to staining with propidium iodide for cell cycle analysis.
BM colony forming unit (CFU) assays were conducted as previously described (Hermans et al, 1998). For ICL induction studies, MMC was added directly to the methylcellulose mix prior to plating. Colonies containing more than 50 cells were scored 1 week after plating. For cell counts, colonies were pooled in PBS containing 20 μg/ml DNAse and rotated at room temperature for 30 min to create a single cell suspension. Statistical significance was determined using the Student's t-test. Cytological preparations were stained with May–Grünwald–Giemsa (Shandon Holland) and observed at × 100 magnification.
Fetal liver cell culture
Livers were removed from E12.5–13.5 embryos isolated from euthanized females. Progenitors were cultured as previously described (von Lindern et al, 2001). Briefly, cells were plated as single cell suspensions in Iscove's modified Dulbecco's medium+Glutamax supplemented with StemPro-34™ (Invitrogen), BSA, antibiotics and 2 mM L-glutamine. Progenitors were separated from differentiated erythrocytes by harvesting the cultures, centrifuging repeatedly, then re-plating to maintain a >90% pure progenitor population. For differentiation studies, cells were washed twice with PBS and re-plated in fresh media supplemented with erythropoietin (Epo, 10 U/ml), iron-saturated human transferrin (1 mg/ml) and insulin (4 × 10−4 IE/ml). Cytological preparations were stained with histological dyes and neutral benzidine. Hemoglobin content was quantified by photometric assay (Kowenz et al, 1987).
For proliferation studies, Epo (0.02 U/ml), mSCF (100 ng/ml) and dexamethasone (10−9 U/ml) were added and cells were maintained at a density of 1.5 × 106/ml. Cells were counted on a CASY/TTC cell counter (Schärfe System) and viability determined by Trypan blue exclusion. Replica platings of the fetal liver progenitors were harvested every 24 h for 8 d and apoptosis detected by TUNEL assay using the Promega Apoptosis detection system according to the manufacturer's instructions. SA β-gal staining was performed as previously described (Dimri et al, 1995).
Acknowledgments
We thank Harry van Steeg (RIVM, Bilthoven, The Netherlands) for the kind gift of Xpa−/− mice and Gijsbertus van der Horst for old wt mice (Erasmus Medical Center, Rotterdam, The Netherlands). This work was supported by the Dutch Cancer Society (Koningin Wilhelmina Fonds). LJN was supported by Postdoctoral Fellowship # PF-99-142 from the American Cancer Society.
References
- Alter BP, Knobloch ME, Weinberg RS (1991) Erythropoiesis in Fanconi's anemia. Blood 78: 602–608 [PubMed] [Google Scholar]
- Bagnara GP, Strippoli P, Bonsi L, Brizzi MF, Avanzi GC, Timeus F, Ramenghi U, Piaggio G, Tong J, Podesta M, Paolucci G, Pegoraro L, Gabutti V, Bacigalupo A (1992) Effect of stem cell factor on colony growth from acquired and constitutional (Fanconi) aplastic anemia. Blood 80: 382–387 [PubMed] [Google Scholar]
- Balducci L, Extermann M (1997) Cancer chemotherapy in the older patient: what the medical oncologist needs to know. Cancer 80: 1317–1322 [PubMed] [Google Scholar]
- Barja G (2002) Rate of generation of oxidative stress-related damage and animal longevity. Free Radic Biol Med 33: 1167–1172 [DOI] [PubMed] [Google Scholar]
- Bauer A, Tronche F, Wessely O, Kellendonk C, Reichardt HM, Steinlein P, Schutz G, Beug H (1999) The glucocorticoid receptor is required for stress erythropoiesis. Genes Dev 13: 2996–3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Yehuda A, Globerson A, Krichevsky S, Bar On H, Kidron M, Friedlander Y, Friedman G, Ben Yehuda D (2000) Ageing and the mismatch repair system. Mech Ageing Dev 121: 173–179 [DOI] [PubMed] [Google Scholar]
- Bender CF, Sikes ML, Sullivan R, Huye LE, Le Beau MM, Roth DB, Mirzoeva OK, Oltz EM, Petrini JHJ (2002) Cancer predisposition and hematopoietic failure in Rad50S/S mice. Genes Dev 16: 2237–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggerstaff M, Szymkowski DE, Wood RD (1993) Co-correction of the ERCC1, ERCC4 and xeroderma pigmentosum group F DNA repair defects in vitro. EMBO J 12: 3685–3692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredberg A, Lambert B, Soderhall S (1982) Induction and repair of psoralen cross-links in DNA of normal human and xeroderma pigmentosum fibroblasts. Mutat Res 93: 221–234 [DOI] [PubMed] [Google Scholar]
- de Laat WL, Appeldoorn E, Jaspers NG, Hoeijmakers JHJ (1998) DNA structural elements required for ERCC1-XPF endonuclease activity. J Biol Chem 273: 7835–7842 [DOI] [PubMed] [Google Scholar]
- de Vries A, van Oostrom CT, Hofhuis FM, Dortant PM, Berg RJ, de Gruijl FR, Wester PW, van Kreijl CF, Capel PJ, van Steeg H, Verbeek SJ (1995) Increased susceptibility to ultraviolet-B and carcinogens of mice lacking the DNA excision repair gene XPA. Nature 377: 169–173 [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J (1995) A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92: 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dronkert M, Kanaar R (2001) Repair of DNA interstrand cross-links. Mutat Res 486: 217–247 [DOI] [PubMed] [Google Scholar]
- Effros RB, Globerson A (2002) Hematopoietic cells and replicative senescence. Exp Gerontol 37: 191–196 [DOI] [PubMed] [Google Scholar]
- Engelhardt M, Kumar R, Albanell J, Pettengell R, Han W, Moore MA (1997) Telomerase regulation, cell cycle, and telomere stability in primitive hematopoietic cells. Blood 90: 182–193 [PubMed] [Google Scholar]
- Epstein C (1967) Cell size, nuclear content, and the development of polyploidy in the mammalian liver. Proc Natl Acad Sci USA 57: 327–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408: 239–247 [DOI] [PubMed] [Google Scholar]
- Globerson A (2001) Haematopoietic stem cell ageing. Novartis Found Symp 235: 85–96, discussion 96–100, 101–104 [DOI] [PubMed] [Google Scholar]
- Hadjur S, Ung K, Wadsworth L, Dimmick J, Rajcan-Separovic E, Scott RW, Buchwald M, Jirik FR (2001) Defective hematopoiesis and hepatic steatosis in mice with combined deficiencies of the genes encoding Fancc and Cu/Zn superoxide dismutase. Blood 98: 1003–1011 [DOI] [PubMed] [Google Scholar]
- Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J (2003) Aging and genome maintenance: lessons from the mouse? Science 299: 1355–1359 [DOI] [PubMed] [Google Scholar]
- Hermans MHA, Ward AC, Antonissen C, Karis A, Löwenberg B, Touw IP (1998) Perturbed granulopoiesis in mice with a targeted mutation in the granulocyte colony-stimulating factor receptor gene associated with severe chronic neutropenia. Blood 92: 32–39 [PubMed] [Google Scholar]
- Ito K, Hirao A, Arai F, Matsuoka S, Takubo K, Hamaguchi I, Nomiyama K, Hosokawa K, Sakurada K, Nakagata N, Ikeda Y, Mak TW, Suda T (2004) Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 431: 997–1002 [DOI] [PubMed] [Google Scholar]
- Kasai H, Iwamoto-Tanaka N, Fukada S (1998) DNA modifications by the mutagen glyoxal: adduction to G and C, deamination of C and GC and GA cross-linking. Carcinogenesis 19: 1459–1465 [DOI] [PubMed] [Google Scholar]
- Koomen M, Cheng NC, van de Vrugt HJ, Godthelp BC, van der Valk MA, Oostra AB, Zdzienicka MZ, Joenje H, Arwert F (2002) Reduced fertility and hypersensitivity to mitomycin C characterize Fancg/Xrcc9 null mice. Hum Mol Genet 11: 273–281 [DOI] [PubMed] [Google Scholar]
- Kowenz E, Leutz A, Doderlein G, Graf T, Beug H (1987) ts-oncogene-transformed erythroleukemic cells: a novel test system for purifying and characterizing avian erythroid growth factors. Haematol Blood Transfus 31: 199–209 [DOI] [PubMed] [Google Scholar]
- Ladd AC, Pyatt R, Gothot A, Rice S, McMahel J, Traycoff CM, Srour EF (1997) Orderly process of sequential cytokine stimulation is required for activation and maximal proliferation of primitive human bone marrow CD34+ hematopoietic progenitor cells residing in G0. Blood 90: 658–668 [PubMed] [Google Scholar]
- Martinez-Jaramillo G, Espinoza-Hernandez L, Benitez-Aranda H, Mayani H (2000) Long-term proliferation in vitro of hematopoietic progenitor cells from children with congenital bone marrow failure: effect of rhGM-CSF and rhEPO. Eur J Haematol 64: 173–181 [DOI] [PubMed] [Google Scholar]
- Masuoka HC, Townes TM (2002) Targeted disruption of the activating transcription factor 4 gene results in severe fetal anemia in mice. Blood 99: 736–745 [DOI] [PubMed] [Google Scholar]
- McHugh PJ, Spanswick VJ, Hartley JA (2001) Repair of DNA interstrand crosslinks: molecular mechanisms and clinical relevance. Lancet Oncol 2: 483–490 [DOI] [PubMed] [Google Scholar]
- McWhir J, Selfridge J, Harrison DJ, Squires S, Melton DW (1993) Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat Genet 5: 217–224 [DOI] [PubMed] [Google Scholar]
- Meng A, Wang Y, Van Zant G, Zhou D (2003) Ionizing radiation and busulfan induce premature senescence in murine bone marrow hematopoietic cells. Cancer Res 63: 5414–5419 [PubMed] [Google Scholar]
- Mu D, Park CH, Matsunaga T, Hsu DS, Reardon JT, Sancar A (1995) Reconstitution of human DNA repair excision nuclease in a highly defined system. J Biol Chem 270: 2415–2418 [DOI] [PubMed] [Google Scholar]
- Murray D, Vallee-Lucic L, Rosenberg E, Andersson B (2002) Sensitivity of nucleotide excision repair-deficient human cells to ionizing radiation and cyclophosphamide. Anticancer Res 22: 21–26 [PubMed] [Google Scholar]
- Niedernhofer LJ, Daniels JS, Rouzer CA, Greene RE, Marnett LJ (2003) Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J Biol Chem 278: 31426–31433 [DOI] [PubMed] [Google Scholar]
- Niedernhofer LJ, Odijk H, Budzowska M, van Drunen E, Maas A, Theil AF, de Wit J, Jaspers NGJ, Beverloo HB, Hoeijmakers JHJ, Kanaar R (2004) The structure-specific endonuclease Ercc1-Xpf is required to resolve DNA interstrand crosslink-induced double-strand breaks. Mol Cell Biol 24: 5776–5787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palis J, Robertson S, Kennedy M, Wall C, Keller G (1999) Development of erythroid and myeloid progenitors in the yolk sac and embryo proper of the mouse. Development 126: 5073–5084 [DOI] [PubMed] [Google Scholar]
- Pamplona R, Barja G, Portero-Otin M (2002) Membrane fatty acid unsaturation, protection against oxidative stress, and maximum life span: a homeoviscous-longevity adaptation? Ann NY Acad Sci 959: 475–490 [DOI] [PubMed] [Google Scholar]
- Parrinello S, Samper E, Krtolica A, Goldstein J, Melov S, Campisi J (2003) Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 5: 741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penninx BW, Guralnik JM, Onder G, Ferrucci L, Wallace RB, Pahor M (2003) Anemia and decline in physical performance among older persons. Am J Med 115: 104–110 [DOI] [PubMed] [Google Scholar]
- Polani PE (1979) DNA repair defects and chromosome instability disorders. Ciba Found Symp 66: 81–131 [DOI] [PubMed] [Google Scholar]
- Rio P, Segovia JC, Hanenberg H, Casado JA, Martinez J, Gottsche K, Cheng NC, Van de Vrugt HJ, Arwert F, Joenje H, Bueren JA (2002) In vitro phenotypic correction of hematopoietic progenitors from Fanconi anemia group A knockout mice. Blood 100: 2032–2039 [PubMed] [Google Scholar]
- Rosti G, Kopf B, Cariello A, Monti M, Dazzi C, Papiani G, Giovanis P, De Giorgi U, Marangolo M (2003) Prevention and therapy of neutropenia in elderly patients. Crit Rev Oncol Hematol 46: 247–253 [DOI] [PubMed] [Google Scholar]
- Rothstein G (2003) Disordered hematopoiesis and myelodysplasia in the elderly. J Am Geriatr Soc 51: S22–S26 [DOI] [PubMed] [Google Scholar]
- Salive ME, Cornoni-Huntley J, Guralnik JM, Phillips CL, Wallace RB, Ostfeld AM, Cohen HJ (1992) Anemia and hemoglobin levels in older persons: relationship with age, gender, and health status. J Am Geriatr Soc 40: 489–496 [DOI] [PubMed] [Google Scholar]
- Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC (2004) Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol 6: 168–170 [DOI] [PubMed] [Google Scholar]
- Sherr CJ, DePinho RA (2000) Cellular senescence: mitotic clock or culture shock? Cell 102: 407–410 [DOI] [PubMed] [Google Scholar]
- Sijbers AM, van der Spek PJ, Odijk H, van den Berg J, van Duin M, Westerveld A, Jaspers NG, Bootsma D, Hoeijmakers JH (1996) Mutational analysis of the human nucleotide excision repair gene ERCC1. Nucleic Acids Res 24: 3370–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura K, Oyaizu N, Pahwa R, Kalyanaraman VS, Pahwa S (1992) Effect of human immunodeficiency virus-1 envelope glycoprotein on in vitro hematopoiesis of umbilical cord blood. Blood 80: 1463–1469 [PubMed] [Google Scholar]
- Timaffy M (1962) A comparative study of bone marrow function in young and old individuals. Gerontol Clin (Basel) 4: 13–18 [DOI] [PubMed] [Google Scholar]
- Udupa KB, Lipschitz DA (1984a) Erythropoiesis in the aged mouse: I. Response to stimulation in vivo. J Lab Clin Med 103: 574–580 [PubMed] [Google Scholar]
- Udupa KB, Lipschitz DA (1984b) Erythropoiesis in the aged mouse: II. Response to stimulation in vitro. J Lab Clin Med 103: 581–588 [PubMed] [Google Scholar]
- Van Zant G, Liang Y (2003) The role of stem cells in aging. Exp Hematol 31: 659–672 [DOI] [PubMed] [Google Scholar]
- Vogel H, Lim DS, Karsenty G, Finegold M, Hasty P (1999) Deletion of Ku86 causes early onset of senescence in mice. Proc Natl Acad Sci USA 96: 10770–10775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Lindern M, Deiner EM, Dolznig H, Parren-Van Amelsvoort M, Hayman MJ, Mullner EW, Beug H (2001) Leukemic transformation of normal murine erythroid progenitors: v- and c-ErbB act through signaling pathways activated by the EpoR and c-Kit in stress erythropoiesis. Oncogene 20: 3651–3664 [DOI] [PubMed] [Google Scholar]
- Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, van Steeg H, Bootsma D, Hoeijmakers JH (1997) Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol 7: 427–439 [DOI] [PubMed] [Google Scholar]
- Wijnhoven SW, van Steeg H (2003) Transgenic and knockout mice for DNA repair functions in carcinogenesis and mutagenesis. Toxicology 193: 171–187 [DOI] [PubMed] [Google Scholar]
- Wilkinson TJ, Warren MR (2003) What is the prognosis of mild normocytic anaemia in older people? Intern Med J 33: 14–17 [DOI] [PubMed] [Google Scholar]
- Wong JC, Buchwald M (2002) Disease model: Fanconi anemia. Trends Mol Med 8: 139–142 [DOI] [PubMed] [Google Scholar]