

Abstract
Histone lysine methylation has been shown to index silenced chromatin regions at, for example, pericentric heterochromatin or of the inactive X chromosome. Here, we examined the distribution of repressive histone lysine methylation states over the entire family of DNA repeats in the mouse genome. Using chromatin immunoprecipitation in a cluster analysis representing repetitive elements, our data demonstrate the selective enrichment of distinct H3-K9, H3-K27 and H4-K20 methylation marks across tandem repeats (e.g. major and minor satellites), DNA transposons, retrotransposons, long interspersed nucleotide elements and short interspersed nucleotide elements. Tandem repeats, but not the other repetitive elements, give rise to double-stranded (ds) RNAs that are further elevated in embryonic stem (ES) cells lacking the H3-K9-specific Suv39h histone methyltransferases. Importantly, although H3-K9 tri- and H4-K20 trimethylation appear stable at the satellite repeats, many of the other repeat-associated repressive marks vary in chromatin of differentiated ES cells or of embryonic trophoblasts and fibroblasts. Our data define a profile of repressive histone lysine methylation states for the repetitive complement of four distinct mouse epigenomes and suggest tandem repeats and dsRNA as primary triggers for more stable chromatin imprints.
Keywords: DNA repeats; double-stranded RNA; histone lysine methylation; H3-K9, H3-K27 and H4-K20 methylation states; mouse epigenome
Introduction
Over the last years, the genome sequencing of several model organisms revealed that mammals, as compared to unicellular organisms or invertebrates, have a highly complex genome organization, largely resulting from the accumulation of repetitive elements and noncoding sequences (Lander et al, 2001; Waterston et al, 2002). In mouse, such elements account for the majority of its DNA content (44% repetitive and 52% noncoding), whereas only 4% encodes for protein function. As a result, most mammalian genes are disrupted with long intervening sequences that often also contain interspersed repeats.
Since the pioneering work of Muller (1930) and McClintock (1951), nonspecific or repetitive sequences have been thought of as ‘epigenetic elements' that can modulate gene expression programmes and also organize heterochromatic domains at centromeres and telomeres (Pardue and Gall, 1970). These repetitive elements range from short interspersed transposable elements to large centromere-associated and telomeric arrays of DNA (Waterston et al, 2002). Although the clustering of repetitive elements in large segments contributes to specialized structures in pericentric heterochromatin, most of the repetitive elements pose an inherent burden to genome stability, as their mobilization facilitates recombination between nonhomologous loci, leading to chromosomal deletions and translocations (Kazazian, 2004).
Multicellular organisms have developed silencing mechanisms to prevent remobilization of transposons. These include RNAi-triggered silencing (Ratcliff et al, 1997; Mette et al, 2000; Vastenhouw and Plasterk, 2004), DNA methylation (Jaenisch and Bird, 2003; Bourc'his and Bestor, 2004) and histone modifications (Jenuwein and Allis, 2001). Current data have given rise to models in which transcription across DNA repeats would induce formation of double-stranded RNA (dsRNA), which in turn recruits repressive chromatin modifications and DNA methylation to the underlying chromatin template (Tamaru and Selker, 2001; Hall et al, 2002; Volpe et al, 2002; Chan et al, 2004; Lippman and Martienssen, 2004; Verdel et al, 2004). Recent mapping studies across large chromosome regions revealed a strong correlation between DNA repeats, noncoding RNA, histone H3 lysine 9 methylation and DNA methylation (Lippman et al, 2004). Similarly, transcription factor mapping along human chromosomes (Cawley et al, 2004) indicated that these factors are not only found at promoters, but also can be identified at noncoding regions.
There are three repressive histone lysine methylation marks (H3-K9, H3-K27 and H4-K20) and three distinct methylation states (mono-, di- and trimethylation). H3-K9 trimethylation (Peters et al, 2003) and H4-K20 trimethylation (Schotta et al, 2004) are concentrated at pericentric and centric heterochromatin (Lehnertz et al, 2003). By contrast, H3-K27 trimethylation is enriched at the inactive X chromosome (Plath et al, 2003; Silva et al, 2003; Kohlmaier et al, 2004). Although this chromosome has the highest density of DNA repeats, the contribution of these repeats in X-chromosome inactivation remains unclear. Similarly, H3-K27 trimethylation has been implicated in Polycomb-dependent gene silencing (Ringrose and Paro, 2004) via Polycomb response elements (PREs), which also contain short repetitive elements (Ringrose et al, 2003). Despite these parallels, it is largely unknown whether the same or different histone lysine methylation marks are recruited to large arrays or to interspersed repeats and whether these epigenetic states are stably inherited across distinct cell types. Chromatin modifications have been shown to be highly dynamic at heterochromatin during differentiation (O'Neill and Turner, 1995), and recent data indicated significant differences in occupancy of transcription factor binding in a genome-wide analysis in Saccharomyces cerevisiae comparing various transcriptional states (Harbison et al, 2004).
Here, we analysed the distribution of all nine repressive histone methylation states (H3-K9, H3-K27, H4-K20 mono-, di- and trimethylation) across the repetitive complement of the mouse genome. Using directed and array-based chromatin immunoprecipitation (ChIP), we compared tandem satellite repeats (pericentric and centric heterochromatin) with distinct families of interspersed repeats including DNA transposons, long terminal repeats (LTRs), long interspersed nucleotide elements (LINEs) and short interspersed nucleotide elements (SINEs). We detect distinct chromatin modification patterns between tandem and the various interspersed repeats, which are further reflected by differences in the noncoding and dsRNAs that are generated from these elements. In addition, the observed repeat-associated histone lysine methylation profiles display significant variability in chromatin of different cell types (e.g. embryonic stem (ES) cells, fibroblasts and trophoblasts). Together, our data provide a representative cluster analysis for repressive chromatin modifications of the repetitive part of four distinct mouse epigenomes.
Results
Cluster analysis of repetitive elements
Mouse chromosomes are acrocentric and contain cytologically visible heterochromatin around their centromeres (Figure 1A). This constitutive heterochromatin can be subdivided into domains of tandem arrays of A/T-rich major satellite repeats that can comprise ⩾10 000 copies of a 234 base-pair (bp) unit per pericentric region. Similarly, centric heterochromatin (the primary constriction) consists of tandem arrays of ∼2000 copies of 123 bp minor satellite repeats. Both major and minor satellite repeats account for ∼3.5% of the mammalian genome (Lander et al, 2001; Waterston et al, 2002) (Figure 1B).
Figure 1.
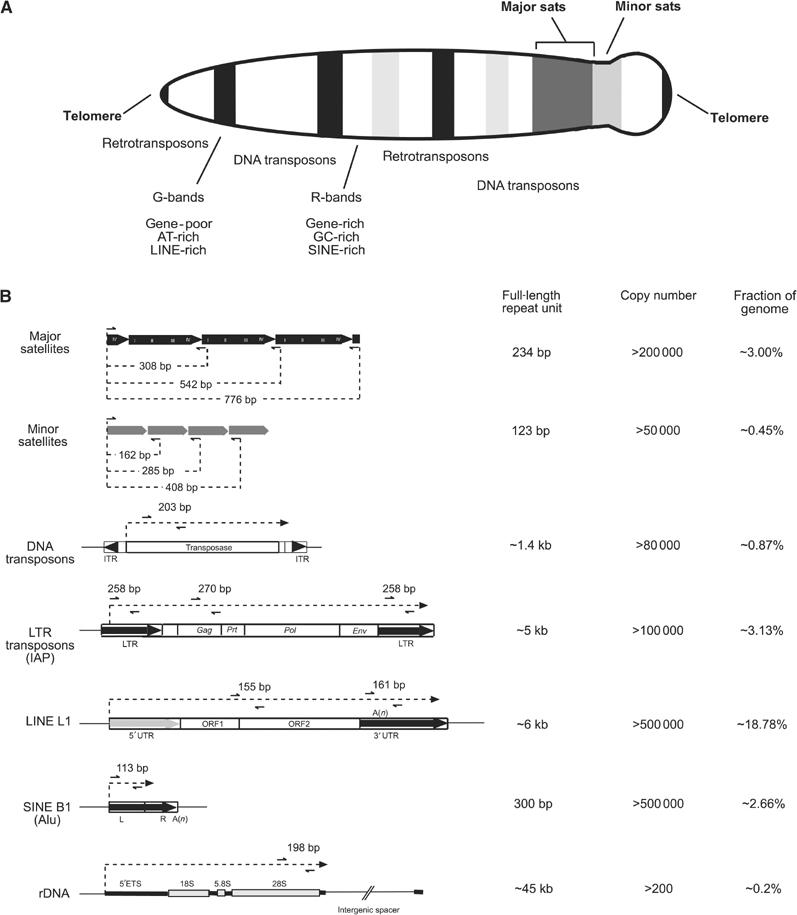
Overview of repetitive elements in the mouse genome. (A) Schematic diagram of a mitotic mouse chromosome illustrating the distribution of major (pericentric) and minor (centromeric) satellite repeats and of the various interspersed repetitive elements. (B) Summary description of repeat classes, highlighting repeat organization, length, copy number and overall abundance in the mouse genome. Specific primers (black arrows) were designed to generate PCR fragments from within the repetitive elements, thereby allowing a cluster analysis that detects the sum of chromatin modifications for a given repeat class. The sequences of these primers are indicated in Supplementary data. Although the entire repeat units are shown, most repetitive elements in the mouse genome are not full length (Waterston et al, 2002).
Interspersed repeats are singular repetitive elements that are integrated over the entire genome. Around 1% of interspersed repeats are DNA transposons (e.g. Mariner, Tigger, URR1 and Charlie), which are inactive remnants of a mutated DNA transposase element (Kazazian, 2004). The most abundant class of interspersed repeats is represented by different subtypes of retro- or RNA transposons, which together account for 25% of the mouse genome. Subclass I are the LTR transposons, including the highly active intracesternal A particle (IAP) elements. These resemble retroviruses and are associated with over 10% of all spontaneous mutations in mice (Waterston et al, 2002). Subclass II are non-LTR transposons or LINE (or L1) elements, which form the single largest fraction (∼19%) of interspersed repeats in the mouse. It has been estimated that between 0.5 and 1% of LINEs are potentially active (Goodier et al, 2001). Subclass III are SINEs, which can be further subdivided into SINE B1, the human counterpart of which are Alu elements, B2 and RSINEs. SINEs are nonautonomous elements and are thought to rely on LINEs for retrotransposition (Smit, 1996).
For all of these distinct repeat classes, specific primers for ChIP analyses were designed, which will generate, on average, 200 bp fragments from within each repetitive unit or over the LTR and UTR segments of the various transposons (see Figure 1B). Although this strategy will not allow measurement of potential differences in repeat-associated chromatin modifications that may be present at various chromosomes and cannot discriminate solitary copies of interspersed repeats, it offers a solid cluster analysis of chromatin at DNA repeats, which gauges the sum of a given histone modification over these distinct repetitive elements.
The profile of repressive histone lysine methylation states at distinct repeat classes
Lysates of crosslinked chromatin from wild-type (wt) and Suv39h double null (dn) mouse ES cells were sonicated to generate fragments of approximately 300–1500 bp. Chromatin fragments were analysed by DNA blot to confirm representation of DNA repeats (see Supplementary Figure S1). The sonicated lysates were then used in ChIP with our panel of H3-K9, H3-K27 and H4-K20 antibodies. Precipitated DNA was analysed by real-time PCR with the repeat-specific primer sets (see Figure 1B). As controls, we included primers for ribosomal DNA, actin and tubulin and also performed ChIP with an active mark, histone H3-K4 trimethylation (Santos-Rosa et al, 2002). Based on these control ChIP, we observed that most modifications show a dispersed, basal level signal, which remained below 0.5% of precipitated material (Figure 2, bottom panels). We therefore applied the 0.5% threshold to evaluate accumulation of distinct histone lysine methylation marks across repetitive elements.
Figure 2.
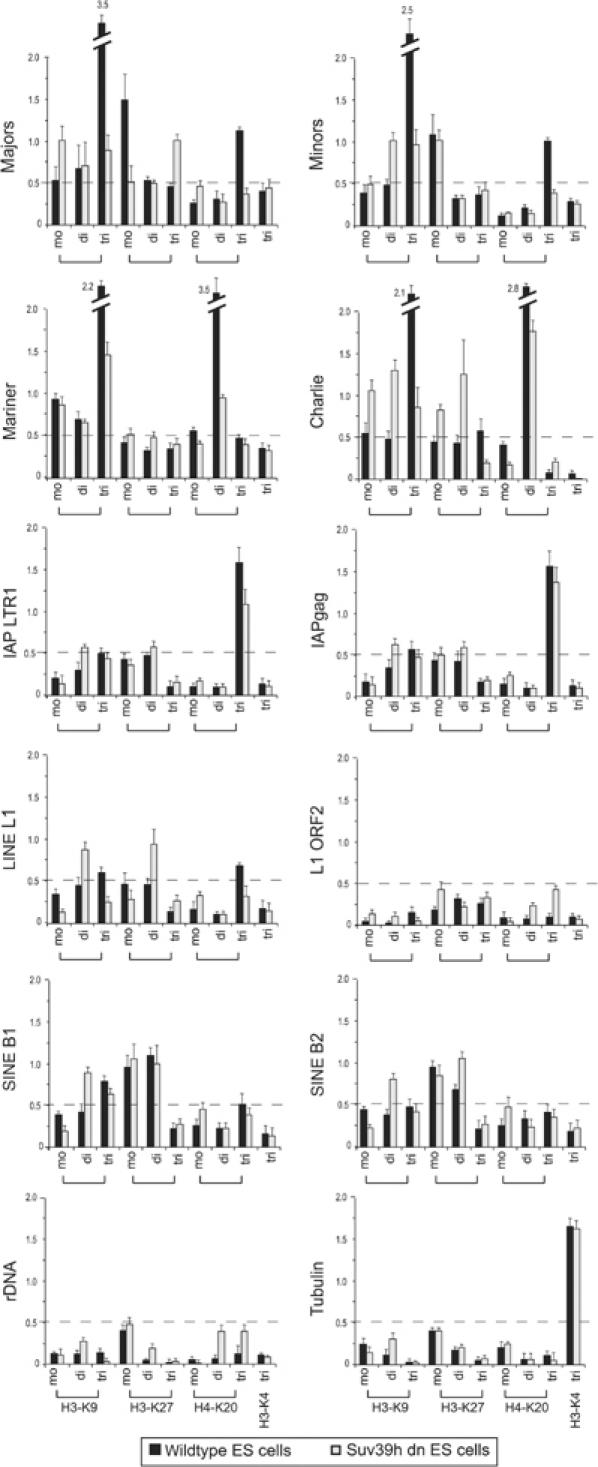
Cluster analysis for repeat-associated histone lysine methylation states. ChIP of wt and Suv39h dn ES cell chromatin with antibodies detecting H3-K9, H3-K27 and H4-K20 mono-, di- and trimethylation. As controls, ChIP was also performed with H3-K4 trimethyl (an active mark) antibodies and for the tubulin gene. Purified DNA from enriched chromatin fragments was amplified by real-time PCR with the repeat-specific primer sets (see Figure 1B), and values are indicated as percentage precipitation relative to the input. The dashed line represents the threshold signal of 0.5%, above which significant enrichment for distinct histone lysine methylation states was scored.
Using this threshold, we observe selective enrichment for H3-K9 tri-, H3-K27 mono- and H4-K20 trimethylation across mouse major and minor satellite repeats as previously reported (Peters et al, 2003; Schotta et al, 2004). This enrichment of H3-K9 trimethylation was not detected at human centromeric chromatin (Sullivan and Karpen, 2004). Similar to the apparent under-representation of H3-K9 trimethylation in Drosophila (Ebert et al, 2004) or Arabidopsis thaliana (Jackson et al, 2004) heterochromatin, species-specific alterations in satellite composition could account for these differences. For both major and minor satellites, the trimethyl marks are significantly impaired in the control ChIP with chromatin from Suv39h dn ES cells (Figure 2). DNA transposons, exemplified by Mariner and Charlie, are also enriched for H3-K9 trimethylation in a Suv39h-dependent manner. In addition, DNA transposons also contain H4-K20 dimethylation as a second signature mark, although this profile is not consistently associated with different members of DNA transposons, such as, for example, Tigger and URR1 (data not shown).
For IAP LTRs, we detect H4-K20 trimethylation as the sole prominent mark. Since only a modest reduction in the level of H4-K20 trimethylation is observed in Suv39h dn chromatin, establishment of this mark is probably independent of the Suv39h enzymes and may occur in a mechanism that is distinct from the induction of H4-K20 trimethylation at pericentric heterochromatin (Schotta et al, 2004). For LINEs, hardly any signals are detectable above threshold (e.g. H3-K9 tri- and H4-K20 trimethylation are marginal). Similarly, although SINEs can be identified by H3-K27 mono- and dimethylation, these marks are significantly less pronounced as compared to the prominent histone lysine methylation imprints at the satellite repeats, the DNA and IAP retrotransposons. Since our cluster analysis gauges the average enrichment of histone methylation marks in a chromatin environment, these low-level signals do not exclude that there may be enriched modifications at some solitary LINE or SINE repeats. It also remained possible that the repeat-associated histone methylation profiles would not reflect their representation over the coding regions of the IAP LTR and LINE elements. We therefore used additional primer pairs that are around 1.6 kb (IAPgag) or 3.1 kb (L1 ORF2) distal from the terminal repeats (see Figure 1B), and confirmed enrichment of H4-K20 trimethylation across IAP LTR transposons or absence of significant methylation marks within the LINE L1 element (Figure 2).
In addition to the directed ChIP, we also generated a custom-made microarray that is specific for all the above repeat classes in the mouse genome. PCR fragments were generated using the primer sets indicated in Figure 1B, and amplified sequences were spotted in quadruplicate on glass slides, which were then probed with ChIP material from wt and Suv39h dn ES cells. These ChIP-on-chip experiments (data not shown) are consistent with the above results of the directed ChIP (Figure 2).
Reduced DNA methylation does not alter repressive histone lysine methylation profiles
To examine whether DNA methylation would affect the observed histone lysine methylation profiles, we repeated the directed ChIP with ES cells that are wt (J1) or mutant for DNA methyltransferase (DNMT) Dnmt1 and double mutant for the DNMTs Dnmt3a and Dnmt3b (Okano et al, 1999). Genomic DNA was cleaved with the restriction enzyme McrBC, which selectively digests methylated DNA. The resulting DNA fragments were used in PCR amplifications with the same primer sets as indicated in Figure 1B to estimate the relative distribution of DNA methylation (Rabinowicz et al, 2003) (Figure 3, lanes indicated 5mC). This analysis reveals high levels of DNA methylation at IAP LTRs, moderate DNA methylation at major and minor satellite repeats and at LINEs, and low DNA methylation at DNA transposons and SINEs. Importantly, all of these DNA methylation levels are significantly reduced in the mutant Dnmt1 and Dnmt3a/Dnmt3b backgrounds.
Figure 3.
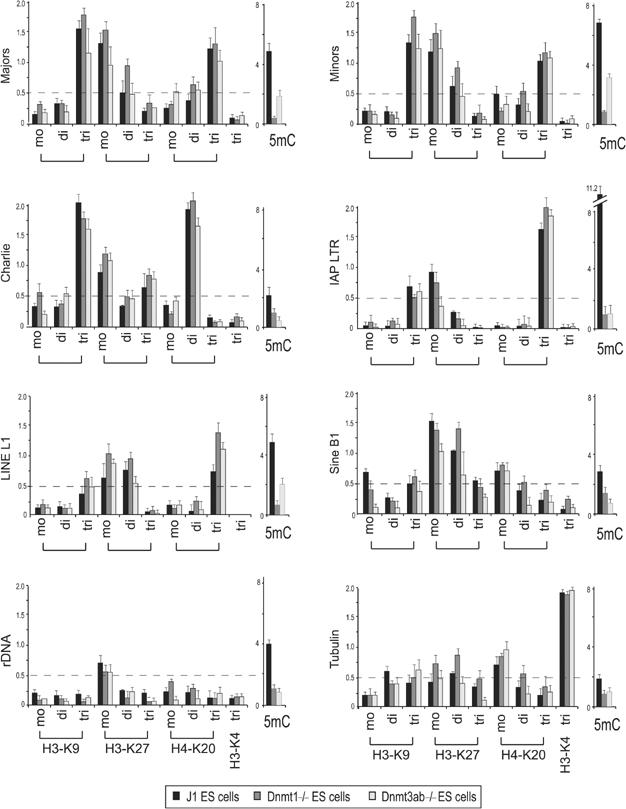
Repeat-associated H3-K9, H3-K27 and H4-K20 methylation states in chromatin of Dnmt-deficient ES cells. ChIP profile of wt (J1), Dnmt1-/- and Dnmt3ab-/- ES cell chromatin as described in Figure 2. Also indicated is the degree of DNA methylation (5mC) that is present at the distinct repeat classes in wt and mutant Dnmt chromatin. For this analysis, genomic DNA was digested with the methylation-specific restriction enzyme McrBC, and relative DNA methylation was measured by the inverse ability of the remaining DNA fragments to generate PCR products (Rabinowicz et al, 2003) with the repeat-specific primer sets (see Figure 1B).
We then analysed the repressive histone lysine methylation profile in chromatin of J1 and the Dnmt mutant ES cells. This indicated a very similar, although slightly less pronounced, pattern of the marks across repetitive elements as compared to the data described in Figure 2. Importantly, we did not observe significant differences between J1 and Dnmt mutant chromatin, suggesting that impaired DNA methylation in ES cells does not significantly alter the observed histone lysine methylation pattern. It is, however, possible that in chromatin fully deficient for all DNMTs, a more dramatic alteration of repeat-associated histone lysine methylation may be induced.
Repeat-associated transcript levels are elevated in Suv39h dn ES cells
To determine the transcriptional status of the different repeat classes, we performed reverse transcription (RT–PCR) on total RNA that was isolated from wt and Suv39h dn ES cells (Figure 4A). Transcriptional activity of satellite repeats has been described (Rudert et al, 1995), and previous studies have indicated transcripts across major and minor satellites at elevated (35) PCR cycles (Lehnertz et al, 2003). Here, real-time PCR with the primer sets indicated in Figure 1B was used to calibrate linear range (22–27) PCR cycles (see Supplementary Figure S2) for visualization of transcripts.
Figure 4.
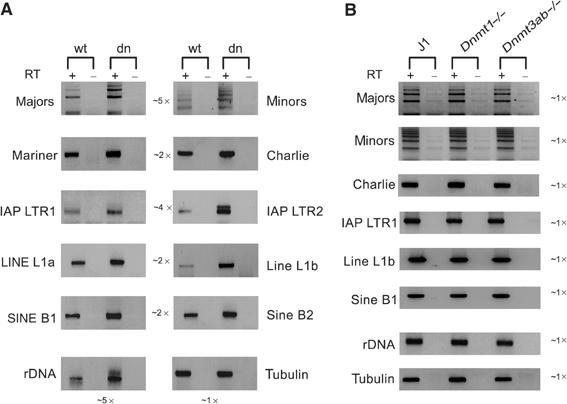
Elevated transcript levels for repetitive DNA elements in Suv39h dn ES cells. RT–PCR analysis for repeat-derived transcript levels in total RNA from wt and Suv39h dn ES cells (A) or from wt (J1) and Dnmt1-/- or Dnmt3ab-/- ES cells (B). cDNA was generated by random priming and then amplified with the repeat-specific primer sets (see Figure 1B). Linear range amplification of cDNA was calibrated by real-time PCR (between 22 and 27 cycles; see Supplementary Figure S2), and PCR products are visualized by inverse images of EtBr-stained 2% agarose gels. As controls, reactions were performed without reverse transcriptase (−RT). The relative average increase in transcription of two distinct elements within a repeat class is indicated in the middle of the two columns shown in panel A.
The data indicate that for all repeat classes, transcripts can be detected (Figure 4A), although the cluster analysis does not allow measurement of transcriptional activity for individual repeats. In Suv39h dn ES cells, transcripts are significantly increased (up to five-fold) for major and minor satellite repeats and for IAP LTRs, and modestly elevated (∼2-fold) for DNA transposons, LINEs and SINEs. Surprisingly, there is also a five-fold increase for rDNA transcripts in the Suv39h dn ES cells. Together, these data reflect higher transcript levels in Suv39h dn ES cells for all of the analysed repeats. By contrast, no apparent difference in the abundance of repeat-associated transcripts was detected in the comparative RNA analysis with total RNA from Dnmt1 and Dnmt3a/Dnmt3b mutant ES cells (Figure 4B).
The tandem satellite repeats generate dsRNA
To examine whether the quality and nature of RNA transcripts may differ between tandem and interspersed repeats, we used enzymes that preferentially cleave either single-stranded (ss) (RNAseONE) or dsRNA (RNAseV1). First, a titration experiment was performed to determine the optimal digestion time. Total RNA from wt ES cells was incubated for different times with RNAseONE or RNAseV1, and the remaining RNA molecules were analysed by real-time RT–PCR with primers for major satellites and tubulin. The generated signal was compared to reactions with undigested RNA, which was set at 100% (see Supplementary Figure S3). Thus, this analysis does not measure total abundance of RNA as shown in Figure 4, but rather indicates the relative presence of ds or ss transcripts in the remaining pool of digested RNA.
A 30 s digest with RNAseONE was used to remove ssRNAs. At this time point, ∼22% of major satellite transcripts are left (see Supplementary Figure S3). This remaining pool of dsRNA was then analysed with the repeat-specific primer sets. We observed significant signal for major and minor satellite repeats, whereas very little or no dsRNA could be detected for any of the interspersed repeats. Importantly, the dsRNAs for major and minor satellite repeats are significantly upregulated in Suv39h dn cells (Figure 5A).
Figure 5.
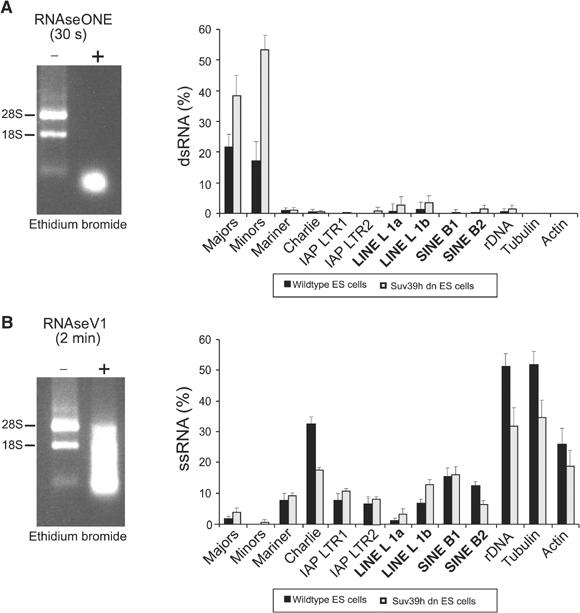
Tandem satellite repeats generate dsRNA. Total RNA from wt and Suv39h dn ES cells was treated for the indicated time points with RNAseONE™ to digest ssRNAs (A) or with RNAseV1 to cleave dsRNAs (B). The remaining pool of RNA molecules was then converted to cDNA and amplified by real-time PCR with the repeat-specific primer sets (see Figure 1B). The histograms indicate the percentages, relative to undigested RNA, of repeat-derived RT–PCR products that can be generated after RNAseONE™ (% dsRNA) or RNAseV1 (% ssRNA) treatment.
A 2 min digest with RNAseV1 was used to remove dsRNA. At this time point, ∼50% of tubulin transcripts are still present, whereas <2% of major satellite RNAs are left (see Supplementary Figure S3). The remaining pool of ssRNA was then analysed with the repeat-specific primer sets. Very low levels of ssRNA for the tandem repeats (major and minor) were detected, but there are significant transcripts for DNA transposons, retrotransposons, LINEs and SINEs (Figure 5B). With the exception of the DNA transposon Charlie, SINE B2 elements and rDNA, the levels of ss transcripts are not reduced for the other interspersed repeats in RNA preparations from Suv39h dn cells.
Together, the results indicate that tandem repeats favour the generation of ds transcripts, whereas interspersed repeats primarily generate ss transcripts. Moreover, since ds transcripts are significantly elevated in Suv39h dn cells, the data further suggest a function for the Suv39h histone methyltransferases (HMTases) in processing or stabilization of ds transcripts complementary to the major and minor satellite repeats.
Expression of repeat-associated transcripts in four distinct epigenomes
So far, we have analysed repeat-associated histone lysine methylation profiles in only one chromatin environment, ES cells. The question arises as to how stable these modifications are in different cell types. To expand our analysis, we treated ES cells with all-trans-retinoic acid (RA) to form embryoid bodies, thereby generating a retinoic-acid differentiated ES cell population (RA-ES). In addition, we also used day 13.5 mouse embryonic fibroblasts (MEFs) and a trophoblast stem (TS) cell line (Tanaka et al, 1998) (Figure 6A, top panels). The different character of these four distinct cell populations was confirmed by RT–PCR marker analysis for Oct-4 (ES cells), Cdx-2 (TS cells) and Annexin-5 (MEFs) (see Supplementary Figure S4). We also examined expression profiles of the major enzymatic systems transducing H3-K9 trimethylation (Suv39h1, Suv39h 2), H4-K20 trimethylation (Suv4-20h1, Suv4-20h2), H3-K27 trimethylation (Ezh1, Ezh2) and ‘euchromatic' H3-K9 dimethylating enzymes, such as G9a, Glp1, Eset and Cll8. All of these HMTases are broadly expressed in ES cells and MEFs, whereas some are downregulated in TS cells (see Supplementary Figure S4).
Figure 6.
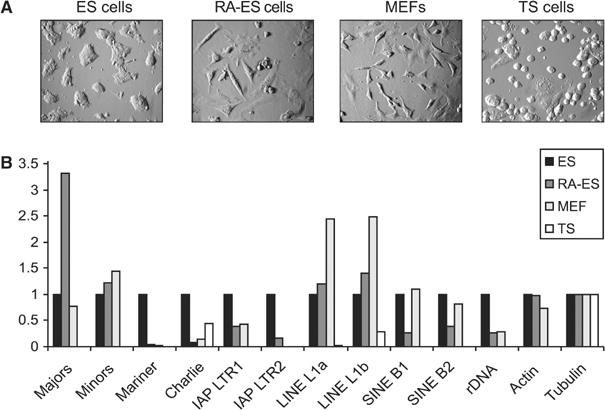
Repeat-associated transcript levels in four distinct mouse epigenomes. (A) Images of cultured mouse ES cells, RA-ES cells, day 13.5 MEFs and TS cells. (B) RT–PCR analyses for repeat-derived transcript levels in the four distinct cell populations. Total RNA was converted into cDNA, which was then amplified by real-time PCR with the repeat-specific primer sets (see Figure 1B). The histogram indicates the relative difference (after normalization for tubulin expression) in the abundance of repeat-derived transcripts with respect to levels observed in wt ES cells (see Figure 4 and Supplementary Figure S2), which were set at 1.
We then extended the RT–PCR analysis and examined the presence of repeat-associated transcripts in total RNA preparations from all four different cell populations. For comparison, wt ES cell transcripts for each given repeat class (data from Figure 4 and Supplementary Figure 2) were set at 1, and their relative increase or decrease in the other cell populations was determined. Transcript levels for major satellite repeats are significantly increased upon RA induction, and levels of both major and minor transcripts are high in ES cells and MEFs (Figure 6B). Transcripts for LINEs are also selectively elevated (∼2.5-fold) in MEFs. Across the various cell populations, major satellite and LINE transcripts are most highly abundant, suggesting a potential role for increased transcription of these repeats during differentiation. By contrast, transcripts from DNA and IAP LTR transposons are downregulated, as is rDNA transcription, while SINE-derived RNA levels vary among the four different cell types. In general, transcript levels for all repeat classes are very low in TS cells. This low abundance has been confirmed with total RNA preparations from a different TS cell line that was generated by outgrowth from Bl6/Sv129 blastocysts (data not shown).
Stability of repressive histone lysine methylation marks at tandem repeats
To compare the repeat-associated histone lysine methylation profile among the various cell types, we spiked samples with Drosophila S2 chromatin and again used a threshold of 0.5%. This internal standard resulted in a different calibration of material precipitated from ES and RA-ES cells (4%) versus MEFs and TS cells (2%), as indicated in Figure 7. Although we used our full panel of antibodies as shown in Figure 2, we only summarize the data where we have observed informative differences and signals surpassing the 0.5% threshold (H3-K9 tri-, H3-K27 mono- and di-, and H4-K20 di- and trimethylation). In chromatin of ES cells, major and minor satellites are enriched for H3-K9 tri-, H3-K27 mono- and H4-K20 trimethylation (Figure 7, top panels). After RA differentiation, the sum of these marks is elevated and both tandem repeats gain significant signal for H4-K20 dimethylation. In chromatin of MEFs and TS cells, the profile of histone lysine methylation at major and minor satellite repeats resembles that of undifferentiated ES cells, with minor differences for reduced definition of H3-K27 mono- and H4-K20 trimethylation in MEFs or of H3-K27 monomethylation in TS cells. These data indicate that the pattern of repressive histone lysine methylation states (particularly the combination of H3-K9 tri- and H4-K20 trimethylation) across tandem satellite repeats is largely maintained in chromatin of different cell types.
Figure 7.
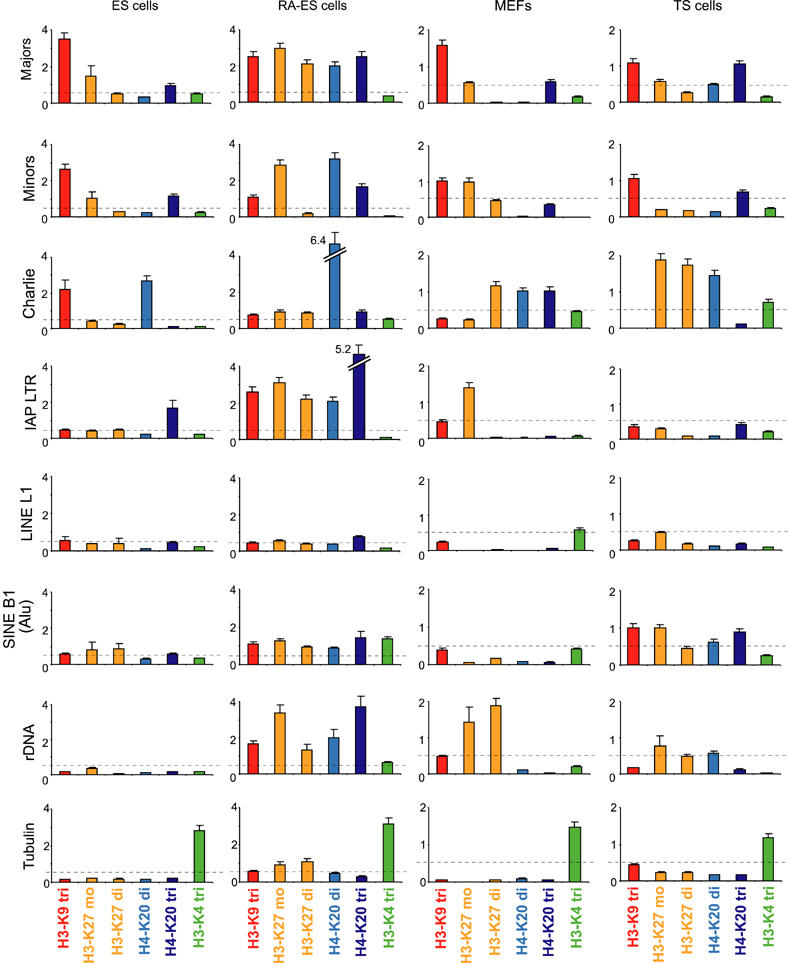
Stability of repressive histone lysine methylation marks at tandem but not at interspersed repeats. ChIP profile for repeat-associated histone lysine methylation states in wt chromatin of ES cells, RA-ES cells, MEFs and TS cells. The dashed line represents the threshold signal of 0.5%, above which significant enrichment for distinct histone lysine methylation states was scored. To adjust for differences in the ChIP efficiency with chromatin of distinct cell populations, samples were spiked with Drosophila chromatin as an internal standard. For clarity, enriched marks are colour-coded.
Variability of repressive histone lysine methylation marks at interspersed repeats
The comparative analysis for interspersed repeats indicates signature modifications for DNA transposons (H3-K9 tri- and H4-K20 dimethylation) and IAP LTRs (H4-K20 trimethylation) in ES cell chromatin (see also Figure 2). However, these marks are not stably maintained in chromatin of the other cell types, where they are either significantly reduced and even lost (e.g. H3-K9 tri-methylation in MEFs and TS cells for Charlie or H4-K20 trimethylation in MEFs and TS cells for IAP LTRs), or greatly increased (e.g. H4-K20 dimethylation for Charlie and H4-K20 trimethylation for IAP LTRs in RA-ES cells) (Figure 7). In addition, H3-K27 mono- and dimethyl signals are variably gained for Charlie in MEFs and TS cells and for IAP LTR in RA-ES cells. For LINEs, we did not detect informative signal above threshold in chromatin of the different cell types and none of the marks was significantly upregulated after RA induction. For SINE B1 elements, a modest increase for most modifications was observed in RA-treated ES cells and in TS cells, whereas MEFs displayed reduced signals as compared to the low-level profile present in ES cells. We have also not detected enriched signal for H3-K9 dimethylation (data not shown), although this mark may be present at Alu repeats (the equivalent of mouse SINE B1) in chromatin of human cells (Kondo et al, 2004). Surprisingly, highest variability was observed for rDNA, with dramatic increases of all repressive marks after RA treatment, prominent signals for H3-K27 mono- and dimethylation in MEFs and modest increases for H3-K27 mono- and dimethylation and H4-K20 dimethylation in TS cells. Together, the results illustrate a surprising variability for the definition of repressive histone lysine methylation patterns at interspersed repeats in distinct chromatin environments of different cell types.
Discussion
Histone lysine methylation and the repetitive complement of the mouse genome
We describe a representative cluster analysis for repressive histone lysine methylation states across the repetitive complement of the mouse genome. Our data indicate that selective patterns of H3-K9, H3-K27 and H4-K20 methylation can discriminate tandem satellite repeats from various classes of interspersed repeats, such as DNA transposons, LTRs, LINEs and SINEs. In chromatin of wt ES cells, the most prominent enrichment for repressive histone modifications was observed over tandem satellite repeats and DNA transposons, where distinct combinations of H3-K9 tri-, H3-K27 mono- and H4-K20 tri- or dimethylation accumulate (Figure 8). In contrast, IAP LTRs are characterized by H4-K20 trimethylation as the sole signature mark. Other repetitive elements, such as SINEs and LINEs, do not display informative signals but comprise diverse repressive imprints at low level (see Figure 7). Since our cluster analysis detects the sum of modifications over a given repeat class, the pronounced accumulation of repressive histone lysine methylation states at some but not other repetitive elements is unlikely to be a mere reflection of differences in their copy numbers. For example, LINEs and SINEs comprise >21% of the mouse genome, whereas DNA transposons account for <1% (Waterston et al, 2002) (see Figure 1B).
Figure 8.

Summary of repeat-associated histone lysine methylation marks in mouse chromatin. Repressive histone lysine methylation marks with prominent enrichment over the 0.5% threshold in wt ES cell chromatin (see Figures 2 and 7) were added to give the sum of these ‘signature' modifications for a distinct repeat class. For example, the major satellite repeats accumulate H3-K9 tri-, H3-K27 mono- and H4-K20 trimethylation at ∼6% of precipitated material, whereas SINEs and LINEs do not display a specific signal. For the satellite repeats and for some DNA transposons, H3-K9 tri-, H3-K27 mono- and H4-K20 trimethylation are significantly reduced in Suv39h dn ES cell chromatin (see Figure 2); these Suv39h-dependent marks are highlighted by yellow-coded Me in the respective methyl hexagons. In chromatin of differentiated cell types, most of the histone lysine methylation profiles at interspersed repeats differ from those observed in ES cell chromatin (see Figure 7), while they appear more stable at tandem satellite repeats. An example for this variability and the accumulation of additional marks is schematically indicated for RA-induced alterations in chromatin of wt ES cells (bottom panel). While these combinations of marks are, on average, enriched as detected by our cluster analysis, these groupings do not necessarily reflect their combined presence over individual repeats in distinct chromosomal locations.
In chromatin of Suv39h dn ES cells, H3-K9 and H4-K20 trimethylation is significantly reduced across major and minor satellite repeats and for some (Charlie and Mariner) (see Figure 2) but not other (e.g. Tigger and URR1) DNA transposons (data not shown). The enzymes for H3-K9 and H4-K20 trimethylation at pericentric heterochromatin are known and were shown to be linked in a silencing pathway, where the activity of the Suv39h enzymes precedes nucleosomal methylation by the Suv4-20h HMTases (Schotta et al, 2004). In contrast, the nature of the HMTase(s) conferring the modest H3-K27 monomethylation at major and minor satellite repeats or of the enzyme inducing H4-K20 dimethylation at DNA transposons is currently not known. Since H4-K20 trimethylation at IAP LTRs remains high in chromatin of Suv39h dn ES cells (see Figure 2), a Suv39h-independent H4-K20 trimethylating enzyme, which is most likely distinct from the described Suv4-20h HMTases, is independently targeted at these highly active retrotransposons.
Stability of epigenetic marks at tandem satellite repeats of constitutive heterochromatin
Some of the most prominent repetitive domains in chromosomes of higher eukaryotes are the tandem satellite repeats, which are often clustered in the vicinity of centromeres. These A/T-rich regions are cytologically visible as DAPI-bright pericentric foci (so-called constitutive heterochromatin) and have been implicated to ensure correct centromere function, thereby protecting chromosome segregation (Karpen and Allshire, 1997). Although there is variability and different evolutionary drive for satellites in higher eukaryotes (Henikoff et al, 2001), they represent crucial sequence elements for the definition of centromeres or neo-centromeres (Amor and Choo, 2002; Rudd et al, 2003).
Based on their important structuring role, satellite repeats appear to represent landmarks for a more stable chromatin architecture, which is further highlighted by the persistence of signature histone lysine methylation marks in chromatin of various cell types (see Figure 7). Satellite sequences, or similar arrays of tandem repeats, are likely to trigger an epigenetic hierarchy that integrates DNA repeats, the RNAi machinery, repressive histone lysine methylation and DNA methylation (see Introduction). Transcription across tandem repeats will favour the generation and self-templating of dsRNA (Jenuwein, 2002; Martienssen, 2003), which is then processed by the RNAi machinery to recruit repressive histone lysine methylation systems to the underlying chromatin template. Consistent with these models, dsRNA is primarily associated with the tandem satellites, but not the interspersed repeats (see Figure 5). Surprisingly, levels of dsRNA for major and minor satellites are significantly elevated in Suv39h dn ES cells, concomitant with pronounced reduction in pericentric and centric H3-K9 and H4-K20 trimethylation. It is likely that these more abundant dsRNAs may not be correctly converted by the RNAi machinery into siRNAs, as has been described for several mutants of heterochromatin components in Schizosaccharomyces pombe (Hall et al, 2002; Volpe et al, 2002; Schramke and Allshire, 2003). Elevated levels of dsRNA accumulate in Suv39h dn cells probably because recruitment of RITS or related RNAi processing complexes is impaired in the absence of H3-K9 methylation at chromatin regions of highly repetitive DNA content (Fukagawa et al, 2004; Noma et al, 2004; Verdel et al, 2004). It is according to this interpretation that the selective increase of satellite transcripts after RA induction (see Figure 6) could also interfere with the efficient generation of siRNAs, since the described genome instabilities and chromosome mis-segregation defects in Suv39h dn mutant mice (Peters et al, 2001) are particularly pronounced during midgestation and not in undifferentiated ES cells.
Variability of epigenetic marks at transposons and interspersed repeats
The robustness of epigenetic imprints at tandem repeats is further illustrated by the relative stability of H3-K9 and H4-K20 trimethylation at major and minor satellites in chromatin of four distinct epigenomes (see Figures 7 and 8). For example, these marks are maintained over the satellite repeats (although additional imprints are gained) after RA induction of ES cells or in fibroblasts and trophoblasts. For all other repeat-associated methylation profiles, there is significant variability and relaxed signal combination in chromatin of diverse cell types (see Figures 7 and 8).
These differences indicate that the various DNA transposons and retrotransposons do not serve a structuring role, rather they may interfere with efficient gene transcription. Indeed, reactivation and transposition of DNA transposons and retrotransposons has been proposed in the reshaping of epigenomes, and recent data support a role for transposed LINE L1 elements in aborting transcriptional elongation (Han et al, 2004) or for SINE B2 transcripts to stall RNA polymerase II (Allen et al, 2004; Espinoza et al, 2004). While DNA transposons are less active in mouse than in human, IAP LTRs are among the most active retrotransposons in mouse (Waterston et al, 2002). Mutants for DNMTs have been shown to increase transcript levels for IAP LTRs in Dnmt1 null embryos (Walsh et al, 1998) and to enhance IAP LTR and LINE transcription in Dnmt3L mutant germ cells (Bourc'his and Bestor, 2004). In Suv39h dn ES cells, we detect significant enrichment of transcripts corresponding to all tested repeat classes, at levels that are much more pronounced than those observed for Dnmt1 and Dnmt3a/3b mutant ES cells (see Figure 4). Although for some repetitive elements, increased expression coincides with reduced levels of repressive histone methylation marks (e.g. major and minor satellites, and Charlie), this is not paralleled for IAP LTRs. This apparent discrepancy between persistent H4-K20 trimethylation and elevated IAP LTR transcription in Suv39h dn cells could possibly be explained by a subpopulation of these repeats to become transcriptionally active or, alternatively, it may reflect enhanced transcription from intronic and adjacent genomic sequences. It will be interesting to analyse how this increased transcription from repetitive elements may modulate gene expression programmes or even facilitate remobilization of some of the DNA or RNA transposons.
Toward a histone modification map of the mouse epigenome
Profiling analyses for histone lysine methylation have been performed on defined chromosomal loci (Litt et al, 2001; Noma et al, 2001), parts of entire chromosomes (Greil et al, 2003; Lippman et al, 2004) and with genome-wide but promoter-biased or EST sequences that were displayed on microarrays (Ren et al, 2002; Santos-Rosa et al, 2002; Schübeler et al, 2004). Although the recent advances in chromatin tiling allow for examination of large chromosomal domains, few studies have investigated the regional distribution of epigenetic modifications in a mammalian genome. The available microarrays (primarily for human chromosomes) have so far only been used for transcription factor mapping (Cawley et al, 2004) or RNA expression profiling (Kapranov et al, 2002; Kampa et al, 2004) where, intriguingly, a widespread occurrence of noncoding RNAs has been observed.
None of the above studies examined the repetitive complement of a mammalian genome or addressed the combinatorial nature of repressive histone lysine methylation states in distinct chromatin environments of various cell types. The profile of repeat-associated histone lysine methylation marks described here is part of the construction of a chromosome-wide epigenetic matrix by high-resolution sequence tiling. Based on our data and from similar analyses in A. thaliana (Lippman et al, 2004), we predict that any chromosomal region that comprises tandem DNA repeats will favour the generation of dsRNA and the accumulation of H3-K9 and H4-K20 trimethylation. While this pathway appears essential for the definition of pericentric heterochromatin, it could also contribute to the structural organization and even the banding patterns of chromosomes. In addition, we also propose that epigenetic modifications at interspersed repeats or single-copy sequences will vary in chromatin of distinct cell types as well as during lineage specification. This epigenetic plasticity may be particularly important for modulating the differentiation potential of cloned embryos (Santos et al, 2003) and progenitor or stem cells (Baxter et al, 2004), or for protecting a normal cell from conversion to a neoplastic or senescent cell (Narita et al, 2003).
It is according to this view that comprehensive and unbiased analyses for histone lysine methylation states in many distinct epigenomes will lead to a better understanding of epigenetic transitions and how alterations in their associated pathways will affect stem cell plasticity, proliferation and differentiation.
Materials and methods
Cell culture
Cell culture of wt and Suv39h dn ES cells, MEFs and wt TS cells was carried out as described (Tanaka et al, 1998; Lehnertz et al, 2003). RA-induced differentiation of wt and Suv39h dn ES cells was performed in ES cell media lacking LIF with 100 nM RA.
Antibodies
Generation and characterization of position- and state-specific methyl-lysine histone antibodies against H3-K9, H3-K27 and H4-K20 has been described (Peters et al, 2003; Schotta et al, 2004). H3-K4 trimethylation antibodies were purchased from Upstate Biotechnology (#07-473).
Chromatin immunoprecipitation
ES cells at 80% confluence were crosslinked with 1% formaldehyde for 10 min. After quenching with 125 mM glycine, whole cell extracts were prepared for ChIP as described (Peters et al, 2003). Purified DNA of the immunoprecipitates and of input DNA was analysed by real-time PCR using the Roche Sybr green quantitation method on an MJ research Lightcycler. Results were normalized and presented as percentage of input DNA. For cell type-specific ChIP, we spiked the sonicated lysates (400 μg) from the four different cell types with 20% (80 μg) of sonicated Drosophila melanogaster S2 chromatin prior to ChIP. Immunoprecipitated DNA was analysed by real-time PCR using primers specific for the different repeat classes (see Figure 1B and Supplementary data) and for Drosophila satellite repeats. Values for ChIP data from different cell types were subsequently adjusted using the Drosophila signal as an internal standard.
DNA methylation analysis
DNA methylation analysis was performed as described (Rabinowicz et al, 2003; Lippman et al, 2004). Genomic DNA was isolated from different cell lines and sonicated to an average size of 3–5 kb. Samples were digested overnight with the methylation-specific restriction enzyme McrBC (# M0272; New England Biolabs), separated on a 1% agarose gel, and DNA fragments larger than 2 kb were gel-purified. These DNA fragments, which are enriched for nonmethylated DNA, were then analysed by real-time PCR and compared with nondigested DNA. Methylated DNA sequences will have decreased amounts of PCR product following McrBC digestion.
Reverse transcription–PCR
Total RNA was extracted from mouse ES cells with TRIzol (Invitrogen). Genomic DNA was removed by shearing in an UltraTurrax homogenizer and digestion with DNase I. First strand cDNA was synthesized from 1 μg total RNA using dN6-hexamer random primers (New England Biolabs) and amplified by PCR and real-time PCR.
RNAseONE™/RNAseV1 treatment
Total RNA was extracted as described above and incubated with RNAseONETM (Promega) or RNAseV1 (Ambion) for various time points (see Supplementary Figure S3). Ribonuclease activity was terminated by addition of SDS and ethanol. The remaining pool of undigested RNA was then precipitated, processed for cDNA synthesis and analysed by real-time PCR.
Supplementary Material
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Data
Acknowledgments
We thank E Li and J Rossant for the Dnmt mutant ES cells and the TS cells, Y Linderson, C Wippo and G Seisenbacher for technical help and A Peters for discussion. JM is supported by an EMBO long-term fellowship, and research in the laboratory of TJ is sponsored by the IMP through Boehringer Ingelheim and by grants from the Vienna Economy promotion fund, the European Union (EU-network HPRN-CT 2000-00078 and NoE network ‘The Epigenome' LSHG-CT-2004-503433) and the Austrian GEN-AU initiative, which is financed by funds from the Austrian Federal Ministry for Education, Science and Culture (BMBWK).
References
- Allen TA, von Kaenel S, Goodrich JA, Kugel JF (2004) The SINE-encoded mouse B2 RNA represses mRNA transcription in response to heat shock. Nat Struct Mol Biol 11: 816–821 [DOI] [PubMed] [Google Scholar]
- Amor DJ, Choo KH (2002) Neocentromeres: role in human disease, evolution, and centromere study. Am J Hum Genet 71: 695–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter J, Sauer S, Peters A, John R, Williams R, Caparros M-L, Arney K, Otte A, Jenuwein T, Merkenschlager M, Fisher A (2004) Histone hypo-methylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J 23: 4462–4472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourc'his D, Bestor TH (2004) Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431: 96–99 [DOI] [PubMed] [Google Scholar]
- Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, Wheeler R, Wong B, Drenkow J, Yamanaka M, Patel S, Brubaker S, Tammana H, Helt G, Struhl K, Gingeras TR (2004) Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of non-coding RNAs. Cell 116: 499–509 [DOI] [PubMed] [Google Scholar]
- Chan SW, Zilberman D, Xie Z, Johansen LK, Carrington JC, Jacobsen SE (2004) RNA silencing genes control de novo DNA methylation. Science 303: 1336. [DOI] [PubMed] [Google Scholar]
- Ebert A, Schotta G, Lein S, Kubicek S, Krauss V, Jenuwein T, Reuter G (2004) Su(var) genes regulate the balance between euchromatin and heterochromatin in Drosophila. Genes Dev 18: 2973–2983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinoza CA, Allen TA, Hieb AR, Kugel JF, Goodrich JA (2004) B2 RNA binds directly to RNA polymerase II to repress transcript synthesis. Nat Struct Mol Biol 11: 822–829 [DOI] [PubMed] [Google Scholar]
- Fukagawa T, Nogami M, Yoshikawa M, Ikeno M, Okazaki T, Takami Y, Nakayama T, Oshimura M (2004) Dicer is essential for formation of the heterochromatin structure in vertebrate cells. Nat Cell Biol 8: 784–791 [DOI] [PubMed] [Google Scholar]
- Goodier JL, Ostertag EM, Du K, Kazazian HH Jr (2001) A novel active L1 retrotransposon subfamily in the mouse. Genome Res 11: 1677–1685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greil F, van der Kraan I, Delrow J, Smothers JF, de Wit E, Bussemaker HJ, van Driel R, Henikoff S, van Steensel B (2003) Distinct HP1 and Su(var)3-9 complexes bind to sets of developmentally coexpressed genes depending on chromosomal location. Genes Dev 17: 2825–2838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall IM, Shankaranarayana GD, Noma K, Ayoub N, Cohen A, Grewal SI (2002) Establishment and maintenance of a heterochromatin domain. Science 297: 2232–2237 [DOI] [PubMed] [Google Scholar]
- Han JS, Szak ST, Boeke JD (2004) Transcriptional disruption by the L1 retrotransposon and implications for mammalian transcriptomes. Nature 429: 268–274 [DOI] [PubMed] [Google Scholar]
- Harbison CT, Gordon DB, Lee TI, Rinaldi NJ, Macisaac KD, Danford TW, Hannett NM, Tagne JB, Reynolds DB, Yoo J, Jennings EG, Zeitlinger J, Pokholok DK, Kellis M, Rolfe PA, Takusagawa KT, Lander ES, Gifford DK, Fraenkel E, Young RA (2004) Transcriptional regulatory code of a eukaryotic genome. Nature 431: 99–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K, Malik HS (2001) The centromere paradox: stable inheritance with rapidly evolving DNA. Science 293: 1098–1102 [DOI] [PubMed] [Google Scholar]
- Jackson JP, Johnson L, Jasencakova Z, Zhang X, Perez-Burgos L, Singh PB, Cheng X, Schubert I, Jenuwein T, Jacobsen SE (2004) Dimethylation of histone H3 lysine 9 is a critical mark for DNA methylation and gene silencing in Arabidopsis thaliana. Chromosoma 112: 308–315 [DOI] [PubMed] [Google Scholar]
- Jaenisch R, Bird A (2003) Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet 33 (Suppl): 245–254 [DOI] [PubMed] [Google Scholar]
- Jenuwein T (2002) An RNA-guided pathway for the epigenome. Science 297: 2215–2218 [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD (2001) Translating the histone code. Science 293: 1074–1080 [DOI] [PubMed] [Google Scholar]
- Kampa D, Cheng J, Kapranov P, Yamanaka M, Brubaker S, Cawley S, Drenkow J, Piccolboni A, Bekiranov S, Helt G, Tammana H, Gingeras TR (2004) Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res 14: 331–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SP, Gingeras TR (2002) Large-scale transcriptional activity in chromosomes 21 and 22. Science 296: 916–919 [DOI] [PubMed] [Google Scholar]
- Karpen GH, Allshire RC (1997) The case for epigenetic effects on centromere identity and function. Trends Genet 13: 489–496 [DOI] [PubMed] [Google Scholar]
- Kazazian HH Jr (2004) Mobile elements: drivers of genome evolution. Science 303: 1626–1632 [DOI] [PubMed] [Google Scholar]
- Kohlmaier A, Savarese F, Lachner M, Martens J, Jenuwein T, Wutz A (2004) A chromosomal memory triggered by Xist regulates histone methylation in X inactivation. PLoS Biol 2: E171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo Y, Shen L, Yan PS, Huang TH, Issa JP (2004) Chromatin immunoprecipitation microarrays for identification of genes silenced by histone H3 lysine 9 methylation. Proc Natl Acad Sci USA 101: 7398–7403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ (International Human Genome Sequencing Consortium) (2001) Initial sequencing and analysis of the human genome. Nature 409: 860–921 [DOI] [PubMed] [Google Scholar]
- Lehnertz B, Ueda Y, Derijck AA, Braunschweig U, Perez-Burgos L, Kubicek S, Chen T, Li E, Jenuwein T, Peters AH (2003) Suv39h-mediated histone H3 lysine 9 methylation directs DNA methylation to major satellite repeats at pericentric heterochromatin. Curr Biol 13: 1192–1200 [DOI] [PubMed] [Google Scholar]
- Lippman Z, Gendrel AV, Black M, Vaughn MW, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD, Carrington JC, Doerge RW, Colot V, Martienssen R (2004) Transposable elements in heterochromatin and epigenetic control. Nature 430: 471–476 [DOI] [PubMed] [Google Scholar]
- Lippman Z, Martienssen R (2004) The role of RNA interference in heterochromatic silencing. Nature 431: 364–370 [DOI] [PubMed] [Google Scholar]
- Litt MD, Simpson M, Gaszner M, Allis CD, Felsenfeld G (2001) Correlation between histone lysine methylation and developmental changes at the chicken beta-globin locus. Science 293: 2453–2455 [DOI] [PubMed] [Google Scholar]
- Martienssen R (2003) Maintenance of heterochromatin by RNA interference of tandem repeats. Nat Genet 35: 213–214 [DOI] [PubMed] [Google Scholar]
- McClintock B (1951) Chromosome organization and genic expression. Cold Spring Harbor Symp Quant Biol 16: 13–47 [DOI] [PubMed] [Google Scholar]
- Mette MF, Aufsatz W, van der Winden J, Matzke MA, Matzke AJ (2000) Transcriptional silencing and promoter methylation triggered by double-stranded RNA. EMBO J 19: 5194–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller HJ (1930) Types of visible variations induced by X-rays in Drosophila. Genetics 22: 299–334 [Google Scholar]
- Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe S (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113: 703–716 [DOI] [PubMed] [Google Scholar]
- Noma K, Allis CD, Grewal SI (2001) Transitions in distinct histone H3 methylation patterns at the heterochromatin domain boundaries. Science 293: 1150–1155 [DOI] [PubMed] [Google Scholar]
- Noma KI, Sugiyama T, Cam H, Verdel A, Zofall M, Jia S, Moazed D, Grewal SI (2004) RITS acts in cis to promote RNA interference-mediated transcriptional and post-transcriptional silencing. Nat Genet 36: 1174–1180 [DOI] [PubMed] [Google Scholar]
- O'Neill LP, Turner BM (1995) Histone H4 acetylation distinguishes coding regions of the human genome from heterochromatin in a differentiation-dependent but transcription-independent manner. EMBO J 14: 3946–3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E (1999) DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99: 247–257 [DOI] [PubMed] [Google Scholar]
- Pardue ML, Gall JG (1970) Chromosomal localization of mouse satellite DNA. Science 168: 1356–1358 [DOI] [PubMed] [Google Scholar]
- Peters AH, Kubicek S, Mechtler K, O'Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, Martens JH, Jenuwein T (2003) Partitioning and plasticity of repressive histone methylation states in mammalian chromatin. Mol Cell 12: 1577–1589 [DOI] [PubMed] [Google Scholar]
- Peters AH, O'Carroll D, Scherthan H, Mechtler K, Sauer S, Schofer C, Weipoltshammer K, Pagani M, Lachner M, Kohlmaier A, Opravil S, Doyle M, Sibilia M, Jenuwein T (2001) Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 107: 323–337 [DOI] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y (2003) Role of histone H3 lysine 27 methylation in X inactivation. Science 300: 131–135 [DOI] [PubMed] [Google Scholar]
- Rabinowicz PD, Palmer LE, May BP, Hemann MT, Lowe SW, McCombie WR, Martienssen RA (2003) Genes and transposons are differentially methylated in plants, but not in mammals. Genome Res 13: 2658–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratcliff F, Harrison BD, Baulcombe DC (1997) A similarity between viral defense and gene silencing in plants. Science 276: 1558–1560 [DOI] [PubMed] [Google Scholar]
- Ren B, Cam H, Takahashi Y, Volkert T, Terragni J, Young RA, Dynlacht BD (2002) E2F integrates cell cycle progression with DNA repair, replication, and G(2)/M checkpoints. Genes Dev 16: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringrose L, Paro R (2004) Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu Rev Genet 38: 413–443 [DOI] [PubMed] [Google Scholar]
- Ringrose L, Rehmsmeier M, Dura JM, Paro R (2003) Genome-wide prediction of Polycomb/Trithorax response elements in Drosophila melanogaster. Dev Cell 5: 759–771 [DOI] [PubMed] [Google Scholar]
- Rudd MK, Schueler MG, Willard HF (2003) Sequence organization and functional annotation of human centromeres. Cold Spring Harb Symp Quant Biol 68: 141–149 [DOI] [PubMed] [Google Scholar]
- Rudert F, Bronner S, Garnier JM, Dolle P (1995) Transcripts from opposite strands of gamma satellite DNA are differentially expressed during mouse development. Mamm Genome 6: 76–83 [DOI] [PubMed] [Google Scholar]
- Santos F, Zakhartchenko V, Stojkovic M, Peters A, Jenuwein T, Wolf E, Reik W, Dean W (2003) Epigenetic marking correlates with developmental potential in cloned bovine preimplantation embryos. Curr Biol 13: 1116–1121 [DOI] [PubMed] [Google Scholar]
- Santos-Rosa H, Schneider R, Bannister AJ, Sherriff J, Bernstein BE, Emre NC, Schreiber SL, Mellor J, Kouzarides T (2002) Active genes are tri-methylated at K4 of histone H3. Nature 419: 407–411 [DOI] [PubMed] [Google Scholar]
- Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T (2004) A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev 18: 1251–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramke V, Allshire R (2003) Hairpin RNAs and retrotransposon LTRs effect RNAi and chromatin-based gene silencing. Science 301: 1069–1074 [DOI] [PubMed] [Google Scholar]
- Schübeler D, MacAlpine DM, Scalzo D, Wirbelauer C, Kooperberg C, van Leeuwen F, Gottschling DE, O'Neill LP, Turner BM, Delrow J, Bell SP, Groudine M (2004) The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev 18: 1263–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J, Mak W, Zvetkova I, Appanah R, Nesterova TB, Webster Z, Peters AH, Jenuwein T, Otte AP, Brockdorff N (2003) Establishment of histone H3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev Cell 4: 481–495 [DOI] [PubMed] [Google Scholar]
- Smit AF (1996) The origin of interspersed repeats in the human genome. Curr Opin Genet Dev 6: 743–748 [DOI] [PubMed] [Google Scholar]
- Sullivan BA, Karpen GH (2004) Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat Struct Mol Biol 11: 1076–1083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamaru H, Selker EU (2001) A histone H3 methyltransferase controls DNA methylation in Neurospora crassa. Nature 414: 277–283 [DOI] [PubMed] [Google Scholar]
- Tanaka S, Kunath T, Hadjantonakis AK, Nagy A, Rossant J (1998) Promotion of trophoblast stem cell proliferation by FGF4. Science 282: 2072–2075 [DOI] [PubMed] [Google Scholar]
- Vastenhouw NL, Plasterk RH (2004) RNAi protects the Caenorhabditis elegans germline against transposition. Trends Genet 20: 314–319 [DOI] [PubMed] [Google Scholar]
- Verdel A, Jia S, Gerber S, Sugiyama T, Gygi S, Grewal SI, Moazed D (2004) RNAi-mediated targeting of heterochromatin by the RITS complex. Science 303: 672–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe TA, Kidner C, Hall IM, Teng G, Grewal SI, Martienssen RA (2002) Regulation of heterochromatic silencing and histone H3 lysine-9 methylation by RNAi. Science 297: 1833–1837 [DOI] [PubMed] [Google Scholar]
- Walsh CP, Chaillet JR, Bestor TH (1998) Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat Genet 20: 116–117 [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, Antonarakis SE, Attwood J, Baertsch R, Bailey J, Barlow K, Beck S, Berry E, Birren B, Bloom T, Bork P, Botcherby M, Bray N, Brent MR, Brown DG, Brown SD, Bult C, Burton J, Butler J, Campbell RD, Carninci P, Cawley S, Chiaromonte F, Chinwalla AT, Church DM, Clamp M, Clee C, Collins FS, Cook LL, Copley RR, Coulson A, Couronne O, Cuff J, Curwen V, Cutts T, Daly M, David R, Davies J, Delehaunty KD, Deri J, Dermitzakis ET, Dewey C, Dickens NJ, Diekhans M, Dodge S, Dubchak I, Dunn DM, Eddy SR, Elnitski L, Emes RD, Eswara P, Eyras E, Felsenfeld A, Fewell GA, Flicek P, Foley K, Frankel WN, Fulton LA, Fulton RS, Furey TS, Gage D, Gibbs RA, Glusman G, Gnerre S, Goldman N, Goodstadt L, Grafham D, Graves TA, Green ED, Gregory S, Guigo R, Guyer M, Hardison RC, Haussler D, Hayashizaki Y, Hillier LW, Hinrichs A, Hlavina W, Holzer T, Hsu F, Hua A, Hubbard T, Hunt A, Jackson I, Jaffe DB, Johnson LS, Jones M, Jones TA, Joy A, Kamal M, Karlsson EK, Karolchik D, Kasprzyk A, Kawai J, Keibler E, Kells C, Kent WJ, Kirby A, Kolbe DL, Korf I, Kucherlapati RS, Kulbokas EJ, Kulp D, Landers T, Leger JP, Leonard S, Letunic I, Levine R, Li J, Li M, Lloyd C, Lucas S, Ma B, Maglott DR, Mardis ER, Matthews L, Mauceli E, Mayer JH, McCarthy M, McCombie WR, McLaren S, McLay K, McPherson JD, Meldrim J, Meredith B, Mesirov JP, Miller W, Miner TL, Mongin E, Montgomery KT, Morgan M, Mott R, Mullikin JC, Muzny DM, Nash WE, Nelson JO, Nhan MN, Nicol R, Ning Z, Nusbaum C, O'Connor MJ, Okazaki Y, Oliver K, Overton-Larty E, Pachter L, Parra G, Pepin KH, Peterson J, Pevzner P, Plumb R, Pohl CS, Poliakov A, Ponce TC, Ponting CP, Potter S, Quail M, Reymond A, Roe BA, Roskin KM, Rubin EM, Rust AG, Santos R, Sapojnikov V, Schultz B, Schultz J, Schwartz MS, Schwartz S, Scott C, Seaman S, Searle S, Sharpe T, Sheridan A, Shownkeen R, Sims S, Singer JB, Slater G, Smit A, Smith DR, Spencer B, Stabenau A, Stange-Thomann N, Sugnet C, Suyama M, Tesler G, Thompson J, Torrents D, Trevaskis E, Tromp J, Ucla C, Ureta-Vidal A, Vinson JP, Von Niederhausern AC, Wade CM, Wall M, Weber RJ, Weiss RB, Wendl MC, West AP, Wetterstrand K, Wheeler R, Whelan S, Wierzbowski J, Willey D, Williams S, Wilson RK, Winter E, Worley KC, Wyman D, Yang S, Yang SP, Zdobnov EM, Zody MC, Lander ES (Mouse Genome Sequencing Consortium) (2002) Initial sequencing and comparative analysis of the mouse genome. Nature 420: 520–562 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Data