

Abstract
Cadherins are critically involved in tissue development and tissue homeostasis. We demonstrate here that neuronal cadherin (N-cadherin) is cleaved specifically by the disintegrin and metalloproteinase ADAM10 in its ectodomain. ADAM10 is not only responsible for the constitutive, but also for the regulated, shedding of this adhesion molecule in fibroblasts and neuronal cells directly regulating the overall levels of N-cadherin expression at the cell surface. The ADAM10-induced N-cadherin cleavage resulted in changes in the adhesive behaviour of cells and also in a dramatic redistribution of β-catenin from the cell surface to the cytoplasmic pool, thereby influencing the expression of β-catenin target genes. Our data therefore demonstrate a crucial role of ADAM10 in the regulation of cell–cell adhesion and on β-catenin signalling, leading to the conclusion that this protease constitutes a central switch in the signalling pathway from N-cadherin at the cell surface to β-catenin/LEF-1-regulated gene expression in the nucleus.
Keywords: ADAM10, cell adhesion, N-cadherin, nuclear signalling, shedding
Introduction
Neuronal cadherin (N-cadherin A-CAM) belongs to a family of intercellular adhesion molecules that mediate calcium-dependent cell–cell adhesion through homophilic interaction. Cadherins are essential for cell recognition and tissue morphogenesis, as well as for the maintenance of solid tissue (Takeichi, 1991; Gumbiner, 1996). Like the other classical type I cadherins, E- and P-cadherin, N-cadherin contains a large N-terminal extracellular region which consists of five tandem repeated domains (EC1–EC5). The conserved cytoplasmic domains of cadherins interact with β-catenin, which in turn is linked to the cytoskeleton. β-Catenin plays a key role in the transduction of the Wnt signals, acting as coactivator for the transcription factor lymphocyte enhancer binding factor-1 (LEF-1). N-cadherin is critically involved in heart tube formation, neurulation and somitogenesis (Radice et al, 1997), and in connective tissue remodelling and wound healing (Gabbiani, 1981; Van Hoorde et al, 1999; Ko et al, 2001).
The release of the extracellular domain, which contains the homophilic binding sites, is functionally of major importance for the regulation of cell adhesion, cell migration and neurite outgrowth (Paradies and Grunwald, 1993; Nakagawa and Takeichi, 1998). This ectodomain cleavage of N-cadherin can be inhibited with MMP inhibitors, including the tissue inhibitors of metalloproteinases (TIMPs). TIMPs promote fibroblast adhesion through stabilisation of focal adhesion contacts, which is correlated with an increase in N-cadherin expression at the cell surface (Ho et al, 2001). The extracellular domain released through metalloproteinase activity has been shown to retain biological function and to promote neuronal cell adhesion and neurite outgrowth (Bixby and Zhang, 1990; Paradies and Grunwald, 1993; Utton et al, 2001). The nature of the N-cadherin ectodomain-generating protease is unknown. This is also of importance since the proteolytic ectodomain cleavage leads to a membrane-bound carboxy-terminal fragment which is a substrate for regulated intramembrane proteolysis (RIP) (Marambaud et al, 2003).
In this process, the carboxy-terminal membrane-bound fragment becomes a substrate for a presenilin/γ-secretase-mediated intramembrane proteolysis, resulting in the release of the cytoplasmic carboxy-terminal fragment. In case of N-cadherin, this intracellular fragment is apparently involved in signal transduction, since it promotes in a not yet clarified way the degradation of the transcriptional coactivator CREB-binding protein (CBP) (Marambaud et al, 2003). The regulation of RIP (e.g. Notch, APP, CD44) is thought to occur at the level of the first ectodomain-shedding processing step.
This observation, especially when combined with the fact that also other RIP substrates are cleaved by similar proteases, suggested to us that a member of the disintegrin and metalloprotease (ADAM) family is involved in the ectodomain cleavage of N-cadherin. The ADAMs are a family of type I transmembrane proteins and combine features of both cell adhesion molecules and proteinases. They play important roles in fertilisation, neurogenesis and angiogenesis (Blobel, 2000), and they are involved in the shedding of various membrane-bound proteins, including cytokines, growth factors and adhesion molecules (Schlondorff and Blobel, 1999; Seals and Courtneidge, 2003). ADAM10 and ADAM17 (TNFα-converting enzyme (TACE)) have been studied in particular in the context of ectodomain shedding. They are involved in the proteolysis of various substrates such as Notch, EGF ligands, APP and fractalkine (Lammich et al, 1999; Hartmann et al, 2002; Hundhausen et al, 2003; Sahin et al, 2004).
In the present study, we focused on the potential role of different ADAMs in N-cadherin shedding. We demonstrate that ADAM10 is the major proteinase responsible for N-cadherin ectodomain cleavage in fibroblasts and neuronal cells. We demonstrate further how ADAM10 activity regulates cell adhesion and importantly affects β-catenin signalling, resulting in the expression of cyclin D1, c-myc and c-jun. Our results link extracellular protein cleavage to the regulation of intracellular processes, thereby establishing the couple ADAM10/N-cadherin as a central switch in mediating signals from the extracellular matrix to the nucleus.
Results
ADAM10 is responsible for ectodomain shedding of N-cadherin
The full-length 135 kDa N-cadherin protein is cleaved in the extracellular domain by metalloproteinase activity, generating a 40 kDa C-terminal fragment termed CTF1, which can be further processed by a γ-secretase-like activity into a soluble 35 kDa CTF2 (Figure 1A). Recently, Marambaud et al (2003) showed that CTF2 is strongly diminished but not absent in fibroblasts deficient for presenilin 1 (PS1), a component of the γ-secretase complex. We confirmed these findings and additionally demonstrated that the remaining proteolytic activity was due to presenilin 2 (PS2) by comparing PS1 and PS1/2 double-deficient mouse embryonic fibroblasts (MEFs) and analysing these cells by Western blot analysis using monoclonal antibodies against the C-terminal part of mouse N-cadherin (Supplementary Figure A).
Figure 1.
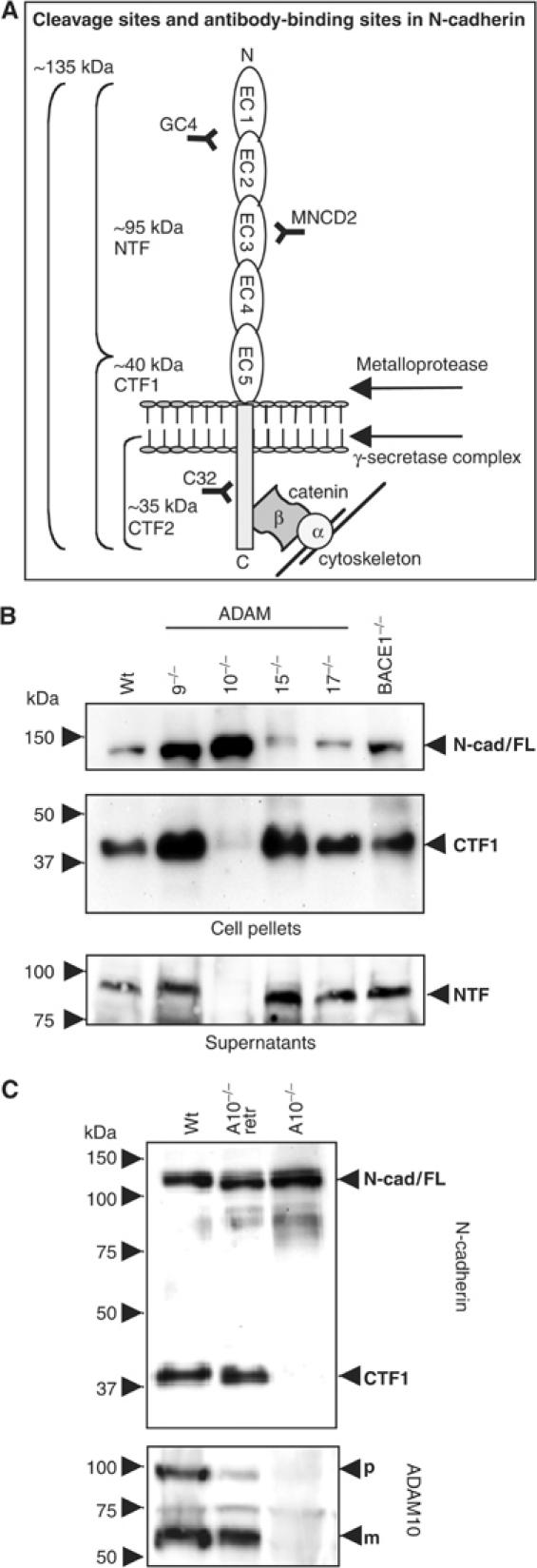
Involvement of ADAM10 in N-cadherin processing. (A) Schematic representation of N-cadherin cleavage sites and antibody-binding regions. Full-length 135 kDa N-cadherin is cleaved by metalloproteinase activity in N-terminal 95 kDa fragments (NTF) and C-terminal 40kDa fragments (CTF1), which can be further processed by γ-secretase-like activity in soluble 35kDa fragments (CTF2). (B) Constitutive N-cadherin cleavage is reduced in ADAM10−/− fibroblasts. MEFs were preincubated with γ-secretase inhibitor (0.5 μM) overnight and harvested. Immunoblot with total cell extracts from ADAM9−/−, ADAM10−/−, ADAM15−/− and ADAM17−/− fibroblasts, BACE1−/− cells and wild-type MEFs stained with C-terminal anti-N-cadherin antibody. Supernatants of these cells were also subjected to Western blot analysis using N-terminal anti-N-cadherin antibodies (MNCD2). N-Cad/FL: full-length N-cadherin; CTF: carboxy-terminal fragment of N-cadherin; NTF: N-terminal fragment of N-cadherin; WT: wild-type MEFs. (C) ADAM10-deficient cells were stably retransfected with wild-type ADAM10 (A10−/−retr) and compared with wild-type and ADAM10-deficient MEFs for N-cadherin expression in the presence of γ-secretase inhibitor (0.5 μM). N-cad/FL: full-length N-cadherin; CTF: C-terminal fragment of N-cadherin; NTF: N-terminal fragment of N-cadherin; p: precursor of ADAM10; m: mature form of ADAM10.
In order to identify the protease responsible for the first, rate-limiting processing step in the RIP of N-cadherin, we compared a panel of ADAM-deficient fibroblasts (Hartmann et al, 2002; Weskamp et al, 2002; Horiuchi et al, 2003) in a similar assay. We also included fibroblasts from BACE1-deficient mice since BACE is a sheddase responsible for the cleavage of APP and PSGL-1 (Lichtenthaler et al, 2003). To avoid the rapid processing of the CTF1 by γ-secretase, we included the γ-secretase inhibitor L-685,458 in our assays. As shown in Figure 1B, only ADAM10-deficient fibroblasts showed a clear reduction in the generation of the N-cadherin CTF1 (Figure 1B, upper panel, lane 3). Accordingly, the proteolytically released ectodomain was detectable in the supernatants of the analysed cell lines, but was absent in the supernatant of ADAM10-deficient cells (Figure 1B, lower panel, lane 3). Considering the minor differences in N-cadherin expression in these different cell lines, we also quantified the amount of CTF1 as percentage of ‘total' N-cadherin (full-length N-cadherin plus N-cadherin/CTF1) by densitometric analysis (Supplementary Figure B), making the reduced shedding in ADAM10-deficient cells even more apparent. We confirmed this observation in three independently derived ADAM10-deficient cell lines (Hartmann et al, 2002), (Supplementary Figure C). Additional strong corroborative evidence came from genetic reconstitution assays: N-cadherin shedding could be completely restored in these cells after transfection of wild-type ADAM10 (Figure 1C). Finally, we confirmed the findings also by pharmacological means using the hydroxamate-based inhibitors GW280623X (blocking ADAM10 and TACE) and GI254023X (preferentially blocking ADAM10) (Hundhausen et al, 2003). Both inhibitors reduced the ectodomain shedding of N-cadherin in wild-type fibroblasts in a dose-dependent manner, confirming the essential role of ADAM10 in this process (Supplementary Figure D). In the absence of γ-secretase inhibitor, ADAM10 inhibition did not only correlate with a decrease of CTF1 but also with a decrease of CTF2 (Supplementary Figure E), illustrating a sequence of proteolytic events.
ADAM10 is also crucial for induced shedding of N-cadherin
In general, shedding of proteins can occur in a constitutive and regulated fashion. ADAM17 has been implied in the regulated shedding of many membrane-bound proteins (Hundhausen et al, 2003; Sahin et al, 2004), and we therefore set out to analyse in more detail regulated shedding of N-cadherin. Stimulation of protein kinase C (PKC) using phorbol ester phorbol-12 myristate 13-acetate (PMA) (Hooper et al, 1997) as well as depletion of cholesterol using cyclodextrin (MCD) strongly induced N-cadherin shedding in wild-type fibroblasts (Figure 2A). Staurosporine (SP), which induces apoptosis and has been previously implicated in the activation of metalloproteinase-mediated cleavage of E-cadherin (Marambaud et al, 2002), also stimulated N-cadherin shedding. Also, ionomycin (IM), an agent that promotes shedding of cadherins through stimulation of calcium influx (Marambaud et al, 2002), increased CTF1 production in wild-type cells. This clearly established that N-cadherin cleavage is regulated by several signalling pathways, including PKC and Ca2+-regulated ones. We next addressed to what extent this inducible cleavage was depending on ADAM10 or on other proteases by analysing the effects of these different compounds in ADAM10-deficient cells. If another protease (e.g. TACE) is responsible for the induction of N-cadherin shedding, we expected to observe a partial restoration of CTF1 generation. This was, however, not the case (Figure 2A), and we therefore conclude that ADAM10 is required for the induction of N-cadherin cleavage. Extracellular calcium influx, which can be induced by mechanical stimulation of cells (Ko et al, 2001), has important physiological relevance during wound healing and tissue remodelling after injury. Therefore, we investigated this aspect in more detail. In wild-type cells, IM induced a rapid accumulation of the CTF1 within minutes (Figure 2B, left panel). This effect was completely blocked with the ADAM10 inhibitor GI254023X (Figure 2B, middle panel) and in ADAM10-deficient cells (Figure 2B, right panel). Taken together, these results demonstrate that ADAM10 is the major proteinase responsible for constitutive, and also for, induced N-cadherin shedding.
Figure 2.
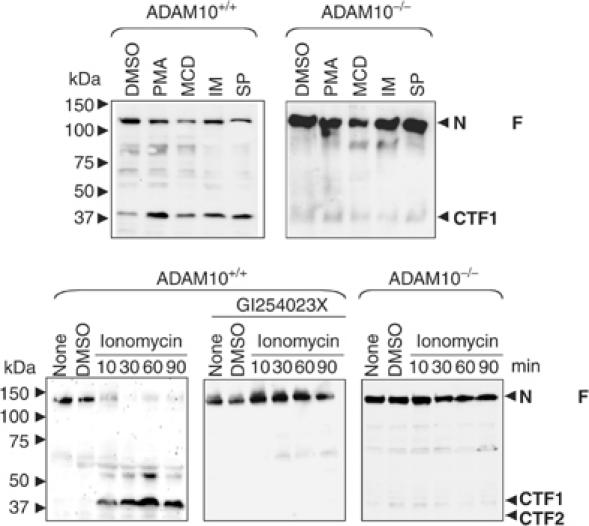
ADAM10 involvement in stimulated N-cadherin shedding. (A) Effect of different stimuli on N-cadherin shedding. Cells were stimulated with PMA (100 ng/ml), MCD (10 mM) or vehicle control (DMSO) for 4 h, with IM (5 μM) for 30 min or with SP (1 μM) for 6 h in the presence of γ-secretase inhibitor (0.5 μM). Subsequently, cell pellets were lysed and subjected to N-cadherin (C-terminal) Western blot analysis. (B) IM-induced N-cadherin shedding occurred in wild-type but not in ADAM10-deficient fibroblasts. Cells were stimulated with 5 μM IM or vehicle control (DMSO) for different periods in the presence or absence of GI254023X. Cell pellets were harvested and analysed by Western blotting using C-terminal anti-N-cadherin antibodies. N-Cad/FL: full-length N-cadherin; CTF: C-terminal fragment of N-cadherin.
ADAM10-dependent N-cadherin cleavage in neuronal cells
N-cadherin, like ADAM10, is predominantly expressed in neuronal cells (Radice et al, 1997; Karkkainen et al, 2000) and promotes possibly axonal outgrowth and regulation of synaptogenesis (Huntley, 2002). Like MEFs, mouse neuroblastoma N2a cells and human neuroglioma H4 cells express both the full-length N-cadherin form (not shown) and the 40 kDa CTF1 proteolytic fragment (Figure 3A). Upon ADAM10 overexpression (Figure 3A, upper panel), an increased level of N-cadherin CTF1 was observed (Figure 3A, lower panel). A similar increase of CTF1 after overexpression of ADAM10 could also be observed in human neuronal H4 cells (not shown). We finally confirmed the role of endogenously expressed ADAM10 in N-cadherin shedding in neuronal cells using vector-based RNA interference (RNAi) (Brummelkamp et al, 2002). After 72 h, ADAM10 levels dropped to 15% as compared to mock-transfected cells (Figure 3B and C). CTF1 was decreased to a similar extent (Figure 3B and C). N-cadherin is physically associated with N-methyl-D-aspartic acid (NMDA) receptors in large, multiprotein complexes (Husi et al, 2000). It has been speculated that NMDA receptor activation may provide a signal that regulates the molecular configuration of synaptic N-cadherin, and therefore the strength of adhesion across the synaptic cleft. Direct stimulation of the NMDA receptor after application of NMDA in H4 neuronal cells resulted in an increased generation of CTF1 and CTF2 N-cadherin fragments. This stimulation was lost after simultaneous addition of the ADAM10 inhibitor GI254023X (Figure 3D), indicating that the ADAM10-mediated N-cadherin ectodomain cleavage can be modulated by the presence of neurotransmitters and might influence synaptic plasticity.
Figure 3.
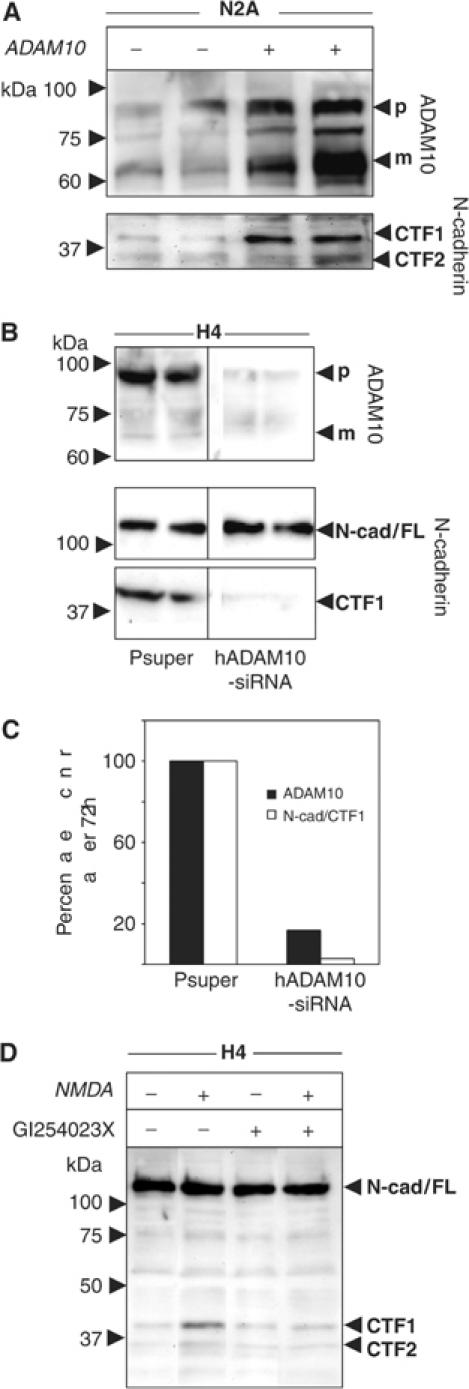
ADAM10-mediated N-cadherin shedding in neuronal cells. (A) Overexpression of ADAM10 protein in mouse N2A cells. Cells were transiently transfected with ADAM10 or empty vector. Subsequently, cells were lysed and analysed by Western blotting using anti-ADAM10 antibodies. The same blot was reprobed with anti-N-cadherin antibodies. (B) H4 cells were transiently transfected with pSUPER-ADAM10 siRNA or empty vector. Cell pellets were harvested 72 h after transfection and analysed for ADAM10 expression. Membranes were reprobed with anti-N-cadherin antibodies. (C) Densitometric analysis of (B). (D) H4 cells were incubated in the presence or absence of NMDA (50 μM) and the ADAM10 inhibitor GI254023X (5 μM) for 30 min.
Cadherin-mediated cell–cell adhesion is upregulated in ADAM10-deficient fibroblasts
The adhesion between ADAM10-deficient cells was much stronger, as indicated by failures to mechanically disrupt cell aggregates, which was easily achieved in wild-type fibroblasts (Figure 4A). Since an increased N-cadherin cell surface expression enhances cell adhesion (Steinberg and Takeichi, 1994; Yap et al, 1997b), we next investigated to what extent ADAM10 could be involved in the regulation of cell surface expression of N-cadherin. For this purpose, cells were stained with antibodies against the extracellular region of N-cadherin (GC4, Figure 4B). In comparison to wild-type cells, ADAM10-deficient fibroblasts showed an increased expression of full-length N-cadherin at the cell surface as determined by flow-cytometric analysis (Figure 4B). To elucidate the relevance of this increase for cell–cell adhesion, calcein-stained wild-type and ADAM10-deficient cells were seeded on top of an unstained monolayer of corresponding cells in the presence or absence of EGTA, the inhibitory N-cadherin antibody GC4 or the corresponding isotype control. After 20 min incubation, nonadherent cells were washed away and the remaining fluorescent cells were quantified (Figure 4C). While the Ca2+-and N-cadherin-independent adhesions were comparable, ADAM10-deficient cells showed an increased N-cadherin-dependent cell–cell adhesion. We confirmed this result in a cell–substrate adhesion assay (Figure 4D). For this assay, 96-well plates were coated with recombinant N-cadherin or buffer overnight, and afterwards the adhesion of wild-type and ADAM10-deficient cells was analysed after 20 min incubation. Likewise, this assay demonstrated an increased N-cadherin-mediated adhesion in ADAM10-deficient cells.
Figure 4.
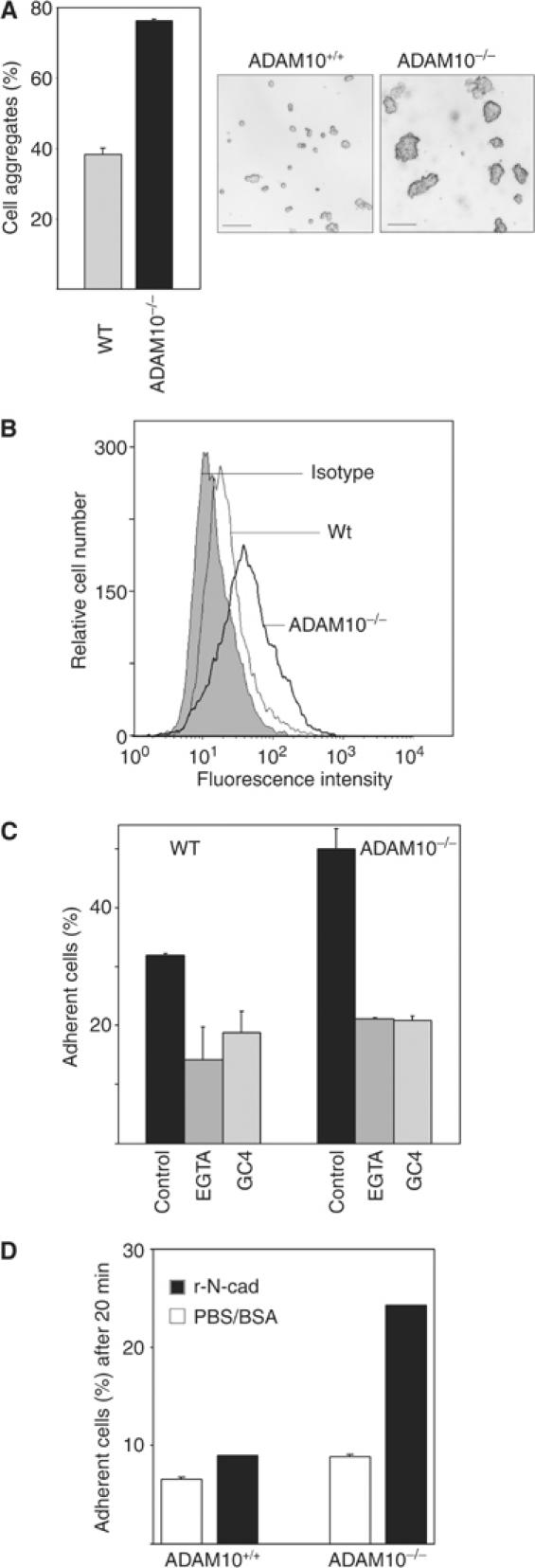
Increased N-cadherin-mediated adhesion of ADAM10-deficient cells. (A) Increased adhesion between ADAM10-deficient cells. Confluent monolayers of ADAM10-deficient and wild-type MEFs were mechanically dissociated though 30-times pipetting with a 5 ml pipette. Afterwards, the cells were photographed and the amount of aggregates was quantified with a Casy TT cell Counter system (Schärfe System, Reutlingen, Germany) (bars=50 μm). (B) Surface expression of N-cadherin. Cells were harvested and investigated for N-cadherin surface expression by flow cytometry with an N-terminal antibody (GC4). Unspecific antibody binding was evaluated with IgG1 control. Since isotype control of wild-type and ADAM10-deficient cells looked identical, only the wild-type control is shown. Results are representative for two independent cell lines. (C) ADAM10 deficiency leads to increased cell–cell adhesion. Calcein-labelled wild-type and ADAM10-deficient fibroblasts were seeded on top of an unlabelled monolayer of the corresponding cells in the presence or absence of EGTA (5 mM), the inhibitory anti-N-cadherin antibody GC4 or the corresponding IgG1 isotype (control). After 20 min of incubation, cells were washed and the remaining fluorescence was calculated as percentage of total fluorescence before washing. The isotype control was also representative for the adhesion of untreated cells (not shown). (D) Cell adhesion on recombinant N-cadherin. Calcein-stained wild-type and ADAM10-deficient MEFs were seeded in N-cadherin- or PBS/BSA-coated wells of a 96-well plate in triplicates. After 20 min incubation, nonadherent cells were washed away with PBS and the fluorescence of the remaining cells was quantified as percentage of total fluorescence.
ADAM10 colocalises with N-cadherin and affects its distribution in fibroblasts
ADAM10 staining has already been described in different cell types to be localised in the Golgi apparatus and at the cell surface (Lammich et al, 1999; Gutwein et al, 2003). This holds also true for wild-type MEFs (Supplementary Figure F). ADAM10 as well as N-cadherin both localise in a region near the nucleus and at the plasma membrane. The analysis of N-cadherin expression in ADAM10-deficient cells showed that N-cadherin surface-bound immunoreactivity was much stronger in ADAM10-deficient cells as compared to the staining in control cells and was distributed in an almost continuous belt along the region of cell–cell contact (Figure 5A, upper panel). Similar results were obtained when wild-type cells were treated with the ADAM10 protease inhibitor GI254023X (Figure 5A, lower panel). In contrast, untreated wild-type cells only showed minimal expression at the cell surface restricted to short segments on cell borders at the cell boundary (Figure 5A, top left). Our finding that full-length N-cadherin co-precipitates with ADAM10 in wild-type fibroblasts in co-immunoprecipitation (IP) experiments (Figure 5B) and the observation that recombinant ADAM10 can cleave recombinant N-cadherin in vitro (data not shown) provides additional evidence for an interaction of these proteins.
Figure 5.
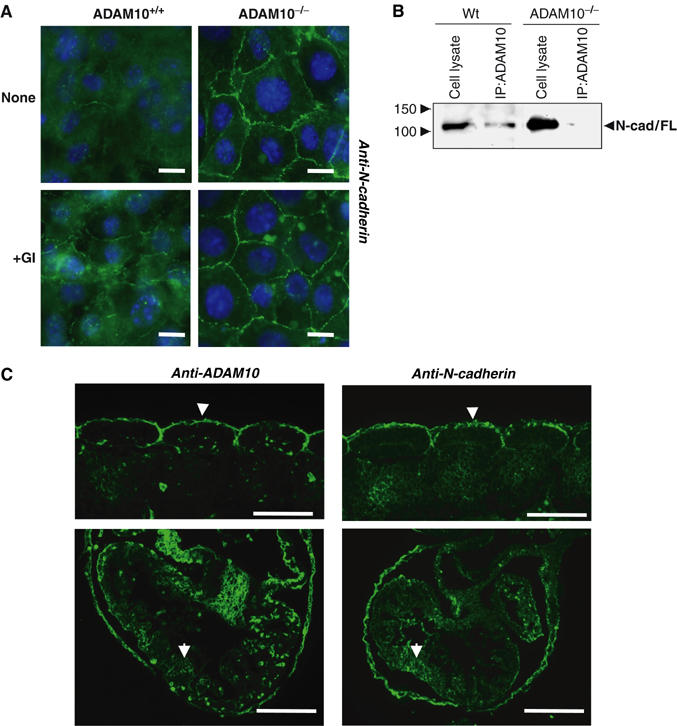
N-cadherin expression in ADAM10-deficient cells and embryos. (A) Subcellular localisation of N-cadherin in ADAM10+/+ and ADAM10−/− cells. Bar=10 μm. GI254023X induces N-cadherin surface expression. Cells were incubated with the inhibitor (3 μM) for 4 h, fixed in methanol and immunostained with anti-N-cadherin antibodies. (B) N-cadherin co-precipitates with ADAM10. Identical amounts of wild-type or ADAM10-deficient cell lysates were immunoprecipitated with anti-ADAM10 antibodies (IP:ADAM10) and loaded onto SDS–PAGE. The immunoblot was stained with anti-N-cadherin antibodies. (C) Partial ADAM10 and N-cadherin colocalisation in E 9.5 wild-type embryos. Upper panel: somites; lower panel: heart. Note the immunostaining along the dermatomyotome surfaces (arrowheads in upper panel) and on cardiomyocyte surfaces (arrows in lower panel). Bars: 100 μm.
In embryonic day (E) 9.5-old embryos, N-cadherin and ADAM10 were colocalised in different regions of the embryo, such as somites (upper panel of Figure 5C), heart (lower panel of Figure 5C) and neural tube (not shown), giving further support that both proteins are functionally linked. Within the heart anlage, both ADAM10 and N-cadherin were present on the membranes of cardiomyocytes, and N-cadherin also featured an intense expression in the surrounding pericardial sac. In somites, both proteins concentrate along the surfaces of the dermatomyotome cells at their interface with the ectoderm, and extend well into the intersomitic clefts.
In order to analyse N-cadherin distribution in the ADAM10-deficient embryo, we performed whole-mount staining with a N-cadherin antibody at embryonic day 9 (Supplementary Figure G). In both embryos, N-cadherin was found to be expressed in the optic and otic pit, in the heart anlage and in the neural tube, like it has also been described by Radice et al (1997). The only difference was that the N-cadherin expression in the somites was apparently lower in the ADAM10 knockout mice, probably related to the developmental differences of the embryos. Since ADAM10 has also been described to cleave several other substrates like EGF or Notch, which are also critically involved in development, the combination of several protein-processing disturbances might be important to understand the altered N-cadherin distribution. These findings highlight the much more complex situation in the embryo and indicate that the ADAM10 knockout phenotype is probably a result of an impaired shedding of different substrates.
β-Catenin recruitment to cell surface leads to impaired signal transduction in ADAM10-deficient cells
β-Catenin and its homolog armadillo from Drosophila are components of the Wnt/wingless signal pathway that plays a key role in embryogenesis (Nelson and Nusse, 2004). β-Catenin exists in two pools in the cell. The levels of the cytoplasmic, soluble pool are regulated by Wnt and adenomatous polyposis coli (APC). A cell membrane-bound pool is linked to cadherins, also affecting the overall β-catenin distribution and β-catenin-dependent signalling. Indeed, in ADAM10-deficient cells, β-catenin immunostaining (Figure 6D) was closely associated with the increased N-cadherin staining (Figure 6B), while in wild-type cells much weaker signals, both for N-cadherin and β-catenin (Figure 6A and C), were observed. β-Catenin immunoreactivity was mainly located in the cytoplasm of these cells.
Figure 6.
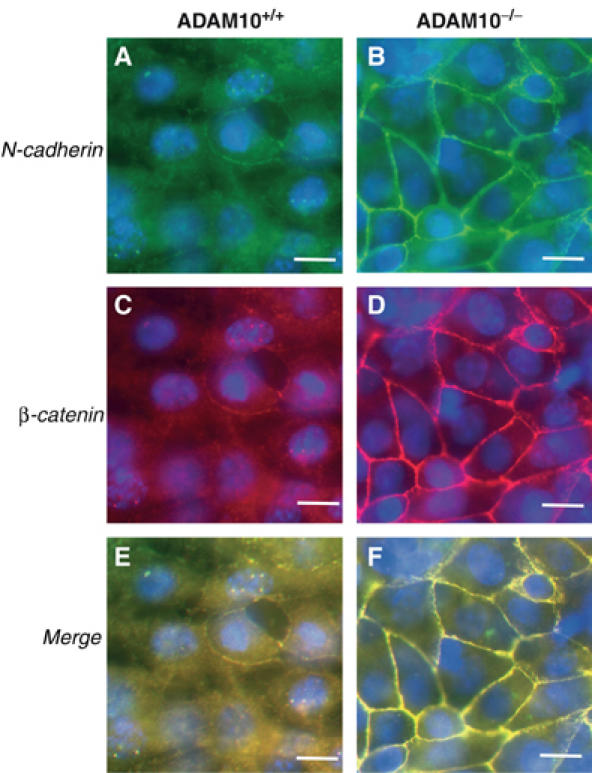
β-Catenin and N-cadherin expression in ADAM10-deficient cells. Subcellular localisation of N-cadherin (green: A, B) and β-catenin (red: C, D) in ADAM10+/+ and ADAM10−/− cells. Double-staining for N-cadherin (C-terminal) and β-catenin. Colocalisation is shown in the merged (E, F) image (yellow). The experiment was repeated three times with similar results. Bar: 10 μm.
We confirmed biochemically the increased association of N-cadherin and β-catenin by co-IP experiments using anti-N-cadherin antibodies as well as anti-β-catenin antibodies (Figure 7A). The suitable antibody concentration was determined by titration and the efficiency of IP was controlled by immunoblots comparing the amount of N-cadherin and β-catenin before and after the IP (Supplementary Figure H). In ADAM10-deficient cells, similar levels of N-cadherin (Figure 7A, upper panel) and β-catenin (lower panel) were immunoprecipitated with N-cadherin, or in the reverse experiment with β-catenin (Figure 7A, lanes 3 and 4). In contrast, less β-catenin immunoreactivity was co-precipitated with N-cadherin antibodies (lane 1) and, vice versa, less N-cadherin was present in the β-catenin immunoprecipitates in wild-type cells compared to ADAM10-deficient cells (lower panel, lanes 1 and 3), even though the β-catenin overall content was comparable (lower panel, lanes 2 and 4), indicating an increased number of N-cadherin/β-catenin complexes in ADAM10-deficient fibroblasts. β-Catenin was shown to associate with transcription factors of the LEF/TCF family, forming a complex that can transactivate genes containing a LEF/TCF-binding sequence. To assess whether the observed alterations in β-catenin distribution in ADAM10-deficient cells were also functionally significant, we transfected these cells and wild-type MEFs with the β-catenin/TCF transcriptional reporter TOP-FLASH or a control reporter with mutated TCF-binding sites (FOP-FLASH). In wild-type cells, we observed a six-fold higher basal expression of the β-catenin reporter as compared to ADAM10-deficient cells (Figure 7B). To further explore the consequences of this reduced β-catenin transcriptional activity, we monitored the transcription of β-catenin target genes such as c-myc and cyclin D1 and c-jun (He et al, 1998; Mann et al, 1999; Shtutman et al, 1999; Tetsu and McCormick, 1999).
Figure 7.
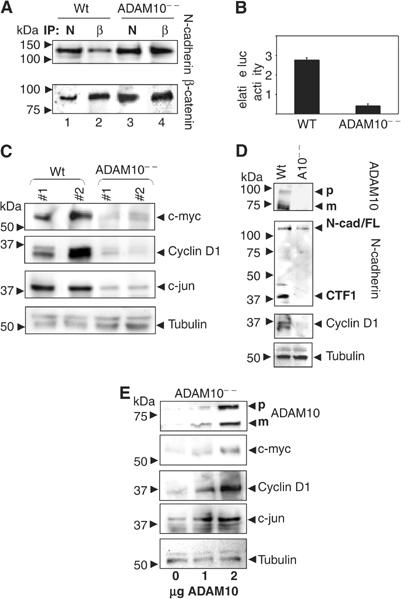
β-Catenin signal transduction in ADAM10-deficient cells. (A) Co-IP of N-cadherin (N) and β-catenin (β). Identical amounts of the same cell lysate were co-immunoprecipitated with either anti-N-cadherin or anti-β-catenin antibodies and loaded onto SDS–PAGE. The immunoblot was first stained with anti-N-cadherin antibodies and afterwards reprobed with anti-β-catenin antibodies. (B) Analysis of β-catenin/TCF activity. Wild-type and ADAM10-deficient MEFs were transfected with TOP-FLASH or the control plasmid FOP-FLASH and transfection control, renilla luciferase. After 48 h, the expression of TOP-FLASH was assayed. Data are expressed as relative light units normalised to the co-transfected Renilla luciferase and show TOP-FLASH activity with FOP-FLASH activity subtracted. The average activities and standard deviations were derived from three independent experiments, each based on duplicate transfected samples. Similar results were obtained from separate experiments with different cell lines. (C) Two wild-type and ADAM10-deficient MEF lines were analysed for the presence of c-myc, cyclin D1 and c-jun by immunoblot analysis. (D) Loss of N-cadherin ectodomain shedding and cyclin D1 expression in ADAM10-deficient E 9.0 embryos was demonstrated by immunoblot analysis of whole embryo extracts. (E) ADAM10-deficient MEFs were transiently transfected with different amounts of ADAM10 expression plasmid and analysed for the re-expression of ADAM10, c-myc, cyclin D1 and c-jun. Tubulin expression was used as loading control in (C), (D) and (E).
Expressions of the proto-oncogene c-jun, a component of the AP-1 transcription factor complex, as well as c-myc were reduced in two independently derived ADAM10-deficient cell lines (Figure 7C). This was also the case for the expression level of cyclin D1, which plays a critical role in cellular proliferation and survival (Brown et al, 1998). In order to verify that cyclin D1 is indeed regulated through β-catenin/TCF signalling in the MEFs, we analysed cyclin D1 expression and activation in a luciferase reporter assay after transfection of β-catenin or a dominant-negative (DN)-TCF-4 construct (Supplementary Figure I). Indeed, DN-TCF-4 decreased cyclin D1 expression and activation. Vice versa, β-catenin overexpression correlated with an increase in the expression of the predicted downstream genes (Supplementary Figure J).
To further elucidate the in vivo relevance of ADAM10 in β-catenin signalling, we analysed lysates of wild-type and ADAM10-deficient embryos by Western blotting. Our immunoblot analysis (Figure 7D) showed that the CTF was also missing in the ADAM10-deficient embryo even though the full-length protein was expressed. Together with the colocalisation of N-cadherin and ADAM10 in wild-type embryos (Figure 5C), this clearly indicates an in vivo relevance of the ADAM10/N-cadherin interaction. Moreover, ADAM10 deficiency was correlated with an absence of N-cadherin CTF1, but also with dramatically reduced cyclin D1 expression level (Figure 7D). We finally confirmed the essential role of ADAM10 in this signalling event by restoring its expression in the fibroblasts in a dose-dependent manner. The ADAM10 expression level correlated with the rescue of expression of c-myc, cyclin D1 and c-jun (Figure 7E). Noteworthy, stimulation of the ADAM10-mediated N-cadherin shedding (e.g. IM) correlated with an increase of cyclin D1 expression in wild-type cells, which could not be observed when these cells were stimulated in the presence of the ADAM10 inhibitor and remained nearly absent in ADAM10-deficient cells (Supplementary Figure K). Thus, our findings suggest that ADAM10 is not only regulating cell–cell adhesion but is also involved in the regulation of crucial intracellular signalling pathways.
Discussion
Here, we have provided multiple lines of evidence that the metalloproteinase ADAM10 is responsible for the initial and crucial proteolytic processing of N-cadherin, leading to the generation of the CTF1 fragment and thereby modulating cell–cell adhesion as well as signal transduction through influencing the cytoplasmic β-catenin pool. Apart from the identification of ADAM10 as the major proteinase responsible for N-cadherin ectodomain cleavage in fibroblasts and neuronal cells, we demonstrate how this cleavage is involved in the regulation of cell adhesion on the one hand and gene transcription on the other hand.
While ADAM10 activity has been implicated in the constitutive shedding of several substrates, many authors attribute stimulated shedding to ADAM17 (Lammich et al, 1999; Sahin et al, 2004). In contrast, we demonstrate here that both constitutive and inducible ectodomain shedding of N-cadherin was lost in ADAM10-deficient cells. Cadherin-mediated adhesion is regulated by a variety of eternal stimuli mostly mediated by tyrosine phosphorylation or PKC activation (Winkel et al, 1990; Yap et al, 1997a). Our findings reveal in addition a time- and ADAM10-dependent stimulation of N-cadherin shedding after IM treatment. This is in accordance with the observations that calcium imbalance disrupted cadherin mediated cell–cell adhesion (Ito et al, 1999; Ko et al, 2001). Thus, activation of ADAM10 via calcium influx may provide an important mechanism for regulating dynamic processes during tissue remodelling and tissue repair after injury.
The strength of cell–cell interaction can be affected by changing the expression level of cadherins on the cell surface (Steinberg and Takeichi, 1994; Yap et al, 1997b). We demonstrate here that ADAM10 is critically involved in regulating N-cadherin-mediated cell–cell adhesion. While this interaction correlates with increased amounts of full-length N-cadherin on the cell surface of ADAM10-deficient fibroblasts, the in vivo situation turned out to be more complicated. Despite the partial similarity of ADAM10-deficient mice and N-cadherin-deficient mice (day of embryonic lethality and impaired development of the heart, neural tube and somites), N-cadherin distribution and expression in ADAM10 knockout embryos can apparently not be directly correlated with the ADAM10 phenotype. Since ADAM10 seems to be involved in the processing of several important proteins, it is much more likely that the phenotype of the embryo as well as N-cadherin distribution is related to various endogenous substrates which are cleaved by ADAM10, and different signalling pathways which are affected by the missing initial ectodomain cleavage. Taking this complex situation into account, it is even more surprising that ADAM10-mediated N-cadherin shedding can apparently not be compensated in the embryo (Figure 7D).
Previous studies have shown that E-cadherin cleavage and the subsequent degradation of the cytoplasmic domain lead to translocation of β-catenin from the cell–cell contacts to the cytoplasm (Ito et al, 1999). β-Catenin is a central component of the Wnt signalling pathway, which is of key importance in development as well as implicated in a variety of human cancers (Nelson and Nusse, 2004). Overexpression of cadherins in the embryos of Xenopus leavis and Drosophila inhibited signal transduction via β-catenin/armadillo (Fagotto et al, 1996; Sanson et al, 1996). When β-catenin is removed from its cytoplasmic pool, it becomes inaccessible for participating in signalling. On the contrary, elevated cytosolic levels are associated with increased β-catenin nuclear accumulation and LEF-1-mediated transcription (Sadot et al, 1998; Simcha et al, 2001). The enhanced N-cadherin surface expression caused by ADAM10 deficiency was directly correlated with a high β-catenin level at the cell membrane. β-Catenin transactivation has been shown to induce a set of genes critical for cell proliferation and cell survival, including cyclin D1 (Shtutman et al, 1999), c-myc (He et al, 1998) and c-jun (Mann et al, 1999), and we show here the decreased expression of these different genes in ADAM10-deficient cells. These findings phenotypically correlate with an extremely retarded proliferation of ADAM10-deficient primary mouse fibroblasts compared to the wild-type cells (data not shown).
The cytoplasmic β-catenin level can be downregulated either by binding to cadherins at the cell surface or through association with a complex containing the APC protein. Conversely, Wnt binding to its receptor results in accumulation of cytoplasmic β-catenin and activation of transcriptional events. Although we cannot formally exclude that ADAM10 also directly or indirectly influences proteins of the APC complex, we propose that the ADAM10-dependent regulation of β-catenin downstream genes is likely related to the cleavage of N-cadherin and possibly also other cadherins, which might also influence β-catenin-mediated target gene expression. Interestingly, the loss of E-cadherin and the gain of N-cadherin expression has been correlated with the transition from a benign tumour to an invasive, metastatic cancer. In this context, regulation of N-cadherin cell surface expression seems to be an important prerequisite for tumour cell migration (Hazan et al, 2000; Li et al, 2001), while β-catenin accumulation could contribute to neoplastic transformation by accumulation of c-myc, c-jun and cyclin D1, leading to uncontrolled progression into the cell cycle (Korinek et al, 1997; Morin et al, 1997). Therefore, inhibition of ADAM10 might not only limit cell migration due to decreased N-cadherin shedding but also control the proliferation of tumour cells due to downregulation of β-catenin signalling.
In conclusion, we have shown that the metalloproteinase ADAM10 is crucial for the constitutive as well as for the induced shedding of N-cadherin. This proteolysis influences cell–cell adhesion as well as cell signalling in physiological, but probably also in pathological, conditions. The coordinated interaction of ADAM10 and N-cadherin may be significant for the coordinated interplay between cell–cell adhesion, cell detachment, cell proliferation and cell survival during embryogenic development, in wound healing and during tumour invasion.
Materials and methods
Primary antibodies and reagents
Primary antibodies against mouse proteins included anti-N-cadherin antibody (BD Bioscience), β-catenin antibody, c-jun and c-myc antibodies from Santa Cruz Biotechnology, Inc. and anti cyclin D1 (Labvision) antibody. β-Tubulin antibody (E7) and N-terminal anti-N-cadherin MNCD2 were from DSHB, Iowa. The monoclonal N-terminal anti-N-cadherin antibody (A-CAM, clone GC-4) was purchased from Sigma (Deisenhofen, Germany) and the corresponding control monoclonal IgG1 clone was from R&D Systems (Wiesbaden, Germany). ADAM10 was detected using a polyclonal antiserum B42.1 described previously (Hartmann et al, 2002). γ-Secretase inhibitor L-685,458 was a kind gift of Wim Anneart, University Leuven. PMA, SP, IM and NMDA were purchased from Sigma. Methyl-β-MCD was obtained from Research Biochemicals International. Hydroxamate-based inhibitors GW280264 and GI254023 were described elsewhere (Hundhausen et al, 2003). Protein G-sepharose beads were purchased from Pierce. CompleteTM EDTA-free proteinase inhibitor mixture was obtained from Roche Molecular Biochemicals (Mannheim, Germany).
Cell culture, stimulation and transfection
Simian virus large T-antigen (SV-T)-immortalised and primary MEF cell lines from ADAM10−/−, ADAM9−/−, ADAM15−/−, ADAM17−/− mice, BACE1−/−, PS1−/−, PS1/2−/− and respective wild-type animals were generated and characterised as described elsewhere (Hartmann et al, 2002; Weskamp et al, 2002; Lichtenthaler et al, 2003; Sahin et al, 2004). All cells were grown in DMEM (high glucose) (PAA Laboratories) supplemented with 10% foetal calf serum and 1% penicillin/streptomycin. For FACS analysis and adhesion assays, cells were harvested under cadherin-saving conditions (Boterberg et al, 2001). The cloning of ADAM10 in pcDNA3.1 vector (Invitrogen, Karlsruhe, Germany) was reported previously (Hundhausen et al, 2003). Human neuronal H4 cells, kindly provided by J Wilfang (Erlangen, Germany), and mouse neuroblastoma cells (N2a) were transfected in six-well tissue culture plates with FuGENE 6 (Roche) according to the manufacturer's instructions.
siRNA transfection
The mammalian expression vector pSUPER, kindly provided by Dr Brummelkamp, was used for expression of siRNA in neuronal H4 cells. The sequence of the human ADAM10 siRNA was as follows: 5′-GACAUUUCAACCUACGAAUTT-3′. The sequence was separated by a nine-nucleotide noncomplementary spacer (tctcttgaa) from the corresponding reverse complement of the same 19-nucleotide sequence. These sequences were inserted into the pSUPER backbone after digestion with BglII and HindIII. H4 cells were transfected with pSuper-ADAM10-siRNA vector with the use of FuGENE 6 (Roche) according to the manufacturer's recommendations.
Flow cytometry
Cells were labelled with anti-N-cadherin (GC4) antibody (1:60) for 1 h, followed by incubation with Cy3-conjugated sheep anti-mouse F(ab)2 antibody (Dianova). As a negative control, the cells were labelled with mouse IgG1. By excluding propidium iodide-permeable cells, only living cells were analysed by flow cytometry (FACScan; Becton Dickinson, Heidelberg, Germany).
Immunoblotting and IP
Briefly, cells were lysed in RIPA buffer. Same amounts of protein were loaded on 10% SDS–PAGE gels and electrotransferred onto PVDF membranes (Hybond-P; Amersham). Primary antibodies were detected using peroxidase-conjugated secondary antibodies. For IP, sepharose-prepared cell lysates were incubated for 2 h at 4°C with the appropriate antibody and protein G sepharose (Pierce). The beads were resuspended in 2 × sample loading buffer and loaded on 10% SDS–PAGE. For Western blot analysis of supernatants, cells were incubated in DMEM overnight and supernatants were concentrated (30-fold) through centrifugation in Microcon Centrifugal Filter Devices (YM-10, Millipore). More detailed information is provided in the supplements.
Immunocytochemistry/immunohistochemistry
Cells were fixed and permeabilised with methanol at −20°C for 10 min, blocked with 3% BSA in PBS for 15 min, and incubated with primary antibody (β-catenin (Santa Cruz); N-cadherin (BD Bioscience)) for 1 h at room temperature, washed three times with PBS containing 0.2%BSA, and incubated with goat anti-mouse Alexa Fluor®488 or donkey anti-goat Alexa Fluor®594 (Molecular Probes). For immunohistochemistry, embryos were fixed by immersion in 2% paraformaldehyde. Paraffin sections were incubated with N-cadherin antibody (BD Bioscience) and detected by the avidin–biotin complex technique (reagents from Vector, Burlingame, USA). Nonspecific control staining was performed using only the secondary antibody.
Luciferase assay
Cells cultured as described previously were transfected 1 day after plating with 1 μg of TOP-FLASH or FOP-FLASH plus 0.1 μg phRG-TK (Renilla-Luciferase) as a cotransfection control. Cells were lysed 48 h after transfection and the luciferase activity was determined with a luminometer (TD-20/20 Luminometer Turner Designs, CA, USA) using the Dual-Luciferase system (Promega). Firefly luciferase activity, indicating TCF-dependent transcription, was normalised to the Renilla luciferase activity of each extract. The cyclin D1 reporter constructs were analysed similar to the TOP-FLASH plasmids.
Whole-mount embryo staining
Embryos were stained according to Radice et al (1997). Briefly, after heating and incubation in cold methanol containing 3% hydrogen peroxide for 30 min, the embryos were rehydrated with PBS containing 0.1% Triton X-100 and incubated with anti-N-cadherin antibody (MNCD2) 1:100 in PBS with 3% BSA. The samples were washed three times in PBS and incubated with HRP-conjugated anti-mouse monoclonal antibody (1:1000 in PBS containing 3% BSA). After washing, the embryos were incubated with diaminobenzidine and 0.015% hydrogen peroxide.
Adhesion assays
Detailed information for all adhesion assays is provided in the supplements.
Briefly, for analysing cell–cell adhesion, MEFs were suspended at 2 × 106/ml in PBS/0.1% BSA and incubated with 2.5 μM fluorescent dye (calcein AM; Molecular Probes, Leiden, the Netherlands) at 37°C for 30 min. After washing, cells were preincubated with the inhibitory N-cadherin antibody GC4 (50 μg/ml) or isotype control (50 μg/ml) for 20 min, with EGTA (5 mM) for 10 min or left untreated. The labelled MEFs were added to a monolayer of unstained cells at 5 × 104 cells per well. The fluorescence signal from the adherent cells was measured after 20 min incubation before and after PBS washing (2 × 200 μl) using a fluorescence plate reader (Lambda Fluoro 230; MWG Biotech, Munich, Germany).
To investigate cell substrate adhesion, 96-well plates (Sarstedt Inc., NC, USA) were coated with 10 μg/ml recombinant human N-cadherin (R&D Systems, Minneapolis, MN) or 10 μg/ml BSA in 100 μl PBS at 4°C overnight. Calcein-labelled MEFs were seeded at a density of 5 × 104 cells/well and incubated for 20 min and analysed as described above.
For determining the adhesion capacity of ADAM10-deficient and wild-type MEFs, confluent monolayers were mechanically dissociated though 30-times pipetting with a 5 ml pipette (Sarstedt) in 5 ml medium, photographed, and the amount of aggregates was quantified with a Casy TT cell Counter system (Schärfe System, Reutlingen, Germany).
Supplementary Material
Supplementary Method Information
Supplementary Figure Legends
Supplementary Figures
Acknowledgments
We thank J Buchholz for his excellent technical assistance, R Kemler for the TOP/FOP-FLASH reporter constructs, and O Tetsu and F McCormick for the DN-TCF-4 plasmid. The cyclin D1 reporter plamid was kindly provided by R Pestell. We are grateful to C Blobel for providing ADAM9 and 15-deficient MEFs. We thank B Rudolph for providing us with the phRGTK construct and also S Rose-John for ADAM17-deficient MEFs. This work was supported by the Deutsche Forschungsgemeinschaft Sonderforschungsbereich 415 to PS and KR, DFG LU869/1-2 to AL, Interuniversity Attraction Poles Program P5/19 of the Belgian Federal Science Policy Office, the Alzheimer Forschungsinititative to KR and the APOPIS EU-network.
References
- Bixby JL, Zhang R (1990) Purified N-cadherin is a potent substrate for the rapid induction of neurite outgrowth. J Cell Biol 110: 1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blobel CP (2000) Remarkable roles of proteolysis on and beyond the cell surface. Curr Opin Cell Biol 12: 606–612 [DOI] [PubMed] [Google Scholar]
- Boterberg T, Bracke M, Bruyneel E, Mareel M (2001) Cell aggregation assays. In Methods in Molecular Medicine, Brooks S, Schumacher U (eds), pp 33–45. Totowa, NJ, USA: Humana Press [DOI] [PubMed] [Google Scholar]
- Brown JR, Nigh E, Lee RJ, Ye H, Thompson MA, Saudou F, Pestell RG, Greenberg ME (1998) Fos family members induce cell cycle entry by activating cyclin D1. Mol Cell Biol 18: 5609–5619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp TR, Bernards R, Agami R (2002) A system for stable expression of short interfering RNAs in mammalian cells. Science 296: 550–553 [DOI] [PubMed] [Google Scholar]
- Fagotto F, Funayama N, Gluck U, Gumbiner BM (1996) Binding to cadherins antagonizes the signaling activity of beta-catenin during axis formation in Xenopus. J Cell Biol 132: 1105–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabbiani G (1981) The myofibroblast: a key cell for wound healing and fibrocontractive diseases. Prog Clin Biol Res 54: 183–194 [PubMed] [Google Scholar]
- Gumbiner BM (1996) Cell adhesion: the molecular basis of tissue architecture and morphogenesis. Cell 84: 345–357 [DOI] [PubMed] [Google Scholar]
- Gutwein P, Mechtersheimer S, Riedle S, Stoeck A, Gast D, Joumaa S, Zentgraf H, Fogel M, Altevogt DP (2003) ADAM10-mediated cleavage of L1 adhesion molecule at the cell surface and in released membrane vesicles. FASEB J 17: 292–294 [DOI] [PubMed] [Google Scholar]
- Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lubke T, Lena IA, von Figura K, Saftig P (2002) The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet 11: 2615–2624 [DOI] [PubMed] [Google Scholar]
- Hazan RB, Phillips GR, Qiao RF, Norton L, Aaronson SA (2000) Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol 148: 779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B, Kinzler KW (1998) Identification of c-MYC as a target of the APC pathway. Science 281: 1509–1512 [DOI] [PubMed] [Google Scholar]
- Ho AT, Voura EB, Soloway PD, Watson KL, Khokha R (2001) MMP inhibitors augment fibroblast adhesion through stabilization of focal adhesion contacts and up-regulation of cadherin function. J Biol Chem 276: 40215–40224 [DOI] [PubMed] [Google Scholar]
- Hooper NM, Karran EH, Turner AJ (1997) Membrane protein secretases. Biochem J 321 (Part 2): 265–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi K, Weskamp G, Lum L, Hammes HP, Cai H, Brodie TA, Ludwig T, Chiusaroli R, Baron R, Preissner KT, Manova K, Blobel CP (2003) Potential role for ADAM15 in pathological neovascularization in mice. Mol Cell Biol 23: 5614–5624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A (2003) The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell–cell adhesion. Blood 102: 1186–1195 [DOI] [PubMed] [Google Scholar]
- Huntley GW (2002) Dynamic aspects of cadherin-mediated adhesion in synapse development and plasticity. Biol Cell 94: 335–344 [DOI] [PubMed] [Google Scholar]
- Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SG (2000) Proteomic analysis of NMDA receptor-adhesion protein signaling complexes. Nat Neurosci 3: 661–669 [DOI] [PubMed] [Google Scholar]
- Ito K, Okamoto I, Araki N, Kawano Y, Nakao M, Fujiyama S, Tomita K, Mimori T, Saya H (1999) Calcium influx triggers the sequential proteolysis of extracellular and cytoplasmic domains of E-cadherin, leading to loss of beta-catenin from cell–cell contacts. Oncogene 18: 7080–7090 [DOI] [PubMed] [Google Scholar]
- Karkkainen I, Rybnikova E, Pelto-Huikko M, Huovila AP (2000) Metalloprotease-disintegrin (ADAM) genes are widely and differentially expressed in the adult CNS. Mol Cell Neurosci 15: 547–560 [DOI] [PubMed] [Google Scholar]
- Ko KS, Arora PD, McCulloch CA (2001) Cadherins mediate intercellular mechanical signaling in fibroblasts by activation of stretch-sensitive calcium-permeable channels. J Biol Chem 276: 35967–35977 [DOI] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H (1997) Constitutive transcriptional activation by a beta-catenin–Tcf complex in APC−/− colon carcinoma. Science 275: 1784–1787 [DOI] [PubMed] [Google Scholar]
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F (1999) Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA 96: 3922–3927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Satyamoorthy K, Herlyn M (2001) N-cadherin-mediated intercellular interactions promote survival and migration of melanoma cells. Cancer Res 61: 3819–3825 [PubMed] [Google Scholar]
- Lichtenthaler SF, Dominguez DI, Westmeyer GG, Reiss K, Haass C, Saftig P, de Strooper B, Seed B (2003) The cell adhesion protein P-selectin glycoprotein ligand-1 is a substrate for the aspartyl protease BACE1. J Biol Chem 278: 48713–48719 [DOI] [PubMed] [Google Scholar]
- Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C (1999) Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci USA 96: 1603–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Shioi J, Serban G, Georgakopoulos A, Sarner S, Nagy V, Baki L, Wen P, Efthimiopoulos S, Shao Z, Wisniewski T, Robakis NK (2002) A presenilin-1/gamma-secretase cleavage releases the E-cadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J 21: 1948–1956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, Robakis NK (2003) A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 114: 635–645 [DOI] [PubMed] [Google Scholar]
- Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW (1997) Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC. Science 275: 1787–1790 [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Takeichi M (1998) Neural crest emigration from the neural tube depends on regulated cadherin expression. Development 125: 2963–2971 [DOI] [PubMed] [Google Scholar]
- Nelson WJ, Nusse R (2004) Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303: 1483–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradies NE, Grunwald GB (1993) Purification and characterization of NCAD90, a soluble endogenous form of N-cadherin, which is generated by proteolysis during retinal development and retains adhesive and neurite-promoting function. J Neurosci Res 36: 33–45 [DOI] [PubMed] [Google Scholar]
- Radice GL, Rayburn H, Matsunami H, Knudsen KA, Takeichi M, Hynes RO (1997) Developmental defects in mouse embryos lacking N-cadherin. Dev Biol 181: 64–78 [DOI] [PubMed] [Google Scholar]
- Sadot E, Simcha I, Shtutman M, Ben Ze'ev A, Geiger B (1998) Inhibition of beta-catenin-mediated transactivation by cadherin derivatives. Proc Natl Acad Sci USA 95: 15339–15344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP (2004) Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol 164: 769–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanson B, White P, Vincent JP (1996) Uncoupling cadherin-based adhesion from wingless signalling in Drosophila. Nature 383: 627–630 [DOI] [PubMed] [Google Scholar]
- Schlondorff J, Blobel CP (1999) Metalloprotease-disintegrins: modular proteins capable of promoting cell–cell interactions and triggering signals by protein-ectodomain shedding. J Cell Sci 112 (Part 21): 3603–3617 [DOI] [PubMed] [Google Scholar]
- Seals DF, Courtneidge SA (2003) The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev 17: 7–30 [DOI] [PubMed] [Google Scholar]
- Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben Ze'ev A (1999) The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA 96: 5522–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simcha I, Kirkpatrick C, Sadot E, Shtutman M, Polevoy G, Geiger B, Peifer M, Ben Ze'ev A (2001) Cadherin sequences that inhibit beta-catenin signaling: a study in yeast and mammalian cells. Mol Biol Cell 12: 1177–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg MS, Takeichi M (1994) Experimental specification of cell sorting, tissue spreading, and specific spatial patterning by quantitative differences in cadherin expression. Proc Natl Acad Sci USA 91: 206–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M (1991) Cadherin cell adhesion receptors as a morphogenetic regulator. Science 251: 1451–1455 [DOI] [PubMed] [Google Scholar]
- Tetsu O, McCormick F (1999) Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature 398: 422–426 [DOI] [PubMed] [Google Scholar]
- Utton MA, Eickholt B, Howell FV, Wallis J, Doherty P (2001) Soluble N-cadherin stimulates fibroblast growth factor receptor dependent neurite outgrowth and N-cadherin and the fibroblast growth factor receptor co-cluster in cells. J Neurochem 76: 1421–1430 [DOI] [PubMed] [Google Scholar]
- Van Hoorde L, Braet K, Mareel M (1999) The N-cadherin/catenin complex in colon fibroblasts and myofibroblasts. Cell Adhes Commun 7: 139–150 [DOI] [PubMed] [Google Scholar]
- Weskamp G, Cai H, Brodie TA, Higashyama S, Manova K, Ludwig T, Blobel CP (2002) Mice lacking the metalloprotease-disintegrin MDC9 (ADAM9) have no evident major abnormalities during development or adult life. Mol Cell Biol 22: 1537–1544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkel GK, Ferguson JE, Takeichi M, Nuccitelli R (1990) Activation of protein kinase C triggers premature compaction in the four-cell stage mouse embryo. Dev Biol 138: 1–15 [DOI] [PubMed] [Google Scholar]
- Yap AS, Brieher WM, Gumbiner BM (1997a) Molecular and functional analysis of cadherin-based adherens junctions. Annu Rev Cell Dev Biol 13: 119–146 [DOI] [PubMed] [Google Scholar]
- Yap AS, Brieher WM, Pruschy M, Gumbiner BM (1997b) Lateral clustering of the adhesive ectodomain: a fundamental determinant of cadherin function. Curr Biol 7: 308–315 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Method Information
Supplementary Figure Legends
Supplementary Figures